

SUMMARY
Rod photoreceptors generate amplified, reproducible responses to single photons via a G protein signaling cascade. Surprisingly, genetic perturbations that dramatically alter the deactivation of the principal signal amplifier, the GPCR rhodopsin (R*), do not much alter the amplitude of single-photon responses (SPRs). These same perturbations, when crossed into a line lacking calcium feedback regulation of cGMP synthesis, produced much larger alterations in SPR amplitudes. Analysis of SPRs from rods with and without feedback reveal that the consequences of trial-to-trial fluctuations in R* lifetime in normal rods are also dampened by feedback regulation of cGMP synthesis. Thus, calcium feedback trumps the mechanisms of R* deactivation in determining the SPR amplitude, attenuating responses arising from longer R* lifetimes to a greater extent than those arising from shorter ones. As a result, rod SPRs achieve a more stereotyped amplitude, a characteristic considered important for reliable transmission through the visual system.
INTRODUCTION
G protein-coupled receptors (GPCRs) are enzymatic amplifiers of extracellular signals, activating multiple copies of heterotrimeric G proteins to generate the intracellular response. The amplitude of the response depends upon the rates of GPCR activation and deactivation and any inherent nonlinearities imposed by spatial compartmentalization or signal saturation (e.g., Ramanathan et al., 2005). In retinal rod photoreceptors, a single activated GPCR, rhodopsin (R*), drives the signaling cascade that decreases cGMP and its associated inward cation current in a manner that is highly reproducible from trial to trial (e.g., Rieke and Baylor, 1998). The amplitude of the single-photon response (SPR) is considered a key factor in overcoming intrinsic cellular noise and thus for reliable transmission through the visual pathway (Field et al., 2005).
There has been much progress in understanding the molecular basis of the amplification and deactivation steps that underlie the rod SPR (reviewed in Burns and Pugh, 2010). In the initial amplifying step, a R* activates G proteins at a rate of several hundred per second (Leskov et al., 2000; Heck and Hofmann, 2001) until R* is deactivated by phosphorylation by GRK1 and arrestin-1 binding (Kühn and Wilden, 1987). The second major amplifying step arises from cGMP hydrolysis by the activated G protein-PDE6 enzyme complex (G*-E*), whose activity persists until deactivation by GTP hydrolysis catalyzed by the RGS9 complex (He et al., 1998; Makino et al., 1999; Hu and Wensel, 2002). Although rapid R* and G*-E* deactivation are required for normal recovery of the SPR, they are not sufficient; the fall in intracellular calcium that accompanies the SPR must activate the synthesis of cGMP through guanylate cyclase activating proteins (GCAPs). Abolishing calcium feedback via GCAPs increases the amplitude and slows the recovery of the SPR (Mendez et al., 2001; Burns et al., 2002). In addition, loss of feedback via GCAPs increases the intrinsic cellular noise in a manner that can impair transmission at the rod-to-rod bipolar synapse and behavioral performance at visual threshold (Okawa et al., 2010).
Genetic perturbations of R* and G*-E* deactivation also produce dramatic changes in the overall time course of the rod photoresponse. Nonetheless, the SPRs of rods with defective deactivation have average peak amplitudes very close to those of wild-type rods. For example, preventing rhodopsin phosphorylation slows the rate of R* deactivation 75-fold, from a normal average lifetime of ~40 ms (Gross and Burns, 2010) to about 3 s (Grk1−/−; Chen et al., 1999), yet the amplitude of the SPR is increased by a factor of only two. Similarly, abolishing expression of the RGS9 complex slows G*-E* deactivation about 10-fold yet has only a subtle effect on the SPR amplitude (Chen et al., 2000; Krispel et al., 2003; Keresztes et al., 2004). Understanding the stability of SPR amplitudes across genotypes that perturb deactivation promises to yield fresh insight into the mechanisms that act to minimize trial-to-trial fluctuations in SPR amplitude in normal mammalian rods and, by comparison, into the processes that may limit physiological responses in other G protein systems.
Several potential mechanisms could contribute to SPR amplitude stability, including local signal saturation (Ramanathan et al., 2005; Caruso et al., 2011). Specific mechanisms for such saturation include depletion of available PDE molecules for activation and response compression arising from extensive local closure of cGMP-gated (CNG) channels in the plasma membrane. Here we have determined the relative contributions of these factors to the stability of SPR amplitudes in wild-type rods and in rods of six additional lines with distinct genetic perturbations to response deactivation and recovery. We find that neither saturation mechanism plays a significant role even when R* lifetime is prolonged ~2-fold. Contrary to current thinking, we find that calcium-dependent feedback to cGMP synthesis through GCAPs stabilizes SPR amplitudes by more strongly attenuating SPRs driven by longer R* lifetimes. With this knowledge, we examine the role of GCAPs-mediated feedback in the trial-to-trial reproducibility of the SPR and provide experimental evidence that such feedback likewise plays a critical role in reducing variation arising from the stochastically varying R* lifetime in normal rods.
RESULTS
Genetic Manipulation of R* Lifetime Has Little Effect on SPR Amplitude
To investigate how the lifetime of R* affects SPR amplitude, we first measured the effective time constant of R* deactivation, defined as the time integral of normalized rhodopsin activity (τReff; Equation 1), in mouse lines with altered rhodopsin kinase expression. Using suction electrodes, we recorded families of saturating flash responses from mice that expressed roughly half the normal level of rhodopsin kinase (Grk1+/−; Chen et al., 1999) and from mice that expressed a high level of a mutant form of rhodopsin kinase predicted to have a higher than normal rate of phosphorylation (Grk1S561L; see Experimental Procedures; Figure S1 available online). For bright flash responses that close all of the cGMP-gated channels, the time that the responses remained in saturation (Tsat) is linearly related to the natural log of the number of R* produced by the flash (Pepperberg et al., 1992) with the slope of this relation reflecting the ~200 ms time constant of G*-E* deactivation in wild-type rods (Krispel et al., 2006). We found no change in the slope of the Tsat relations for either Grk1+/− or Grk1S561L rods (Figures 1A and 1B), consistent with no change in the rate of G*-E* deactivation. Because the normal R* lifetime (τReff = 40 ms) is much shorter than the time constant for G*-E* deactivation (τE = 200 ms), modest changes in the effective R* lifetime do not alter the slope of the relation, but rather change the magnitude of Tsat across all values of R* produced, resulting in a vertical offset, ΔTsat (Gross and Burns, 2010). Indeed, the vertical offsets in saturation times of Grk1+/− and Grk1S561L rods are consistent with slower and faster than normal R* deactivation, respectively (Figure 1B). The ΔTsat shift was +180 ms (upward) for Grk1+/− responses compared to wild-type, and −250 ms (downward) for Grk1S561 responses. Assuming downstream signaling is the same in WT, Grk1+/−, and Grk1S561L rods, the lifetimes of R*(τReff) can be calculated from these ΔTsat values (Gross and Burns, 2010):
| (1) |
Figure 1. Saturating Flash Responses Reveal Perturbed R* Lifetimes.
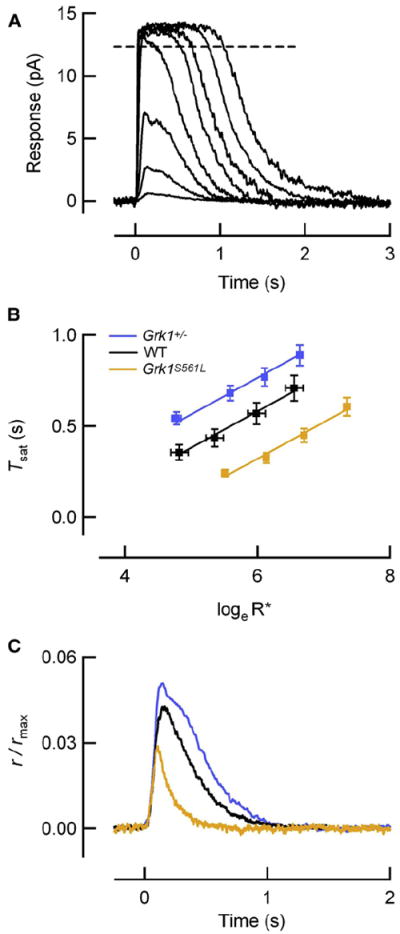
(A) Family of flash responses from a wild-type (WT) mouse rod showing the determination of the time in saturation (dashed line). Flashes ranged from 4 to 3,000 photons/μm2, corresponding to 1–630 R*/flash in this rod with collecting area of 0.21 μm2. Translational invariance of the falling phases reflects the same underlying rate processes as the flash strength increases.
(B) The time that the response to a bright flash remains in saturation is plotted against the loge of the number of R* produced by the flash for WT (n = 8), Grk1S561L (n = 12), and Grk1+/− (n = 22) rods. Straight lines through the data have a slope of 200 ms, consistent with similar means of τD values measured individually in each rod (Table 1). The vertical displacements (ΔTsat) of the Grk1S561L and Grk1+/− lines from the WT line were −250 ms and +180 ms, respectively, and yield the values of the altered effective R* lifetimes (τReff) according to Equation 1. Error bars reflect S.E.M.
(C) Population average SPRs of rods with effective R* lifetimes (τReff) that span a 5-fold range (WT, black, Table 1; Grk1+/−, blue, Table 1; Grk1S561L, orange, n = 11, ID = 14.8 ± 0.6) have peak amplitudes that span only a 2-fold range.
Assuming the lifetime of R* in normal rods (τReff,wt) is 40 ms (Gross and Burns, 2010), the values of τReff for Grk1+/− and Grk1S561L rods calculated with Equation 1 are 76 ms and 15 ms, respectively. Thus, modifying the expression level or catalytic activity of rhodopsin kinase tunes the effective lifetime of R* and the vertical offset of the Tsat relation, while the slower, G*-E* deactivation governs the slope of the relation.
To examine the consequences of shorter and longer R* effective lifetimes for the SPR, we recorded the responses of Grk1+/− and Grk1S561L rods to very dim flashes and found little change in the amplitude of the SPR (Figure 1C; Table 1). Grk1+/− rods with ~2-fold longer effective R* lifetime (τReff = 76 ms versus 40 ms for WT rods) had only a modest, 23% increase in SPR amplitude. Rods expressing transgenic Grk1S561L, with a more than 2-fold shorter effective R* lifetime (τReff = 15 ms), had only a 24% decrease in SPR amplitude. Overall, while the effective R* lifetimes of the three genetic lines span a 5-fold range with ratios of about 1:2.7:5, the normalized average SPR amplitudes span a much smaller range, with ratios of 1:1.3:1.6. These results establish that SPR amplitude does not vary in proportion to R* lifetime.
Table 1.
Measured Parameters of Rods with Perturbations in R* Phosphorylation
| GCAPs+/+ Background (Feedback)
|
GCAPs−/− Background (No Feedback)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Dark Current (rmax) (pA) | Single-Photon Response Amplitude (pA) | Dim Flash Time to Peak (ms) | Dominant Time Constant of Recovery τD (ms) | ΔTsat (ms) | Dark Current (rmax) (pA) | Single-Photon Response Amplitude (pA) | Dim Flash Time to Peak (ms) | Dominant Time Constant of Recovery τD (ms) | ΔTsat (ms) |
| WT | 13.3 ± 0.7 (19) | 0.61 ± 0.06 (19) | 130 ± 10 (19) | 204 ± 17 (8) | — | 16.7 ± 0.9 (17) | 2.3 ± 0.2 (17) | 300 ± 20 (17) | 200 ± 15 (14) | — |
|
| ||||||||||
| Grk1+/− | 11.7 ± 0.7 (25) | 0.65 ± .06 (25) | 130 ± 10 (25) | 212 ± 10 (22) | +180 | 17.6 ± 1.5 (17) | 3.4 ± 0.4 (17) | 450 ± 30 (14) | 208 ± 17 (17) | +180 |
|
| ||||||||||
| Grk1S561L | 13.6 ± 0.6 (19) | 0.47 ± 0.07 (11) | 110 ± 10 (11) | 190 ± 14 (12) | −250 | 15.3 ± 1.0 (20) | 0.96 ± 0.13 (18) | 240 ± 10 (20) | 182 ± 13 (16) | −220 |
PDE Depletion and Local Channel Saturation Do Not Contribute to SPR Amplitude Stability
In principle, R* molecules with longer lifetimes should activate more PDE molecules on the disc membrane and result in larger decreases in cGMP, locally closing a greater fraction of CNG channels. Because the density of PDE is only about 150 holoenzymes per disc face (1:300 ratio to rhodopsin, Pentia et al., 2006), it is conceivable that the rate of G*-E* production may decrease as available PDE molecules are depleted by longer-lived R* molecules. Thus, we calculated the average number of G*-E* molecules active during the SPR and compared this to the total number of PDE molecules on the disc (see Experimental Procedures). Assuming a maximal rate of 300 s−1 for R* activation of the G protein (Leskov et al., 2000; Heck and Hofmann, 2001) and our measured R* and G*-E* lifetimes, only ~7 G*-E* complexes are predicted at the peak of the SPR in normal rods (τReff = 40 ms). For Grk1+/− rods (τReff = 76 ms), the maximum number of G*-E* units active during the SPR is only ~10 (7% of the total number; Figure 2A, dashed line plotted against righthand ordinate). Thus, even if the maximal rate of G protein activation is 2-fold higher than current estimates, PDE depletion makes negligible contribution to the SPR amplitude stability over the range of R* lifetimes extending well beyond 76 ms.
Figure 2. Depletion of Available PDE Molecules and Local Channel Saturation Do Not Explain Amplitude Stability for Long R* Lifetimes.
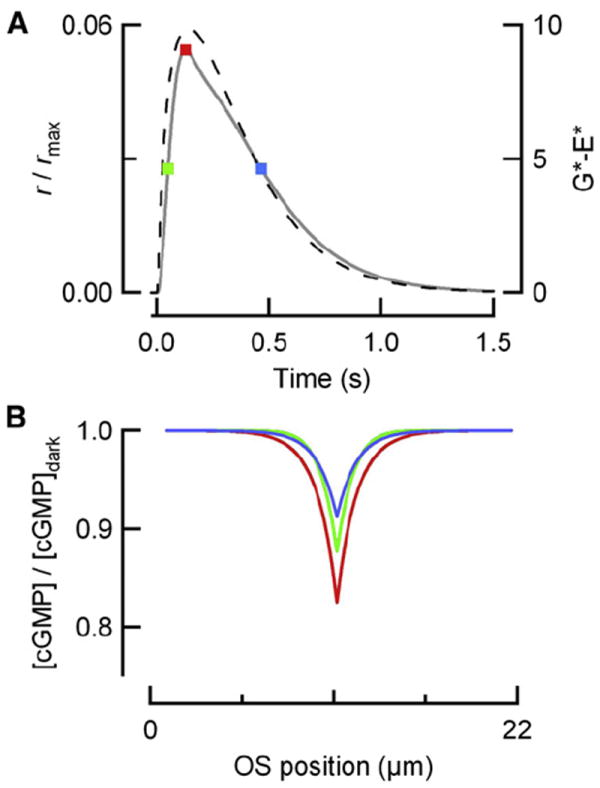
(A) Simulated SPR of Grk1+/− rods (solid) superimposed upon the calculated number of G*-E* (dashed) produced as a function of time, assuming that R* activates G*-E* at a maximal rate of 300 s−1, with an effective lifetime τReff = 76 ms, and a G*-E* lifetime of 200 ms.
(B) Spatial profile of cGMP concentration at three time points corresponding to the colored circles in (A). The fall in cGMP at the local site of photoisomerization is maximal at the SPR peak but does not exceed 18% of the dark cGMP concentration. Thus, local channel saturation contributes only slightly to the large degree of amplitude stability observed when R* lifetime is increased 2-fold beyond its WT level.
Another possible mechanism that could cause SPR amplitudes to tend to saturate is extensive closure of CNG channels over a spatially restricted portion of the outer segment. If the maximal fractional decrease in cGMP concentration is small, it will produce a directly proportional fractional change in CNG current, with a proportionality or gain factor corresponding to the Hill coefficient of 3 (Hodgkin and Nunn, 1988; Pugh and Lamb, 1993). In contrast, if the local change in cGMP concentration is relatively large, the gain factor contributed by the channels will be reduced and the SPR amplitude attenuated. To test this idea, we utilized a spatiotemporal model of cGMP dynamics in mouse rods (Gross et al., 2012; Experimental Procedures) to calculate the spatial profiles of cGMP at time points corresponding to the rising phase, the peak, and the recovery of the SPR for Grk1+/− rods (colored dots in Figure 2A correspond to colored spatial profiles in 2B). We compared the fractional change in the spatially integrated cGMP concentration predicted for the Grk1+/− SPR to the fractional change in CNG current that we measured. The channel gain factor was reduced only slightly, from its maximal possible value of 3 to 2.7 in Grk1+/− rods. Thus, extensive local closure of channels makes negligible contribution to the observed SPR amplitude stability, even when τReff = 76 ms.
GCAPs-Mediated Feedback More Strongly Attenuates SPRs Arising from Long R* Lifetimes
In normal rods, the closure of cGMP-gated channels causes a fall in intracellular free calcium, and this fall in calcium leads to an activation of cGMP synthesis by guanylate cyclase (reviewed in Stephen et al., 2008). The increased rate of cGMP synthesis rapidly opposes the fall in cGMP caused by G*-E*, thereby reducing the amplitude of SPRs (Mendez et al., 2001; Burns et al., 2002; Okawa and Sampath, 2007). To test the idea that feedback to cGMP synthesis can stabilize the SPR amplitude against perturbations to R* deactivation, we crossed the Grk1+/− and Grk1S561L mice with mice lacking calcium-dependent feedback to guanylate cyclase (GCAPs−/−; Figure 3A; Mendez et al., 2001). Despite the fact that the flash responses were much longer lasting than those of wild-type rods, the vertical shift ΔTsat associated with each genotype was very nearly the same in the GCAPs−/− background (Figure 3B; +180 ms for GCAPs−/−Grk1+/− and −220 ms for GCAPs−/−S561L). These results further confirm the assignments of the effective R* lifetimes determined above for these GRK perturbations (compare to Figure 1B). However, the SPRs of rods with altered R* lifetimes showed a larger spread in the peak amplitudes in the absence of GCAPs-mediated feedback (Figure 3C). While the ratios of R* lifetimes estimated from the Tsat data remain 1:2.7:5 as in the GCAPs+/+ background, the normalized SPR amplitudes in the GCAPs−/− background have ratios 1:2.2:3. Thus, GCAPs-mediated feedback contributes to the observed stabilization of SPR amplitudes when R* is altered.
Figure 3. GCAPs-Mediated Feedback Stabilizes the SPR Amplitude against Genetic Perturbations of R* Lifetime.
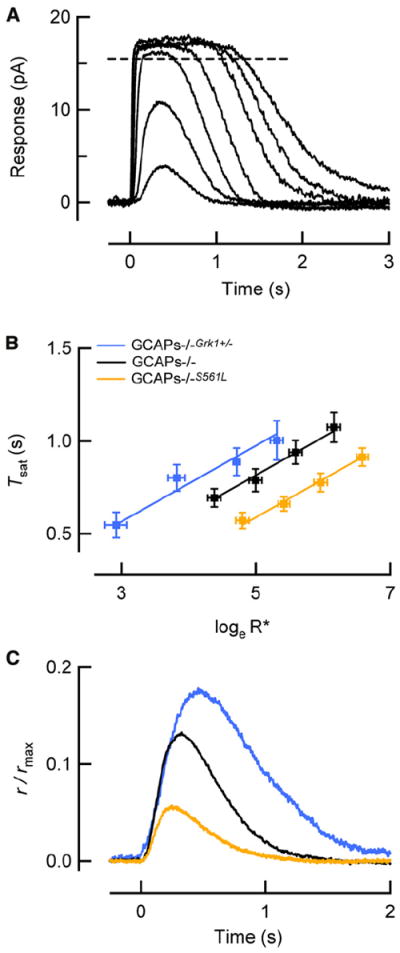
(A) Family of flash responses from a GCAPs−/− mouse rod showing the determination of the time in saturation (adapted from Gross et al., 2012). Flashes ranged from 4 to 1,800 photons/μm2, corresponding to 1–510 R*/flash in this rod with collecting area of 0.29 μm2.
(B) The time that the response to a bright flash remains in saturation versus the natural log of the number of R* produced by the flash for wild-type (n = 8) Grk1S561L (n = 12), and Grk1+/− (n = 22) rods lacking GCAPs-mediated feedback (GCAPs−/− background), fitted by straight lines with a slope of 200 ms, consistent with the means of τD values measured individually in each rod (Table 1). The vertical displacements (ΔTsat) of the Grk1S561L and Grk1+/− lines from the WT line were −220 ms and +180 ms, respectively, consistent with measurements of τReff in the GCAPs+/+ background (Figure 1B). Error bars reflect S.E.M.
(C) SPRs of rods lacking calcium feedback to cGMP synthesis via GCAPs (GCAPs−/−), but with identical changes in τReff, have substantially larger differences in peak amplitudes than the corresponding responses in the GCAPs+/+ background in Figure 1C.
Having found that feedback regulation of cGMP synthesis could diminish the effects of changes in R* lifetime, we used a spatiotemporal model of cGMP and calcium dynamics (Gross et al., 2012) to test whether amplitude stability is expected, given the rates of R* and G*-E* deactivation for each of the mouse lines. Using parameters optimized within 10% of the canonical values of Table 2, the predictions of the tightly constrained model were found to be in excellent agreement with the experimental SPRs of each genotype in both the wild-type and the GCAPs−/− backgrounds (Figures 4A and 4B). Thus, amplitude stability is an inherent feature of a model of phototransduction that incorporates measured lifetimes of R* and G*-E*, an experimentally determined diffusion coefficient for cGMP (Gross et al., 2012), and parameters of calcium feedback determined by biochemical measurements.
Table 2.
Parameters of the Spatiotemporal Model of Mouse Rod Phototransduction
| Parameter | Description | Value | Source | Reference/Method |
|---|---|---|---|---|
| kR | Rate of R* deactivation (s−1) | Genotype-dependent | Measured | Figure 1; also Gross and Burns (2010) |
| kE | Rate of G*-E* deactivation (s−1) | 5 | Measured | Figure 1; also Gross and Burns (2010) |
| νRE | Max rate of G*-E* activation per R* (s−1) | 300 | Literature | Leskov et al., 2000; Heck and Hofmann, 2001 |
| βidv | Rate of cGMP hydrolysis per G*-E* (s−1) | 43 | Literature | Gross et al. (2012) |
| βdark | Rate of spontaneous cGMP hydrolysis (s−1) | 4.1 | Literature | Gross et al. (2012) |
| DcG | Longitudinal diffusion coefficient of cGMP (μm2 s−1) | 40 | Literature | Gross et al. (2012) |
| fCa | Fraction of current carried by calcium | 0.12 | Literature | Gross et al. (2012) |
| αdark | Dark cGMP synthesis rate (μM s−1) | 16.7 | — | αmax/9; Gross et al. (2012) |
| Cadark | Dark adapted Ca2+ concentration (nM) | 320 | — | Set by αdark |
| cGdark | Dark adapted cGMP concentration (μM) | 4.1 | — | Set by αdark and βdark |
| BCa | Calcium buffer capacity | 50 | Fitted | ±15% of the values in Lagnado et al. (1992) and Nikonov et al. (1998) |
| ncyc | Hill coefficient for Ca2+ dependence of cGMP synthesis | 1.5 | Fitted | ±15% of the values in Makino et al. (2008) |
| Kcyc | K1/2 for Ca2+ dependence of cGMP synthesis (nM) | 80 | ||
| αmax | Max rate of cGMP synthesis (μM s−1) | 150 | ||
| Kex | K1/2 for NCKX activation (μM) | 1.1 | Fitted | ±15% of the values in Schnetkamp et al. (1991) |
| Jexsat | Maximum NCKX current (pA μm−1) | 0.21 | — | Set by Kex, Cadark, fCa, L |
Figure 4. A Spatiotemporal Model of Phototransduction Captures the Enhanced Amplitude Stability in the Presence of GCAPs-Mediated Feedback.
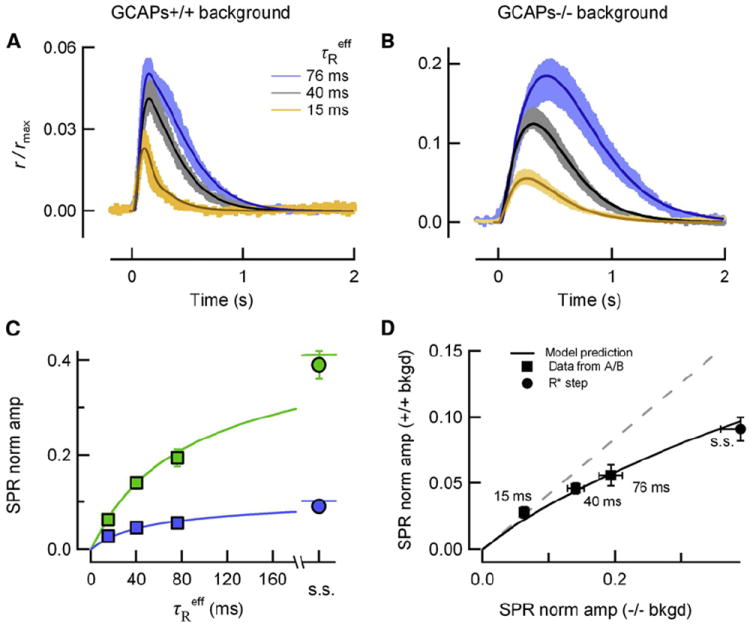
(A) Simulated SPRs (thin traces) closely match the SPR (means +/− SEM, thick traces) experimentally determined from populations of wild-type (black/gray), Grk1+/− (blue), and Grk1S561L (orange) SPRs with feedback (A, GCAPs+/+ background) and without feedback (B, GCAPs−/− background). Parameters were optimized individually for each simulated response within 10% of the canonical values in Table 2.
(C) Normalized mean SPR peak amplitudes (r/rmax at the time of the peak in A and B) are plotted against τReff for GCAPs+/+ and GCAPs−/− backgrounds (blue and green boxes, respectively). Mean steady state (s.s.) amplitudes of SPRs arising from R*s that do not deactivate are plotted for both genetic backgrounds (blue circle corresponds to SPR amplitude of Grk1−/− (Song et al., 2009 supplement); green circle corresponds to amplitude of GCAPs−/− “rogues” (Gross et al., 2012). Theoretical green and blue traces were simulated using the parameters in Table 2. Error bars reflect S.E.M. (error bars are smaller than the points in most cases).
(D) GCAPs-mediated feedback more strongly attenuates SPRs arising from long-lived R*s. GCAPs+/+ and GCAPs−/− SPR amplitude data from (C) are plotted against each other (black squares; numbers indicate τReff), as are the corresponding theory traces (black trace). The dashed gray line is the relationship that would be expected if rods in GCAPs+/+ and GCAPs−/− backgrounds exhibited the same degree of stability. Error bars reflect S.E.M.
To better understand the specific mechanisms contributing to stability, we used the model to calculate SPR amplitudes for theoretical effective R* lifetimes (τReff) ranging from a few milliseconds to several seconds, which is adequately long to approximate a step of R* activity and for the SPR to achieve steady state (Figure 4C). The model (solid curves) accurately predicts the average SPR amplitudes of rods of both GCAPs−/− (green symbols) and GCAPs+/+ backgrounds (blue symbols), including the steady-state amplitudes of SPRs produced by R*s that remain fully active for several seconds (Gross et al., 2012). Notably, both data and theory differ strongly from the intuitive notion that the SPR amplitude would increase in proportion to R* lifetime, except for τReff < 20 ms.
Our results establish that GCAPs-mediated feedback makes a distinct contribution to SPR amplitude stability. To characterize this contribution, we plotted the SPR amplitudes for GCAPs+/+ and GCAPs−/− backgrounds (blue and green symbols in Figure 4C) for each value of τReff against each other (Figure 4D). For τReff > 40 ms, the amplitudes of the SPRs of the GCAPs+/+ and GCAPs−/− backgrounds significantly deviate from proportionality (dashed gray line). For longer R* lifetimes, the relative increase in SPR amplitude is systematically greater for rods of the GCAPs−/− background than for rods of GCAPs+/+ background. This reveals that GCAPs-mediated feedback reduces the amplitudes of SPRs driven by longer R* lifetimes to a greater extent than those driven by shorter R* lifetimes.
Ramping Activation of cGMP Synthesis Underlies SPR Amplitude Stability
To understand how SPR amplitude stability is conferred by GCAPs-mediated feedback, it is instructive to separately consider the time courses of light-driven cGMP hydrolysis and synthesis, integrated over the length of the outer segment. The spatially integrated rates of cGMP hydrolysis are illustrated for SPRs corresponding to three different values of τReff (15, 40, and 76 ms) in the GCAPs+/+ (Figure 5A, orange, black, and blue traces) and GCAPs−/− (gray dotted lines) backgrounds. All six hydrolysis rate functions follow a common initial trajectory (pink area) but peel off at times that depend on τReff. For the SPRs of GCAPs+/+ and GCAPs−/− backgrounds with the same value of τReff, the hydrolysis rates reach nearly identical maximal magnitudes. The hydrolysis rates in rods of the GCAPs+/+ background level out into plateaus that continue until the SPR peak (~120 ms; indicated by the transition from solid to dashed colored lines, Figure 5). In contrast, the hydrolysis rates in rods of the GCAPs−/− background begin to decline shortly after reaching their peaks. Analysis of the spatiotemporal profiles reveals that the decline in hydrolysis rate arises because of substrate depletion: the absence of calcium-activated synthesis causes depletion of cGMP in the regions flanking the disc where R* and G*-E*s reside, thereby lowering the local hydrolysis rate (βdark cG) (Figure S3).
Figure 5. The Rate of cGMP Synthesis Increases More Rapidly for Longer R* Lifetimes, Constraining the Time to Peak and Conferring SPR Amplitude Stability.
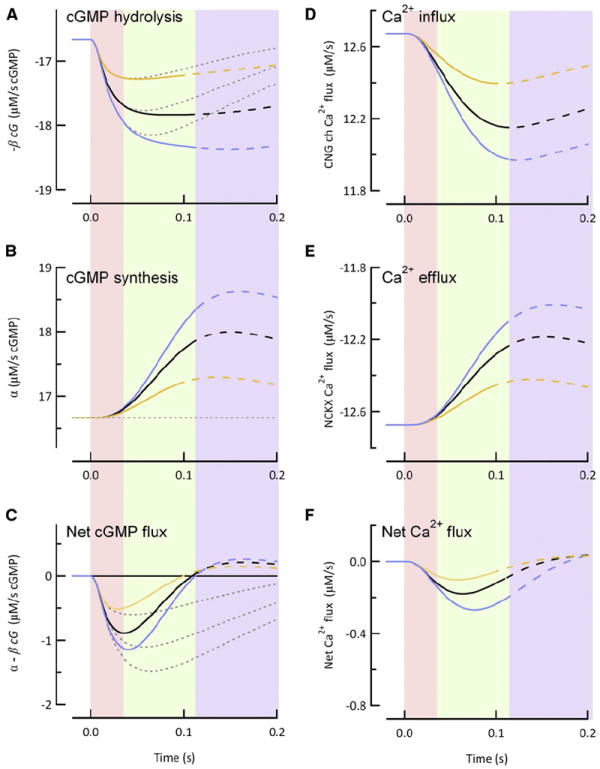
(A) Time course of the spatially integrated cGMP hydrolysis rate calculated for mean effective R* lifetimes of 15 ms (orange curve), 40 ms (black curve), and 76 ms (blue curve) with (solid traces) or without (gray dotted lines) GCAPs-mediated feedback.
(B) Spatially integrated cGMP synthesis rates for the R* lifetimes listed in (A). Thin dotted black line indicates the constant (dark) rate of cGMP synthesis in the absence of GCAPs-mediated feedback. Note that the larger G*-E* activities in (A) correspond to steeper rates of change in the cGMP synthesis rates.
(C) Spatially integrated rate of change of cGMP concentration (differences of the time courses in A and B).
(D) Calculated influx of calcium through CNG channels and (E) calculated efflux of calcium via Na2+/Ca2+-K+ transporter for the same effective R* lifetimes of (A)–(C) in the presence of GCAPs-mediated feedback.
(F) Time course of net calcium flux (differences of the time courses in D and E). In all panels, the solid curves transition to dashed curves at times corresponding to the peak of the SPRs. Shaded regions indicate distinct epochs during the SPR: Pink: cGMP synthesis remains near its dark level; green: rate of cGMP synthesis increases as a ramp with slope proportional to the constant cGMP hydrolysis rate, so that synthesis and hydrolysis cancel each other at the SPR peak; blue: SPR recovery phase.
Unlike the step-like rates of steady hydrolysis in the GCAPs+/+ background, the light-driven increases in the cGMP synthesis rates (Figure 5B) rise on delayed ramps whose slopes are in approximately the same ratios (1:2:3) as the hydrolysis plateau magnitudes. To determine the underlying causes of the delayed ramps of synthesis activity, we examined the space-averaged changes in calcium influx and efflux (Figures 5D and 5E). In the dark, calcium influx and efflux are perfectly balanced. During the initial 35 ms of the SPR (pink region), the calcium influx decreases as CNG channels close, but there is little change in the rate of calcium efflux at this early time. From about 35 ms onward, the fall in free calcium causes its efflux to slow (Figure 5E). As a result, the net calcium flux for each genotype is a fairly symmetric bell-shaped curve (Figure 5F). Given a constant calcium buffer power, the change in free calcium (not shown) is directly proportional to the integral of the bell-shaped curve, giving rise to ramping decreases in calcium. As a consequence, the time course of cGMP synthesis (Figure 5B), which is approximately proportional to the decrease in free calcium (Equation 4), is also ramp-like.
To complete the picture detailing the mechanism of GCAPs-mediated feedback contribution to SPR amplitude stability, we now consider the net rate of change of cGMP (i.e., the rate of synthesis minus that of hydrolysis) for each genotype (Figure 5C). The three color-coded rate functions share a common initial trajectory from which they diverge as the ramping synthesis overtakes the step-like hydrolysis time course. Consequently, the predicted times of the SPR peaks (given by the zero-crossings, the times at which cGMP synthesis balances hydrolysis) are nearly identical for the three genotypes, as observed in the experimental SPRs (Table 1). In order to achieve the similarity in time to peak for different R* lifetimes, the cGMP synthesis rate must rise in proportion to the steady hydrolysis rates. Calculation of calcium influx and efflux rates show that it is the integrating effect of the calcium dynamics (the ramping decline in calcium) that allows the delayed ramps of cGMP synthesis to overtake the hydrolysis plateaus at similar times for SPRs whose R* lifetimes range from 15 to 76 ms.
The Role of GCAPs-Mediated Feedback in Suppressing Response Variability in Normal Rods
Having established how GCAPs-mediated feedback stabilizes the amplitude of the average SPR across genotypes with differing average R* lifetimes, we now consider whether it also contributes to reduction of the trial-to-trial variability of SPR amplitudes in an individual rod, i.e., contributes to SPR reproducibility.
SPR reproducibility has long been deemed something of a biophysical mystery: despite being driven by individual stochastically deactivating R* molecules, SPRs have highly invariant amplitudes, with coefficient of variation (c.v.; standard deviation divided by the mean) of ~0.2 in amphibian rods (Baylor et al., 1979; Rieke and Baylor, 1998) and ~0.3 in mammalian rods (Baylor et al., 1984; Figure 6F). In recent years, empirical and theoretical studies have led to general agreement that multiple phosphorylations of R* smooth its stochastic deactivation (Rieke and Baylor, 1998; Mendez et al., 2000; Field and Rieke, 2002; Hamer et al., 2003; Doan et al., 2006). Theoretical simulations have suggested that stochastic R* shutoff is nonetheless the primary source of SPR variability and also that the limited diffusion of cGMP acts to suppress the variability associated with R* deactivation (Bisegna et al., 2008; Caruso et al., 2010, 2011). These same theoretical studies have also concluded that calcium-mediated feedback plays little role in the reproducibility of the SPR (Caruso et al., 2011), a conclusion at odds with what might now be expected, given our current results with GCAPs-mediated feedback and SPR amplitude stability.
Figure 6. Single-Photon Response Amplitude Is Less Reproducible in the Absence of GCAPs-Mediated Feedback.
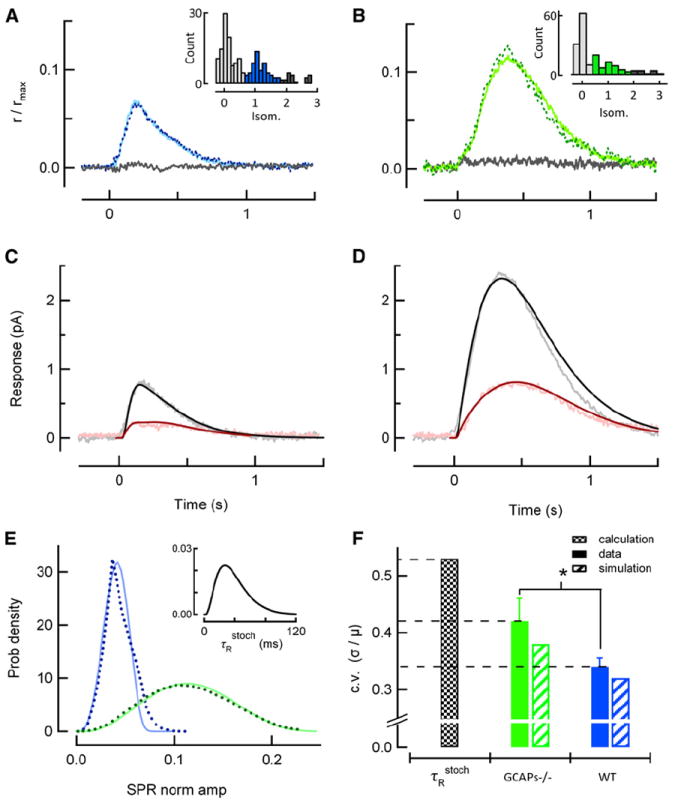
(A) Average of identified singletons in a representative WT rod, determined by histogram analysis of 200 dim flash trials (inset; identified singletons highlighted in blue, failures in light gray, amplitudes from multiple R* in dark gray). This average singleton (solid) closely resembles the average SPR determined independently in this rod by variance-to-mean analysis for the entire set of dim flashes (dotted). The gray trace is the average of the responses yielding failures in the histogram.
(B) Same analysis as in (A), but from a representative GCAPs−/− rod in response to 188 repeated dim flashes. Solid green trace plots the average of the 63 identified singletons, corresponding to the green portion of the inset histogram, and closely resembles the average SPR determined independently by variance-to-mean analysis (dotted).
(C and D) Population mean singleton response (gray) and time-dependent standard deviation (pink) calculated from five WT (C) and four GCAPs−/− (D) rods, with mean dark currents of 15.5 ± 1.4 and 16.1 ± 1.2 pA, respectively. The smooth curves are the mean (black) and time-dependent standard deviations (dark red) derived from model simulations of ensembles of SPRs for each genotype.
(E) Probability distributions of calculated stochastic R* lifetimes (inset) and singleton amplitudes (dotted lines) determined from the simulation of 100,000 WT and GCAPs−/− SPRs using the stochastic R* deactivation scheme (Figure S2). Blue and green solid curves are probability density functions of singleton amplitudes predicted by transforming the τRstoch probability density by the theoretical stability curves in Figure 4C and reveal that the mechanisms that generate amplitude stability underlie reproducibility.
(F) Filled bars indicate the experimentally determined c.v. of the GCAPs−/− (green) and WT (blue) singleton amplitudes calculated from rods whose averages are shown in (C) and (D) (asterisk denotes p = 0.02 in Student’s t test comparison). Black checked bar is the c.v. of the calculated τRstoch distribution shown in the inset in (E), and hatched bars indicate coefficients of variation of simulated singleton amplitude distributions shown in (E). Error bars reflect S.E.M.
To directly assess whether calcium feedback to cGMP synthesis contributes to SPR reproducibility, we recorded hundreds of dim flash responses from wild-type (Figure 6A) and GCAPs−/− (Figure 6B) rods and calculated the mean and time-dependent standard deviation of the ensembles of isolated SPRs (“singletons”; gray and pink traces, Figures 6C–6D; Experimental Procedures). In addition to being larger, the response peaks of isolated GCAPs−/− singletons were more variable in amplitude and were more broadly distributed in time. As a result, the time-dependent standard deviation of GCAPs−/− singletons had a larger, broader peak than that of WT singletons (note difference in both x- and y-scaling, Figures 6C–6D). The increase in the GCAPs−/− singleton standard deviation relative to that of WT was larger than the relative increase in singleton mean amplitude, resulting in a larger c.v. of the response amplitude (c.v. = 0.34 ± 0.01, n = 5 for WT and 0.42 ± 0.02, n = 4 for GCAPs−/− rods; p = 0.02; Figure 6F, solid green and blue bars). Thus, although R* and G*-E* deactivation are the same for WT and GCAPs−/− rods, reproducibility is impaired in the absence of GCAPs-mediated feedback.
To determine the relative contributions of multistep stochastic deactivation of R* and GCAPs-mediated feedback to the observed SPR reproducibility, we implemented a stochastic, multistep R* deactivation model with a mean R* lifetime of 40 ms (Experimental Procedures; Figure S2). The probability distribution of stochastic R* lifetimes (τRstoch) predicts that the vast majority of R* lifetimes are less than 80 ms, with a mode of 33 ms (Figure 6E, inset). The c.v. of this distribution is 0.52 (Figure 6F, black checked bar), smaller than that of a first-order deactivation process (c.v. = 1) but significantly larger than the experimentally measured c.v. of WT and GCAPs−/− SPR amplitudes (Figure 6F, solid green and blue bars).
To predict the SPR amplitude c.v. associated with the multistep R* deactivation scheme, 100,000 stochastic R* trajectories were simulated (Figure S2) and each trajectory was used as the driving input for the spatiotemporal model under conditions with and without feedback to cGMP synthesis. The amplitude distributions of the two ensembles of simulated SPRs (Figure 6E, dark blue and dark green dotted lines) have coefficients of variation that are close to the measured values for both WT and GCAPs−/− rods (hatched green and blue bars in Figure 6F). These simulated amplitude distributions were nearly identical to those obtained by simply transforming the τRstoch distribution by the compressive relations describing the stability data (Figure 4C), emphasizing that GCAPs-mediated feedback contributes to reproducibility in the same way that it confers amplitude stability (Figure 6E, solid blue and green curves). In addition to calculating the c.v. of these distributions, we also calculated the time-dependent mean and standard deviations of the ensembles of simulated SPRs. The theoretical mean SPRs and standard deviations agree both in magnitude and time course with the experimental population average SPR data of both genotypes (black and red smooth curves in Figures 6C and 6D). Thus, even fairly noisy, stochastic deactivation of R* can yield SPRs with the experimentally observed degree of reproducibility, owing to the compensatory effects of calcium feedback to cGMP synthesis. Notably, the c.v. of the SPR amplitudes produced by the very same ensemble of R* deactivation trajectories is predicted to be lower in WT than in GCAPs−/− rods. The analysis shows this lowered c.v. is achieved mainly by the ramping cGMP synthesis in WT rods (Figure 5B), which effectively removes variation that would otherwise arise from the occasionally slower stochastic R* deactivations of the ensemble.
DISCUSSION
Mechanisms Underlying the Stability of SPR Amplitudes against Perturbations in R* Lifetime
We have found that GCAPs-mediated feedback to cGMP synthesis helps to stabilize the SPR amplitude against variation in R* lifetime (Figure 4). Although this stabilization is clearly more potent for longer R* lifetimes, the “power” of the calcium feedback does not arise from a decline in calcium concentration disproportionate to R* lifetime, nor because the feedback has greater cooperativity than estimated in biochemical experiments, as previously suggested (Burns et al., 2002). Rather, GCAPs-mediated feedback stabilizes the SPR amplitude because the increase in cGMP synthesis is initially delayed and then increases at a rate proportional to the magnitude of the steady hydrolysis rate. As a result, the delayed, ramping synthesis of cGMP overtakes hydrolysis at nearly the same time independent of τReff (Figure 5).
A significant degree of amplitude stability persists in the absence of GCAPs-mediated feedback (Figure 4C). Most of this residual stability appears to come from the time course of PDE activity. The maximum cGMP hydrolysis rates in rods with and without GCAPs-mediated feedback are nearly the same and stand in the ratio 1:2:3 for R* lifetimes in the ratio 1:2.7:5 (Figure 5A). This reduction in direct proportionality of the maximum hydrolysis rate to R* lifetime arises in part from the imperfect integration of R* activity by G*-E* with its 200 ms lifetime (Equation 13), as well as from the fall in cGMP, which reduces the hydrolysis rate.
Reproducibility Reconsidered
Multistep deactivation of R* activity by phosphorylation and arrestin binding has been considered by many investigators as a mechanism that reduces the SPR variability relative to that which would occur were R* deactivation a first-order, stochastic event (Rieke and Baylor, 1998; Mendez et al., 2001; Burns et al., 2002; Field and Rieke, 2002; Hamer et al., 2003, 2005; Doan et al., 2006; Caruso et al., 2010). We agree with this view. The multistep scheme based on known biochemistry employed here reduced the c.v. of R* lifetimes from 1 (first-order) to 0.5. However, our results show that both the measured and theoretical coefficients of variation of the SPRs are larger when calcium feedback to cGMP synthesis is abolished (Figure 6F). Thus, we have reached the surprising conclusion that even a fairly “noisy” distribution of R* lifetimes can be compensated for by calcium feedback to cGMP synthesis, which more strongly attenuates SPRs that are driven by longer R* lifetimes.
Our conclusions may seem to conflict with those of others who have investigated SPR variability and concluded that calcium feedback plays no role. For example, Rieke and Baylor (1998) and Field and Rieke (2002) found that slowing intracellular calcium dynamics by introducing exogenous calcium buffer (BAPTA) or interfering with Na/Ca exchange increased the amplitude and duration of the SPRs but caused no significant increase in the c.v. of their amplitudes or areas. Such similarity in the coefficients of variation might arise if the slower, larger responses measured in those experiments produce a greater degree of local signal saturation than occurs in normal rods. In this context, it should be noted that Whitlock and Lamb, when analyzing the rising phases of amphibian rod SPRs (i.e., early times when the fall in cGMP is small), found that BAPTA incorporation was associated with a broadening of the distribution of singleton amplitudes (c.v. 0.35 in BAPTA versus 0.20 in control) (Whitlock and Lamb, 1999). It is also the case that in the absence of GCAPs-mediated feedback, the time course, and thus the area, of the response is severely filtered by the slow restoration of cGMP (Gross et al., 2012), and thus may be less sensitive to fluctuations in R* lifetime than the peak amplitude (see also the discussion in Hamer et al., 2003).
It has been claimed that the diffusion of cGMP and/or of calcium plays a central role in SPR reproducibility, acting as a “variability suppressor” (Bisegna et al., 2008; Caruso et al., 2010; Shen et al., 2010; Caruso et al., 2011). In WT rods, the experimentally determined longitudinal diffusion coefficient for cGMP (Gross et al., 2012) is large enough that the maximal decrease in cGMP concentration is small (~15%) even when R* deactivation is slowed ~2-fold (Figure 2). Thus, the limited diffusion of cGMP does not contribute to reduction of SPR amplitude variability through saturating local channel closure. Furthermore, the spatial profile of calcium is not determined by the diffusion coefficient of calcium, but rather by the spatial profile of cGMP, which governs calcium influx (Gross et al., 2012). However, the fall in cGMP can reduce the rate of cGMP hydrolysis in the absence of GCAPs-mediated feedback (compare gray and colored traces in Figure 5A), producing compression of PDE activity relative to R* lifetime, as noted above. In this sense, the local fall in cGMP tends to self-limit the PDE activity, a phenomenon that can contribute to SPR reproducibility (“cGMP hydrolysis saturation effects”; Caruso et al., 2011).
Summary of the Role of Calcium Feedback in SPR Stability and Reproducibility
With the lifetimes of R* and G*-E* measured from the ΔTsat data (Figures 1 and 3; Table 1), a remarkably accurate account can be given of the SPRs of rods with genetic manipulations of R* deactivation, both with and without calcium feedback to cGMP synthesis (Figures 4A and 4B). The diffusion of cGMP is sufficiently rapid to insure maximal amplification (Gross et al., 2012), while the delayed decline in calcium drives cGMP synthesis more strongly for longer R* lifetimes (Figure 5) in rods with normal GCAPs expression. As a consequence, the amplitude of the mean SPR is stabilized against genetic perturbations to R* lifetime (Figure 4), and the trial-to-trial SPR amplitude is more reproducible in rods with functional calcium feedback (Figure 6). In general, then, these results reveal how a fast feedback mechanism, operating at a downstream stage in a GPCR cascade, can sharpen the timing of a signal and reduce its variability while maintaining high signal amplification.
EXPERIMENTAL PROCEDURES
Animals
Mice were cared for and handled following an approved protocol from the Institutional Animal Care and Use Committee of the University of California, Davis and in compliance with the National Institutes of Health guidelines for the care and use of experimental animals. Mice were reared in 12 hr cyclic lighting conditions and euthanized by CO2 narcosis followed by decapitation. All mice were between 1 and 6 months of age when used for experiments.
Novel data are presented from rods of Grk1S561, Grk1+/−, GCAPs−/−, GCAPs−/−S561L, and GCAPs−/−Grk1+/− mice. Additional data are republished from (Krispel et al., 2006; 4-fold RGS9-ox) and (Gross and Burns, 2010; RGS9-ox and c57/B6). Transgenic Grk1S561L mouse was created and provided by C.K. Chen (Virginia Commonwealth University). This mutation alters the C-terminal prenylation sequence (CaaX) from one directing farnesylation (C15) to geranylgeranylation (C20), as found in cone opsin kinase (Inglese et al., 1992; Zhao et al., 1995). Quantitative western blotting revealed that there was also 8.7 ± 1.0-fold higher expression (n = 17) of the protein over wild-type expression levels (Figure S1).
Grk1+/− mice were obtained by breeding C57B/6 mice with Grk1−/− mice (Chen et al., 1999). Transgenic Grk1S561, transgenic RGS9-ox, and Rgs9−/− mice were bred into the GCAPs−/− background (Mendez et al., 2001). The GCAPs−/− mice used in these experiments were transgene-negative littermates of GCAPs−/−S561L of GCAPs−/−RGS9-ox mice.
Electrophysiology and Analysis
Mice were dark-adapted overnight prior to recordings, with all dissection and cell selection procedures performed under infrared illumination with the aid of infrared-visible converters. Retinas were isolated in L-15 media supplemented with BSA and glucose, and stored on ice. Suction electrode recordings of the outer segment membrane current were made from intact rods at 37°C in oxygenated, bicarbonate-buffered Locke’s solution, as previously described (Krispel et al., 2006). Brief calibrated flashes (10 ms, 500 nm) were used to elicit light-evoked responses.
The average single-photon response amplitude and effective collecting area of each rod were estimated by variance-to-mean analysis (Baylor et al., 1979) from an ensemble of dim flash responses (at least 25 responses with amplitudes less than 20% of the dark current).
For determination of the vertical Tsat offsets used to calculate τReff, the time in saturation (Tsat) for bright flash responses was measured between the time of the flash and 10% recovery from saturation. This 10% threshold was used because the invariance of response shape was less reliable at late times. The Tsat values were plotted as a function of the natural log of the number of R* produced by the flash, and not the flash strength expressed in photon density, in order to normalize for differences in sensitivity arising from small differences in outer segment length or unavoidable occasional shadows in the recording chamber. For each cell, the average number of R* produced by a given flash was determined by multiplying the calibrated flash strength (photons/μm2) by the effective collecting area determined for that cell. Calculation of τReff from vertical Tsat offsets (Equation 1) is valid assuming that the integrated R* activity (Equation 12) is short relative to G*-E* lifetime and that all other cascade elements are the same.
Throughout, error bars reflect ± standard error of the mean (SEM).
Isolation of Singletons and Assessment of Reproducibility
Singletons were isolated for reproducibility analysis by first identifying failures from an ensemble of a large number of dim flash responses. A response was deemed a failure if the integrated response area after the flash was less than 64% greater than the integral of the absolute value of the baseline-corrected trace before the flash; the time window for integration was 400 and 600 ms in wild-type and GCAPs−/− rods, respectively. This 64% difference criterion was selected for its empirical robustness: in one test rod, the difference in failure count between using a 50% criterion and a 90% criterion corresponded to a difference in 38 versus 42 failures out of 155 total responses. The 64% criterion counted 41.
The failure count provided an estimate of the number of singletons (ns), using Poisson statistics. We then assumed our singleton population to be the set of ns nonfailure responses having the smallest amplitudes. The mean singleton and the amplitude c.v. did not vary greatly when the singleton population was altered by the inclusion of 1–2 additional responses or the exclusion of 1–2 of the largest responses. In addition, the average singleton determined from the set of ns was indistinguishable from that average single photon response calculated from variance-to-mean analysis. Likewise, the average of the responses that were deemed failures showed no time-dependent changes in current (Figures 6A-6B).
We elected not to use matched-filter analysis to identify singletons, as this approach assumes that all SPRs have an effectively identical shape combined with broadband noise—an assumption that begs the question of how the SPR shape would change with variation in R* lifetime. Nonetheless, we compared the method used here to the traditional matched filter analysis-histogram method (e.g., Field and Rieke, 2002). We found that the matched filter method had a tendency to produce lower coefficients of variation for both WT and GCAPs−/− singletons, though this difference was not significant (GCAPs−/− c.v.: 0.40 ± 0.02 using matched filter analysis versus 0.42 ± 0.02 using smallest nonfailures; WT: 0.31 ± 0.03 using matched filter analysis versus 0.34 ± 0.01 using smallest nonfailures). The matched filter analysis identified 246 total wild-type singletons from 5 rods, while the number of singletons expected from the number of identified failures is 263. The corresponding numbers for 4 GCAPs−/− rods are 142 and 152. In sum, matched filter analysis tended to identify fewer responses as singletons, in particular excluding more of the large amplitude responses on the fringe that would escape the boundaries imposed on the singles peak. When these responses are included in the statistical expectation method employed in the paper, they tend to increase the standard deviation.
For each rod used for measuring reproducibility, the identified singletons were used to generate the time-dependent average. The time-dependent standard deviation for each set was determined for each cell by taking the square root of the time-dependent variance after subtracting the offset variance in the second before the flash was delivered. For the traces shown in Figures 6C and 6D, the average singletons and standard deviations were averaged from a population of 5 wild-type and 4 GCAPs−/− rods.
Spatiotemporal Model of cGMP and Calcium Dynamics
Our simulations of rod photoreceptor responses were generated using a spatiotemporal model of second-messenger dynamics described in detail elsewhere (Gross et al., 2012). Briefly, we used a generally accepted pair of coupled partial differential equations (Equations 2 and 3) describing Ca2+ and cGMP concentrations in the rod outer segment, and ancillary equations (Equations 4-6) relating these two variables to cGMP-sensitive current density (JcG) and Na/Ca-K exchange current density (Jex):
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
Table 2 provides definitions and units of the variables and parameters. Equations 2-6 were solved subject to the initial condition E*(t) = 0, zero-flux boundary conditions for cGMP and calcium at the tip and base of the outer segment, and a boundary condition that applies at the locus of the R*:
| (7) |
This boundary condition assumes that the R* is created by a photon capture on a disc whose position (x0) is near the middle of the outer segment with disc spacing δ and identifies the hydrolytic flux (righthand side of Equation 7) with the bidirectional diffusional flux (left hand side) at the same position. The system of Equations 2-7 were solved numerically with the numerical method of lines (Schiesser, 1991) using original scripts written in Matlab.
The model predicts the spatiotemporal changes in cGMP concentration and corresponding current density (J(x,t)) caused by the activation of rhodopsin on an outer segment disc. The simulated rod response is defined in terms of this current density:
| (8) |
where ID is the rod dark current J = JcG + Jex, and the integral is carried out over the length of the outer segment. The model parameters used for simulating amplitude stability (Figures 4C and 4D), second-messenger dynamics (Figures 2 and 5), and SPR amplitude c.v. (Figures 6E and 6F) were unchanged from the original description (Gross et al., 2012; see also Table 2). In order to best fit the data in Figures 4A, 4B, 6C, and 6D model parameters were optimized by least-squared fitting within ± 10% of the canonical parameter values for each SPR. The only significant difference in the implementation of the model from that previously described (Gross et al., 2012) is the adoption of a multistep model for R* deactivation in place of simple exponential decay, as now described.
Multistep Model of R* Deactivation
We employed a biochemically motivated scheme of R* deactivation for all model calculations (Figures 2 and 4-6). This model is similar to those developed previously by other groups (e.g., Hamer et al., 2003; Caruso et al., 2010). However, unlike previous simulations, we included no initial delay before phosphorylation (Caruso et al., 2010) nor did our scheme make any assumptions about competition between G protein, Grk1, and arrestin, or incorporate any mechanism for feedback via recoverin (Hamer et al., 2003). We emphasize that the details of the multistep scheme were not selected in order to make any novel claim about the actual mechanism of rhodopsin deactivation or its actual degree of reproducibility; instead, it was used to test whether transduction operating with variable rhodopsin lifetimes (c.v. ~0.5) could generate reproducible single-photon responses, and whether the degree of reproducibility was improved with GCAPs-mediated feedback. We also found that this multistep model provided a better account of the rising phase of the response than the single exponential decay function for R* deactivation (Figure S2).
In this scheme, it is assumed that R* reaches maximal catalytic efficiency within the first millisecond following isomerization, and that as long as it remains unphosphorylated, its affinities for the G protein and the kinase (GRK1) are maximal, while its affinity for arrestin is negligible (Gibson et al., 2000; Vishnivetskiy et al., 2007). Sequential phosphorylations of rhodopsin by GRK1 decrease the rate of subsequent phosphorylation (Kennedy et al., 2001), while increasing the rate of irreversible Arr1 binding sharply after three phosphorylations (Vishnivetskiy et al., 2007). These biochemical features are embodied in the phosphorylation dependence of the rate constants for transitions between phosphorylation states and the arrestin-bound state:
| (9) |
| (10) |
Here kph(p) is the transition rate constant (s−1) of R* from the state with p to that with p+1 phosphates (p = 0, 1, …, 6), the maximum phosphorylation rate (which applies when R* is not yet phosphorylated, p = 0), karr(p) is the rate constant for arrestin binding, and its maximum (Figure S2). According to Equation 10, the rate of arrestin quench is a sigmoidal function of phosphorylation state p with midpoint at p0 = 2.9 and a steepness factor θ = 0.1. As implemented, this sigmoid approximates a step function (Figure S2B), a feature consistent with previous conclusions (Vishnivetskiy et al., 2007), and employed in previous models that incorporate stochastic R* deactivation (e.g., Hamer et al., 2003, 2005; Bisegna et al., 2008; Caruso et al., 2010, 2011).
The transition rates defined in Equations 9 and 10 correspond to a continuous-time Markov process for the decay of R* activity and determine the probability Pr{p∣t} that an R* molecule has p phosphates or has been quenched by arrestin at time t after photoisomerization. Because phosphorylation of R* reduces its catalytic activity (Xu et al., 1997; Gibson et al., 2000; Burns et al., 2006), the mean normalized activity of R* ( ) was calculated by multiplying the probability of R* occupying a particular state by a term representing the phosphorylation-driven decline in R* activity and summing over p:
| (11) |
Here, the second convolved term Π(t) is a 10 ms step function of unit area representing the measured stimulus duration. For simulating the average SPRs of rods of Grk1+/−, WT, and Grk1S561L genotypes, only the maximum phosphorylation rate was adjusted: the values were , 81, and 243 s−1, respectively. These values were determined by matching the theoretical effective R* lifetime, with the values of τReff obtained from the Tsat offset analysis (Figure 1):
| (12) |
where τReff = 76, 40, and 15 ms respectively. Similarly, the model prediction of amplitude stability as a function of τReff (Figures 4C and 4D) was produced by continuously varying .
Characterization of Variability in Stochastic R* Deactivation Trajectories
The multistep deactivation model was also used to assess the trial-to-trial variability of R* lifetimes resulting from the stochastic nature of individual phosphorylation and arrestin binding events (Figure S2). The stochastic R* lifetime (τRstoch) is defined analogously to Equation 12 as the time integral of an individual R* activity trajectory (time course). We constructed the frequency distribution of τRstoch (Figure 6E, inset) directly from the state-transition rate constants (Equations 9 and 10) by calculating the probability and time integral of all likely R* trajectories. This frequency distribution precisely matched that obtained from the simulation of 100,000 random R* trajectories (scatterplot of simulated τRstoch provided in Figure S2). For these simulations, state transitions were determined by checking the transition rate constants (kph(p) and karr(p)) multiplied by the time interval (1 ms) at each time point against a random variable distributed over the unit interval. Each simulated R* trajectory was run through the phototransduction model using the canonical parameter set (Table 2) to generate ensembles of simulated responses; the SPR amplitude frequency distributions (Figure 6E, dashed lines) were constructed from these ensembles. An analogous set of simulations were generated to obtain the mean SPRs of GCAPs+/+ and GCAPs−/− rods used for reproducibility analysis (Figures 6C and 6D) using optimized parameters that remained within ± 10% of the canonical values.
G*-E* Production by R* Activity
The average time course of R* activity, , was used to obtain the average time course of the number of active PDE molecules, E*(t), by integrating the following rate equation:
| (13a) |
whose general solution is
| (13b) |
Here νRE is the rate of G*-E* activation by a fully active R*, kE = 1/τE is the rate constant of deactivation of G*-E*, and the initial condition is E*(0) = 0. The function E*(t) was then applied to Equation 7, which was solved along with Equations 2-8 to generate simulated mean SPRs. An analogous method was applied to generate stochastic SPRs, with the mean R* time course replaced with a stochastic R* trajectory for each simulation.
Supplementary Material
Acknowledgments
We thank Dr. Ching-Kang Chen for the Grk1S561L transgenic mice and Denis Baylor for helpful comments and discussions about the spatiotemporal model and reproducibility. This work was supported by award number R01EY14047 (to MEB) and vision training fellowship (to OPG) from the National Eye Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2012.07.029.
References
- Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- Baylor DA, Nunn BJ, Schnapf JL. The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis. J Physiol. 1984;357:575–607. doi: 10.1113/jphysiol.1984.sp015518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisegna P, Caruso G, Andreucci D, Shen L, Gurevich VV, Hamm HE, DiBenedetto E. Diffusion of the second messengers in the cytoplasm acts as a variability suppressor of the single photon response in vertebrate phototransduction. Biophys J. 2008;94:3363–3383. doi: 10.1529/biophysj.107.114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns ME, Pugh EN., Jr Lessons from photoreceptors: turning off g-protein signaling in living cells. Physiology (Bethesda) 2010;25:72–84. doi: 10.1152/physiol.00001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns ME, Mendez A, Chen J, Baylor DA. Dynamics of cyclic GMP synthesis in retinal rods. Neuron. 2002;36:81–91. doi: 10.1016/s0896-6273(02)00911-x. [DOI] [PubMed] [Google Scholar]
- Burns ME, Mendez A, Chen CK, Almuete A, Quillinan N, Simon MI, Baylor DA, Chen J. Deactivation of phosphorylated and non-phosphorylated rhodopsin by arrestin splice variants. J Neurosci. 2006;26:1036–1044. doi: 10.1523/JNEUROSCI.3301-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso G, Bisegna P, Lenoci L, Andreucci D, Gurevich VV, Hamm HE, DiBenedetto E. Kinetics of rhodopsin deactivation and its role in regulating recovery and reproducibility of rod photoresponse. PLoS Comput Biol. 2010;6:e1001031. doi: 10.1371/journal.pcbi.1001031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso G, Bisegna P, Andreucci D, Lenoci L, Gurevich VV, Hamm HE, DiBenedetto E. Identification of key factors that reduce the variability of the single photon response. Proc Natl Acad Sci USA. 2011;108:7804–7807. doi: 10.1073/pnas.1018960108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- Doan T, Mendez A, Detwiler PB, Chen J, Rieke F. Multiple phosphorylation sites confer reproducibility of the rod’s single-photon responses. Science. 2006;313:530–533. doi: 10.1126/science.1126612. [DOI] [PubMed] [Google Scholar]
- Field GD, Rieke F. Mechanisms regulating variability of the single photon responses of mammalian rod photoreceptors. Neuron. 2002;35:733–747. doi: 10.1016/s0896-6273(02)00822-x. [DOI] [PubMed] [Google Scholar]
- Field GD, Sampath AP, Rieke F. Retinal processing near absolute threshold: from behavior to mechanism. Annu Rev Physiol. 2005;67:491–514. doi: 10.1146/annurev.physiol.67.031103.151256. [DOI] [PubMed] [Google Scholar]
- Gibson SK, Parkes JH, Liebman PA. Phosphorylation modulates the affinity of light-activated rhodopsin for G protein and arrestin. Biochemistry. 2000;39:5738–5749. doi: 10.1021/bi991857f. [DOI] [PubMed] [Google Scholar]
- Gross OP, Burns ME. Control of rhodopsin’s active lifetime by arrestin-1 expression in mammalian rods. J Neurosci. 2010;30:3450–3457. doi: 10.1523/JNEUROSCI.5391-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross OP, Pugh EN, Jr, Burns ME. Spatiotemporal cGMP dynamics in living mouse rods. Biophys J. 2012;102:1775–1784. doi: 10.1016/j.bpj.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer RD, Nicholas SC, Tranchina D, Liebman PA, Lamb TD. Multiple steps of phosphorylation of activated rhodopsin can account for the reproducibility of vertebrate rod single-photon responses. J Gen Physiol. 2003;122:419–444. doi: 10.1085/jgp.200308832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer RD, Nicholas SC, Tranchina D, Lamb TD, Jarvinen JL. Toward a unified model of vertebrate rod phototransduction. Vis Neurosci. 2005;22:417–436. doi: 10.1017/S0952523805224045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Cowan CW, Wensel TG. RGS9, a GTPase accelerator for phototransduction. Neuron. 1998;20:95–102. doi: 10.1016/s0896-6273(00)80437-7. [DOI] [PubMed] [Google Scholar]
- Heck M, Hofmann KP. Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J Biol Chem. 2001;276:10000–10009. doi: 10.1074/jbc.M009475200. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Nunn BJ. Control of light-sensitive current in salamander rods. J Physiol. 1988;403:439–471. doi: 10.1113/jphysiol.1988.sp017258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Wensel TG. R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein, RGS9-1. Proc Natl Acad Sci USA. 2002;99:9755–9760. doi: 10.1073/pnas.152094799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J, Koch WJ, Caron MG, Lefkowitz RJ. Isoprenylation in regulation of signal transduction by G-protein-coupled receptor kinases. Nature. 1992;359:147–150. doi: 10.1038/359147a0. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Lee KA, Niemi GA, Craven KB, Garwin GG, Saari JC, Hurley JB. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron. 2001;31:87–101. doi: 10.1016/s0896-6273(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Keresztes G, Martemyanov KA, Krispel CM, Mutai H, Yoo PJ, Maison SF, Burns ME, Arshavsky VY, Heller S. Absence of the RGS9.Gbeta5 GTPase-activating complex in photoreceptors of the R9AP knockout mouse. J Biol Chem. 2004;279:1581–1584. doi: 10.1074/jbc.C300456200. [DOI] [PubMed] [Google Scholar]
- Krispel CM, Chen CK, Simon MI, Burns ME. Prolonged photoresponses and defective adaptation in rods of Gbeta5-/- mice. J Neurosci. 2003;23:6965–6971. doi: 10.1523/JNEUROSCI.23-18-06965.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen CK, Burns ME. RGS expression rate-limits recovery of rod photoresponses. Neuron. 2006;51:409–416. doi: 10.1016/j.neuron.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Kühn H, Wilden U. Deactivation of photoactivated rhodopsin by rhodopsin-kinase and arrestin. J Recept Res. 1987;7:283–298. doi: 10.3109/10799898709054990. [DOI] [PubMed] [Google Scholar]
- Lagnado L, Cervetto L, McNaughton PA. Calcium homeostasis in the outer segments of retinal rods from the tiger salamander. J Physiol. 1992;455:111–142. doi: 10.1113/jphysiol.1992.sp019293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskov IB, Klenchin VA, Handy JW, Whitlock GG, Govardovskii VI, Bownds MD, Lamb TD, Pugh EN, Jr, Arshavsky VY. The gain of rod phototransduction: reconciliation of biochemical and electrophysiological measurements. Neuron. 2000;27:525–537. doi: 10.1016/s0896-6273(00)00063-5. [DOI] [PubMed] [Google Scholar]
- Makino ER, Handy JW, Li T, Arshavsky VY. The GTPase activating factor for transducin in rod photoreceptors is the complex between RGS9 and type 5 G protein beta subunit. Proc Natl Acad Sci USA. 1999;96:1947–1952. doi: 10.1073/pnas.96.5.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino CL, Peshenko IV, Wen XH, Olshevskaya EV, Barrett R, Dizhoor AM. A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J Biol Chem. 2008;283:29135–29143. doi: 10.1074/jbc.M804445200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci USA. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov S, Engheta N, Pugh EN., Jr Kinetics of recovery of the dark-adapted salamander rod photoresponse. J Gen Physiol. 1998;111:7–37. doi: 10.1085/jgp.111.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa H, Sampath AP. Optimization of single-photon response transmission at the rod-to-rod bipolar synapse. Physiology (Bethesda) 2007;22:279–286. doi: 10.1152/physiol.00007.2007. [DOI] [PubMed] [Google Scholar]
- Okawa H, Miyagishima KJ, Arman AC, Hurley JB, Field GD, Sampath AP. Optimal processing of photoreceptor signals is required to maximize behavioural sensitivity. J Physiol. 2010;588:1947–1960. doi: 10.1113/jphysiol.2010.188573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentia DC, Hosier S, Cote RH. The glutamic acid-rich protein-2 (GARP2) is a high affinity rod photoreceptor phosphodiesterase (PDE6)-binding protein that modulates its catalytic properties. J Biol Chem. 2006;281:5500–5505. doi: 10.1074/jbc.M507488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperberg DR, Cornwall MC, Kahlert M, Hofmann KP, Jin J, Jones GJ, Ripps H. Light-dependent delay in the falling phase of the retinal rod photoresponse. Vis Neurosci. 1992;8:9–18. doi: 10.1017/s0952523800006441. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- Ramanathan S, Detwiler PB, Sengupta AM, Shraiman BI. G-protein-coupled enzyme cascades have intrinsic properties that improve signal localization and fidelity. Biophys J. 2005;88:3063–3071. doi: 10.1529/biophysj.103.039321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Baylor DA. Origin of reproducibility in the responses of retinal rods to single photons. Biophys J. 1998;75:1836–1857. doi: 10.1016/S0006-3495(98)77625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiesser WE. The Numerical Method of Lines: Integration of Partial Differential Equations. New York: Elsevier Academic Press; 1991. [Google Scholar]
- Schnetkamp PP, Basu DK, Li XB, Szerencsei RT. Regulation of intracellular free Ca2+ concentration in the outer segments of bovine retinal rods by Na-Ca-K exchange measured with fluo-3. II. Thermodynamic competence of transmembrane Na+ and K+ gradients and inactivation of Na(+)-dependent Ca2+ extrusio. J Biol Chem. 1991;266:22975–22982. [Google Scholar]
- Shen L, Caruso G, Bisegna P, Andreucci D, Gurevich VV, Hamm HE, DiBenedetto E. Dynamics of mouse rod phototransduction and its sensitivity to variation of key parameters. IET Syst Biol. 2010;4:12–32. doi: 10.1049/iet-syb.2008.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Enhanced arrestin facilitates recovery and protects rods lacking rhodopsin phosphorylation. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen R, Filipek S, Palczewski K, Sousa MC. Ca2+ -dependent regulation of phototransduction. Photochem Photobiol. 2008;84:903–910. doi: 10.1111/j.1751-1097.2008.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock GG, Lamb TD. Variability in the time course of single photon responses from toad rods: termination of rhodopsin’s activity. Neuron. 1999;23:337–351. doi: 10.1016/s0896-6273(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Zhao X, Palczewski K, Ohguro H. Mechanism of rhodopsin phosphorylation. Biophys Chem. 1995;56:183–188. doi: 10.1016/0301-4622(95)00031-r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.