

Abstract
Yersinia pestis causes bubonic and pneumonic plague in humans. The pneumonic infection is the most severe and invariably fatal if untreated. Because of its high virulence, ease of delivery and precedent of use in warfare, Y. pestis is considered a potential bioterror agent. No licensed plague vaccine is currently available in the US. Laboratory research with virulent strains requires appropriate biocontainment (i.e., Biosafety Level 3 (BSL-3) for procedures that generate aerosol/droplets) and secure facilities that comply with federal select agent regulations. To assist in the identification of promising vaccine candidates during the early phases of development, we characterized mouse models of systemic and pneumonic plague infection using the Y. pestis strain EV76, an attenuated human vaccine strain that can be rendered virulent in mice under in vivo iron supplementation. Mice inoculated intranasally or intravenously with Y. pestis EV76 in the presence of iron developed a systemic and pneumonic plague infection that resulted in disease and lethality. Bacteria replicated and severely compromised the spleen, liver and lungs. Susceptibility was age dependent, with younger mice being more vulnerable to pneumonic infection. We used these models of infection to assess the protective capacity of newly developed Salmonella-based plague vaccines. The protective outcome varied depending on the route and dose of infection. Protection was associated with the induction of specific immunological effectors in systemic/mucosal compartments. The models of infection described could serve as safe and practical tools for identifying promising vaccine candidates that warrant further potency evaluation using fully virulent strains in BSL-3 settings.
Keywords: Yersinia pestis, plague infection model, EV76, plague vaccines
1. Introduction
Yersinia pestis is a highly infective organism that causes bubonic, septicemic and pneumonic plague in humans. Bubonic plague is the most common and benign form of this disease. It occurs naturally, develops gradually and can be treated with antibiotics. The reported mortality rate is 50-60% (or greater) if untreated [1]. Pneumonic plague, on the other hand, is the most severe and feared form of infection [2]. It can be transmitted easily from person to person through contaminated droplets, progresses rapidly and is invariably fatal [1] unless antibiotics are administered immediately, and despite treatment, ~15% fatality occurs [3]. Because of its high infectivity and ease of release (i.e., via aerosol), Y. pestis is regarded as one of the organisms most likely to be deployed in bioterror warfare [3;4]. In fact, Y. pestis has been developed and used as a biological weapon on multiple occasions throughout history [2-4]. The Centers for Disease Control and Prevention (CDC) lists Y. pestis among the Category A organisms recognized as the highest threat to national security [5] and as a select agent of bioterrorism [6]. There is presently no licensed vaccine to protect against plague in the US. A number of vaccine candidates have been proposed [Reviewed in [7-9]]. These vaccines, however, have been shown to induce only partial protection when tested in multiple animal models, and none of them can protect against all forms of the disease [8;9]. A recombinant F1/V vaccine was tested in humans with limited success [10], but improved rF1/V formulations are being investigated in Phase 1 and Phase 2 clinical studies [11].
The demonstration of protective efficacy is a critical step during the process of vaccine development and typically involves challenge of vaccinated and control animals with virulent strains that reproduce disease. The manipulation of virulent Y. pestis strains during laboratory procedures that may create aerosols and droplets requires BSL-3 containment [12]. Because the plague bacterium is also a potential bioterror agent, research with this organism requires the availability of secure laboratory facilities and bio-containment that meet security standards in compliance with existing federal select agent regulations. Experiments using select agents are highly restricted and involve many regulatory and administrative hurdles. In the US, investigators must be approved by multiple government agencies to work with select agents. BSL-3 and Animal BSL-3 (ABSL-3) facilities are infrequent, and access is highly restricted, requiring specially trained personnel and the use of dedicated equipment. In addition, the use of fully virulent infectious strains poses a serious biohazard for the operator. Research under these conditions is therefore cumbersome, time-consuming and expensive. During the early stages of vaccine development, when promising vaccine candidates must initially be identified and further refined, the risk and complexity associated with the use of virulent challenge strains in potency tests could be alleviated by using less virulent strains in reliable animal models that reproduce infection under BSL-2, rather than BSL-3, containment.
In previous work from our group, we reported that the attenuated Y. pestis pigmentation negative (pgm-) strain Y. pestis EV76 induced a lethal systemic infection in mice when administered intravenously in the presence of iron [13;14]. Several pgm- plague strains with attenuated phenotypes have been described [15-17]. The pigmentation locus, which contains the two established virulence-related gene clusters, the high-pathogenicity island and the haemin storage (hms) system (pigmented colony formation in Congo red media), is impaired in these mutants [18-20]. Consequently, these strains are unable to scavenge iron from the host, which is necessary for the successful establishment of disease [21], and pathogenesis is abrogated unless an external source of iron is provided [20]. We chose the pgm- Y. pestis EV76 strain for our studies because of its safety profile, having been used extensively as a plague vaccine in humans [22;23].
In this work, we established and characterized mouse models of systemic and pulmonary plague infection using the strain EV76 with the purpose of assessing the protective capacity of vaccine candidates during the early stages of development. We propose the use of these models as practical and safer tools to identify promising vaccine candidates that could be further tested for potency using fully virulent challenge strains.
2. Materials and methods
2.1. Preparation of the challenge inoculum
Y. pestis EV76 was kindly provided by Dr. H. Wolf-Watz (Umeå University, Umeå, Sweden), and stock cultures were maintained at –80 °C. Prior to challenge, bacteria were streaked onto animal-product free Luria Bertani Lennox broth (APF-LB; Athena Enzyme Systems, Baltimore, MD) agar plates for 72 h at 30 °C. The day before the challenge, 2 ml aliquots of APF-LB broth were inoculated and incubated for 6-8 h at 30 °C. Optical density at 600 nm (OD600nm) was measured and adjusted to 0.1 with APF-LB containing 2.5 mM CaCl2 and incubated at 37 °C for 16-18 h. Aliquots (0.5 ml) were centrifuged, and pellets were resuspended and screened for flocculency (indicative of F1 expression). Selected cultures were adjusted to the desired concentration with sterile phosphate-buffered saline (PBS) for the systemic challenge or saline solution for the pulmonary challenge. The inoculum dose was verified by serial dilution on APF-LB agar plates incubated at 30 °C for 48-72 h.
2.2. Systemic and pulmonary infection procedures
Systemic infection: 8- to 23-week-old female BALB/c mice (Charles River, Wilmington, MA) were injected intravenously (i.v.) in the lateral tail vein with either 2.3×104 or 2.3×105 CFU of Y. pestis EV76 in a 200 µl volume (doses and challenge conditions are summarized in Table 1). FeCl2 was administered via intraperitoneal (i.p.) injection immediately before challenge as previously reported [24]; each mouse received 40 µg of FeCl2 (Fluka, Steinheim, Germany) in 100 µl of water (from a freshly prepared sterile solution). Pulmonary infection: 8- to 16-week-old female BALB/c mice were inoculated intranasally (i.n.) with either 6.2×106 or 1.5×108 CFU of Y. pestis EV76 in a 15-20 µl volume admixed with 15-20 µl of a freshly prepared FeCl2 solution containing 40 µg of FeCl2 or saline. Control groups received bacteria or FeCl2 alone. Intranasal inoculation was performed under Isoflurane anesthesia (Abbott Laboratories, North Chicago, IL); half of the inoculum was delivered with a pipette into each nare. All animal studies were approved by the University of Maryland Animal Care and Use Committee.
Table 1.
Immunization protocols to validate the Y. pestis EV76 i.v. and i.n. infection models
| Age at (weeks old) |
Route of administration |
Challenge dose |
||||
|---|---|---|---|---|---|---|
| Groups | Prime | Boost | Challenge | Iron | Y. pestis EV76 | |
| Adults (n=5) |
8 | 11 | 17 | i.p. | i.v. | 3,150 MLD50 (2.3×105CFU) |
| Adults (n=5) |
8 | 11 | 17 | i.n. | i.n. | 15.8 MLD50 (1.5×108CFU) |
| Neonates (n=5-6) |
1 | 3 | 7 | i.p. | i.v. | 1,150 MLD50 (2.3×104CFU) |
| Neonates (n=5-6) |
1 | 3 | 8 | i.n. | i.n. | 4.7 MLD50 (6.2×106CFU) |
2.3. Post-challenge health assessment
Animals were monitored daily (or twice a day) for two weeks after challenge; health status, weight loss and deaths were recorded. A scoring system with a scale of 1 to 4 was used for physical health assessment that was performed by two independent investigators. A score of 1 was assigned for normal posture, shiny coats and no signs of dehydration; a score of 2 for early stage of piloerection but alert with normal posture and mild dehydration; a score of 3 for mild piloerection with mice reacting slowly and with dull coats, hunched posture, difficult ambulation and moderate dehydration; and a score of 4 (very sick) for severe piloerection with nonreactive, dull, dirty, hunched and squinting mice with severe tenting and weight loss > 20% of initial weight. Animals with a score of 4 were promptly euthanized. Survivors were euthanized at the end of the monitoring period.
2.4. Bacterial colonization
Blood, liver, spleen and lungs were collected from controls and from mice infected with sublethal doses of Y. pestis EV76 on days 0-4 after infection. Organs were weighed and homogenized in 2 ml of PBS using 70 µm tissue grinders (BD Falcon, Bedford, MA). Blood samples and tissue homogenates were serially diluted and plated on CIN agar base (Remel, Lenexa, KS) [14]. Plates were incubated for 72 h at 30 °C, and colonies were counted. All colonies exhibited the bull’s-eye morphology characteristic of Yersinia when grown on this selective media [25].
2.5. Histology
The liver, spleen and lungs were collected from infected and control mice as described above. Organs were fixed with 10% buffered formalin for 48 h, processed and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (H&E) and evaluated by a board certified pathologist (C.B.D.).
2.6. Construction of recombinant Salmonella vaccine strains expressing Y. pestis antigens
Two live attenuated Salmonella enterica serovar Typhi (S. Typhi) vaccine strains expressing Y. pestis F1 or LcrV were used to establish the usefulness of our infection model to evaluate protective efficacy. The strain expressing F1 (Sal-F1) had been previously described by our group [13]. A new strain expressing LcrV was constructed for this work as follows. The gene encoding LcrV was PCR amplified from Y. pestis EV76 (pgm–) genomic DNA using Taq DNA polymerase (New England BioLabs, Ipswich, MA) with primers Lloyd113 (5′-GCC GGA TCC GCG GCC GCA GGA GGA ATT AAC CAT GAT TAG AGC CTA CGA ACA AAA C-3’) and Lloyd72 (5’-GCC GTC GAC ACG CGT TCA TTT ACC AGA CGT GTC ATC-3’). The Lloyd113 upstream primer incorporated an optimal Shine-Dalgarno site (based on the Yersinia yopD Shine-Dalgarno sequence) [26] for increased translation of LcrV. Following amplification, the PCR product was digested with BamHI and SalI and cloned into the same sites of the kanamycin-resistant, medium copy number plasmid pSEC91 [27], replacing the clyA and tetA genes to create pSEC91-LcrV. The identity of the plasmid was verified by DNA sequence analysis. Plasmid pSEC91-LcrV was electroporated into S. Typhi vaccine strain ACAM948CVD (Acambis) following standard techniques. ACAM948CVD is a reconstruction of the clinically acceptable S. Typhi vaccine strain CVD 908-htrA (an aroC, aroD, htrA mutant) [28], which is derived from virulent Ty2 and passaged only on animal-product-free medium to comply with regulatory requirements. The resulting vaccine strain, ACAM948CVD (pSEC91-LcrV), is hereafter referred to as Sal-LcrV.
Vaccine inocula were prepared from overnight cultures of bacteria grown in APF-LB broth supplemented with 0.0001% (w/v) 2,3-dihydroxybenzoic acid (DHB; Sigma-Aldrich, Co., St. Louis, MO) and 50 μg/ml kanamycin (Sigma-Aldrich). Overnight cultures were subcultured in 250 ml of fresh medium for ~4 h at 37 °C (OD600nm of ~1.0; late-log phase). Bacteria were then harvested, washed and resuspended in sterile PBS to ~1×109 CFU in 5 μl or 10 μl for immunization of newborn and adult mice, respectively. Viability was determined by plating serial dilutions of the inoculum onto APF-LB agar supplemented with DHB and kanamycin as needed. Aliquots (1 ml) of each subculture were centrifuged at 4500×g for 2 min, and pellets were stored at –20 °C until analyzed for protein expression.
2.8. Analysis of F1 and LcrV expression by western blot
A selection of Salmonella vaccine pellets were thawed and resuspended in sample buffer (Bio-Rad, Hercules, CA) with 5% 2-mercaptoethanol. A subculture of Y. pestis EV76 was incubated in APF-LB broth supplemented with 2.5 mM CaCl2 (Fisher Scientific Company, Fair Lawn, NJ) for 16-18 h at 37 °C. After incubation, supernatants were precipitated with trichloroacetic acid (American Bioanalytical, Natick, MA) as previously described [29] and resuspended in 8 M urea (Sigma-Aldrich), Laemmli sample buffer and 2-mercaptoethanol. Proteins in Salmonella vaccine pellets and Y. pestis culture supernatants were separated by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked and incubated with mouse anti-F1 and -LcrV monoclonal antibodies (Abcam, Cambridge, MA) followed by HRP-labeled anti-mouse IgG1 (Roche Diagnostics, Indianapolis, IN). Reactive bands were visualized by chemiluminescence (PerkinElmer, Inc.; Boston, MA). Recombinant F1 and LcrV (1 µg/lane) were used as positive controls.
2.9. Immunization of newborn and adult mice
Neonatal BALB/c mice were bred as previously described [30]. Newborn (7 days old) or adult mice (8 weeks old) were immunized twice by the i.n. route with ~1×109 CFU of each Salmonella vaccine contained in a 5-10 µl volume (half of which administered into each nostril). Detailed information on immunization and challenge procedures are presented in Table 1. Serum samples were collected from the retro-orbital sinus under Isoflurane anesthesia. Samples were stored at -20 °C until use.
2.10. Specimen collection
Nasal washes and fecal pellets were obtained from mice immunized as newborns as previously described [31] with modifications. To collect nasal washes, mice were euthanized, the lower jaw was removed, and 1 ml of PBS was flushed from the posterior nares into the exposed nasal cavity. The liquid expelled from the anterior openings was collected (kept on ice) and cleared of cellular debris by centrifugation (1500×g for 5 min). Fresh fecal pellets were collected, weighed and adjusted to 100 mg per mouse, and resuspended in 1 ml of PBS containing 0.2% sodium azide by vortexing for 15 min. The suspension was centrifuged twice (13,600×g for 5 min), and the supernatants were transferred to a clean tube. Phenylmethylsulfonyl fluoride was added to both nasal washes and stool supernatants to a final concentration of 1 mM. The samples were stored at -20 °C until use.
2.11. Measurement of F1 and LcrV antibodies
Serum IgG and mucosal IgA specific for Y. pestis F1 and LcrV were measured by ELISA as previously described [13;14]. HRP-labeled goat anti-mouse Fc-γ and Fc-α (KPL, Gaithersburg, MD) were used as secondary antibodies. End point titers were calculated through linear regression equations as the reciprocal of the serum dilution that produced an OD450nm value of 0.2 above the blank and were reported in ELISA units (EU ml–1).
2.12. Statistical analysis
Serological measurements were log transformed to calculate the geometrical mean titers. Differences in antibody titers among groups were compared using Student’s t test when the data were normally distributed and Mann-Whitney U test when normality failed. Mouse lethal dose 50% (MLD50) values were calculated by the method of Reed and Muench [32]. Survival curves were analyzed by the Log-rank (Mantel-Cox) Test using GraphPad Prism® version 5.01, GraphPad Software (San Diego, CA). Health scores and weight were analyzed by Mann-Whitney Rank Sum Test using SigmaStat® 3.5 (Systat Software, Inc., Chicago, IL). A p value <0.05 was considered significant at the 95% confidence interval.
3. Results
3.1. The effect of iron in Y. pestis EV76 virulence
Early studies had shown that pgm- attenuated mutants of Y. pestis and Y. pseudotuberculosis, which are deficient in their capacity to obtain iron from biological fluids, regained virulence in mice and guinea pigs when parenterally supplemented by haemin or inorganic iron before inoculation [20;33]. To establish reproducible infection models for evaluating vaccine efficacy, we examined the virulence of the Y. pestis EV76 strain when administered to mice through different routes in the presence of iron supplementation. The i.v. route of injection had been used to infect mice with Y. pestis strains [34]. On the other hand, i.p. iron supplementation had been shown to restore virulence of Y. pestis pgm- mutants administered by the same route [24]. To establish a systemic infection, we combined both approaches and examined the development of disease and survival rates in mice inoculated i.v. with 1×104 CFU of Y. pestis EV76 that also received 40 µg of iron i.p. at the time of infection (Fig. 1A, left). Control mice received bacteria alone or iron alone. Mice that received the organisms along with iron became sick and started to die on day 5 after inoculation, and 100% mortality was reached on day 8. Mice that received either iron or bacteria alone remained healthy.
Figure 1.

Survival curves and mouse lethal dose 50% (MLD50) values in mice infected systemically or via the respiratory tract with Y. pestis EV76 supplemented with iron. (A) Virulence of Y. pestis EV76 restored by iron supplementation. Mice (5-6 per group) were administered 1×104 CFU of Y. pestis EV76 intravenously (i.v.) with 40 μg FeCl2 i.p. or 3×107 CFU combined with 40 μg of FeCl2 delivered intranasally (i.n.). Control groups received iron or bacteria alone. Mice were monitored for 14 days after challenge as described in Section 2. (B) MLD50 following systemic and pulmonary infection. Mice (5-6 per group) were inoculated i.v. or i.n. with increasing doses of bacteria in the presence of iron and monitored for survival for 14 days as described above. (C) Age-dependent susceptibility of infection. Mice of different ages (8 to 24 weeks old) were infected i.v. or i.n. with Y. pestis EV76 plus iron as described above, and MLD50 values were determined. The data shown are representative from three experiments.
To establish a productive respiratory infection, a series of preliminary experiments were performed first to identify optimal conditions (Supplementary Figure 1). In these experiments, the organisms were administered intranasally (1×105-7×107 CFU) admixed with increasing amounts of iron (5, 10, 20, 40 and 80 μg) by the same route (i.n.) in different inoculum volumes (30 to 40 µl) or were administered separately by different routes (i.n. and i.p., respectively). The volume of inoculum was found to be important to reproduce pneumonic disease; pulmonary infection and lethality were achieved when the organisms were administered in 30-40 µl but not when using smaller volumes, which might have prevented the bacteria from reaching the lungs (where the infection begins). Another critical observation was that the microorganisms had to be combined with iron and delivered by the same route to produce a consistent pulmonary infection; no disease was observed when the bacteria were given i.n. and the iron was given i.p., suggesting that iron must be available in vivo at the site of the infection to produce pneumonic disease. The amount of iron delivered was also critical to obtain a reproducible infection. Mice inoculated with the organisms in the presence of 5, 10 and 20 µg of FeCl2 had inconsistent survival rates (i.e., between 33 and 100%), and the time to death was delayed compared to mice that received higher amounts of iron. Additionally, results were difficult to reproduce. All mice that received the organisms admixed with 80 μg of iron died within 4 to 7 days post-inoculation. These animals, however, became very sick immediately after infection and exhibited significant distress. Mice that received 80 µg of iron alone also became sick, indicating that this amount of iron was excessive and toxic. Forty micrograms of iron were well tolerated and associated with consistent and reproducible disease, and this dose was used in subsequent experiments. Fig. 1A, right, shows the mortality curves from our optimized pulmonary model in which mice were inoculated with 3×107 CFU admixed with 40 µg of FeCl2 in a total volume of 30-40 μl. Death started to occur 2 days after inoculation and reached 100% mortality by day 4.
Supplementary material related to this article found, in the online version, at http://dx.doi.org/10.1016/j.cimid. 2012.10.005.
3.2. Determination of MLD 50
To determine the MLD50 for each route of infection, naïve 9- to 11-week-old mice were inoculated with increasing doses of Y. pestis EV76 via i.v. (3×100 to 3×103 CFU) or i.n. (3×105 to 3×108 CFU) co-administered with 40 µg of FeCl2 i.p. or i.n., respectively (Fig. 1B). A dose as low as 3×101 CFU resulted in 83% mortality in mice infected i.v., with deaths observed 6 days post-inoculation. Higher dosage levels (3×102 and 3×103 CFU) caused 100% lethality ~1 week after inoculation. In contrast, a larger number of organisms were required for pulmonary infection; 3×106 CFU was the first dose that led to significant morbidity, but this dose only reached 40% mortality. Doses of 3×107 and 1×108 CFU resulted in 100% mortality starting 48 h after inoculation. The calculated MLD50 for systemic and respiratory Y. pestis EV76 challenge in adult mice were 20 CFU (Fig. 1B, left) and 4.8×106 CFU (Fig. 1B, right), respectively.
3.3 Susceptibility to infection at different ages
One of our main research interests is the development of vaccines suitable for different age groups, particularly the pediatric population, and one of our primary goals is to investigate vaccine-induced protection at different stages of life. To evaluate the effect of age in susceptibility to infection with EV76, mice of varying ages, from 8 to 24 weeks old, were challenged i.v. or i.n. with an increasing number of organisms in the presence of iron. These ages were selected as the corresponding ages to assess protection following neonatal and adult immunization. The MLD50 values for systemic infection increased from 20 CFU in young adult mice (8±1 weeks old) to 73 CFU in older adults (23±1 weeks old); the difference representing a 3.6-fold rise for a 16-week age increase. The MLD50 for pulmonary infection had a sharper increase, from 1.3×106 in 8±1-week-old mice to 9.5×106 CFU in 16±1-week-old mice, corresponding to a 7.3-fold increase in an even shorter age interval (8 weeks vs. 16 weeks). The trend of higher susceptibility of younger mice to pneumonic plague was confirmed when we compared the lethal doses corrected by body weight (Fig. 1C). The MLD50 values were 3 times higher (1.08 to 3.2 CFU/g) in 23-week-old mice infected i.v. and 7 times higher (6.7×104 to 4.6×105 CFU/g) in 16-week-old mice infected i.n., compared with mice at 8 weeks of age (Fig. 1C). The MLD100 for 23- and 16-week-old mice were 6 and 9 times higher than those of 8-week-old adults for systemic and respiratory infection, respectively.
3.4. Characteristics of the disease
To establish the characteristics of the disease, health status and weight were monitored daily for 4 days in mice challenged i.v. or i.n. with sublethal doses of Y. pestis EV76 in the presence of iron (Fig. 2). Signs of disease, i.e., decreased activity, dehydration and ruffled coats, appeared within 24 h of infection regardless of the route of inoculation (Fig. 2A and B). Mice infected i.v. exhibited a sharp decrease in weight during the 72-96 h following challenge (Fig. 2C), whereas those infected i.n. had only a moderate and more progressive weight loss (Fig. 2D). Despite maintaining weight, mice inoculated i.n. appeared sicker than those inoculated i.v. during the first 3 days after challenge (Fig. 2B). In both cases, disease progressed until the mice reached a moribund state.
Figure 2.

Physical health assessment of mice infected with Y. pestis EV76 plus iron. Mice (25 per group, 10-11 weeks old) were challenged i.v. or i.n. with sublethal doses of Y. pestis EV76 administered with FeCl2 (160 CFU corresponding to 8 MLD50 and 1.6×107 CFU corresponding to 3.3 MLD50, respectively). The data represent mean health scores and percent of initial weight ±SE during days 1-4 after challenge.
3.5. Organ distribution and colonization in vivo
We next examined the distribution of the organisms in vivo following systemic and pulmonary infection, as described above. Y. pestis EV76 replicated rapidly and efficiently in the spleen and liver of mice infected i.v. and in the spleen of mice infected i.n. (Fig. 3). Organisms were found in the lungs of mice inoculated by either route, but the replication pattern was very different. A moderate and progressive replication, up to 1×104 CFU per g of lung, was observed in the i.v.-infected mice. In contrast, mice infected i.n. had a considerable bacterial load 24 h after inoculation, which receded significantly thereafter. A similar high number of organisms reached the spleen 24 h after i.n. challenge, which persisted within the 1 log range until day 4. Although the bacteria were recovered from various organs, they were rarely detected in blood; the animals that had positive blood cultures were generally injected i.v. and at late time points (3-4 days after infection). Spleen, liver, lungs and blood obtained from mice that received saline or iron alone had negative cultures (data not shown).
Figure 3.

Bacterial colonization in mice infected with Y. pestis EV76 plus iron. Mice were infected i.v. or i.n. with sublethal doses of Y. pestis EV76 as described in Fig 2. The blood, spleen, liver and lungs were collected, homogenized, and plated (in serial dilutions) on agar CIN base plates, and the plates were incubated to determine the number of colonizing organisms. The results are expressed as individual CFU/g of organ or ml of blood. Horizontal lines indicate the mean bacterial counts; the dotted line shows detection limit.
3.6. Histology
The severity of disease was also assessed through histological analysis. The spleen, liver and lungs from infected and control mice were examined for tissue architecture and signs of inflammation or overt pathology. Organs were collected during the first 4 days after infection. Sections corresponding to day 2 (i.n.) and day 4 (i.v.) are shown (Fig. 4). Mice infected i.v. exhibited confluent foci of necrosis and abscess formation in the spleen (Fig. 4B, arrowheads) with numerous microabscesses (~0.2 mm) in the liver (Fig. 4D, arrowheads), indicating typical manifestations of bloodborne bacterial dissemination (septicemia). The lungs of these animals showed generalized congestion and focal margination of neutrophils in septal capillaries, which are indicative of early pneumonia (Fig. 4F, arrows). Macroscopic observations in this group included splenomegaly with foci of necrosis and a soft, friable and pale discolored liver correlating with the severe necrosis observed in histological analysis. The spleen, liver and lungs from mice treated with iron i.p. exhibited normal tissue structure (Fig. 4A, C and E). Mice infected i.n. presented bronchopneumonia consisting of necrosis and massive neutrophil infiltrates around the airways (Fig. 4H and J). The spleen revealed mild congestion, while the liver was normal (data not shown). Macroscopic observations included mild splenomegaly with few foci of necrosis and severe damage in the lungs. Mice that received only iron i.n. had a mild peribronchial inflammation consistent with chemical injury but no signs of pneumonia (Fig. 4G and I), while the spleen and the liver were normal. Conversely, mice inoculated i.n. with bacteria alone induced mild pneumonia at day 6 post-infection, but it did not lead to death (data not shown).
Figure 4.

Representative histological analysis of the spleen, liver and lungs from mice infected with Y. pestis EV76 plus iron as described in Fig. 2. Mice treated with iron i.p. or i.n. were included as controls. Histological sections from spleen (A and B), liver (C and D), and lungs (E-J) were fixed, processed and stained with H&E to visualize tissue architecture on days 2 and 4 for i.n. and i.v.-infected mice, respectively. Mice infected i.v. with Y. pestis EV76 exhibited necrotic areas in the spleen (B), a microabscess in the liver (D), and very early pneumonia in the lung (F) and are indicated with arrowheads (B and D) and arrows (F). Mice infected i.n. showed signs of bronchopneumonia (H and higher magnification in J) with a massive neutrophil infiltration in the lungs (J, insert). Mice that received only iron i.n. had a mild transient peribronchial inflammation consistent with chemical injury (G, higher magnification in J). Magnifications: 20× (A-D, G and H), 40× (E, F, I and J) and 60× (J insert).
3.7. F1 and LcrV expression by recombinant S. Typhi vaccine and Y. pestis challenge strains
To test the usefulness of our models of infection, we selected two recently developed recombinant S. Typhi vaccine strains expressing Y. pestis F1 and LcrV and investigated their capacity to induce protective immunity. We first examined the levels of expression of F1 and LcrV by our S. Typhi vaccine strains (Fig. 5A) vs. F1 and LcrV expressed by the Y. pestis EV76 challenge strain through western blot analysis of bacterial lysates and culture supernatants (Fig. 5B). F1 produced by EV76 or Sal-F1 had the expected molecular mass of ~17 kDa, whereas LcrV produced a band of ~39 kDa. Bands of the equivalent molecular weight were produced by rF1 and rLcrV using the same specific antibodies (positive controls). No signals were detected in lysates or supernatants from Salmonella alone (not expressing any antigen).
Figure 5.
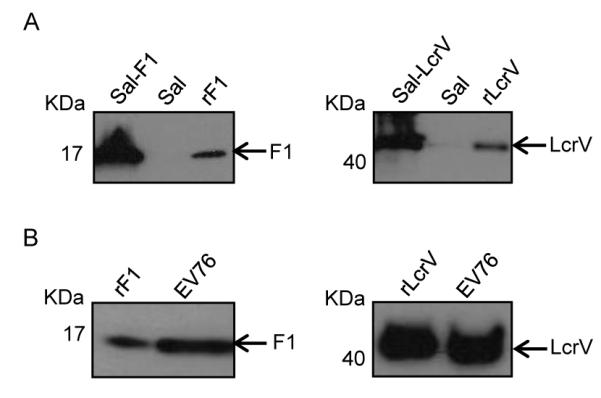
Expression of Y. pestis F1 and LcrV in S. Typhi recombinant vaccine strains (A) and Y. pestis strain EV76 (B) determined by western blot. Lysates from log late-phase cultures of S. Typhi recombinant strains were separated by SDS-PAGE, transferred to nitrocellulose membranes and treated with monoclonal antibodies anti-F1 (left) or anti-LcrV (right). To test the expression of antigens in EV76, overnight culture supernatants were precipitated with trichloroacetic acid; proteins were separated and detected by western blot as described above.
3.8. Validation of the infection models in adult immunized mice
We next examined the usefulness of our systemic and pulmonary mouse infection model to determine the protective capacity of vaccine candidates in early stages of development. Interested in vaccines that could be safe and effective in different age groups, we evaluated vaccine potency in mice that had been vaccinated as newborns or as adults. In both cases, mice were immunized twice with S. Typhi expressing F1 or LcrV via the nasal route, and they were subsequently challenged i.v. or i.n. with Y. pestis EV76 supplemented with iron (Table 1). A summary of the immune responses and protection outcomes in adult mice is shown in Fig. 6. Mice immunized with S. Typhi expressing F1 or LcrV developed high levels of F1 and LcrV-specific serum IgG antibodies (Fig. 6A). Only antibodies to the vaccine-delivered antigen (F1 or LcrV) were detected in each group, and no responses were observed in mice that received S. Typhi alone (not expressing any antigen). A subgroup of 5-6 mice was challenged i.v. with 3,150 MLD50 (2.3×105 CFU) of Y. pestis EV76; survival curves are shown in Fig. 6B. All mice immunized with Sal-F1 survived the challenge. In contrast, none of the mice immunized with Sal-LcrV were protected. Naïve mice and controls that received Salmonella alone all succumbed to the disease. The health score (Fig. 6D) and body weight assessments (Fig. 6F) correlated with the survival outcome; the group immunized with Sal-F1 was the healthiest. Another subgroup of mice was challenged i.n. with 15.8 MLD50 (1.5×108 50 CFU) of Y. pestis EV76. Interestingly, we did not see protection against respiratory infection under these conditions; regardless of the vaccine treatment, all mice died by day 5 (Fig. 6C). The health scores (Fig. 6E) and weight curves (Fig. 6G) were all in agreement with the survival results.
Figure 6.

Immunogenicity and protective efficacy of S. Typhi-based plague vaccine candidates. Live attenuated S. Typhi strains expressing F1 and LcrV (1×109 CFU) were administered to adult mice via the nasal route in 2 occasions, 3 weeks apart (days 0 and 21). A control group received Salmonella alone. Six weeks after the second immunization, they were challenged with Y. pestis EV76 via i.v. or i.n. (A) F1 and LcrV-specific serum IgG titers were measured by ELISA. The data represent individual titers measured 1 month after the second vaccination and geometric mean titers for each group (horizontal line). Survival curves are presented following systemic (B) and pulmonary (C) infection; mice were administered 2.3×105 CFU (3,150 MLD50) and 1.5×108 CFU (15.8 MLD50.) i.v. and i.n., respectively (D and E). Health scores (mean values) following i.v. or i.n. infection are shown. (F and G) Weight assessment after i.v. or i.n. infection; results are expressed as percent of initial weight (mean values). The dotted line indicates the time point of the last death in the control group, which was used for group comparisons (*p<0.05).
3.9. Immune responses and validation of the infection models in mice immunized as newborns
The next set of experiments to validate the infection models were performed in mice immunized as newborns with Sal-F1 and Sal-LcrV. We also examined the immune responses in more detail to better interpret the protection outcome. Fig. 7A shows the kinetics of serum IgG responses to F1 and LcrV in mice immunized at day 7 after birth and boosted at day 21. For both vaccines, antibody responses to the delivered antigens were first detected on day 28, which was 1 week after the second immunization. The levels of IgG specific for F1 produced in this experiment were similar to those reported previously [13] and ~1 log lower in magnitude than those measured in adults (2.5×103 vs. 2.1×104, 4-6 weeks after the second immunization). No responses were observed in the negative control group (Sal). We also investigated antibody responses in mucosal tissues and found robust IgA titers against F1 in nasal lavages from mice immunized with Sal-F1 but no responses in mice that received Sal-LcrV (Fig. 7B). Similarly, most of the mice immunized with Sal-F1 produced IgA antibodies in stool, but only a few of them responded with modest titers against LcrV (Fig. 7C). These mice were challenged i.v. or i.n. with Y. pestis EV76 in the presence of iron 4-5 weeks after the boost. All of the animals that had been immunized with either Sal-F1 or Sal-LcrV survived an i.v. challenge with 1,150 MLD50 (2.3×104 CFU) (Fig. 8A); 50% of mice immunized with Salmonella alone survived as well, but none of the unvaccinated mice survived. Despite the lack of statistical significance in the survival rate between recipients of Sal-F1 or Sal-LcrV and Salmonella alone, there were clear differences in the magnitude and severity of the disease between the groups. Mice that received Sal-F1 or Sal-LcrV appeared healthy throughout the study, whereas those treated with Salmonella alone either died or became visibly sick (p<0.05) showing signs of recovery only after day 10 (Fig. 8C). The body weight curves showed the same trend (Fig. 8E). Newborns immunized with the Sal-F1 or Sal-LcrV were challenged i.n. with Y. pestis EV76 (4.7 MLD50; 6.2×106 CFU). Immunization with Sal-F1 conferred the highest level of protection, i.e., 60% (p<0.05), whereas 16% protection was afforded by Sal-LcrV (p<0.17). All mice that received Salmonella alone or naïve controls died (Fig. 8B). The results from health status score and weight showed a similar trend, although the differences among the groups did not reach statistical significance (Fig. 8 D and F).
Figure 7.

Serum and mucosal antibody responses in mice immunized as newborns with S. Typhi expressing F1 or LcrV. Newborn (7-day-old) mice were immunized i.n. on days 7 and 21 after birth (arrows) with 1×109 CFU of Sal-F1 or Sal-LcrV (A) F1 and LcrV-specific serum IgG titers measured by ELISA; the results shown are geometric mean titers±SE. (B) F1 and LcrV-specific IgA antibodies were measured in nasal lavages; the data shown are individual titers from a subset of 7 mice measured on day 35 after birth. (C) F1 and LcrV-specific IgA titers were measured in fecal pellets; the data represent individual titers from 15 mice per group measured on day 35.
Figure 8.

Protection of mice immunized as newborns with Sal-F1 and Sal-LcrV. Mice were immunized as described in Fig. 7 and challenged i.n. or i.v. with Y. pestis EV76 4-5 weeks after the last immunization. (A) Survival curves from mice challenged with 2.3×104 CFU (1,150 MLD50) i.v. or (B) with 6.2×106 CFU (4.7 MLD50) i.n. (C and D) Health scores (mean values) following i.v. or i.n. infection. (E and F) Weight assessment after i.v. or i.n. infection; results are expressed as the mean percent of initial weight (mean values). The dotted line indicates the time point of the last death in the control group, which was used for group comparisons (*p<0.05).
4. Discussion
Murine models have been widely used to study the pathogenesis of Y. pestis and to evaluate immunogenicity and protective efficacy of plague vaccine candidates [9;34-36]. Mice are natural hosts for Y. pestis and develop a disease that is specific for the route of inoculation, similar to what occurs in humans infected with the organism [35]. Our group has been interested in the development of plague vaccines, and a key parameter to ascertain the potential value of vaccine candidates is the demonstration of protective efficacy. This typically involves in vivo challenge with virulent strains. Because of its high infectivity and lethality when inhaled, laboratory work with Y. pestis that may generate aerosols or droplets is best conducted following BSL-3 practices [12]. Furthermore, because Y. pestis is a potential bioterror agent (a select agent), research with virulent strains requires laboratory facilities with heightened security that complies with federal regulations. Thus, challenge experiments using such strains are complex and beset with biosecurity risks.
To identify efficacious plague vaccines in a safer, expedited and practical manner during the early stages of development, we characterized mouse models of systemic and pulmonary plague infection using the attenuated Y. pestis pgm- strain EV76 administered via i.v. or i.n. This plague strain has been extensively used as a human vaccine and is exempt from the regulatory requirements of fully virulent select agent strains and can be safely handled under BSL-2 containment [37]. Although the Y. pestis EV76 does not cause overt disease in mice, it can be rendered virulent when supplemented with an external source of iron (conditional virulence) with presumably no increased risk to investigators. Precaution should be exercised, however, in situations where personnel may be exposed to iron overload from excessive consumption, prolonged iron supplementation [38] or disorders such as genetic hemochromatosis [39;40] or other iron-loading anemias.
The influence of iron on the growth and virulence of pgm- plague strains was first reported by Jackson and Burrows in the mid-1950s while studying mechanisms of pathogenesis [20]. Infection of mice using Y. pestis pgm- KIM strains supplemented with iron has been described in two recent studies [41;42]. The first paper by Lee-Lewis et al. reports a lethal infection in BALB/c mice inoculated i.n. with the KIM D27 strain and a notably large amount of iron (up 500 µg) given i.p. Under this condition, the investigators observed bacterial replication and damage of multiple organs but no signs of pneumonia or lesions characteristic of pneumonic plague. Protection against pneumonic plague is required of any new vaccine to prevent epidemic spread [43] and can be only demonstrated in animal models that faithfully recapitulate this form of the disease. The second study by Galvan et al. reports pneumonic lesions using KIM5 infection of C57BL/6 mice. This model required even larger amounts of iron (4-10 mg) given i.p. on multiple occasions, before and after challenge. A problem arising from the use of large amounts of iron in vivo is toxicity. This is demonstrated in our study and has long been recognized by others [44]. The highest dose of FeCl2 that we were able to administer safely to naïve mice using the nasal route was 40 µg; larger amounts caused obvious discomfort and side effects, including eye dryness, ruffled hair and hunched posture, which required 1-3 days to resolve. These non-specific adverse effects not only impose undue stress on the animals but also confound the assessment of “true” disease. The use of iron-dextran mitigates this toxicity, as it released slowly from carbohydrate complexes, although even larger amounts and multiple dosing are required. In the study described by Galvan et al. mice were given daily injections of dextran-iron 2-3 h before and up to 10 days after challenge [41]. This procedure is impractical and adds stress to the already sick animals. The need to administer iron daily for the organisms to grow and replicate in vivo creates another set of variables that could affect reproducibility.
Unlike previous studies, we were able to establish a pulmonary infection model in which the typical signs of pneumonia were observed after inoculating mice i.n. with conditionally virulent organisms admixed with a small amount of iron. The amount of iron, the route of inoculation and the volume of the inoculum were found to be critical to the robustness of our model. Smaller amounts of iron proved to be sufficient in our studies, most likely because it was given admixed with the bacteria through the same (i.n.) route. In previously published studies, the iron was administered i.p., and the levels that reached the lung may have been insufficient for a productive pulmonary infection [41;42]. Iron homeostasis is finely regulated to protect the host from infection. Host innate immune mechanisms retain iron within macrophages and reduce free iron available to invading pathogens. However, high levels of intracellular iron exacerbate disease susceptibility and reduce cell-mediated immune responses to infection [45]. Iron overload has been shown to reduce macrophage activity and pro-inflammatory responses (e.g., antagonizing IFN-γ-mediated expression of TNF-α, MHCII and iNOS), thus impairing the killing of intracellular pathogens [46]. Hemochromatosis, a genetic disorder that results in excess of iron, has been associated with deficient recruitment of pulmonary neutrophils [47] and increased susceptibility to respiratory Y. pestis infection both in mice [40] and in humans [39]. In addition to restoring the capacity of the bacteria to infect and replicate within the host cells, the iron administered in our models may enhance infection by interfering with the innate immune defenses, preventing bacterial clearance through phagocytic cells. Lung neutrophils play a key role in the early control of pneumonic plague [48], and the iron supplementation likely facilitated infection by impairing neutrophil-mediated killing. Additionally, the lung tissue directly exposed to iron in our pulmonary challenge displayed signs of chemical injury, which most likely further compromised immune cell function and enhanced infection.
To obtain additional and more stringent protective efficacy data, we also established a model of systemic plague infection. As expected, lethality was achieved with a much smaller number of organisms (1×103 CFU for 8-week-old mice) when injected into the bloodstream, as opposed to administration through the airways (3×107 CFU for the same age), where bacteria would encounter multiple physical and immunological barriers [49]. The in vivo distribution of organisms and tissue damage following i.n. and i.v. infection with the Y. pestis EV76 strain was similar to that described for virulent plague strains [36]. Even though the organisms did not persist in the lungs as described for virulent strains such as Y. pestis CO92 [35;50], they spread and eventually reached the spleen where we observed multiple foci that presumably led to necrosis and breakdown of tissue structure as described for CO92 [34;51;52]. The bacteria also localized in the liver and generated microabscesses with distinct signs of necrosis in the absence of an inflammatory response.
Interestingly, the pneumonic infection was more severe in younger mice, which succumbed to doses ~ 10 times lower compared to those given to older mice (106 vs. 107 CFU). Only a hint of increased susceptibility was observed in the younger mice when the organisms were given i.v. (20 to 70 CFU). These findings were corroborated when the lethal doses required for i.n. and i.v. infection were normalized to body weight at the different ages. Establishing age-dependent lethal dose values was important so that the models could be applied to the study of vaccines that could be effective at different life stages.
We demonstrated the usefulness of our plague infection models for assessing vaccine potency by testing two Salmonella-based plague vaccine candidates expressing F1 and LcrV in mice immunized i.n. as we previously described [13]. Live vectors are regarded as promising vaccine candidates because they can prime robust mucosal and systemic immunity using a practical route of immunization (orally for humans). Other groups have successfully reported the use of Salmonella expressing Y. pestis antigens as potential plague vaccines [reviewed in [7]]. For the most part, these studies examined immune responses and protection after multiple immunizations via the orogastric or intranasal route. We envision using our vaccines as mucosal priming agents to stimulate an immune response (potentially through a single-dose) that could be rapidly expanded in levels and quality when followed by subunit vaccine candidates in “prime-boost” combinations [13]. Interestingly, Sal-F1 and Sal-LcrV exhibited different protective capacity depending on the route of infection and the amount of virulent organisms used for challenge. Although both vaccines induced significant levels of serum IgG specific for the encoded antigen, only Sal-F1 had a protective effect in adult mice, which was observed against systemic but not pulmonary infection. Protection against systemic vs. pulmonary infection involves different immunological mechanisms and effectors that may or may not be induced, depending on the nature and characteristics of the vaccine. The lack of protection in the respiratory challenge likely reflects insufficient immunity within the lungs and airways to block the overwhelming infectious dose administered. The challenge dose may also account for the differences observed. Although lethal doses were used in each model, they may not have represented the same severity of challenge; it is difficult to match the stringency of infection when using different routes (and a supplemental nutrient). Nonetheless, these results highlight the importance of using infection models that employ different routes of infection to thoroughly assess the potency and overall performance of candidate vaccines.
Results from the adult mouse studies also indicated that F1 appears to be a dominant virulent factor in our challenge model. Although F1 and LcrV were both expressed by the challenge strain, F1-specific antibodies alone afforded full protection.
Interestingly, the immunogenicity and protection outcomes were different when the same vaccines were administered to newborns. They fully protected them against systemic infection and partially protected them against pulmonary infection (Sal-F1 showing the highest survival rates). Despite eliciting higher levels of antigen-specific serum IgG than Sal-F1, Sal-LcrV failed to elicit a mucosal response. In contrast, high IgA responses were produced by the Sal-F1-immunized newborns.
As with adult mice, all naïve mice in the newborn study succumbed to the Y. pestis EV76 challenge, regardless of the route of infection used. We observed, however, that half of the mice immunized as newborns with Salmonella alone and challenged i.v. were protected in a non-specific manner, most likely through innate immunity or some vector-induced response. Similar findings were reported by others using Salmonella Typhimurium, alone or as a carrier for LcrV, when examined for protection against Y. enterocolitica or Y. pseudotuberculosis [53]. A higher challenge dose could have increased the death rate in this control group, as was observed in adults when challenged with a much larger number of organisms (1.5×108 vs. 6.2×106 CFU). Nevertheless, the agreement between survival, health scores and body weight loss argues in favor of a real protective effect of our neonatal vaccination against systemic disease, although regrettably, it did not reach statistical significance. In a previous study from our group, we successfully used the systemic EV76 infection model to demonstrate LcrV-mediated protection in mice immunized as newborns with a probiotic-based mucosal vaccine [14].
Unlike in the adults, it was possible to discriminate potency of our vaccines when we used the respiratory challenge in mice immunized as newborns. Notably, 60% of the newborns immunized with Sal-F1 were protected, which is in agreement with the efficacy reported for other F1 recombinant vaccines in adults [54;55]. The partial protection despite substantial systemic and mucosal immunity suggests that other immunological effectors and/or immune responses to other bacterial components (e.g., YadC and Yops) might be required [33;56]. Another possibility is that responses of a higher magnitude might be required to protect against the large number of organisms used in the pulmonary challenge [50;57;58]. Newborns were vaccinated on day 7 and 14 after birth to capture immune responses early in life [13]. They were also challenged slightly earlier (4-5 weeks after the boost vs. 6 weeks for adults) to ascertain protective status at a still young age (i.e., 7-8 weeks). These differences are unlikely to dramatically affect the vaccine protective outcome but should be taken into consideration when comparing vaccine performance in both age groups.
A limitation of our studies is the lack of side-by-side comparison of vaccine protective capacity using the EV76 mutant vs. fully virulent strains. The models described herein, however, are not meant to replace but to complement fully virulent challenges by prioritizing potential lead vaccine candidates for further assessment. Conceivably, the use of these models, which includes both routes of infection, offers a comprehensive tool to rapidly and safely evaluate candidate vaccines and distinguish those that appear promising for further potency testing under BSL-3 conditions.
Supplementary Material
Supplementary Figure. Optimization of the pulmonary challenge model. (A) Survival curves of mice (5 per group) infected intranasally (i.n.) with increasing numbers of Y. pestis EV76 (inoculum volume 20μl). Iron was administered i.p. (40 μg FeCl2). A control group received the bacterium alone (3×105 CFU). Mice were monitored for 14 days as described in the Materials and Methods. (B) Survival curves in mice infected i.n. with 1×107 CFU of Y. pestis EV76 admixed with 80 μg FeCl2 in 40 μl of saline solution. Control groups received the bacteria or iron alone. (C) Effect of iron on health status. Mice (3 per group) were given increasing amounts of iron i.n. in 40 μl of saline. The data represent the mean piloerection scores. All animals showed normal activity, hydration and posture. (D) Survival curves with different levels of iron supplementation. Mice (3 per group) were inoculated i.n. with 1×107 CFU of Y. pestis EV76 admixed with increasing amounts of iron (FeCl2) in a total volume of 40 μl. Control groups received the bacteria or iron alone. (E) Preliminary experiments to determine MLD50 values in mice (5 per group) infected i.n. with Y. pestis EV76 admixed with 40 μg FeCl2 in 40 μl of saline solution.
Acknowledgments
The authors acknowledge Dr. Scott A. Lloyd for the construction of plasmids, expertise and critical discussions and Mr. Cesar Paz, Ms. Kitty Davis and personnel from the Applied Immunology Section for exceptional technical assistance. G.M. received a post-doctoral fellowship (2008-2009) from the National Council of Science and Technology, México. This work was supported, in part, by National Institute of Health grants U19-AI-56578 (to J.P.N.), U01-AI077911 (to J.P.N. and J.E.G.) and R01-AI065760 (to M.F.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Poland JD, Barnes AM. Plague. In: Steele J, editor. Handbook of Zoonoses. CRC Press; Boca Raton, FL: 1979. pp. 515–59. [Google Scholar]
- [2].Pohanka M, Skladal P. Bacillus anthracis, Francisella tularensis and Yersinia pestis. The most important bacterial warfare agents - review. Folia Microbiol (Praha) 2009;54(4):263–72. doi: 10.1007/s12223-009-0046-1. [DOI] [PubMed] [Google Scholar]
- [3].Riedel S. Plague: from natural disease to bioterrorism. Proc (Bayl Univ Med Cent ) 2005 Apr;18(2):116–24. doi: 10.1080/08998280.2005.11928049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Inglesby TV, Dennis DT, Henderson DA, et al. Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 2000 May 3;283(17):2281–90. doi: 10.1001/jama.283.17.2281. [DOI] [PubMed] [Google Scholar]
- [5].Centers for Disease Control and Prevention (CDC) Emergency preparedness and response. 2012 http://www.bt.cdc.gov/agent/agentlist-category.asp.
- [6].Centers for Disease Control and Prevention (CDC) Possession, use, and transfer of select agents and toxins. Final rule. Fed Regist. 2008 Oct 16;73(201):61363–6. [PubMed] [Google Scholar]
- [7].Sun W, Roland KL, Curtiss R., III Developing live vaccines against plague. J Infect Dev Ctries. 2011 Sep;5(9):614–27. doi: 10.3855/jidc.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Smiley ST. Current challenges in the development of vaccines for pneumonic plague. Expert Rev Vaccines. 2008 Mar;7(2):209–21. doi: 10.1586/14760584.7.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rosenzweig JA, Jejelowo O, Sha J, et al. Progress on plague vaccine development. Appl Microbiol Biotechnol. 2011 Jul;91(2):265–86. doi: 10.1007/s00253-011-3380-6. [DOI] [PubMed] [Google Scholar]
- [10].Williamson ED, Flick-Smith HC, Lebutt C, et al. Human immune response to a plague vaccine comprising recombinant F1 and V antigens. Infect Immun. 2005 Jun;73(6):3598–608. doi: 10.1128/IAI.73.6.3598-3608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hart MK, Saviolakis GA, Welkos SL, House RV. Advanced Development of the rF1V and rBV A/B Vaccines: Progress and Challenges. Adv Prev Med. 2012;2012:731604. doi: 10.1155/2012/731604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Centers for Disease Control and Prevention (CDC) Biosafety in microbiological and biomedical laboratories. 2009 http://www.cdc.gov/biosafety/publications/bmbl5/bmbl.pdf, HHS Publication No. (CDC)21-112.
- [13].Ramirez K, Capozzo AV, Lloyd SA, Sztein MB, Nataro JP, Pasetti MF. Mucosally delivered Salmonella Typhi expressing the Yersinia pestis F1 antigen elicits mucosal and systemic immunity early in life and primes the neonatal immune system for a vigorous anamnestic response to parenteral F1 boost. J Immunol. 2009 Jan 15;182(2):1211–22. doi: 10.4049/jimmunol.182.2.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramirez K, Ditamo Y, Rodriguez L, et al. Neonatal mucosal immunization with a non-living, non-genetically modified Lactococcus lactis vaccine carrier induces systemic and local Th1-type immunity and protects against lethal bacterial infection. Mucosal Immunol. 2010 Mar;3(2):159–71. doi: 10.1038/mi.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Okan NA, Mena P, Benach JL, Bliska JB, Karzai AW. The smpB-ssrA mutant of Yersinia pestis functions as a live attenuated vaccine to protect mice against pulmonary plague infection. Infect Immun. 2010 Mar;78(3):1284–93. doi: 10.1128/IAI.00976-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Congleton YH, Wulff CR, Kerschen EJ, Straley SC. Mice naturally resistant to Yersinia Accepted Manuscript pestis Delta pgm strains commonly used in pathogenicity studies. Infect Immun. 2006 Nov;74(11):6501–4. doi: 10.1128/IAI.00597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Welkos S, Pitt ML, Martinez M, Friedlander A, Vogel P, Tammariello R. Determination of the virulence of the pigmentation-deficient and pigmentation-/plasminogen activator-deficient strains of Yersinia pestis in non-human primate and mouse models of pneumonic plague. Vaccine. 2002 May 22;20(17-18):2206–14. doi: 10.1016/s0264-410x(02)00119-6. [DOI] [PubMed] [Google Scholar]
- [18].Iteman I, Guiyoule A, de Almeida AM, Guilvout I, Baranton G, Carniel E. Relationship between loss of pigmentation and deletion of the chromosomal iron-regulated irp2 gene in Yersinia pestis: evidence for separate but related events. Infect Immun. 1993 Jun;61(6):2717–22. doi: 10.1128/iai.61.6.2717-2722.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou D, Qin L, Han Y, et al. Global analysis of iron assimilation and fur regulation in Yersinia pestis. FEMS Microbiol Lett. 2006 May;258(1):9–17. doi: 10.1111/j.1574-6968.2006.00208.x. [DOI] [PubMed] [Google Scholar]
- [20].Burrows TW, Jackson S. The virulence-enhancing effect of iron on nonpigmented mutants of virulent strains of Pasteurella pestis. Br J Exp Pathol. 1956 Dec;37(6):577–83. [PMC free article] [PubMed] [Google Scholar]
- [21].Bearden SW, Perry RD. The Yfe system of Yersinia pestis transports iron and manganese and is required for full virulence of plague. Mol Microbiol. 1999 Apr;32(2):403–14. doi: 10.1046/j.1365-2958.1999.01360.x. [DOI] [PubMed] [Google Scholar]
- [22].Meyer KF. Effectiveness of live or killed plague vaccines in man. Bull World Health Organ. 1970;42(5):653–66. [PMC free article] [PubMed] [Google Scholar]
- [23].Titball RW, Williamson ED. Vaccination against bubonic and pneumonic plague. Vaccine. 2001 Jul 20;19(30):4175–84. doi: 10.1016/s0264-410x(01)00163-3. [DOI] [PubMed] [Google Scholar]
- [24].Brubaker RR, Beesley ED, Surgalla MJ. Pasteurella pestis: Role of Pesticin I and Iron in Experimental Plague. Science. 1965 Jul 23;149(3682):422–4. doi: 10.1126/science.149.3682.422. [DOI] [PubMed] [Google Scholar]
- [25].Schiemann DA. Synthesis of a selective agar medium for Yersinia enterocolitica. Can J Microbiol. 1979 Nov;25(11):1298–304. doi: 10.1139/m79-205. [DOI] [PubMed] [Google Scholar]
- [26].Bergman T, Hakansson S, Forsberg A, et al. Analysis of the V antigen lcrGVH-yopBD operon of Yersinia pseudotuberculosis: evidence for a regulatory role of LcrH and LcrV. J Bacteriol. 1991 Mar;173(5):1607–16. doi: 10.1128/jb.173.5.1607-1616.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Galen JE, Zhao L, Chinchilla M, et al. Adaptation of the endogenous Salmonella enterica serovar Typhi clyA-encoded hemolysin for antigen export enhances the immunogenicity of anthrax protective antigen domain 4 expressed by the attenuated live-vector vaccine strain CVD 908-htrA. Infect Immun. 2004 Dec;72(12):7096–106. doi: 10.1128/IAI.72.12.7096-7106.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tacket CO, Sztein MB, Losonsky GA, et al. Safety of live oral Salmonella Typhi vaccine strains with deletions in htrA and aroC aroD and immune response in humans. Infect Immun. 1997;65(2):452–6. doi: 10.1128/iai.65.2.452-456.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Second ed Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- [30].Capozzo AV, Ramirez K, Polo JM, et al. Neonatal immunization with a Sindbis virus-DNA measles vaccine induces adult-like neutralizing antibodies and cell-mediated immunity in Accepted Manuscript the presence of maternal antibodies. J Immunol. 2006 May 1;176(9):5671–81. doi: 10.4049/jimmunol.176.9.5671. [DOI] [PubMed] [Google Scholar]
- [31].Kurono Y, Yamamoto M, Fujihashi K, et al. Nasal immunization induces Haemophilus influenzae-specific Th1 and Th2 responses with mucosal IgA and systemic IgG antibodies for protective immunity. J Infect Dis. 1999 Jul;180(1):122–32. doi: 10.1086/314827. [DOI] [PubMed] [Google Scholar]
- [32].Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. The American Journal of Hygiene. 1938;27:493–7. [Google Scholar]
- [33].Ivanov MI, Noel BL, Rampersaud R, Mena P, Benach JL, Bliska JB. Vaccination of mice with a Yop translocon complex elicits antibodies that are protective against infection with F1- Yersinia pestis. Infect Immun. 2008 Nov;76(11):5181–90. doi: 10.1128/IAI.00189-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bubeck SS, Cantwell AM, Dube PH. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect Immun. 2007 Feb;75(2):697–705. doi: 10.1128/IAI.00403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lathem WW, Crosby SD, Miller VL, Goldman WE. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A. 2005 Dec 6;102(49):17786–91. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Agar SL, Sha J, Foltz SM, et al. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology. 2008 Jul;154(Pt 7):1939–48. doi: 10.1099/mic.0.2008/017335-0. [DOI] [PubMed] [Google Scholar]
- [37].Zilinskas RA. The anti-plague system and the Soviet biological warfare program. Crit Rev Microbiol. 2006;32(1):47–64. doi: 10.1080/10408410500496896. [DOI] [PubMed] [Google Scholar]
- [38].Geissler C, Singh M. Iron, meat and health. Nutrients. 2011 Mar;3(3):283–316. doi: 10.3390/nu3030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].CDC Fatal Laboratory-Acquired Infection with an Attenuated Yersinia pestis Strain --- Chicago, Illinois, 2009. MMWR, Morb Mortal Wkly Rep. 2011 Feb;2560:201–205. Available from: URL: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6007a1.htm. [PubMed] [Google Scholar]
- [40].Quenee LE, Hermanas TM, Ciletti N, et al. Hereditary hemochromatosis restores the virulence of plague vaccine strains. J Infect Dis. 2012 Oct;206(7):1050–8. doi: 10.1093/infdis/jis433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Galvan EM, Nair MK, Chen H, Del PF, Schifferli DM. Biosafety level 2 model of pneumonic plague and protection studies with F1 and Psa. Infect Immun. 2010 Aug;78(8):3443–53. doi: 10.1128/IAI.00382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee-Lewis H, Anderson DM. Absence of inflammation and pneumonia during infection with nonpigmented Yersinia pestis reveals a new role for the pgm locus in pathogenesis. Infect Immun. 2010 Jan;78(1):220–30. doi: 10.1128/IAI.00559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Williamson ED. Plague. Vaccine. 2009 Nov 5;27(Suppl 4):D56–D60. doi: 10.1016/j.vaccine.2009.07.068. [DOI] [PubMed] [Google Scholar]
- [44].Zager RA, Johnson AC, Hanson SY, Wasse H. Parenteral iron formulations: a comparative toxicologic analysis and mechanisms of cell injury 1. Am J Kidney Dis. 2002 Jul;40(1):90–103. doi: 10.1053/ajkd.2002.33917. [DOI] [PubMed] [Google Scholar]
- [45].Wessling-Resnick M. Iron homeostasis and the inflammatory response. Annu Rev Nutr. 2010 Aug 21;30:105–22. doi: 10.1146/annurev.nutr.012809.104804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol. 2010 Dec;12(12):1691–702. doi: 10.1111/j.1462-5822.2010.01529.x. [DOI] [PubMed] [Google Scholar]
- [47].Benesova K, Vujic SM, Schaefer SM, et al. Hfe deficiency impairs pulmonary neutrophil recruitment in response to inflammation. PLoS ONE. 2012;7(6):e39363. doi: 10.1371/journal.pone.0039363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Laws TR, Davey MS, Titball RW, Lukaszewski R. Neutrophils are important in early control of lung infection by Yersinia pestis. Microbes Infect. 2010 Apr;12(4):331–5. doi: 10.1016/j.micinf.2010.01.007. [DOI] [PubMed] [Google Scholar]
- [49].Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. 2000 Feb;173:39–51. doi: 10.1034/j.1600-065x.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- [50].Anderson DM, Ciletti NA, Lee-Lewis H, et al. Pneumonic plague pathogenesis and immunity in Brown Norway rats. Am J Pathol. 2009 Mar;174(3):910–21. doi: 10.2353/ajpath.2009.071168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sebbane F, Gardner D, Long D, Gowen BB, Hinnebusch BJ. Kinetics of disease progression and host response in a rat model of bubonic plague. Am J Pathol. 2005 May;166(5):1427–39. doi: 10.1016/S0002-9440(10)62360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sha J, Agar SL, Baze WB, et al. Braun lipoprotein (Lpp) contributes to virulence of yersiniae: potential role of Lpp in inducing bubonic and pneumonic plague. Infect Immun. 2008 Apr;76(4):1390–409. doi: 10.1128/IAI.01529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Branger CG, Torres-Escobar A, Sun W, et al. Oral vaccination with LcrV from Yersinia pestis KIM delivered by live attenuated Salmonella enterica serovar Typhimurium elicits a protective immune response against challenge with Yersinia pseudotuberculosis and Yersinia enterocolitica. Vaccine. 2009 Aug 27;27(39):5363–70. doi: 10.1016/j.vaccine.2009.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Heath DG, Anderson GW, Jr., Mauro JM, et al. Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine. 1998 Jul;16(11-12):1131–7. doi: 10.1016/s0264-410x(98)80110-2. [DOI] [PubMed] [Google Scholar]
- [55].Quenee LE, Cornelius CA, Ciletti NA, Elli D, Schneewind O. Yersinia pestis caf1 variants and the limits of plague vaccine protection. Infect Immun. 2008 May;76(5):2025–36. doi: 10.1128/IAI.00105-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Murphy BS, Wulff CR, Garvy BA, Straley SC. Yersinia pestis YadC: a novel vaccine candidate against plague. Adv Exp Med Biol. 2007;603:400–14. doi: 10.1007/978-0-387-72124-8_37. [DOI] [PubMed] [Google Scholar]
- [57].Zauberman A, Cohen S, Levy Y, et al. Neutralization of Yersinia pestis-mediated macrophage cytotoxicity by anti-LcrV antibodies and its correlation with protective immunity in a mouse model of bubonic plague. Vaccine. 2008 Mar 20;26(13):1616–25. doi: 10.1016/j.vaccine.2008.01.033. [DOI] [PubMed] [Google Scholar]
- [58].Vernazza C, Lingard B, Flick-Smith HC, Baillie LW, Hill J, Atkins HS. Small protective fragments of the Yersinia pestis V antigen. Vaccine. 2009 May 11;27(21):2775–80. doi: 10.1016/j.vaccine.2009.03.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. Optimization of the pulmonary challenge model. (A) Survival curves of mice (5 per group) infected intranasally (i.n.) with increasing numbers of Y. pestis EV76 (inoculum volume 20μl). Iron was administered i.p. (40 μg FeCl2). A control group received the bacterium alone (3×105 CFU). Mice were monitored for 14 days as described in the Materials and Methods. (B) Survival curves in mice infected i.n. with 1×107 CFU of Y. pestis EV76 admixed with 80 μg FeCl2 in 40 μl of saline solution. Control groups received the bacteria or iron alone. (C) Effect of iron on health status. Mice (3 per group) were given increasing amounts of iron i.n. in 40 μl of saline. The data represent the mean piloerection scores. All animals showed normal activity, hydration and posture. (D) Survival curves with different levels of iron supplementation. Mice (3 per group) were inoculated i.n. with 1×107 CFU of Y. pestis EV76 admixed with increasing amounts of iron (FeCl2) in a total volume of 40 μl. Control groups received the bacteria or iron alone. (E) Preliminary experiments to determine MLD50 values in mice (5 per group) infected i.n. with Y. pestis EV76 admixed with 40 μg FeCl2 in 40 μl of saline solution.