

INTRODUCTION
Science is a game of successive approximations. Our current state of “understanding” is transient, and dogmas almost always require revision and/or refinement. Certainly, much of the underlying physics and chemistry of molecular recognition have been routinely minimized in order to force problems of atomic interactions within our paradigm/computer. Oversimplification is especially dangerous when the systems under study are too complex to easily validate experimentally. Unfortunately, biological systems are both complex and important, and much effort is spent to rationalize their known behavior at low resolution with inadequate methodology at atomic resolution. A case in point is the common use of force fields utilizing monopole electrostatics. Special-purpose computers have been constructed to tackle the complexity of biological processes such as protein folding at the molecular level [2,3]. Three recent examples of MD simulations of complex systems (Src kinase, Abl kinase, and beta(1)/beta(2)-adrenergic receptors) illustrate this approach [4–6]. While these long simulations provide results that were interpreted as consistent with the limited experimental observations available, the complexity of the systems under study preclude any significant validation of the details of molecular dynamics during the simulation. Nevertheless, insight into protein folding has been derived by long MD simulations using a specialized supercomputer [11]. Obviously, the computational methodology used must be validated on well-chosen experimental systems before lending any credibility to the details of the MD results on more complex systems. Unfortunately, most MD simulations of complex biological systems, including those by the special-purpose supercomputer Anton [11], have incorporated monopole electrostatics without polarizability in the force fields used (AMBER, CHARMM, OPLS, etc.) that limits their accuracy.
One reason we do molecular modeling and simulations is to gain access to molecular events that are difficult to observe experimentally. These approaches provide a way to extrapolate between experimental observations. In order to simulate molecular recognition and intermolecular interactions, one simplifies the underlying physics and chemistry due to their inherent complexity. The question one must face is whether the introduced simplifications produce results with adequate resolution for the problem being studied. Obviously, adequate resolution means the ability to distinguish between alternative hypotheses; unfortunately, structure-based drug design requires accurate results if one is to predict binding affinities due to the frustrated potential surface. On the other hand, many of the observed properties of folded proteins can be correlated with simplified lattice models as demonstrated by Dill and co-workers [12,13]. The parameterization of CHARMM [14–16], OPLS-AA [17] and AMBER [18] and the monopole water models demonstrated that many intensive and colligative molecular properties are adequately modeled with monopole electrostatics, including solvation free energies [19]. To reproduce the dynamical behavior of molecules, however, would appear to require more accurate force fields.
Monopole versus Multipole Electrostatics
Molecular mechanics attempts to represent intermolecular interactions in terms of classical physics. Initial efforts assumed a point charge located at the atom center and coulombic interactions. It has been recognized over multiple decades that simply representing electrostatics with a charge on each atom failed to reproduce the electrostatic potential surrounding a molecule as estimated by quantum mechanics. Molecular orbitals are not spherically symmetrical, an implicit assumption of monopole electrostatics. Despite the recognition of its inadequacies [20,21] and efforts to overcome them by the Darden group, [22–26] and others in the modeling community [27–32], the more computationally efficient monopole approximation is still used in order to model more complex systems of biological interest [33,34]; on detailed analysis when there is robust experimental characterization of the system, however, one finds that the computational results do not accurately predict the observed experimental data.
To illustrate the error associated with the monopole approximation, the case of water published in 1988 is shown (Fig. 1). An RMS error of more than 8% for the electrostatic potential sampled at 363 grid points surrounding the water molecule was the best possible fit of a monopole model to the quantum calculation. Addition of a dipole moment reduced the error to 1% and further addition of a quadrupole moment reduced the error to less than 0.1%. Consider the interactions of two waters, each with an error of 8% in their electrostatic fields; unfortunately, such errors do not cancel and lead to significant deviations from the correct geometry of interaction. The inability of monopole electrostatics to reproduce the experimentally determined geometry of water clusters has been shown [35]. One might assume that implicit solvent models would have overcome this limitation of explicit monopole models of water; unfortunately, they have been calibrated primarily with results from explicit calculations with monopole force fields. One can understand the rise in popularity of statistically based potentials derived from experimental atomic proximities that inherently avoid energetic dichotomies and focus on free energy per se [36,37].
Figure 1.

Comparison of best fits of monopole, dipole and quadrupole models to electrostatic potentials calculated by quantum mechanics. Modified from D. E. Williams [1].
Another illustrative example (Fig. 2) was published by Prof. Anthony Stone in his article on intermolecular potentials in Science [8]. This graphical example of the errors in electrostatic potential with the monopole approximation, and its attenuation by inclusion of higher multipoles is compelling. Williams [1,38], Hunter [39–42], Stone [8,43], Price [43,44] and others showed that reproduction of the electrostatic potential required a more complex representation of electrostatics, including dipole and quadrupole (simply four alternating charges at the corners of a parallelogram) moments as well as monopoles. The XED (extended electron distribution) force field developed by Vinter [45] also recognized the limitation of monopole force fields, and was the first to move toward a second-generation force field that reproduced aromatic interactions [46] and other complex interactions, such as cation-pi [47], much better [48]. This led to a relevant method for comparing molecules [49] based on the extrema (Fig. 3) in their electrostatic potentials [9,50]. In many ways, this approach is philosophically similar to the field comparisons available from CoMFA [51] and GRID [52] leading to subsequent approaches such as COMBINE [53,54] and COMBINEr [55,56].
Figure 2.
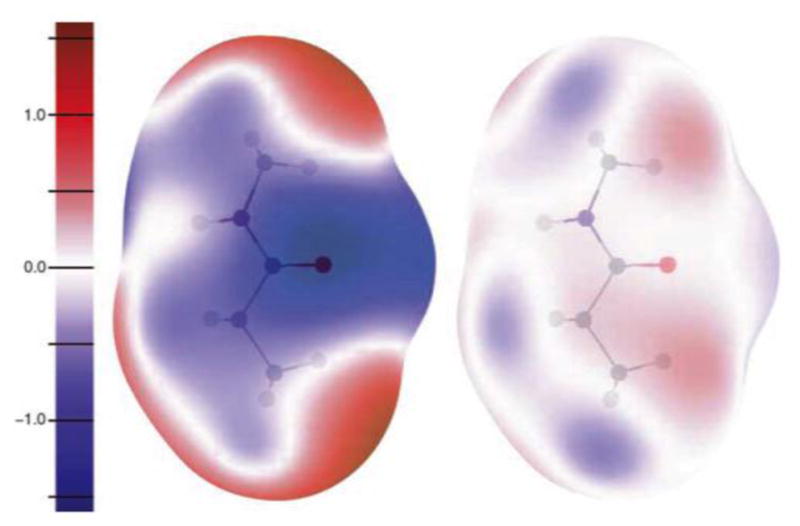
Errors (V) vs. QM for electrostatic potential on a surface at 1.8 times van der Waals radii around N-methyl propanamide for two models. (Left) Point charges; (right) point charge, dipole, and quadrupole on C, N, and O; charge and dipole on H. The errors are much reduced by inclusion of higher multipoles [8].
Figure 3.

Examples of electrostatic extrema used for molecular comparisons [9].
In a recent analysis of helices in the Protein Data Base, Kuster et al. found that the classical view of a- and 310-helices disappeared as one examined high-resolution crystallographic data that was able to generate protein models without constraints from monopole-based force fields (Kuster et al., unpublished). Instead of a bifurcated distribution between α- and 310-helical torsion angles, a smooth, single-minimum distribution was found with intermediate backbone torsional angles. The helical parameters remained essentially identical with the a-helix due to a crankshaft-like motion of the amide bonds to support three-membered backbone hydrogen bonding. A major assumption by Pauling, Donohue, etc. was linear hydrogen bonds between amide groups in protein helices; this has been reinforced by model building programs that use monopole electrostatics as the minimum energy orientation of two interacting dipoles is linear. This association of monopole electrostatics with linear hydrogen bonding is not new. Halgren and Damm pointed out in 2001 in their seminal review of polarizable force fields [57] that “… that a proper account of hydrogen-bond directionality and, in cases like those examined here, hydrogen-bond energetics requires a representation of the permanent charge distribution that goes beyond the simple framework of atom-centered charges used in traditional force fields.” A more recent example, Morozov et al. [58] stated in 2004 that “Current molecular mechanics force fields widely used in biomolecular simulations essentially model hydrogen bonding as a purely electrostatic interaction: positive partial charges are placed on the proton and the acceptor base and negative partial charges, on the acceptor and donor atoms … The hydrogen bond modeled in this way is dominated by dipole–dipole interaction and the energy of two dipoles is at a minimum when all four atoms are collinear.”
Electrostatic Anisotropy and Polarizability - To adequately represent the interaction and orientation between a carbonyl oxygen and an amide hydrogen, multipole electrostatics are essential. Furthermore, non-bonded interatomic interactions require polarizability to describe mutually induced charge perturbations [57]. One must distinguish, however, between electrostatic anisotropy requiring multipoles and polarizability that are different physical interactions. In particular, aromatic/aromatic and charge/aromatic interactions require more sophisticated electrostatics than found in the force fields (AMBER, CHARMM, OPLS, etc.) in common usage. Truchon et al. have shown that aromatic/charge interactions are dominated by polarization, and can be approximated by an internal continuum model that does not include multipoles per se [59].
Fortunately, Ponder recognized the limitations of monopole force fields over a decade ago [60] and started the development of AMOEBA, a second-generation force field based on multipole electrostatics that includes polarizability [61–63]. AMOEBA is freely available online as part of the TINKER package [64]. Recent papers attest to the ability of AMOEBA to reproduce experimental thermodynamics [65,66]. Methodology for deriving parameters for AMOEBA to allow incorporation of novel ligands has recently been published [67]. The intrinsic improvements associated with AMOEBA calculations have prompted others to incorporate it in model building from experimental electron density [27,68]. A comparison of ligand binding of benzamidine-like inhibitors of trypsin used both explicit and implicit solvation with a polarizable force field [69]. The binding free energies calculated from explicit-solvent simulations were well within the accuracy of experimental measurements.
The opportunity to compare the ability of AMOEBA to reproduce the dynamics of intermolecular interactions with monopole force fields presented itself with the NMR experimental studies of Rieman and Waters [10] on a series of four -hairpin peptides (Fig. 4). Stabilization of the hairpins occurred through cation/pi and aromatic/aromatic interactions between two tryptophan residues and a lysine -amino group with variations in the degree of methylation of the -ammonium group. The biological relevance of methylated lysine arises from its role in epigenetic control of gene expression [70], and from the homology with acetylcholine that traverses a deep gorge lined by aromatic residues to reach the active site of acetylcholinesterase [71]. Long molecular dynamics simulations (100 ns) of model peptides, Ac-R-W-V-W-V-N-G-Orn-K(Me)n -I-L-Q-NH2, where n = 0, 1, 2, or 3, were conducted in explicit solvent with AMBER, CHARMM, OPLS and AMOEBA to determine the ability to predict the experimentally observed NOE patterns that differed depending on the degree of methylation [7]. AMOEBA was able to predict over 80% of the observed NOEs from the MD simulation (Fig. 5); the three monopole force fields did not predict the same NOEs for any of the four peptides emphasizing the differences in their internal parameterizations (for an example of optimization efforts of monopole force fields to fit experimental data, See Macias and Mackerrell [72] and Mackerrell et al. [73]). While the summary of the agreement between the NOEs predicted by the MD simulations with AMOEBA are impressive (Fig. 5) and clearly demonstrate the necessity for more complex electrostatics, the lack of full agreement raises a question. Is this an indication of a need for further improvement in the parameterization of AMOEBA, or simply some minor experimental error in the NMR experiments? Will accurate prediction of robust experimental data from dynamic systems require inclusion of many-body interactions as well?
Figure 4.

Diagnostic NOEs observed by Rieman and Waters [10] in the series of four peptides differing by the degree of lysine -N-methylation.
Figure 5.

Summary of the number of experimental NOEs predicted by 100 nsec MD simulations in water for the four hairpin peptides by the four force fields [7].
Implications for other studies
Validation of force fields has been done primarily by comparison with static crystal structures or by estimation of intensive properties, such as solvent density and radial distribution function that are largely dependent on potential minima. A much more significant question is the ability of a force field to reproduce the dynamics of molecular systems that requires reproduction of the potentials surrounding the minima. Biology does not occur at zero degrees Kelvin, and kinetic energy explores the potential surface beyond the minima.
AMOEBA has been shown to reproduce the geometry of water clusters where monopole force fields give linear hydrogen bonding [35]. The quantitative agreement between AMOEBA predictions and experimental measurements on water is good in general for density, heat of vaporization, radial distribution functions, magnetic shielding, self-diffusion, and static dielectric constant [74]. A new water potential DMIP based on AMOEBA has been developed to improve computational performance [75]. Kramer et al. have suggested a method by which atomic multipoles can be rigorously implemented into common biomolecular force fields using monopole electrostatics [76]. Since biology is aqueous in nature, the ability of a force field to reproduce the properties of the solvent is absolutely essential. Another example of the necessity for high resolution in force fields, the bifurcated hydrogen bonding of the amide bond in protein helices, such as crambin, seen in high-resolution structures requires multipole electrostatics to be preserved in MD simulations (Kuster et al., unpublished).
Conclusions
What often appears trivial on first evaluation becomes more difficult as the complexity of the problem is revealed in all its glory. Often the past decades, computer-aided drug design has progressed toward a more complete understanding of the complexities of molecular recognition by attempting to design ligands for protein-binding sites. While numerous success stories are found in the literature, there remain numerous failures, often unreported. Optimization by focused combinatorial chemistry would not be so common if our computational methodologies were truly predictive.
What is clear, however, is that the monopole-electrostatics approximation is inadequate for certain problems requiring accurate molecular modeling. Second-generation force fields, such as AMOEBA, that incorporate multipole electrostatic models and include polarizability are essential for reliably predicting thermodynamic observables, but require considerable effort to develop parameters for novel molecules [67]. XED (freely distributed by Cresset (http://www.cresset-group.com/) to academics) is certainly much more realistic than monopole force fields in its ability to reproduce molecular geometries of complexes at minimal computational expense. One may question whether the inclusion of electrostatic energy generated by monopole force fields is not sometimes misleading? The abundance of aromatic/aromatic [77–79] and aromatic/charge [80,81] interactions in biological systems makes this issue problematic. It clearly remains to be shown for the problem under consideration that the energies calculated including monopole interactions are relevant; certainly, the numerous efforts to modulate electrostatic interactions with distance-dependent dielectrics, for example, question the utility of monopole force fields. Historically, I first encountered this problem when attempting to reproduce minimization results obtained with zwitterionic amino-acid crystals [82]. Regardless of the constant dielectric we tried, the crystals would either expand or contract; a dielectric function was required to reproduce the experimental data. On reflection, lack of polarizability was the probable culprit. The prior results we were trying to reproduce had solved the problem by simply fixing the dimensions of the unit cell of the crystals (reference omitted on purpose). The ability of second-generation force fields to reproduce crystal structures of such systems characterized by high charge density remains to be shown.
Nevertheless, absolute truth is not essential in setting priorities in drug discovery. Often, the essential component in the competitive pharmaceutical world is speed. Despite the evidence that multipole electrostatics and polarizability are essential for accurate predictions, there is a price of increased complexity of computation (approximately 10-fold) to be paid. Force fields with monopole electrostatics can be useful in exploring a problem to determine where a more sophisticated approach is warranted. Certainly, molecular modeling provides a useful framework for hypothesis generation, regardless of the level of atomic resolution. The seductive models produced by modeling, however, must be subjected to validation by prediction and experimental tests. Predictive calculations of affinities, however, require both an accurate force field and exploration of the entropy of binding [83] – still a daunting task.
Nevertheless, it is difficult to ignore the obvious. When we limit the physical basis of molecular recognition to those we can conveniently incorporate into computational structure-based design, then we can expect to have success only when our limiting assumptions are compatible with the system under study. Unfortunately, electrostatics plays an essential role in molecular recognition, in the dynamics of protein folding, and in protein/ligand interactions. Until molecular modeling routinely includes multipole electrostatics and polarizability through the use of more sophisticated, second-generation force fields such as AMOEBA, we must anticipate significant errors in predictions to occur. Results from MD simulations using force fields with monopole electrostatics may, in fact, be adequate at a given level of resolution, but how does one judge unless the system has been validated with robust experimental data? Fortunately, access to computational power sufficient to enable application of more complex force fields, such as AMOEBA, is available by distributed computing (example = http://folding.stanford.edu, for a comparison of the Markov State Model approach with ANTON, see the article by Lane et al. [84]) and the ever increasing power of CPUs available in clustered arrays.
Acknowledgments
Many have contributed to what we have collectively attempted in computer-aided molecular design; my thanks for sharing the dream. In particular I need to thank my colleagues, Drs. Xiange Zheng, for her MD simulations comparing monopole force fields with AMOEBA, and Dan Kuster for his analysis of high-resolution protein helices. Many (too numerous to mention) have generously pointed out critical mistakes along my ultimate path to humility. This includes the referees of the first draft of this manuscript. A long association with Prof. Andy Vinter (a founding editor of JCAMD) opened my eyes to the problems with monopole electrostatics, and generated a noticeable avoidance of modeling nucleic acids and membranes on my part due to their high charge density. In particular, however, my proximity to Prof. Jay Ponder during his development of AMOEBA has taught me the necessity to swim upstream, i. e. to do what is scientifically justified without concern for the myopia of the field. Hopefully, the validation of AMOEBA has reached an acceptable level of maturity, and the molecular-modeling community can reap the benefits in its application. Discussion on the problems of representing electrostatics with Prof. Anthony Stone of Cambridge University has clarified many of the issues. My thanks also to Prof. Rino Ragno and the Sapienza Università di Roma for being my host during the preparation of this perspective. Finally, my thanks to the Editors of JCAMD for providing me this opportunity to pontificate, so appropriate considering my location in Rome.
References
- 1.Williams DE. Representation of the molecular electrostatic potential by atomic multipole and bond dipole models. J Comput Chem. 1988;9(7):745–763. [Google Scholar]
- 2.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330(6002):341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 3.Dror RO, Jensen MO, Borhani DW, Shaw DE. Exploring atomic resolution physiology on a femtosecond to millisecond timescale using molecular dynamics simulations. J Gen Physiol. 2010;135(6):555–562. doi: 10.1085/jgp.200910373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and mechanism of drug binding to g-protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(32):13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shan Y, Kim ET, Eastwood MP, Dror RO, Seeliger MA, Shaw DE. How does a drug molecule find its target binding site? J Am Chem Soc. 2011;133(24):9181–9183. doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MO, Dror RO, Kuriyan J, Shaw DE. A conserved protonation-dependent switch controls drug binding in the abl kinase. Proc Natl Acad Sci U S A. 2009;106(1):139–144. doi: 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng X, Wu C, Ponder JW, Marshall GR. Molecular dynamics of beta-hairpin models of epigenetic recognition motifs. J Am Chem Soc. 2012;134(38):15970–15978. doi: 10.1021/ja306803v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone AJ. Intermolecular potentials. Science. 2008;321(5890):787–789. doi: 10.1126/science.1158006. [DOI] [PubMed] [Google Scholar]
- 9.Apaya RP, Lucchese B, Price SL, Vinter JG. The matching of electrostatic extrema: A useful method in drug design? A study of phosphodiesterase iii inhibitors. J Comput Aided Mol Des. 1995;9(1):33–43. doi: 10.1007/BF00117276. [DOI] [PubMed] [Google Scholar]
- 10.Riemen AJ, Waters ML. Design of highly stabilized beta-hairpin peptides through cation-pi interactions of lysine and n-methyllysine with an aromatic pocket. Biochemistry. 2009;48(7):1525–1531. doi: 10.1021/bi801706k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borhani DW, Shaw DE. The future of molecular dynamics simulations in drug discovery. J Comput Aided Mol Des. 2012;26(1):15–26. doi: 10.1007/s10822-011-9517-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yue K, Fiebig KM, Thomas PD, Chan HS, Shakhnovich EI, Dill KA. A test of lattice protein folding algorithms. Proc Natl Acad Sci U S A. 1995;92(1):325–329. doi: 10.1073/pnas.92.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dill KA. Polymer principles and protein folding. Protein science: a publication of the Protein Society. 1999;8(6):1166–1180. doi: 10.1110/ps.8.6.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. Journal of computational chemistry. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas JL, Tobias DJ, Mackerell AD., Jr Direct comparisons of experimental and calculated neutron structure factors of pure solvents as a method for force field validation. The journal of physical chemistry B. 2007;111(45):12941–12944. doi: 10.1021/jp076501p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. The journal of physical chemistry B. 1998:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the opls all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. [Google Scholar]
- 18.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Bayly CI, Cooper MD, Pande VS. Predicting small-molecule solvation free energies: An informal blind test for computational chemistry. Journal of medicinal chemistry. 2008;51 (4):769–779. doi: 10.1021/jm070549+. [DOI] [PubMed] [Google Scholar]
- 20.Buckingham AD, Fowler PW. Do electrostatic interactions predict structures of van der waals molecules? The Journal of chemical physics. 1983;76:6426–6428. [Google Scholar]
- 21.Volkov A, Gatti C, Abramov Y, Coppens P. Evaluation of net atomic charges and atomic and molecular electrostatic moments through topological analysis of the experimental charge density. Acta crystallographica Section A, Foundations of crystallography. 2000;56(Pt 3):252–258. doi: 10.1107/s0108767300001628. [DOI] [PubMed] [Google Scholar]
- 22.Cisneros GA, Piquemal JP, Darden TA. Generalization of the gaussian electrostatic model: Extension to arbitrary angular momentum, distributed multipoles, and speedup with reciprocal space methods. The Journal of chemical physics. 2006;125(18):184101. doi: 10.1063/1.2363374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cisneros GA, Piquemal JP, Darden TA. Quantum mechanics/molecular mechanics electrostatic embedding with continuous and discrete functions. The journal of physical chemistry B. 2006;110(28):13682–13684. doi: 10.1021/jp062768x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cisneros GA, Piquemal JP, Darden TA. Intermolecular electrostatic energies using density fitting. The Journal of chemical physics. 2005;123(4):044109. doi: 10.1063/1.1947192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagui C, Pedersen LG, Darden TA. Towards an accurate representation of electrostatics in classical force fields: Efficient implementation of multipolar interactions in biomolecular simulations. J Chem Phys. 2004;120(1):73–87. doi: 10.1063/1.1630791. [DOI] [PubMed] [Google Scholar]
- 26.Sagui C, Darden TA. Molecular dynamics simulations of biomolecules: Long-range electrostatic effects. Annual review of biophysics and biomolecular structure. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 27.Fenn TD, Schnieders MJ, Brunger AT, Pande VS. Polarizable atomic multipole x-ray refinement: Hydration geometry and application to macromolecules. Biophysical journal. 2010;98(12):2984–2992. doi: 10.1016/j.bpj.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenn TD, Schnieders MJ, Mustyakimov M, Wu C, Langan P, Pande VS, Brunger AT. Reintroducing electrostatics into macromolecular crystallographic refinement: Application to neutron crystallography and DNA hydration. Structure. 2011;19(4):523–533. doi: 10.1016/j.str.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elking D, Darden T, Woods RJ. Gaussian induced dipole polarization model. J Comput Chem. 2007;28(7):1261–1274. doi: 10.1002/jcc.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elking DM, Perera L, Duke R, Darden T, Pedersen LG. Atomic forces for geometry-dependent point multipole and gaussian multipole models. J Comput Chem. 2010;31(15):2702–2713. doi: 10.1002/jcc.21563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elking DM, Perera L, Duke R, Darden T, Pedersen LG. A finite field method for calculating molecular polarizability tensors for arbitrary multipole rank. J Comput Chem. 2011;32(15):3283–3295. doi: 10.1002/jcc.21914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dominiak PM, Volkov A, Dominiak AP, Jarzembska KN, Coppens P. Combining crystallographic information and an aspherical-atom data bank in the evaluation of the electrostatic interaction energy in an enzyme-substrate complex: Influenza neuraminidase inhibition. Acta crystallographica Section D, Biological crystallography. 2009;65(Pt 5):485–499. doi: 10.1107/S0907444909009433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muddana HS, Gilson MK. Calculation of host-guest binding affinities using a quantum-mechanical energy model. Journal of chemical theory and computation. 2012;8(6):2023–2033. doi: 10.1021/ct3002738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muddana HS, Gilson MK. Prediction of sampl3 host-guest binding affinities: Evaluating the accuracy of generalized force-fields. Journal of computer-aided molecular design. 2012;26(5):517–525. doi: 10.1007/s10822-012-9544-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren P, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107:5933–5947. [Google Scholar]
- 36.Miyazawa S, Jernigan RL. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. Journal of molecular biology. 1996;256(3):623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 37.Zimmermann MT, Leelananda SP, Kloczkowski A, Jernigan RL. Combining statistical potentials with dynamics-based entropies improves selection from protein decoys and docking poses. J Phys Chem B. 2012;116(23):6725–6731. doi: 10.1021/jp2120143. [DOI] [PubMed] [Google Scholar]
- 38.Williams DE. Net atomic charge and multipole models for the ab initio molecular electric potential. In: Lipkowitz KB, Boyd DB, editors. Reviews in computational chemistry. Vol. 2. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2007. [Google Scholar]
- 39.Hunter CA, Sanders JKM. The nature of pi-pi interactions. J Am Chem Soc. 1990;112(14):5525–5534. [Google Scholar]
- 40.Hunter CA. Meldola lecture. The role of aromatic interactions in molecular recognition. Chem Soc Rev. 1994;23:101–109. [Google Scholar]
- 41.Hunter CA, Low CM, Rotger C, Vinter JG, Zonta C. Substituent effects on cation-pi interactions: A quantitative study. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):4873–4876. doi: 10.1073/pnas.072647899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cockroft SL, Hunter CA. Chemical double-mutant cycles: Dissecting non-covalent interactions. Chem Soc Rev. 2006;36:172–188. doi: 10.1039/b603842p. [DOI] [PubMed] [Google Scholar]
- 43.Stone AJ, Price SL. Some new ideas in the theory of inter molecular forces - anisotropic atom atom potentials. J Phys Chem. 1988;92(12):3325–3335. [Google Scholar]
- 44.Price SL, Harrison RJ, Guest MF. An ab initio distributed multipole study of the electrostatic potential around an undecapeptide cyclosporine derivative and comparison with point-charge electrostatic models. J Comput Chem. 1988;10(4):552–567. [Google Scholar]
- 45.Vinter JG. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J Comput-Aided Mol Des. 1994;8:653–668. doi: 10.1007/BF00124013. [DOI] [PubMed] [Google Scholar]
- 46.Sherrill CD. Energy component analysis of pi interactions. Accounts of chemical research. 2012 doi: 10.1021/ar3001124. [DOI] [PubMed] [Google Scholar]
- 47.Quinonero D, Garau C, Frontera A, Ballester P, Costa A, Deya PM. Structure and binding energy of anion-pi and cation-pi complexes: A comparison of mp2, ri-mp2, dft, and df-dft methods. The journal of physical chemistry A. 2005;109(20):4632–4637. doi: 10.1021/jp044616c. [DOI] [PubMed] [Google Scholar]
- 48.Chessari G, Hunter CA, Low CM, Packer MJ, Vinter JG, Zonta C. An evaluation of force-field treatments of aromatic interactions. Chemistry. 2002;8 (13):2860–2867. doi: 10.1002/1521-3765(20020703)8:13<2860::AID-CHEM2860>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 49.Cheeseright TJ, Mackey MD, Melville JL, Vinter JG. Fieldscreen: Virtual screening using molecular fields. Application to the dud data set. J Chem Inf Model. 2008;48(11):2108–2117. doi: 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- 50.Vinter JG, Trollope KI. Multiconformational composite molecular potential fields in the analysis of drug action. I. Methodology and first evaluation using 5-ht and histamine action as examples. J Comput-Aided Mol Des. 1995;9:297–307. doi: 10.1007/BF00125171. [DOI] [PubMed] [Google Scholar]
- 51.Cramer RD, 3rd, Patterson DE, Bunce JD. Recent advances in comparative molecular field analysis (comfa) Prog Clin Biol Res. 1989;291:161–165. [PubMed] [Google Scholar]
- 52.Reynolds CA, Wade RC, Goodford PJ. Identifying targets for bioreductive agents: Using grid to predict selective binding regions of proteins. J Mol Graph. 1989;7(2):103–108. 100. doi: 10.1016/s0263-7855(89)80013-x. [DOI] [PubMed] [Google Scholar]
- 53.Perez C, Pastor M, Ortiz AR, Gago F. Comparative binding energy analysis of hiv-1 protease inhibitors: incorporation of solvent effects and validation as a powerful tool in receptor-based drug design. J Med Chem. 1998;41 (6):836–852. doi: 10.1021/jm970535b. [DOI] [PubMed] [Google Scholar]
- 54.Lozano JJ, Pastor M, Cruciani G, Gaedt K, Centeno NB, Gago F, Sanz F. 3d-qsar methods on the basis of ligand-receptor complexes. Application of combine and grid/golpe methodologies to a series of cyp1a2 ligands. J Comput Aided Mol Des. 2000;14(4):341–353. doi: 10.1023/a:1008164621650. [DOI] [PubMed] [Google Scholar]
- 55.Ballante F, Musmuca I, Marshall GR, Ragno R. Comprehensive models of wild-type and mutant hiv-1 reverse transciptases. J Comp-Aided Mol Design. 2012;26(8):907–919. doi: 10.1007/s10822-012-9586-6. [DOI] [PubMed] [Google Scholar]
- 56.Silvestri L, Ballante F, Mai A, Marshall GR, Ragno R. Histone deacetylase inhibitors: Structure-based modeling and isoform-selectivity prediction. J Chem Inf Model. 2012;52(8):2215–2235. doi: 10.1021/ci300160y. [DOI] [PubMed] [Google Scholar]
- 57.Halgren TA, Damm W. Polarizable force fields. Current opinion in structural biology. 2001;11(2):236–242. doi: 10.1016/s0959-440x(00)00196-2. [DOI] [PubMed] [Google Scholar]
- 58.Morozov AV, Kortemme T, Tsemekhman K, Baker D. Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc Natl Acad Sci U S A. 2004;101(18):6946–6951. doi: 10.1073/pnas.0307578101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Truchon JF, Nicholl’s A, Grant JA, Iftimie RI, Roux B, Bayly CI. Using electronic polarization from the internal continuum (epic) for intermolecular interactions. Journal of computational chemistry. 2010;31(4):811–824. doi: 10.1002/jcc.21369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dudek MJ, Ponder JW. Accurate modeling of the intramolecular electrostatic energy of proteins. J Computational Chem. 1995;16(7):791–816. [Google Scholar]
- 61.Grossfield A, Ren P, Ponder JW. Ion solvation thermodynamics from simulation with a polarizable force field. J Am Chem Soc. 2003;125(50):15671–15682. doi: 10.1021/ja037005r. [DOI] [PubMed] [Google Scholar]
- 62.Ponder JW, Case DA. Force fields for protein simulations. Adv Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 63.Ren P, Ponder JW. Consistent treatment of inter- and intramolecular polarization in molecular mechanics calculations. Journal of computational chemistry. 2002;23(16):1497–1506. doi: 10.1002/jcc.10127. [DOI] [PubMed] [Google Scholar]
- 64.Ponder JW. Tinker 6.0. 2011. [Google Scholar]
- 65.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, DiStasio RA, Jr, Head-Gordon M, et al. Current status of the amoeba polarizable force field. The journal of physical chemistry B. 2010;114(8):2549–2564. doi: 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi Y, Wu C, Ponder JW, Ren P. Multipole electrostatics in hydration free energy calculations. Journal of computational chemistry. 2011;32(5):967–977. doi: 10.1002/jcc.21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ren P, Wu C, Ponder JW. Polarizable atomic multipole-based molecular mechanics for organic molecules. J Chem Theory Comput. 2011;7(10):3143–3161. doi: 10.1021/ct200304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schnieders MJ, Fenn TD, Pande VS, Brunger AT. Polarizable atomic multipole x-ray refinement: Application to peptide crystals. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 9):952–965. doi: 10.1107/S0907444909022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiao D, Zhang J, Duke RE, Li G, Schnieders MJ, Ren P. Trypsin-ligand binding free energies from explicit and implicit solvent simulations with polarizable potential. Journal of computational chemistry. 2009;30(11):1701–1711. doi: 10.1002/jcc.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dvir H, Silman I, Harel M, Rosenberry TL, Sussman JL. Acetylcholinesterase: From 3d structure to function. Chem Biol Interact. 2010;187(1–3):10–22. doi: 10.1016/j.cbi.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Macias AT, Mackerell AD., Jr Ch/pi interactions involving aromatic amino acids: Refinement of the charmm tryptophan force field. Journal of computational chemistry. 2005;26(14):1452–1463. doi: 10.1002/jcc.20281. [DOI] [PubMed] [Google Scholar]
- 73.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 74.Ren P, Ponder JW. Temperature and pressure dependence of the amoeba water model. J Phys Chem B. 2004;108:13427–13437. [Google Scholar]
- 75.Walsh TR, Liang T. A multipole-based water potential with implicit polarization for biomolecular simulations. Journal of computational chemistry. 2009;30(6):893–899. doi: 10.1002/jcc.21111. [DOI] [PubMed] [Google Scholar]
- 76.Kramer C, Gedeck P, Meuwly M. Atomic multipoles: Electrostatic potential fit, local reference axis systems, and conformational dependence. Journal of computational chemistry. 2012;33(20):1673–1688. doi: 10.1002/jcc.22996. [DOI] [PubMed] [Google Scholar]
- 77.Chourasia M, Sastry GM, Sastry GN. Aromatic-aromatic interactions database, a2id: An analysis of aromatic [pi]-networks in proteins. Int J Biol Macromol. 2011;48(4):540–552. doi: 10.1016/j.ijbiomac.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 78.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed Engl. 2003;42 (11):1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 79.Salonen LM, Ellermann M, Diederich F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew Chem Int Ed Engl. 2011;50(21):4808–4842. doi: 10.1002/anie.201007560. [DOI] [PubMed] [Google Scholar]
- 80.Burley SK, Petsko GA. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 81.Crowley PB, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins. 2005;59(2):231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 82.Greenberg DA, Barry CD, Marshall GR. Investigation and parameterization of a molecular dielectric function. J Am Chem Soc. 1978;100:4020–4026. [Google Scholar]
- 83.Marshall GR. Limiting assumptions in structure-based design: Binding entropy. Journal of computer-aided molecular design. 2012;26(1):3–8. doi: 10.1007/s10822-011-9494-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lane TJ, Bowman GR, Beauchamp K, Voelz VA, Pande VS. Markov state model reveals folding and functional dynamics in ultra-long md trajectories. J Am Chem Soc. 2011;133(45):18413–18419. doi: 10.1021/ja207470h. [DOI] [PMC free article] [PubMed] [Google Scholar]