

Abstract
Recent studies have uncovered sterile alpha motif and HD domain 1 (SAMHD1) as the restriction factor that blocks HIV-1 replication in myeloid cells. In contrast to previously identified HIV-1 restriction factors, SAMHD1 does not meet a countermeasure developed by HIV-1. However, HIV-2 and certain simian immunodeficiency virus (SIV) strains express the auxiliary protein Vpx that potently blocks SAMHD1. It is therefore perplexing why this function has been lost or not acquired during the course of lentiviral evolution. This article summarizes the similarities and differences between SAMHD1 and other HIV-1 restriction factors, while highlighting the new questions that are emerging about the crosstalk between restriction factors and innate immune responses.
Intrinsic cellular defenses and the viral arsenal
HIV was identified as a human pathogen 28 years ago [1]. As a result of the inability of the human host to mount a coordinated immune response to the virus, infection by HIV usually results in chronic activation of the immune system that paradoxically allows for poorly controlled viral replication and immune exhaustion: the hallmark of AIDS. HIV-1 has evolved from SIVcpz, which causes AIDS in its natural chimpanzee host, whereas HIV-2 has evolved from SIVsm, which replicates to high levels in its natural simian host without causing disease [2] (Box 1). Hence, it is possible to speculate that lentiviruses and their host immune systems undergo an evolutionary co-adaptation to establish an equilibrium that will permit efficient spread without killing the host. However, tolerance to the infection is a multifactorial process that requires a fertile ground in the host as much as specific features of the virus. Here, we review the recent advances related to how host restriction factors may influence immune sensing and response against HIV. In particular, we examine the recent identification that the immune modulator SAMHD1 is the HIV restriction factor operating in myeloid cells, which are key players in the immune response during viral infection.
Box 1. About the origins of HIV-1-associated pathogenicity.
There is a correlation between the ‘time of presence’ of lentiviral strains in hosts and pathogenicity: despite high viral titers and lack of immune control, infection by SIVsm in Sooty mangabeys and by SIVagm in African green monkeys fail to cause simian AIDS (reviewed in [76,77]). Recent exposition to SIV, as for SIVcpz in chimpanzees, causes symptoms close to AIDS. Probably, transmission of SIV from monkeys to humans is very recent. The use of molecular clocks has allowed the dating of the first event of cross-species transmission between monkey hosts to humans to the early 20th century in regions of central Africa where socioeconomical changes catalyzed the initial spread of the pandemic (reviewed in [78]). Transmission must have occurred on several occasions, followed by rapid diversification, to give rise to all the clades, subtypes and recombinant forms of viruses now infecting humans [78]. Indeed HIV-1 and HIV-2 have distinct origins: the probable progenitor of pandemic HIV-1 is SIVcpz [79], whereas less pathogenic HIV-2 originated independently from transmission from SIVsm [80,81]. The lessened pathogenicity of HIV-2 is characterized by its non-pandemicity and the decline in its prevalence as compared to the rise of HIV-1 [78].
A not so fertile ground?
Upon entry into the human host, viruses are confronted with numerous blocks that oppose their replication (Figure 1). The first line of defense to be triggered is the so-called ‘intrinsic’ immune system. The intrinsic immune system includes proteins that detect the presence of the assailant pattern recognition receptors (PRRs), [Box 2] and which initiate a subsequent immune response, as well as proteins referred to as restriction factors that are directly devoted to arresting the replication cycle of the virus. To date, four restriction factors have been identified that specifically block HIV-1 replication: tripartite motif (TRIM)5α, apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G (APOBEC3G) (A3G), bone marrow stromal cell antigen 2 (BST-2) (also known as tetherin) and SAMHD1 [3-7].
Figure 1.

The hostile host. Cells that are targeted by HIV-1 possess numerous barriers that oppose viral replication. On this schematic representation of the viral replication cycle, restriction factors are in red, sensors are in purple, and facilitating factors are in orange. Signaling pathways are indicated in blue. Bold arrows highlight steps of the viral replication cycle. The classical entry route of HIV-1 in target cells is through attachment (1) at the cell surface and fusion (2). The viral core is delivered in the cytoplasm (3) where its recognition by hTRIM5α can activate the transcription factors NF-κB and AP-1. In simian cells, TRIM5α interacts with the viral core and disturbs uncoating (4). After or concomitantly with uncoating (4) and nuclear import steps, reverse transcription (5) occurs. This generates defective byproducts that can be recognized by TREX1 that causes their degradation, thereby preventing eliciting IFN responses. SAMHD1 operates at a related step and inhibits reverse transcription by either targeting the dNTP pool or reverse transcription products. When reverse transcription and nuclear import (5) are completed, the viral genome is integrated into the host’s genome (6). The provirus is transcribed (7), exported (8) and translated (9) to give rise to the viral proteins that assemble (10) at the plasma membrane, to give rise to new virions that can bud from (11) the cell. After a further maturation step (12), the viral particles can infect new target cells. CA neosynthesis in DCs (9) elicits a signaling cascade via a cryptic sensor, leading to IFN production, maturation of the cells, and therefore, ameliorated antigen presentation. Thus, this stimulates better immune responses. Upon assembly (10), A3G is incorporated in viral particles and subsequently delivered in newly infected cells where it blocks the replication cycle. BST-2 tethers viral particles at the cell surface and thereby blocks budding (11). Another route of entry into target cells is uptake of viruses through endocytosis (1′). The early steps of the viral replication cycle can then lead to delivery of ssRNA in endosomes, activating TLR7 that will lead to IFN production.
Box 2. PRRs constitute the first line of defense against pathogens.
PRRs recognize molecules that are specifically associated with pathogens [82]. In the case of viral infections, these receptors can recognize the viral genome, which is shared by all viruses and not easily mutated to avoid recognition. PRRs can be roughly divided into two classes based on their subcellular localization. Indeed, they are usually strategically positioned within the cell at the entry sites of pathogens in the cytoplasm (cytosolic receptors including the RIG-I-like receptors) or endosomal/lysosomal compartments (Toll-like receptors, TLRs). Upon recognition of foreign elements, they elicit the production of cytokines that protect noninfected cells and attract effector cells to the site of infection. HIV-1 infection perturbs innate antiviral signaling through specific disruption of both TLR and RIG-I signaling [83,84]. The determinants of this inhibition lie in the Pol polyprotein; the viral protease causes inhibition of IFN regulator factor-3 (IRF-3) phosphorylation, which results in a decrease of IFN and IFN-stimulated genes transcription [84]. The virus has evolved other means of circumventing the PRR pathway. During the early steps of the viral life cycle, abortive replication intermediates are produced that could accumulate in the cytoplasm of infected cells. These abortive products could be recognized by PRRs [55], thereby eliciting a signaling cascade leading to antiviral IFN production. The cellular protein TREX-1 recognizes these defective reverse transcription products and causes their specific degradation [55]. In cells that are devoid of TREX-1, the accumulation of such DNA species results in the signaling events that initiate the innate immune response and inhibition of viral replication and spread.
Host restriction factor characteristics
TRIM5α interacts with the viral capsid (CA), disrupts the lattice that forms the viral core, and thereby disturbs the uncoating step (Figure 1). The TRIM, which is composed of Really Interesting New Gene (RING), B-box 2 and coiled-coil domains, has an E3-ligase activity that could target CA to proteasome-dependent degradation. However, the implication of the proteasome machinery in TRIM5α-mediated restriction of HIV-1 replication remains controversial because treatment of HIV-1-infected simian cells with proteasome inhibitors does not fully restore the ability of HIV-1 to establish a productive infection [8]. TRIM5α restriction specificity is determined by the C-terminal B30.2/SPRY domain through CA recognition. Human TRIM5α (hTRIM5α) harbors an amino acid substitution in the B30.2/SPRY domain that disrupts interaction with HIV-1 CA, abrogating its restriction potential against HIV-1 infection [9].
APOBEC3G, a member of the apolipoprotein mRNA-editing enzyme catalytic polypeptide-like editing complex 3 (A3G) family of cytidine deaminases is a host restriction factor involved in the early steps of the viral life cycle (Figure 1). It is needed in virus-producing cells for restriction to be witnessed in newly infected cells. Indeed, during the assembly and budding of viral particles, A3G is incorporated into nascent virions [10-12]. Upon infection of target cells, A3G catalyzes the deamination of cytidine residues in the viral genome, which leads to incorporation of adenosines in place of original guanines during DNA plus-strand synthesis [13], resulting in G to A hypermutations. The hypermutated viral genome is highly unstable and in most cases fails to integrate into the host genome; in cases where integration into the host cell genome occurs, the mutated provirus fails to give rise to functional viral proteins and therefore the production of infectious viral particles is abrogated. Deaminase activity-independent A3G-mediated restriction of HIV-1 has also been reported but this remains controversial [14]. Deaminase activity targeting HIV-1 has also been reported for other members of the APOBEC3 family [15].
The host restriction factor BST-2 is a type II membrane-associated protein – with a cytoplasmic N-terminal region followed by an extracellular coiled-coil extracellular domain and a C-terminal GPI anchor – that acts at the latest stage of the viral life cycle (Figure 1). BST-2 blocks the release of nascent viral particles by tethering them to the plasma membrane. This leads to the formation of viral aggregates that fail to infect new target cells [5].
SAMHD1 is the most recently discovered restriction factor, and is highly expressed and functional in myeloid cells [6,7] (Figure 1). SAMHD1 was initially identified as the human ortholog of the mouse gene Mg11, which is induced by interferon (IFN) treatment of macrophages and dendritic cells (DCs) [16]. SAMHD1 expression is also induced in human macrophages following IFN-stimulatory DNA treatment [17] in an IFN-dependent and Toll-like receptor (TLR)-independent fashion [17,18]. This 626 amino acid protein is the only human protein in which a sterile α motif (SAM) and an HD domain occur in tandem (Figure 2). SAMs are protein–protein interaction domains that can also bind RNA. HD domains are characterized by a motif of doubletcation-coordinating His and Asp residues (H …HD …D). They are evolutionary conserved domains that play crucial roles in nucleic acid metabolism and signaling. SAMHD1 acts at early stages of the viral life cycle but its precise mode of action is yet to be uncovered. It has, however, been described that depletion of SAMHD1 leads to accumulation of full-length HIV DNA, arguing for a potential action around the reverse transcription step [6].
Figure 2.
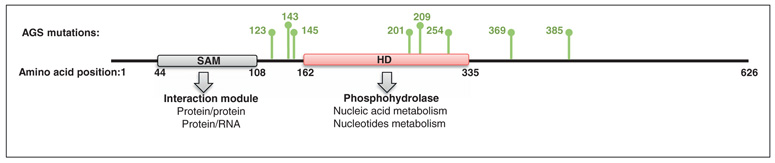
SAMHD1 primary structure. SAMHD1 is a 626-amino acid protein that harbors an SAM and HD domain in tandem. The SAM domain is a protein–protein or protein–RNA interaction module. The HD domain possesses putative hydrolase activity. In green are indicated the amino acids that are found to be mutated in the case of AGS [17].
Viral counteraction of immune responses
Despite the numerous cellular mechanisms to prevent viral replication, HIV-1 is able to replicate efficiently in humans. Not only has the virus developed strategies to avoid triggering of PRRs (Box 2), but also has elaborated ways to counteract host restriction factors.
Essential accessories
In addition to the information required for the production of structural and enzymatic proteins essential for mature viral particles production, lentiviral genomes also encode auxiliary proteins that regulate viral fitness in hosts. Although these auxiliary proteins are mostly unnecessary for viral replication in permissive cells in vitro, disruption of open reading frames (ORFs) corresponding to individual viral auxiliary proteins results in inefficient viral spread ex vivo in non-permissive cells and in vivo in hosts. Most accessory proteins are instrumental in the counteraction of cellular restriction factors – indeed A3G as well as BST-2 and SAMHD1 were identified in attempts to decipher the function of viral auxiliary proteins (Vif, Vpu, Vpx and Vpr). In elegant experiments in which heterokaryons were generated from permissive and non-permissive cells, it was evident that the block in viral replication was caused by the presence of a dominant negative cellular factor that directly hindered the replication cycle [19-21]. The exception to this rule is TRIM5α, which is not counteracted by a viral accessory protein. The observation in simian cells that HIV-1, despite being able to fuse at the cell surface, is not able to proceed through the reverse transcription step has led to the screening of a simian cell-derived cDNA library that allows the identification of TRIM5α as being responsible for this block [4].
Similar screening strategies have allowed the discovery of A3G and BST-2. A3G was initially identified through mRNA profiling of cells that were permissive or not to the replication of a Vif-deficient virus [3]. Vif interacts with A3G in infected cells, and induces its polyubiquitination and subsequent degradation by the proteasome [22]. Vif thereby prevents the incorporation of A3G in nascent viral particles. Some reports have indicated that there are additional proteasome-independent mechanisms by which Vif can counteract A3G [23]. Microarray analysis of membrane-associated proteins in cells in which Vpu is required for viral release led to the identification of BST-2 [5]. Vpu interacts with BST-2 and targets this protein to the trans-Golgi network or to the early endosomes, thereby sequestering it away from its site of action. Whether this leads to preoteasome/lysosome-dependent degradation of BST-2 remains debated [24-28]. Vpu expression is limited to HIV-1 and closely related SIV strains [29,30]. In viral strains in which BST-2 counteraction is not fulfilled by Vpu, this function is carried by other viral proteins [2]; the conservation of this function throughout evolution highlights its crucial importance to the viral life cycle. Accordingly, HIV-1 from the non-pandemic strains O and P lacks anti-BST-2 activity [31].
SAMHD1 as an exception?
In contrast to the other restriction factors, HIV-1 has no means to counteract SAMHD1. This restriction factor was identified based on studies showing that certain strains of lentiviruses that encode the auxiliary protein Vpx can bypass the block to viral replication in myeloid cells[21,32]. SAMHD1 was recovered in biochemical purification of Vpx and its associated cofactors and was identified as the restriction factor operating in myeloid cells [6,7]. Vpx binds and targets SAMHD1 to proteasomal degradation. Vpx is expressed by HIV-2 and its closely related SIV strains but is absent from SIV strains related to HIV-1 as well as from HIV-1 itself. Providing Vpx in trans to DCs and monocytes facilitates their infection by HIV-1. Of note, Vpx from certain SIV strains does not have the ability to cause human SAMHD1 degradation and fails to facilitate HIV-1 infection of DCs [6]. Therefore, because HIV-1 cannot counteract SAMHD1, it seems that, in contrast to other restriction factors, overcoming SAMHD1 is dispensable for efficient HIV-1 spread. This is in contrast, for example, to BST-2, because BST-2 nonspecifically targets the lipidic outlayer of enveloped viruses and has a broad impact on viral release [33-41]. Consequently, viruses have evolved distinct means to counteract this hurdle to viral budding. Therefore, it can be questioned whether there is any advantage for HIV-1 and the related SIV strains to counteract SAMHD1 and to infect DCs and monocytes. The non-permissiveness instead could be instrumentalized by the virus as an escape strategy from the immune system.
The intricate interplay between restriction factors and immune signaling
Recent studies have highlighted that restriction factors keep a more complex relation with the immune system than initially anticipated. The TRIM family of proteins comprises both bona fide restriction factors that directly counteract viral replication, as well as other factors that regulate immune sensing [42]. TRIM5α can act as both a restriction factor and a PRR, depending on the retrovirus with which it is challenged [43,44]. Recent work has described the involvements of TRIM5α in innate immune signaling. Interaction of TRIM5α with the CA lattice activates the kinase TAK1, which leads to downstream activation of the transcription factors nuclear factor (NF)-κB and activator protein-1 (AP-1) (Figure 1). Thus, TRIM5α might act as a PRR by modulating proinflammatory cytokine secretion [43,44]. Moreover, the TRIM family of proteins has been involved in autoimmune and autoinflammatory disorders [45]. Similarly, SAMHD1 is an immune modulator involved in inflammation and type1 IFN production [17,18]. Indeed, patients harboring mutations in SAMHD1 (Figure 2) experience a rare autoimmune disease (with an autosomal recessive mode of transmission), the Aicardi–Goutière syndrome (AGS), with symptoms mimicking those of congenital infections that are characterized by increased IFNα production and chronic lymphocytosis [17]. AGS patients, due to the dysregulation of their IFN homeostasis also experience symptoms reminiscent of the autoimmune disease systemic lupus erythematous. This posits SAMHD1 as a key negative regulator of the IFN pathway and of acquired autoimmune diseases. Moreover, like previously identified restriction factors, SAMHD1 is induced by IFN [17], supporting a potential role of SAMHD1 as an effector of the IFN pathway. This suggests a role for SAMHD1 in immune responses. SAMHD1 may act as a negative regulator of the IFN pathway [17], therefore, Vpx-induced degradation of SAMHD1 could be expected to cause increased IFN secretion; the consequence of which would be an escalation of the immune response. Consistent with this, recent work has shown that Vpx treatment of DCs increases IFN production after HIV-1 infection [46]. However, it is hypothesized that increased IFN production is caused by activation of a cryptic sensor by newly synthesized CA, because Vpx alone does not cause IFN secretion. Limitations of the experimental systems might account for the failure to detect IFN production as a result of SAMHD1 degradation. Alternatively, additional signals may be required to trigger the IFN response. Moreover, it has also been shown that Vpx delivery into IFN-treated DCs relieves the block to HIV-1 replication [47]. Whether SAMHD1 plays a role in this process is yet to be determined. It will be important to determine whether the restriction activity of SAMHD1 and its function in AGS are linked. This can be achieved by exploring the restriction activity of SAMHD1 mutants associated with AGS.
SAMHD1 and nucleic acids
Interestingly, other mutations that play a prominent role in AGS pathogenesis occur in genes that encode the 3′ exonuclease three prime repair exonuclease 1 (TREX1) (which degrades ssDNA) and components of the endonuclease complex RNase H2 (which degrades RNA within RNA/DNA hybrids) [48,49] (Figure 3). Both these factors are involved in nucleic acid metabolism, implying a probable role of SAMHD1 in a similar pathway. Consistently, mutations in the HD domain of SAMHD1 disrupt its ability to restrict HIV-1 infection [6]. Whether SAMHD1 mediates its antiviral activity by targeting viral RNA, DNA or RNA/DNA hybrids is unknown. Both structure-based analyses and nucleic acid binding assays show that SAMHD1 is unable to bind a variety of nucleic acid ligands such as ssRNA, RNA stem-loop, ssDNA or dsDNA [17], possibly arguing against a nuclease activity within SAMHD1. Alternatively, phosphohydrolase and phosphodiesterase activities of HD domain-containing factors play crucial roles in nucleotide metabolism and signaling. Accordingly, using recombinant SAMHD1 protein and in vitro assays, two recent reports have shown that SAMHD1 possesses an intrinsic dGTP-regulated deoxynucleoside triphosphase triphosphohydrolase [50,51]. Thus, it is tempting to speculate that SAMHD1 may mediate its restriction activity by ensuring low intracellular levels of nucleotides, creating an unfavorable cellular environment for viral DNA synthesis. In support of this hypothesis, SAMHD1 is active in differentiated noncycling cells, such as DCs and macrophages [6] that have low intracellular dNTP concentrations compared to proliferating cells [52] in which SAMHD1 is inefficient [7]. Further work is required to decipher the mechanism by which SAMHD1 mediates its restriction activity.
Figure 3.

Genetic causes of AGS and its consequences. AGS, an autosomal recessive genetic disorder, results from mutations in the genes encoding cytoplasmic TREX1, RNase H2 or nuclear SAMHD1. These proteins are regulators of innate immune response through their involvement in nucleic acids metabolism. However, their impact on HIV replication is divergent: TREX1 and RNase H2 appear to facilitate viral replication, whereas SAMHD1 is a restriction factor. *For review [87]; **putative activity.
In contrast to SAMHD1 degradation, TREX1 and RN-ase H2 deficiencies have a deleterious effect on HIV-1 replication [53]. For example, TREX1 is part of the reticulum-associated DNase complex SET, which inhibits auto-integration of the viral genome after completion of reverse transcription [54] and causes the degradation of abortive reverse transcription products, thereby avoiding accumulation of these DNA products and subsequent recognition by TLRs [55]. Therefore, TREX1, which is usually involved in cellular homeostasis by inhibiting the accumulation of abortive replication DNA species from endogenous retroviruses [56], acts as a facilitating factor of HIV-1 replication, indicating that SAMHD1 and TREX1 most probably act on divergent pathways. In this context, it remains debatable whether the degradation of SAMHD1 through Vpx is beneficial to the virus or that viral evolution has eliminated this function as part of an escape strategy from the immune system.
Regulation of SAMHD1 activity
Like the other restriction factors, SAMHD1 is expressed in all tissues. However, SAMHD1 restriction activity is confined to differentiated noncycling cells [6]. Accordingly, in a overexpression screening approach, performed in dividing cells, SAMHD1 does not significantly affect infection of the range of tested viruses, including HIV [57]. This is an intriguing feature because the expression of the other restriction factors is sufficient to confer resistance regardless of the cell type and their cell cycle state. The confinement of SAMHD1 restriction activity to noncycling cells opens the possibility that SAMHD1 may act in a broad spectrum of noncycling cells. It will be interesting to assess whether SAMHD1 can restrict HIV in quiescent CD4+ T cells. This is important because it has recently been shown that abortive HIV infection of quiescent CD4+ T cells, due to incomplete reverse transcription, is responsible for their massive depletion and for inflammation [58]. Exploring the role of SAMHD1 in CD4+ T lymphocyte depletion in lymphoid tissue and inflammation may help understanding the immunopathogenic effect of HIV. How SAMHD1 expression and activity is regulated is unknown. The expression of restriction factors is often modulated in several ways. For example, their transcription is induced in response to IFN [59]. At the post-transcriptional level, several splice variants of TRIM5α have been described [4,60]. At the post-translational level, A3G, BST-2 and TRIM5α are targeted by post-translational modifications that regulate their stability [61]. SAMHD1 restriction activity may be regulated by post-translational modification or may require additional factors expressed only in differentiated noncycling cells. The regulation of SAMHD1 activity remains an important area of study. Indeed, targeting SAMHD1 activity can be used as way to confer resistance to HIV-1 or to initiate innate immune sensing of the incoming virus in vivo.
SAMHD 1 and lentiviral adaptation
Broad spectrum viral counteraction?
In most cases, restriction factors are not active solely against HIV [62]. It has been shown that A3G, as well as other members of the APOBEC family, is active against a broad range of retroviruses [63]. Similarly, BST-2 is promiscuous in its ability to counteract enveloped viruses[64,65]. Interestingly, restriction of murine leukemia virus at the reverse transcription step is abrogated by Vpx in monocytes and macrophages [66,67], suggesting that SAMHD1 may exert its restriction activity against viruses other than lentiviruses. Additionally, if the deoxynucleotide triphosphohydrolase activity is found to be involved in SAMHD1-mediated HIV restriction, then SAMHD1 would be expected to act as restriction factor of viruses that require DNA synthesis during their life cycle. Thus, SAMHD1 may act as the guardian of differentiated noncycling cells from invading viruses. A3G action is also exerted on endogenous retroviruses [68]. As SAMHD1 belongs to a family of proteins that have been involved in the recognition and degradation of endogenous retroelements, it is possible that SAMHD1 plays a housekeeping role in clearing up the byproducts of endogenous retrovirus biology. As far as hTRIM5α is concerned, it has been shown that, despite being ineffective at restricting HIV-1, this human variant is able to counteract a 4-million-year-old endogenous virus that was resurrected from the chimpanzee’s genome [69]. In the course of evolution, TRIM5α has lost its potency against lentiviruses (probably because of lack of genetic conflict), leaving the door open to cross-species transmission. This work demonstrates the evolutionary pressure that acts on cellular restriction factors.
Genetic conflict-dependent adaptation of the immune system
Genetic conflict between host restriction factors and viruses is predicted to lead to rapid selection of mutations that alter amino acid composition of both factors, especially at positions that affect protein–protein interaction. This process, called positive selection, occurs during A3G interaction with Vif [70], TRIM5α interaction with CA, and BST-2 interaction with Vpu, Nef and Env [28,71]. Paleovirological studies are believed to contribute to the better understanding of how viral infections over time have guided the evolution of the host immune system [72]. As available data suggest an intricate relation between SAMHD1 and the innate immune response, studying the evolutionary pressures that have acted upon this protein could improve our understanding of its function. The fact that not all viruses have the ability to degrade this restriction factor adds to a probably complicated evolutionary story that is waiting to be deciphered. vpx has been proposed to have originated by duplication of the common vpr gene present in primate lentiviruses (Box 3); therefore, it will be important to determine, through functional and evolutionary studies, whether Vpx-mediated degradation of SAMHD1 has been acquired after the acquisition of vpx, or if it was lost from vpr and conserved within vpx. Finally, evolutionary studies will also help address whether the restriction activity of SAMHD1 acts on more ancient viruses.
Box 3. Is there a parallel between Vpr and Vpx?
Whether vpx arose from gene duplication of vpr or through acquisition via recombination of viral strains remains debated and recent reviews detail their similarities and discrepancies [85,86]. Modeling of Vpx and Vpr from HIV-2 and HIV-1 origin reveals a highly similar 3D structure and high sequence homology. These two proteins share several features at the functional level. They are both actively incorporated in nascent viral particles and delivered in the cytoplasm of target cells upon infection, where they influence early steps of viral replication. When vpr or vpx ORFs are disrupted, viral replication is decreased in monocyte-derived macrophages but the impact of vpx disruption is more profound than that of vpr. The best characterized function of Vpr, from HIV-1 and HIV-2, is the ability to arrest the cell cycle at the G2/M transition which is not shared by Vpx. Vpr-associated cytopathy has only been reported for Vpr of HIV-1, but not for Vpr or Vpx of HIV-2. HIV-1 Vpr is also involved in nuclear import of the viral preintegration complex. This function has been suggested to be carried by Vpx of HIV-2.Vpr from HIV-2 and HIV-1 have both been reported to have an impact on cellular and viral gene expression. Other Vpr functions such as DNA-damage response induction and IRF-3 degradation have not yet been investigated in HIV-2. Stimulation of reverse transcription appears to be a unique feature of Vpx. Finally, Vpr fails to facilitate infection of DCs, suggesting that the ability to counteract SAMHD1 restriction is absent from Vpr [6].
Concluding remarks
The study of restriction factors and of the viral proteins counteracting their action contributes to our understanding of how our immune system has evolved to counteract viral invasions. The recent discovery of SAMHD1 as a barrier to HIV-1 replication in myeloid cells unravels new possibilities to understand the intricacies of the relation between lentiviral evolution and primate hosts. Indeed, SAMHD1 is the only HIV-1 restriction factor against which the virus has not evolved a neutralizing strategy. Thus, deciphering SAMHD1 function in HIV-1 acquisition, spread and pathogenicity may unravel new ways of combating the virus. Crosstalk between restriction factors and immune response should be considered in the development of effective vaccines. Indeed, correlation between the expression of TRIM5α-SIV-restrictive alleles and protection of vaccinated monkeys has been recently reported [73,74]. This may be achieved through Trim5α-mediated limitation of virus replication and/or viral diversity. Additionally, the discovery of the immune modulator SAMHD1 as the restriction factor operating in DCs opens new areas of investigation in the DC-target vaccination strategies [75].
Acknowledgments
We thank Rosemary Kiernan, Ahmad Yatim, Bijan Sobhian, Nadia Rahm and Amalio Telenti for helpful discussions and critical reading of the manuscript. N.L. is recipient of SIDACTION fellowship. Work in M.B.’s laboratory was supported by ERC (250333), ANRS, SIDACTION and FRM ‘Equipe labéllisée FRM’ to M.B.
References
- 1.Barre-Sinoussi F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Kirchhoff F. Is the high virulence of HIV-1 an unfortunate coincidence of primate lentiviral evolution? Nat. Rev. Microbiol. 2009;7:467–476. doi: 10.1038/nrmicro2111. [DOI] [PubMed] [Google Scholar]
- 3.Sheehy AM, et al. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 4.Stremlau M, et al. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 5.Neil SJ, et al. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 6.Laguette N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrecka K, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Towers GJ. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007;4:40. doi: 10.1186/1742-4690-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap MW, et al. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 2005;15:73–78. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Sheehy AM, et al. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 11.Stopak K, et al. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 12.Mariani R, et al. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 13.Conticello SG, et al. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 2005;22:367–377. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- 14.Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wissing S, et al. HIV-1 Vif versus the APOBEC3 cytidine deaminases: an intracellular duel between pathogen and host restriction factors. Mol. Aspects Med. 2010;31:383–397. doi: 10.1016/j.mam.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li N, et al. Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol. Lett. 2000;74:221–224. doi: 10.1016/s0165-2478(00)00276-5. [DOI] [PubMed] [Google Scholar]
- 17.Rice GI, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao W, et al. Dendritic cell-derived interferon-gamma-induced protein mediates tumor necrosis factor-alpha stimulation of human lung fibroblasts. Proteomics. 2008;8:2640–2650. doi: 10.1002/pmic.200700954. [DOI] [PubMed] [Google Scholar]
- 19.Munk C, et al. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13843–13848. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varthakavi V, et al. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15154–15159. doi: 10.1073/pnas.2433165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharova N, et al. Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008;4:e1000057. doi: 10.1371/journal.ppat.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu X, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 23.Dang Y, et al. APOBEC3G is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J. Biol. Chem. 2008;283:13124–13131. doi: 10.1074/jbc.M708728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Douglas JL, et al. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 2009;83:7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goffinet C, et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Gupta RK, et al. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009;5:e1000443. doi: 10.1371/journal.ppat.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangeat B, et al. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009;5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNatt MW, et al. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009;5:e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strebel K, et al. Molecular and biochemical analyses of human immunodeficiency virus type 1 vpu protein. J. Virol. 1989;63:3784–3791. doi: 10.1128/jvi.63.9.3784-3791.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottlinger HG, et al. Vpu protein of human immunodeficiency virus type 1 enhances the release of capsids produced by gag gene constructs of widely divergent retroviruses. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7381–7385. doi: 10.1073/pnas.90.15.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang SJ, et al. Lack of adaptation to human tetherin in HIV-1 Group O and P. Retrovirology. 2011;8:78. doi: 10.1186/1742-4690-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goujon C, et al. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J. Virol. 2008;82:12335–12345. doi: 10.1128/JVI.01181-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dietrich I, et al. Feline tetherin efficiently restricts release of feline immunodeficiency virus but not spreading of infection. J. Virol. 2011;85:5840–5852. doi: 10.1128/JVI.00071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jouvenet N, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009;83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaletsky RL, et al. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mansouri M, et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2009;83:9672–9681. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radoshitzky SR, et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 2010;84:10569–10580. doi: 10.1128/JVI.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakuma T, et al. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 2009;83:2382–2385. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weidner JM, et al. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010;84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu F, et al. Tetherin inhibits prototypic foamy virus release. Virol. J. 2011;8:198. doi: 10.1186/1743-422X-8-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang SJ, et al. Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology. 2010;7:13. doi: 10.1186/1742-4690-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNab FW, et al. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011;23:46–56. doi: 10.1016/j.coi.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 43.Pertel T, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao G, et al. Rhesus TRIM5alpha disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 2011;7:e1002009. doi: 10.1371/journal.ppat.1002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jefferies C, et al. Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat. Rev. Immunol. 2011;11:617–625. doi: 10.1038/nri3043. [DOI] [PubMed] [Google Scholar]
- 46.Manel N, et al. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pertel T, et al. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology. 2011;8:49. doi: 10.1186/1742-4690-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat. Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 49.Crow YJ, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 50.Powell RD, et al. The Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011 doi: 10.1074/jbc.C111.317628. DOI: 10.1074/jbc.C111.317628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldstone DC, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011 doi: 10.1038/nature10623. DOI: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 52.Rampazzo C, et al. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat. Res. 2010;703:2–10. doi: 10.1016/j.mrgentox.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 53.Genovesio A, et al. Automated genome-wide visual profiling of cellular proteins involved in HIV infection. J. Biomol. Screen. 2011;16:945–958. doi: 10.1177/1087057111415521. [DOI] [PubMed] [Google Scholar]
- 54.Yan N, et al. The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog. 2009;5:e1000327. doi: 10.1371/journal.ppat.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan N, et al. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stetson DB, et al. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doitsh G, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. 2010;143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kirchhoff F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe. 2010;8:55–67. doi: 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 60.Passerini LD, et al. Retroviral restriction factors Fv1 and TRIM5alpha act independently and can compete for incoming virus before reverse transcription. J. Virol. 2006;80:2100–2105. doi: 10.1128/JVI.80.5.2100-2105.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strebel K, et al. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009;7:48. doi: 10.1186/1741-7015-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008;42:709–732. doi: 10.1146/annurev.genet.42.110807.091501. [DOI] [PubMed] [Google Scholar]
- 63.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 64.Martin-Serrano J, Neil SJ. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 2011;9:519–531. doi: 10.1038/nrmicro2596. [DOI] [PubMed] [Google Scholar]
- 65.Evans DT, et al. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010;18:388–396. doi: 10.1016/j.tim.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaushik R, et al. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe. 2009;6:68–80. doi: 10.1016/j.chom.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jarrosson-Wuilleme L, et al. Transduction of nondividing human macrophages with gammaretrovirus-derived vectors. J. Virol. 2006;80:1152–1159. doi: 10.1128/JVI.80.3.1152-1159.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Esnault C, et al. Restriction by APOBEC3 proteins of endogenous retroviruses with an extracellular life cycle: ex vivo effects and in vivo ‘traces’ on the murine IAPE and human HERV-K elements. Retrovirology. 2008;5:75. doi: 10.1186/1742-4690-5-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaiser SM, et al. Restriction of an extinct retrovirus by the human TRIM5alpha antiviral protein. Science. 2007;316:1756–1758. doi: 10.1126/science.1140579. [DOI] [PubMed] [Google Scholar]
- 70.Sawyer SL, et al. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004;2:E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ortiz M, et al. Evolutionary trajectories of primate genes involved in HIV pathogenesis. Mol. Biol. Evol. 2009;26:2865–2875. doi: 10.1093/molbev/msp197. [DOI] [PubMed] [Google Scholar]
- 72.Emerman M, Malik HS. Paleovirology—modern consequences of ancient viruses. PLoS Biol. 2010;8:e1000301. doi: 10.1371/journal.pbio.1000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Letvin NL, et al. Immune and genetic correlates of vaccine protection against mucosal infection by SIV in monkeys. Sci. Transl. Med. 2011;3:81ra36. doi: 10.1126/scitranslmed.3002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim SY, et al. Contributions of Mamu-A*01 status and TRIM5 allele expression, but not CCL3L copy number variation, to the control of SIVmac251 replication in Indian-origin rhesus monkeys. PLoS Genet. 2010;6:e1000997. doi: 10.1371/journal.pgen.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 76.Keele BF, et al. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature. 2009;460:515–519. doi: 10.1038/nature08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paiardini M, et al. Lessons learned from the natural hosts of HIV-related viruses. Annu. Rev. Med. 2009;60:485–495. doi: 10.1146/annurev.med.60.041807.123753. [DOI] [PubMed] [Google Scholar]
- 78.Tebit DM, Arts EJ. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 2011;11:45–56. doi: 10.1016/S1473-3099(10)70186-9. [DOI] [PubMed] [Google Scholar]
- 79.Bailes E, et al. Hybrid origin of SIV in chimpanzees. Science. 2003;300:1713. doi: 10.1126/science.1080657. [DOI] [PubMed] [Google Scholar]
- 80.Hirsch VM, et al. SIV adaptation to human cells. Nature. 1989;341:573–574. doi: 10.1038/341573a0. [DOI] [PubMed] [Google Scholar]
- 81.Wertheim JO, Worobey M. Dating the age of the SIV lineages that gave rise to HIV-1 and HIV-2. PLoS Comput. Biol. 2009;5:e1000377. doi: 10.1371/journal.pcbi.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barbalat R, et al. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 83.Doehle BP, et al. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 2009;83:10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Solis M, et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011;85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ayinde D, et al. Limelight on two HIV/SIV accessory proteins in macrophage infection: is Vpx overshadowing Vpr? Retrovirology. 2010;7:35. doi: 10.1186/1742-4690-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fujita M, et al. Multifaceted activity of HIV Vpr/Vpx proteins: the current view of their virological functions. Rev. Med. Virol. 2010;20:68–76. doi: 10.1002/rmv.636. [DOI] [PubMed] [Google Scholar]
- 87.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet. 2009;18:R130–R136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]