

Abstract
AMPA receptors (AMPARs) mediate synaptic transmission and plasticity during learning, development, and disease. Mechanisms determining subsynaptic receptor position are poorly understood but are key determinants of quantal size. We used a series of live-cell, high-resolution imaging approaches to measure protein organization within single postsynaptic densities in rat hippocampal neurons. By photobleaching receptors in synapse subdomains, we found that most AMPARs do not freely diffuse within the synapse, indicating they are embedded in a matrix that determines their subsynaptic position. However, time lapse analysis revealed that synaptic AMPARs are continuously repositioned in concert with plasticity of this scaffold matrix rather than simply by free diffusion. Using a fluorescence correlation analysis, we found that across the lateral extent of single PSDs, component proteins were differentially distributed, and this distribution was continually adjusted by actin treadmilling. The C-terminal PDZ ligand of GluA1 did not regulate its mobility or distribution in the synapse. However, glutamate receptor activation promoted subsynaptic mobility. Strikingly, subsynaptic immobility of both AMPARs and scaffold molecules remained essentially intact even after loss of actin filaments. We conclude that receptors are actively repositioned at the synapse by treadmilling of the actin cytoskeleton, an influence which is transmitted only indirectly to receptors via the pliable and surprisingly dynamic internal structure of the PSD.
Introduction
At excitatory synapses, the nanometer-scale organization of presynaptic and postsynaptic structures defines the functional coupling of synaptic transmission. Though the PSD and the presynaptic active zone are tightly apposed and typically well matched in size, glutamate diffusion from sites of exocytosis and the low affinity of AMPA receptor (AMPAR) activation create a steep dependence on the spatial relationship between the point of glutamate release and the postsynaptic receptors (Lisman et al., 2007). Thus, not all synaptic receptors are activated by every release event (Liu et al., 1999; McAllister and Stevens, 2000; Yamashita et al., 2009), and release at different sites throughout the active zone may initiate opening of different subsets of postsynaptic receptors (Lisman and Raghavachari, 2006). For these reasons, the distribution of AMPARs within a synapse is broadly thought to influence synaptic efficacy (Xie et al., 1997; Franks et al., 2003; Raghavachari and Lisman, 2004; Lisman et al., 2007). Nevertheless, mechanisms of receptor positioning within the synapse are poorly understood, and whether these processes are dynamic in living synapses is unknown (MacGillavry et al., 2011).
An important consideration is whether receptors can move freely within the confines of the synapse (as in a corral), or remain immobilized (Ritchie et al., 2003; Bressloff and Earnshaw, 2009; Opazo and Choquet, 2011). This has been addressed most directly by tracking the motion of single receptors bound to antibodies carrying quantum dots or other tags (Triller and Choquet, 2008). Such observations have indicated that extrasynaptic AMPARs are essentially freely mobile (Choquet, 2010), whereas synaptic AMPARs are nearly immobilized for times ranging from seconds to minutes, remaining confined within nm-scale subdomains of the synapse (Bats et al., 2007; Ehlers et al., 2007). However, because antibody labeling preferentially samples receptors that exit the synapse to the plasma membrane, where antibody access is higher, it is not clear whether these observations apply to all receptors, and the results have been difficult to confirm with other techniques. AMPAR lateral diffusion has also been measured via Fluorescence Recovery After Photobleaching (FRAP) (Ashby et al., 2004). This approach has revealed that ∼20–60% of the cell surface AMPAR population in spines is labile, exchanging on a time scale of 5–20 min (Ashby et al., 2006; Sharma et al., 2006; Bats et al., 2007; Heine et al., 2008; Frischknecht et al., 2009; Martin et al., 2009; Arendt et al., 2010). The disparate time scales of results from FRAP and single-particle tracking experiments leave open many questions of synaptic AMPAR regulation. Notably, the degree of intrasynaptic mobility of stably incorporated receptors has not been reported, and FRAP has not been used to examine receptor mobility within the synapse.
Here, using high-resolution photobleaching we determined that motion of resident AMPARs across the synapse is rare, suggesting that a postsynaptic matrix stably positions receptors. Exploring the molecular mechanisms of this matrix, however, we find that the distributions of the major scaffold molecules underlying the receptors are continually adjusted by actin treadmilling, revealing an unforeseen level of dynamics within the dense PSD complex.
Materials and Methods
Neuron culture, cDNA, and transfection.
Dissociated hippocampal neuron cultures were prepared from embryonic day 18 male and female rats as previously described (Frost et al., 2010). Cells were grown 3–4 weeks in culture and transfected ∼36–48 h before experiments. cDNAs were obtained or produced as follows (with original sources): SEP-GluA1, SEP-GluA2, and SEP-GluA2 1–880 (R. Huganir, Johns Hopkins University School of Medicine, Baltimore, MD); mCherry (mCh) or PATagRFP-tagged PSD-95 by subcloning PSD-95-GFP (D. Bredt, Eli Lilly and Company, Indianapolis, IN) at HindIII-EcoR1 into N1 versions of the fusion protein constructs; Membrane-mCh by subcloning mCh into Membrane-YPF (Clontech); GKAP-mCh by subcloning from GKAP-GFP (SAPAP2, G. Feng, Massachusetts Institute of Technology, Cambridge, MA) into mCh-C1; Shank3-mCh by subcloning Shank3 (P. Worley, Johns Hopkins University School of Medicine, Baltimore, MD) into mCh-N1 at the HindIII-EcoRI sites; Homer1c-mCh by subcloning from Homer1c-GFP (P. Worley) to NheI-AgeI sites of mCh-N1; mCh (R.Y. Tsien, University of California, San Diego, La Jolla, CA); PA-TagRFP (V. Verkhusha, Albert Einstein College of Medicine of Yeshiva University, Bronx, NY); Ruby-Lifeact (R. Wedlich-Soldner, Max Planck Institute of Biochemistry, Martinsried, Germany). Individual coverslips were transfected with 0.5–0.75 μg of cDNA for each expression construct using Lipofectamine 2000 (Invitrogen).
Live-cell confocal microscopes.
Live-cell imaging was performed on two confocal microscopes. Experiments for morphology quantification (see Figs. 2, 5A–E, 6) were performed on a spinning disk confocal system (Andor Technology). This system consisted of a CSU-22 confocal (Yokagawa) with Orca-ER CCD camera detector (Hamamatsu) mounted on the side port of an Olympus IX-81 inverted microscope with laser excitation provided by a 5W KrAr laser (Coherent, Inc.) and emission filters (Semrock) positioned by a motorized filter wheel (Sutter Instrument Co.). Imaging was performed with a 60× 1.42 numerical aperture (NA) oil-immersion objective with additional 1.6× magnification in the light path and 1.2× magnification placed between the confocal and the camera, yielding a final effective pixel size of 56 nm. In some cases the 1.6× magnification was omitted, yielding a pixel size of 90 nm. Z-stacks of images were collected at each time point with z step size of 0.5 μm. Acquisition was controlled by iQ software. Photobleaching and subsynaptic protein distribution measurements (Fig. 1; see Figs. 3, 4, 5F–J, 7) were performed using the point-scanning confocal of an LSM 5Duo on an inverted Zeiss AxioObserver microscope with a 63× 1.4 NA oil-immersion objective and software acquisition control by Zen. Laser output powers were (in mW): 488 nm (75), 561 nm (150), and 405 nm (100). Pinhole size was set to 1 Airy unit, and a 4× optical zoom was used, yielding a pixel size of 70 nm.
Figure 2.

Morphology of AMPAR clusters is continuously dynamic and coordinated with morphology of the PSD. A, Confocal image of a region of dendrite (left) from a neuron expressing Membrane-mCh and both SEP-GluA1 and SEP-GluA2. Example synapse (right) is from the spine marked with an arrow. Scale bar, 1 μm. B, Receptor cluster EF (EF = length/breadth) for 4 example synapses imaged every 2 min for 60 min. C, SEP-GluA1,2 fluorescence intensity of the synapses in B. D, CV of EF and of integrated SEP-GluA1,2 fluorescence intensity for all imaged synapses on 1 neuron. The best linear fit to the data (black line) revealed no correlation (r2 = 0.0089, p = 0.25; n = 37 synapses). Group mean r2 = 0.0393, p = 0.11; n = 67 synapses, 4 neurons (data not shown). E, Time-lapse images of a synapse from a neuron expressing PSD-95-mCh and SEP-GluA1. Dashed lines represent regions used for intensity profiles in F. Scale bar, 1 μm. F, Line profiles measuring fluorescence intensity across three different axes from the synapse shown in E. Fluorescence normalized to peak intensity value in that dimension. Red, PSD-95-mCh; green, SEP-GluA1. G, EF over time for PSD (red) and AMPAR cluster (green) measured in three synapses. H, EF ratio (the quotient of PSD EF and receptor cluster EF) calculated at single synapses and averaged for each neuron (n = 4 neurons). I, EF of PSD and AMPAR cluster averaged over time (20 min) for each synapse in a neuron. Average EF values were strongly correlated (group mean r2 = 0.85, n = 4 neurons). J, CV of the EF of PSD and AMPAR cluster calculated for individual synapses (>20 min). The two measures also strongly correlated on a synapse-by-synapse basis (group mean r2 = 0.48, n = 4 cells).
Figure 5.

Actin acutely regulates synapse shape and subsynaptic AMPAR distribution. A, Confocal images of an AMPAR cluster (SEP-GluA1,2) over time. Arrow indicates application of 5 μm jasplakinolide. Scale bar, 1 μm. B, Confocal images of an AMPAR cluster (SEP-GluA1,2) over time. Arrow indicates application of 20 μm latrunculin. Scale bar, 1 μm. C, CV of AMPAR cluster EF analyzed in 10 min running bins. Jasplakinolide was added and remained in the bath as indicated by the thick black bar. The thin dashed bar indicates points before the time of drug application that are affected because the binned analysis includes some time points from after the application. Thin dashed line indicates CV of EF measured in fixed cells. n = 6 neurons. D, CV of AMPAR cluster EF analyzed in 10 min running bins. Latrunculin was added and remained in the bath as indicated by the bar. Bars as in C. n = 8 neurons. E, Percentage decrease in CV of EF between 5 and 15 min and 25–35 min bins. F, RF values over time at single synapses from neurons expressing GKAP-mCh + GKAP-GFP (triangle) or GKAP-mCh + SEP-GluA1,2 (square), or GKAP-mCh + SEP-GluA1,2 after application of jasplakinolide (circle). G, H, Cumulative frequency plots of the SD of RF calculated over 10 min (6 time points) at synapses in neurons treated as indicated. For jasplakinolide (5 μm) and latrunculin (20 μm) groups, the treatment was applied 5–10 min before image acquisition so that their effects were complete before t = 0. GKAP-mCh:SEP-GluA1,2: control n (synapses/neurons) = 137/19; jasp n = 73/9, lat n = 41/10, GKAP-mCh:GKAP-GFP n = 98/7. For G, p < 0.05 for latrunculin-treated (dot) compared with control (solid) and p < 0.01 for jasplakinolide-treated (dash) compared with control; for H, p ≪ 0.001 of randomized data compared with experimental data, Kolmogorov–Smirnov test. I, J, SD (I) or mean (J) RF for the groups in G and H. Kruskal–Wallis ANOVA (p ≪ 0.001) post hoc pairwise comparisons by Mann–Whitney U test *p < 0.05, n.s. p ≫ 0.05.
Figure 6.

Synaptic AMPAR retention is primarily actin-independent. A, Region from a neuron expressing SEP-GluA1 and SEP-GluA2 along with the probe Lifeact-Ruby that binds F-actin. Latrunculin (20 μm) was applied just before the image stack was acquired for t = 0. Scale bar, 5 μm. B, Spine marked in A by arrowhead imaged at the indicated times. Scale bar, 1 μm. C, D, Normalized fluorescence intensity over time for a group of experiments performed with the indicated treatments of either 0.1% DMSO (C) or 20 μm latrunculin (D). Red traces are Lifeact-Ruby fluorescence measured in spines (circles) or dendrites (triangles). Green traces are SEP-GluA1 fluorescence measured in spines (circles) or dendrites (triangles). n = 8 lat, 6 DMSO. E, Normalized fluorescence intensity measurements for mCh-tagged PSD scaffold proteins or Lifeact-Ruby with 20 μm latrunculin treatment as indicated. Neurons were grown for either 14–17 DIV (left) or 7–10 DIV (right) before imaging. n = 6 neurons for PSD-95, 6 GKAP, 4 Homer, 3 Lifeact (DIV 14–17), and n = 5 PSD-95, 6 GKAP, 7 Shank, 5 Homer, 3 Lifeact (DIV 7–10). For DIV 7–10, p values Mann–Whitney U test comparing baseline to mean of the two values flanking 10 min were 0.83 PSD-95, 0.03 GKAP, 0.007 Shank, and 0.012 Homer.
Figure 1.
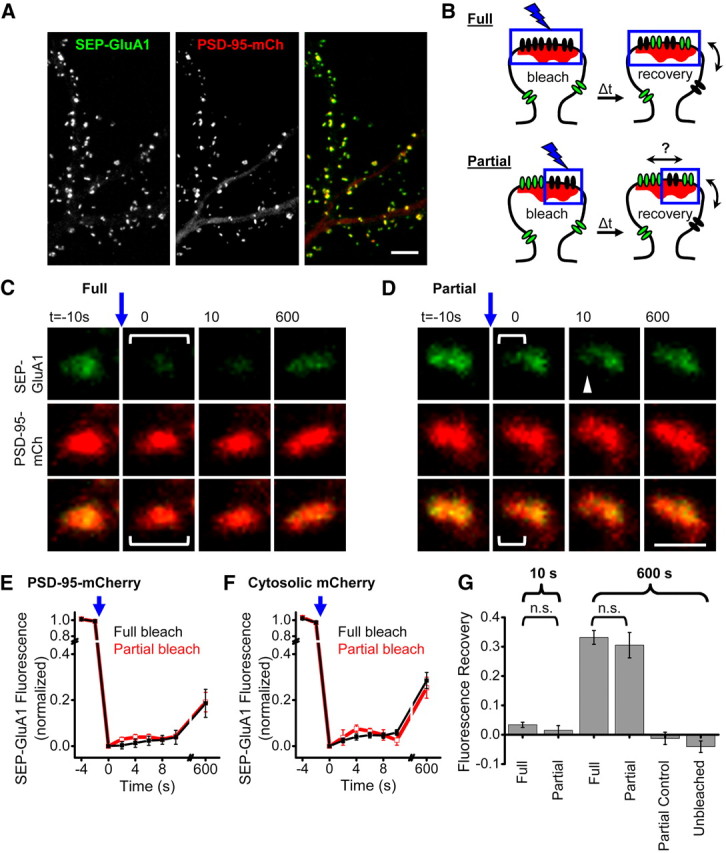
Synaptic AMPARs are confined to subsynaptic domains. A, Confocal image of cultured hippocampal neuron expressing PSD-95-mCh + SEP-GluA1. Scale bar, 10 μm. B, Experimental design for comparing partial and full synapse photobleaching to measure AMPAR intrasynaptic mobility. Fully photobleached synapses (top) will only recover fluorescent receptors via exchange of extrasynaptic receptors. Partially photobleached synapses (bottom) have an additional unbleached population of receptors that will speed recovery in the bleached subregion if they are mobile. C, D, Example synapses expressing SEP-GluA1 and PSD-95-mCh where all or part of the synaptic SEP-GluA1 was photobleached. Brackets represent the region targeted for photobleaching just before t = 0. Scale bar, 1 μm. E, F, SEP-GluA1 fluorescence recovery after either full or partial synapse photobleaching. Experiments were interleaved in neurons coexpressing either PSD-95-mCh or cytosolic mCh. n = 7–19 synapses from 7 to 9 neurons. G, SEP-GluA1 fluorescence recovery in synapses targeted for full or partial synapse photobleaching, and control synaptic regions at the indicated times following photobleaching. Number of synapses/neurons: At 10 s: full 34/9, partial 20/7. At 600 s: full 37/9, partial 10/4. Unbleached region of partially bleached synapses: 10/4. Control, unbleached synapses 10/4. n.s., p ≫ 0.05.
Figure 3.
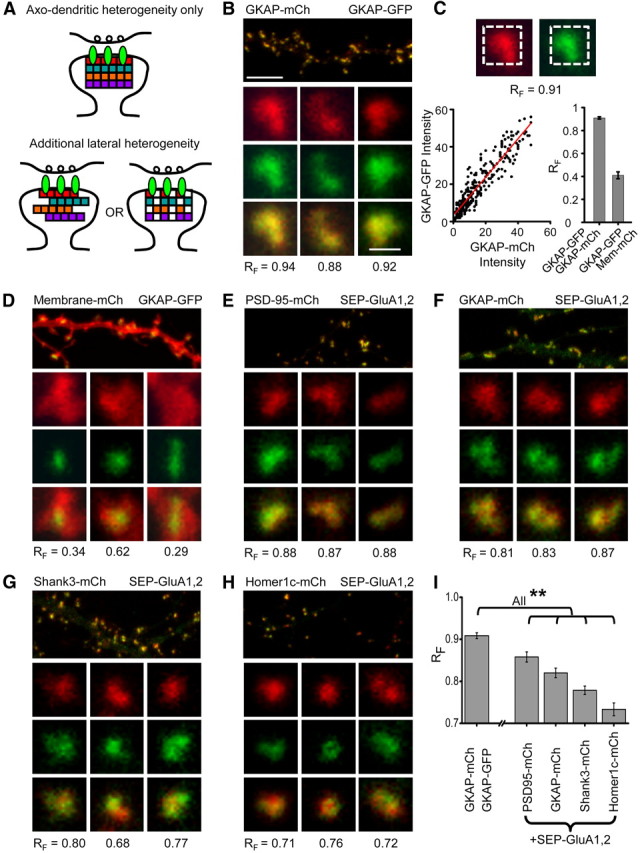
Subsynaptic AMPAR distribution patterns are diverse and differentially related to individual scaffold components. A, Model summarizing the known lamination of PSD proteins in the axo-dendritic axis (top) and two potential organizations of scaffold proteins that are in addition heterogeneous across the lateral extent of the synapse (bottom). B, Neuron expressing two different GKAP constructs, one tagged with mCh and the other with GFP. For each synapse, RF value shown was calculated as in C. Scale bars, 5 μm (top) and 1 μm (bottom), apply to all panels. C, Procedure for calculating the pixel-wise fluorescence correlation coefficient (RF). Dashed line on the images indicates the region in which correlation was calculated. Red line is the result of regression analysis to determine the best linear fit (bottom left graph). The bounds of RF measurements in this assay were empirically determined (bottom right graph) with interleaved measurements of synapses from neurons expressing GKAP-GFP and GKAP-mCh (upper bound) and GKAP-GFP:Membrane-mCh (lower bound). D, Neuron expressing membrane-mCh and GKAP-GFP. Insets show example spines with PSDs; the spines were generally as large as the cropped image, highlighting that membrane-mCherry is found in synapses but not targeted there specifically. E–H, Pairwise coexpression of the indicated scaffold components and SEP-GluA1,2. I, Mean RF for neurons expressing the indicated constructs. (n = 5, 7, 7, 7, 8 neurons in the order shown). Statistics: Kruskal–Wallis ANOVA (p ≪ 0.001) post hoc pairwise comparisons by Mann–Whitney U test, **p < 0.05.
Figure 4.

GluA1 PDZ ligand deletion does not alter receptor positioning or mobility. A, B, Dendrite regions from neurons expressing either full-length SEP-GluA1 (A) or SEP-GluA1 1–880, lacking the PDZ ligand (B), and PSD-95-mCh. Scale bar, 10 μm. C, Mean RF for neurons expressing the indicated constructs. (n = 4, 9, 12, 6 neurons in the order shown). Statistics: Kruskal–Wallis ANOVA (p ≪ 0.001) post hoc pairwise comparisons by Mann–Whitney U test, *p < 0.05. D, E, SEP-GluA1 fluorescence recovery after either full or partial synapse photobleaching. Experiments were interleaved in neurons coexpressing either SEP-GluA1 or SEP-GluA1 1–880. n = 9–31 synapses from 5–10 neurons.
Figure 7.
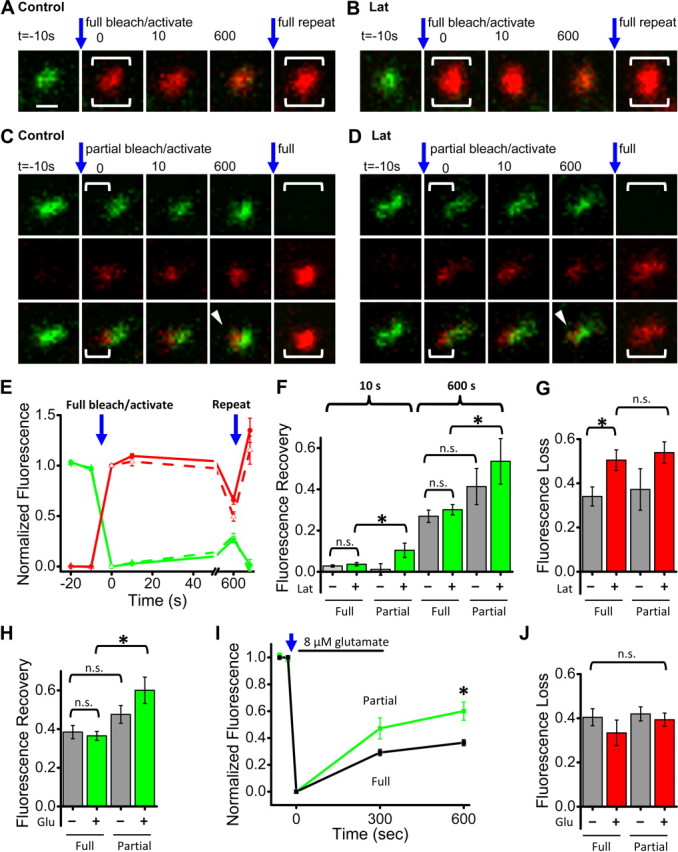
Subsynaptic AMPAR confinement persists even after actin filament depolymerization. A, B, Synapses from neurons expressing SEP-GluA1 and PSD-95-PATagRFP. After acquiring baseline images, a brief 405 nm laser pulse was applied to the full synapse to simultaneously bleach SEP-GluA1 fluorescence and activate PSD-95-PATagRFP fluorescence. A final bleach/activation step was applied to the full synapse after t = 600 s (large bracket). In B and D, neurons were treated with latrunculin (20 μm) for at least 5 min before and during imaging. Scale bar (in A), 500 nm, applies to A–D. C, D, Synapses from experiments conducted as in A and B, except that only a portion of the synapse was targeted (brackets). E, Quantification of normalized SEP-GluA1 (green) and PSD-95-PATagRFP (red) fluorescence over time, for synapses targeted for full photoactivation/photobleaching. Control (solid line, n = 27) or latrunculin treated (dashed line, n = 21). F, SEP-GluA1 fluorescence recovery measured at the indicated time points for either full or partial synapse photobleaching. Gray bars are from untreated neurons and green bars from latrunculin treated. Number of synapses/neurons: Full photobleaching: control 27/11, latrunculin 21/8, Partial photobleaching: control n = 10/6, latrunculin 12/6. *p < 0.05, n.s. not significant. G, PSD-95-PATagRFP fluorescence loss 600 s after photoactivation in either full or partial synapse targeting. Same synapses as in F. Gray bars are from untreated neurons and red bars from latrunculin-treated. *p < 0.05, n.s. not significant. H, SEP-GluA1 fluorescence recovery measured at 600 s for either full or partial synapse photobleaching in control conditions (gray) or with 5 min of 8 μm glutamate application (green). Full photobleaching: control n = 25/15, glutamate 31/12. Partial photobleaching: control 16/10, glutamate 15/7. Kruskal–Wallis ANOVA (p ≪ 0.001) post hoc pairwise comparisons by Mann–Whitney U test *p = 0.001, n.s. not significant. I, SEP-GluA1 fluorescence recovery measured at the indicated time points after full synapse photobleaching in control conditions (black) or with 5 min of 8 μm glutamate application (green). *p = 0.001, as in H. J, PSD-95-PATagRFP fluorescence loss 10 min after photoactivation for full or partial synapse targeting as indicated; gray bars, control, and red bars, glutamate treated. Kruskal–Wallis ANOVA n.s. not significant p = 0.44.
Imaging conditions.
Coverslips were placed in custom-made chambers (Four Hour Day) and cells were warmed from below using a heated objective collar so that the bath solution was ∼34°C. In most cases, imaging solution was perfused throughout the experiment at a rate of 1 ml/min through an inline solution heater (Warner Instruments). Experiments on the LSM 5Duo used a humidified, semiclosed chamber without perfusion. Extracellular imaging solution contained the following (in mm): 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, 10 HEPES, pH adjusted to 7.35 with NaOH. For experiments where jasplakinolide (Calbiochem), latrunculin (Sigma-Aldrich), glutamate (Sigma-Aldrich) or DMSO (American Bioanalytical) was applied, at the indicated time, 0.5 ml of 2× treatment solution was added to the 0.5 ml bath. Jasplakinolide and latrunculin were diluted from 1000× stock solutions in DMSO at 5 and 20 mm, respectively. Each condition thus resulted in a final concentration of 0.1% DMSO.
Fluorescence recovery after photobleaching.
Photobleaching of synaptic AMPARs was achieved using 488 nm laser excitation. Total bleaching time did depend on region number and size but was executed in all cases with 1.61 μs pixel dwell time (equal to the acquisition scan rate), and 4–5 iterations at 30–40% of the available 488 nm laser power. Bleaching parameters were adjusted within this range at the beginning of each experiment to provide near total bleaching of SEP fluorescence while minimizing mCherry bleaching. Time = 0 image acquisition followed directly after photobleaching. For full photobleaching a square or rectangular bleaching region was placed around an entire synapse. In contrast, a small region was placed at the edge of synapses for partial synapse photobleaching. This resulted in a bleaching spot nearly the size of the microscope point spread function with minimal affect at the opposite side of the synapse. On average 35% of total synaptic AMPAR fluorescence was lost during partial synapse bleaching. Each experiment contained some synapses that were targeted for partial synapse photobleaching while others were fully bleached. Synapses targeted for partial photobleaching that inadvertently resulted in decreased fluorescence over >3/4 of the synapse area were excluded.
Simultaneous photoactivation of PSD-95-PATagRFP and photobleaching of SEP-GluA1.
This procedure was similar to the SEP photobleaching above except that the 405 nm laser was used for simultaneous photoactivation and photobleaching in targeted regions. Ten to 12 rapid iterations were used at 70–80% of the available 405 nm laser power. On average, 49% of total synaptic AMPAR fluorescence was bleached. PATagRFP was submaximally (∼2/3) photoactivated. This was necessary because maximal photoactivation of PATagRFP led to spatially unrestricted SEP photobleaching in partially targeted synapses.
Image analysis.
Image processing and analysis was performed in MetaMorph (Molecular Devices) after export from iQ. Values measured in MetaMorph were output to Excel (Microsoft) and further processing, statistics, and graphing were performed in Origin (OriginLab). Images of single synapses were interpolated 4× in MetaMorph before export and figure assembly in PowerPoint. Images were routinely background subtracted before analysis and display. Brightness and contrast were linearly adjusted. In cases where red and green intensity were not compared channels were adjusted separately to maximize clarity of the individual channels. In cases where red and green intensity were compared directly, brightness and contrast were adjusted in an identical fashion for red and green images though they were optimized for individual experiments.
Synapse morphology was quantified essentially as described previously (Blanpied et al., 2008) using custom-written analysis journals in MetaMorph. Image z-stacks at each time point were maximally projected in the z dimension and digitally interpolated 4×. Images of single synapses were segmented by thresholding to delineate the AMPAR cluster or PSD from the surrounding background image. This threshold was generally set as 0.5 times the maximum fluorescence intensity within the synapse. Elliptical form (EF) measurements over time were 3-point smoothed, and coefficient of variation (CV) was calculated with a running 10 min bin. The degree of morphological change was also analyzed by measuring unsmoothed, frame-to-frame differences in EF, which had a higher time resolution but is considerably more noisy than the calculation of CV as above (data not shown). Most importantly, this analysis clearly supported the kinetic conclusion in Figure 5, C and D (see below), that elimination of actin treadmilling suppressed morphological changes of the synapse in <2 min. Fluorescence intensity was integrated within oval regions drawn tightly around synapses.
To measure subsynaptic protein distribution, we first systematically corrected images for imperfection in chromatic alignment in the LSM 5Duo. To match the imaging conditions of experiments, 0.1 μm microspheres (Invitrogen, Invitrogen) were placed onto cells in culture and incubated overnight so that they would become internalized or affixed to the cell surface. The XY displacement measured between red and green images of the microspheres was corrected in each biological image by a sub-pixel XY shift. Images were then background subtracted, and rectangular regions of interest were drawn around each synapse, minimizing the number of included background pixels. For each region, the pixel-wise fluorescence correlation coefficient RF was calculated in MetaMorph. An average RF value was calculated for all synapses on a single neuron and then group averages were calculated from these values.
SEP-GluA FRAP.
For full synapse photobleaching, a single region was drawn around the entire synapse using the PSD-95-mCherry (PSD-95-mCh) image. Integrated fluorescence intensity of SEP-GluA1was quantified at each time. For partial synapse photobleaching, two regions were drawn. The first “bleached” region was drawn based on where the original bleaching region of interest was targeted. An additional “control” region of equal size was placed on the opposite side of the synapse. These two regions sometimes had to be moved for analysis of later time points; in these cases, the PSD-95-mCh image was used to track obvious PSD features that allowed region placement to be updated to track the proper region of the synapse matched to previous time points. In several cases, synapse morphology changed substantially within the 10 min experiment and we were not able to reposition the analysis regions with confidence, and these synapses were discarded from further analysis. Synapses or partial bleaching regions that were <70% bleached with respect to baseline fluorescence intensity were excluded from further analysis. Baseline fluorescence intensity was normalized to 1.0 and FRAP recovery was calculated as the fluorescence increase between t = 0 (immediately after SEP photobleaching) and the indicated time point. Fractional bleaching was also assessed for partial synapse photobleaching. This was calculated by placing a region around the entire synapse and calculating the fraction of total fluorescence lost during photobleaching. We calculated this because the area affected by photobleaching was not equal for each synapse studied. We reasoned that the fraction of unbleached receptors could influence the recovery within the bleached region. However, we found no correlation between the fractional photobleaching and the magnitude of FRAP in the bleached region (data not shown), suggesting that this effect did not complicate interpretation of our results, and consistent with minimal intrasynaptic exchange.
Simultaneous SEP-GluA1 photobleaching and PSD-95-PATagRFP photoactivation.
Analysis of SEP-AMPAR FRAP was performed exactly as described above. To quantify mobility and loss of synaptic PSD-95-PATagRFP following photoactivation, we normalized the fluorescence in the targeted region to 1.0 at t = 10 s. Then, fractional fluorescence was measured at subsequent time points. Normalization to the 10 s time point was necessary because PATagRFP exhibits a nonfluorescent state, which can persist for seconds, following photoactivation but before stochastically entering the excitable state (Subach et al., 2010; data not shown; V. Verkhusha, personal communication). Thus, accurate quantification of PSD-95 content in the targeted region was best achieved by normalizing to this time point. Finally, in all synapses we reactivated PSD-95-PATagRFP which permitted visualization of the entire synapse morphology at the end of the experiment and confirmed the presence of PSD-95 on the initially untargeted regions of partially targeted synapses.
Statistics.
Where means are presented, the accompanying “error” is the SEM. Pairwise statistical tests were performed using the Mann–Whitney U test. For experiments with greater numbers of conditions, Kruskal–Wallis one-way ANOVA was used to test for overall differences among group means. Then, pairwise comparisons were performed with Mann–Whitney U test. Kolmogorov–Smirnov tests were applied in Figure 5, G and H (see below). In all cases, means were considered significantly different if the test reported p < 0.05.
Results
Intrasynaptic mobility of synaptic AMPA receptors is minimal
To distinguish between the corral and matrix-type mechanisms constraining AMPARs at synapses, we assayed the intrasynaptic mobility of receptors by measuring FRAP of SEP-tagged GluA1. Hippocampal neurons grown 21–27 d in culture and transfected with SEP-GluA1 were imaged using time-lapse confocal microscopy (Fig. 1A). We compared recovery in synapses that were entirely photobleached to those in which only a subdomain (approximately half) was bleached, an approach we refer to as partial synapse photobleaching. Partial synapse photobleaching provides a measure of intrasynaptic mobility, because the bleached subregion can recover fluorescence by exchange of receptors within the synapse or with extrasynaptic regions. Thus, if there is exchange within the synapse, these subregions will recover SEP-GluA1 fluorescence more quickly than a fully bleached synapse (where there is only access to the extrasynaptic unbleached pool) (Fig. 1B). Prior work suggests that photobleaching of a whole spine will reveal substantial exchange of the AMPAR population within 10 min (Sharma et al., 2006; Heine et al., 2008; Frischknecht et al., 2009). In contrast, modeling predicts that receptors, if mobile, would disperse within the synapse on a time scale as short as hundreds of milliseconds (Blanpied et al., 2008; Heine et al., 2008; Bressloff and Earnshaw, 2009; Santamaria et al., 2010). Thus, we first examined recovery 10 s after photobleaching but also measured recovery at 10 min in the same experiments.
Following full synapse photobleaching, we detected little fluorescence recovery after 10 s (3.4 ± 0.9%; n = 34 synapses in 9 neurons; Fig. 1C,G), suggesting that the population of rapidly exchanging receptors in these synapses is very small. After 10 min, 33.2 ± 2.4% of synaptic receptor fluorescence was recovered (n = 37 synapses in 9 neurons; Fig. 1C,G), consistent with previous reports (Sharma et al., 2006; Heine et al., 2008; Frischknecht et al., 2009). Following partial synapse photobleaching (performed during the same image series on neighboring synapses), recovery was no different after 10 s than in fully bleached synapses (1.5 ± 1.6% for partial, n = 20 synapses in 7 neurons, p = 0.53 compared with fully bleached synapses; Fig. 1D,G), suggesting a very small fraction of receptors are able to move within the synapse on this time scale. Remarkably, recovery after 10 min was also similar following partial and full synapse photobleaching (30.5 ± 4.3% for partial, n = 10 synapses on 4 neurons, p = 0.51 compared with full; Fig. 1C,D,G). The image acquisition itself caused minimal photobleaching, as fluorescence intensity in neighboring unbleached synapses remained nearly constant (96 ± 2% of baseline after 10 min). We also measured fluorescence in the unbleached region of each synapse that was partially photobleached, and detected no change (−1.2 ± 2%) in fluorescence intensity there after 10 min. Thus, if any unbleached receptors did diffuse into the bleached portion of the synapse, this rate was no faster than the overall rate of exchange between the synaptic and extrasynaptic compartments.
In these experiments, PSD-95-mCh expression was beneficial for targeting photobleaching regions specifically to small subsynaptic AMPAR domains and for tracking the targeted and untargeted regions during analysis. Because PSD-95 is thought to be a key component of mechanisms to capture and stabilize AMPARs in the synapse (Schnell et al., 2002; Bats et al., 2007; Newpher and Ehlers, 2008; Opazo and Choquet, 2011), we tested whether PSD-95-mCh overexpression affected either the rate of receptor exchange or intrasynaptic mobility. In an additional series of experiments interleaving neurons coexpressing SEP-GluA1 with either PSD-95-mCh or free, cytosolic mCherry, we found no difference in the fractional fluorescence recovery (Fig. 1E,F). In neither case did the rate of recovery following partial synapse photobleaching differ from the rate of recovery in fully bleached synapses. Interestingly, there was a trend toward slower recovery in PSD-95-mCh-expressing neurons (Fig. 1E,F; p = 0.090), consistent with the idea that this scaffold, which increases synapse strength, size, and complexity (El-Husseini et al., 2000; Stein et al., 2003; Nikonenko et al., 2008), regulates receptor retention. Nevertheless, intrasynaptic mobility by these measures was minimal even in the absence of PSD-95 overexpression, consistent with the model that the PSD forms a matrix that severely restricts receptor movement within the synapse.
Morphological dynamics of synaptic AMPA receptor clusters
The positional stability of receptors observed within the PSD raises the possibility that the overall structure of the receptor cluster is quite rigid for long periods. However, such rigidity would contrast with previous observations of PSD flexibility (Blanpied et al., 2008). To investigate potential flexibility of the overall receptor cluster in more detail, we performed extended time-lapse imaging for neurons transfected with SEP-GluA1 and SEP-GluA2. Coexpression of mCh-membrane, a construct targeted to the plasma membrane via inclusion of a palmitoylation motif, confirmed that spines contained synaptic clusters of SEP-labeled AMPARs that could be demarcated from the sparse receptors on the dendrite surface (Fig. 2A). Receptor clusters under study were in fact synaptic, as they colocalized with PSD scaffold markers within spines, were quenched by cell-impermeant acidic solution but not affected by a cell-permeant basic solution (Miesenböck et al., 1998; Ashby et al., 2004; Kopec et al., 2006), and colocalized with presynaptic terminals immunopositive for Bassoon (data not shown) (tom Dieck et al., 1998).
Using this time-lapse imaging approach, we found that individual synaptic clusters of AMPARs exhibited substantial and continuous changes in morphology (Fig. 2A). We quantified the shapes of individual synapses at any single time point by measuring their EF (EF = length/breadth), a simple but robust and sensitive measure of morphology (Blanpied et al., 2008). Synapse EF varied continuously over time, was independent for individual, even neighboring synapses, and changed with various magnitude and directionality (Fig. 2B). Because synapse morphology is related to synapse strength, we tested whether synaptic AMPAR number increased or decreased during morphology transitions. For individual synapses we quantified both AMPAR cluster morphology and integrated SEP fluorescence intensity. In this analysis, even though most receptor clusters underwent profound changes in morphology, such changes were not accompanied by clear changes in SEP fluorescence intensity (Fig. 2B,C). To further quantify this, we tested whether the CV (CV = SD/mean) of receptor cluster EF correlated with CV of SEP fluorescence intensity, as would be predicted if morphology changes were accompanied by addition or removal of synaptic AMPARs. Consistent with the stable SEP fluorescence shown in Figure 2C, we found the CV of receptor cluster intensity to be substantially smaller than the CV of EF (mean CVintensity = 0.018 ± 7.8E-4, CVEF = 0.076 ± 0.0047, p < 0.01). We also found no correlation between these two measures for either a single neuron or a larger population from a group of neurons (r2 = 0.0089, p = 0.25; n = 37 synapses from representative neuron analyzed in B-C; r2 = 0.0393, p = 0.11; n = 67 synapses on 4 neurons) (Fig. 2D). From this, we conclude that synapse morphology can change even when a principal postsynaptic determinant of synaptic strength, receptor number, remains constant. Thus, although the overall topology of the postsynaptic complex remains flexible, few receptors appear freely mobile over large regions of the synapse, suggesting that the pattern of receptor distribution within the synapse is controlled by characteristics of the PSD.
If changes in the shape of the PSD determine the shape of the receptor cluster at the synapse, the structures of the two should be highly correlated over time. To test this, we directly compared PSD and AMPAR cluster morphology in neurons expressing both PSD-95-mCh and SEP-GluA1. At single synapses, time-dependent changes in AMPAR distribution directly matched those of the PSD-95-labeled scaffold (Fig. 2E). Line profiles through single synapses clearly demonstrated the fine-scale similarity between fluorescent AMPARs and PSD-95 protein distribution (Fig. 2F). To compare morphology, the EF of PSDs and AMPAR clusters was measured at each time point. Absolute differences between the two EF values were small and the measurements covaried over time (Fig. 2G). We calculated for each synapse the quotient of the EF of its AMPAR cluster and the EF of its PSD (the EF ratio) at each time point. The EF ratio remained close to 1.0 throughout the imaging (Fig. 2H, n = 4 neurons). To examine the correlation in shape across many synapses, we averaged the EF measured once per minute for 20 min and compared this to the averaged EF of the receptor cluster (Fig. 2I). The two averaged EF values were highly correlated across synapses (r2 = 0.85 ± 0.020, n = 4 neurons). To examine this correlation over time, we calculated the CV of EF at single synapses. The CV of PSD EF was well correlated with the CV of the AMPAR cluster EF (Fig. 2J; r2 = 0.48 ± 0.018, n = 4 cells) whereas randomized pairings were uncorrelated (r2 = 0.017 and 0.020 for EF and the CV of EF, respectively). Thus, in a population of synapses with widely varying shapes and sizes, and during substantial, rapid temporal variation, the shape of the PSD predicts the shape of the AMPAR cluster. We note also that the CV of the AMPAR cluster EF correlated only very weakly with either the synapse size or the intensity of SEP-GluA fluorescence (r2 = 0.04 ± 0.01, N = 12 neurons), and was not different in cells expressing only receptors from those also overexpressing mCh-tagged scaffold proteins (Membrane-mCh coexpression CV of EF = 0.086 ± 0.01, GKAP-mCh coexpression CV of EF = 0.090 ± 0.008 p = 0.67). Thus, while overexpression of scaffold proteins likely alters synapse strength or morphology, synapse structural dynamics appear independent of these effects. A straightforward conclusion from these results is that reshaping of the PSD drives global changes in the structure of the receptor cluster, even while confinement of resident AMPARs within very local PSD subdomains is preserved.
Subsynaptic AMPA receptor distribution patterns are differentially correlated with individual components of the PSD scaffold
The preceding observations indicate that malleable global morphology of the PSD controls the overall shape of the synaptic receptor cluster. We next considered mechanisms within single PSDs that might influence receptor positioning at that synapse. Numerous proteins are known to regulate synaptic AMPAR number (Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Elias and Nicoll, 2007), and in particular interactions between the canonical, multidomain scaffold PSD-95 and the transmembrane AMPAR binding proteins (TARPs, typified by Stargazin) are essential for integration of extrasynaptic AMPARs into the PSD (Chen et al., 2000; Bats et al., 2007; Opazo et al., 2010). However, it is unclear whether the distribution of receptors within the PSD is established via continued linkage to PSD-95 (MacGillavry et al., 2011). Even in this case it is not clear what, in turn, would establish the distribution of PSD-95 (Chen et al., 2008, 2011; Swulius et al., 2010). Indeed, the organization of scaffolds within the PSD has been difficult to assess, particularly in living synapses. Immunological labeling of PSD scaffold proteins has revealed a lamination of PSD components along the axo-dendritic axis of the PSD (Valtschanoff and Weinberg, 2001; Dani et al., 2010). However, measuring the distribution across the lateral, en face dimension of single synapses, the one that is likely to actually determine receptor position, has been difficult to achieve in part because antibody labeling is typically kept far from saturation to maintain specificity. Thus, we developed an approach to measure PSD scaffold organization relative to receptor distribution within single synapses that does not rely on antibody labeling.
To investigate PSD scaffold organization in live cells, we used high-resolution confocal microscopy on neurons expressing SEP-GluA1,2 paired with expression of one of four scaffold proteins tagged with mCh. We could then quantify the relative subsynaptic distribution of synaptic AMPARs with respect to particular scaffold protein types by calculating the pixel-wise fluorescence correlation coefficient, which we termed RF. PSDs are thin and the axo-dendritic displacement of proteins is only 20–30 nm (Valtschanoff and Weinberg, 2001). In width, however, they can be >20 times as large (Harris and Stevens, 1989). Therefore, the axo-dendritic offset will contribute little to the overall RF. Thus, if scaffold types differ from one another in their distribution with respect to receptors, we would expect their RF values to differ (Fig. 3A). On the other hand, if the PSD scaffolds differ from one another only along the axo-dendritic axis, we would predict that pairwise relation of each scaffold type to the receptor will be equal.
To assess the dynamic range of this approach, we first coexpressed two different GKAP constructs, one tagged with mCh and the other with GFP, because we expect two differently tagged versions of the same scaffold protein should provide a maximal RF. As expected, the pattern of expression of these constructs essentially completely overlapped within the cell (Fig. 3B) and at individual synapses (Fig. 3B,C). Because pixel intensity contains shot noise proportional to the square root of the number of collected photons, the correlation between the two channels will inevitably depend on signal strength. Accordingly, we adjusted the scanning laser power (over a ∼2-fold range) between cells that expressed different amounts of tagged proteins so as to maintain consistent image intensity across cells and constructs. The voltage of the photomultiplier detector was held constant to minimize differences in instrumentation noise between samples. For single synapses, the individual pixel intensity values of the GFP and mCh channels were plotted as in Figure 3C, and the RF between the two was calculated. The result of this analysis was an RF of 0.91 ± 0.01 (n = 5 neurons) between GKAP-GFP and GKAP-mCh, providing the maximum correlation for identically distributed synaptic proteins achievable in practice given instrumentation and acquisition noise. (Note that this is within the image of the PSD not including intersynaptic regions of low expression.) The minimum RF in this assay would likely be between an exclusively PSD-resident protein and one localized nonspecifically at the synapse. Therefore, we measured RF between GKAP-GFP and membrane-mCh, a marker that diffuses freely over the plasma membrane but also enters the synaptic membrane with a distribution presumably unrelated to the distribution of GKAP (Fig. 3C,D). Between these two proteins, the mean RF within the PSD was 0.41 ± 0.03 (n = 8 neurons).
Given these bounds for the assay, we then measured RF for each of four canonical PSD scaffold components with respect to AMPAR fluorescence (Fig. 3E–H). Three observations were apparent. First, the correlation of receptor with each scaffold was less than the maximum detectable (GKAP-mCh:GKAP-GFP). PSD-95-mCh had the highest correlation coefficient of the scaffolds tested (0.86 ± 0.012, n = 7 neurons), indicating not surprisingly that its subsynaptic distribution was very similar to the AMPARs at the same synapses. However, this was lower than the GKAP to GKAP correlation (p = 0.0094), potentially because some PSD-95 molecules are bound not to AMPARs but to NMDA receptors, neuroligin, or other synapse components (Irie et al., 1997; Kornau et al., 1997; Chen et al., 2000). On the other hand, even the lowest correlation coefficient value measured for Homer (0.73 ± 0.020, n = 8 neurons) was substantially above the low end of the dynamic range of our assay (p = 9.4E-4), reflecting close association of this protein with AMPARs despite linkages to other receptor types and other proteins throughout the spine (Tu et al., 1998; Lu et al., 2007).
Second, the RF values of the different scaffolds were each different from one another. The variety and range of correlation coefficients support the idea that the different types of scaffolds play distinct roles in positioning AMPARs. Third, the correlation values varied systematically across the set of scaffolds. The rank order of correlation coefficients (PSD-95 > GKAP > Shank > Homer) was well predicted by both the mean physical distance (Valtschanoff and Weinberg, 2001; Dani et al., 2010) and the number of expected intermolecular links (Kim and Sheng, 2004) between the scaffold and receptor (Fig. 3I). These experiments require scaffold overexpression, which might be expected to increase size of some PSDs or perturb synapse organization. However, our analysis includes PSDs of nearly any size, and there was only a very weak correlation (r2 ≪ 0.1) between synapse size and RF measured for GKAP-mCh and receptors (data not shown). In addition, PSDs in neurons expressing each of the scaffolds covered a nearly identical range of sizes, and we excluded cells with ectopic or unusually high levels of mCh fluorescence. This suggests that the order we observed is insensitive to scaffold overexpression level.
Role of the GluA1 C-terminal PDZ ligand
Subsynaptic positioning and restricted movement of AMPARs could be mediated by interactions of the receptor C terminus with intracellular partners. Deletion of the C terminus of GluA2 does not affect its mobility in the synapse as measured by antibody tracking (Bats et al., 2007). Similarly, GluA1 contains a PDZ domain-binding ligand capable of binding SAP97 (Leonard et al., 1998; Shepherd and Huganir, 2007). Although deletion of this ligand does not affect basal transmission or induction of plasticity (Hayashi et al., 2000; Kim et al., 2005), the role of this interaction in positioning or retention of synaptic receptors is not known. To determine this, we examined mobility of GluA1 lacking the terminal amino acids (SEP-GluA1 1–880). Truncated receptors trafficked to the surface and accumulated at synapses in a manner indistinguishable from the full-length receptor (Fig. 4A,B) consistent with previous report (Lin et al., 2009). RF values (measured with respect to GKAP-mCh) for synapses expressing SEP-GluA1 1–880 were not different from those measured for full-length SEP-GluA1 (SEP-GluA1 1–880 RF = 0.78 ± 0.01, SEP-GluA1 RF = 0.77 ± 0.01 p = 0.80; Fig. 4C). This suggests that deletion of the PDZ ligand was not sufficient to alter AMPAR distribution within synapses. Next we tested whether this deletion was sufficient to alter AMPAR mobility. Full synapse photobleaching revealed no difference in recovery between full-length and truncated receptors (41.4 ± 5.6% SEP-GluA1, n = 9 synapses in 5 neurons, 37.7 ± 3.0% SEP-GluA1 1–880, n = 31 synapses in 10 neurons p > 0.9 at 600 s; Fig. 4D). Comparing recovery after full or partial synapse photobleaching, we did not detect additional intrasynaptic mobility for SEP-GluA1 1–880 (33.7 ± 8.6% for partial, n = 8 synapses in 5 neurons; p = 0.54 compared with full at 600 s; Fig. 4E). Thus, we conclude that the direct association between GluA1 and SAP-97 is dispensable for the strong immobilization within the PSD matrix.
Actin-dependent restructuring of the synaptic receptor cluster and internal protein organization
Rearrangement of the actin cytoskeleton is required for many forms of synaptic structural and functional plasticity (Kim and Lisman, 1999; Krucker et al., 2000; Zhou et al., 2004; Blanpied et al., 2008; Gu et al., 2010). We thus hypothesized that the ongoing plasticity of synapse organization we observed would be actin-dependent. First, we asked whether constitutive reshaping of the synaptic AMPAR cluster required ongoing actin turnover. Following a 20 min baseline, 5 μm jasplakinolide was added to stabilize existing actin filaments, and this treatment rapidly stopped ongoing morphology changes of the synaptic AMPAR cluster (Fig. 5A). Indeed, CV of EF was abruptly reduced within 1–2 min to levels seen in fixed cells (Fig. 5C and as indicated by a quickly reduced magnitude of frame-to-frame variation in EF, see Materials and Methods). Strikingly, application of 10 μm latrunculin A to depolymerize filaments resulted in similar, rapid stabilization of receptor cluster dynamics (Fig. 5B,D). DMSO vehicle had no effect (Fig. 5E). Thus, ongoing remodeling of the actin cytoskeleton drives continual changes in the shape of the synaptic receptor cluster.
This strong, actin-dependent synapse reshaping seemed likely to drive reorganization of proteins within the PSD. On the other hand, the density of the PSD and the limited mobility of receptors within it suggests that PSD internal organization might be time-invariant. Little is known about internal organization of live synapses, so to resolve this issue, we probed whether the spatial scaffold-receptor correlation is maintained over time. For these experiments, we chose GKAP because of its mid-range RF value. At individual synapses, the spatial correlation between GKAP-mCh and SEP-GluA1,2 varied substantially over time (Fig. 5F). Synapses from neurons coexpressing GKAP-mCh + GKAP-GFP showed much smaller changes in RF over time (Fig. 5F), as did GKAP-mCh + SEP-GluA1,2 synapses from fixed cells (data not shown).
To test the role of actin remodeling in controlling intrasynaptic reorganization, we measured RF after treatment with jasplakinolide or latrunculin. Disrupting actin remodeling did not increase but in fact reduced variation of RF over time (Fig. 5F). We quantified the magnitude of variation by measuring the SD of six RF measurements spaced at 2 min intervals. Treatment with either latrunculin or jasplakinolide promoted a leftward shift in the cumulative distribution of this measure (Fig. 5G, latrunculin p = 0.023, jasplakinolide p < 0.001). In contrast, randomizing the measured RF values in sets of 6, then measuring the SD, resulted in a rightward shift of the curve, indicating that chance would in fact drive these values higher (Fig. 5H, p < 0.001). Synapses from neurons coexpressing GKAP-mCh + GKAP-GFP also had a left-shifted distribution, offering further evidence that intrasynaptic organization varied less in the absence of actin remodeling. Analysis of the mean SD of RF confirmed these observations (Fig. 5I). Application of latrunculin or jasplakinolide reduced the SD of RF between GKAP-mCh and SEP-GluA1,2 (latrunculin p = 0.024, jasplakinolide p = 0.0067). Importantly, momentary changes in RF were not due to imprecision in fluorescence intensity measurements, because the SD but not the mean of RF was significantly lower when fixed cells were repeatedly measured under identical conditions (Fig. 5I,J; p = 0.0029 for SD of RF control compared with fixed). In addition, the larger SD of GKAP-mCh + SEP-GluA1,2 in live cells was not due to a higher mean RF, because measured RF values for GKAP-mCh + GKAP-GFP were also consistently greater than those measured for GKAP-mCh + SEP-GluA1,2 (Fig. 5J). Thus, the subsynaptic distribution of core PSD scaffold proteins and AMPARs is actively regulated by the spine actin cytoskeleton.
Synaptic AMPA receptor retention and subsynaptic confinement are primarily actin-independent
Depolymerization of the actin cytoskeleton has been previously reported to cause a decrease in synaptic AMPAR content over tens of minutes to tens of hours (Allison et al., 1998; Kim and Lisman, 1999; Zhou et al., 2001; Gu et al., 2010) leading to the notion that the actin cytoskeleton “anchors” synaptic AMPARs (Halpain, 2000; Cingolani and Goda, 2008). To test whether the actin-based mechanism responsible for AMPAR cluster dynamics was distinct from receptor maintenance, we measured loss of AMPARs from the synapse over the 10 min following latrunculin application. In contrast to the rapid elimination of morphological dynamics during this time, SEP-GluA1,2 intensity showed only a very modest decrease (Fig. 6A–D; 7.2 ± 1.3%; p = 0.0081 latrunculin vs DMSO), similar to previous reports (Gu et al., 2010). Latrunculin was highly and rapidly effective in disrupting spine F-actin as reported by low-affinity actin probe Lifeact (Riedl et al., 2008) (Fig. 6A–D). We confirmed in fixed cells that the loss of Lifeact signal parallels the loss of phalloidin staining following latrunculin exposure (data not shown).
These data strongly suggest that receptor retention on this acute time scale was primarily actin-independent. It has been reported that a subset of scaffold molecules are lost from the PSD following latrunculin application (Kuriu et al., 2006), and so we examined the effect of latrunculin on synaptic scaffold content in our cultures. In our neurons cultured 14–17 d, we found that latrunculin lead to loss of <10% of mCh-tagged PSD-95, GKAP, or Homer1c (Fig. 6E). Nevertheless, in neurons cultured only 7–10 d, latrunculin application prompted loss of Homer 1c, GKAP, and Shank, with no effect on PSD-95 (Fig. 6E), suggesting that the developmental regulation of synapse stability (Zhang and Benson, 2001) is reflected in enhanced actin-resistance of the scaffold matrix.
The slow decrement of receptors does not support a simple anchoring role for actin. To explore whether actin regulated either overall receptor exchange rate or positional stability within the synapse (Rust et al., 2010), we again used a subsynaptic receptor photobleaching approach, now along with simultaneous photoactivation of PSD-95-PATagRFP to detect potential actin-dependent alteration of scaffold mobility (Fig. 7A,B). Depolymerization of actin filaments with latrunculin treatment did not affect the overall rate of synaptic receptor turnover (Fig. 7C,D,F; Full p = 0.34 at 10 s, p = 0.39 at 600 s). Surprisingly, latrunculin also did not strongly affect stability within the synapse: intrasynaptic receptor mobility barely increased from undetectable to a few percent (Fig. 7D,F; 10 s full 0.0369 ± 0.008, 10 s partial 0.10 ± 0.03, p = 0.049; 600 s full 0.30 ± 0.03 600 s, partial 0.54 ± 0.11, p = 0.045). Nevertheless, the large majority of receptors (∼76%) remained within their initial domain of the synapse, even after 10 min (Fig. 7D,F).
Thus, actin filaments are not acutely necessary for confinement of most receptors in synapse subdomains. An alternative explanation for the continued stabilization of receptors is that PSD-intrinsic mechanisms prevent receptor mobility and remain essentially intact after application of latrunculin. To assay the influence of the cytoskeleton on scaffold dynamics in the PSD, we measured PSD-95 mobility following its partial synapse photoactivation. This mobility was nearly undetectable under control conditions, because the extent of recovery in subsynaptically and fully activated synapses was the same (Fig. 7C,G; p = 0.64). Following latrunculin, the overall rate of PSD-95 dissociation did slightly increase (Fig. 7G, Full p = 0.0069), but loss out of activated subregions still matched that in fully activated synapses (Fig. 7G; p = 0.84), indicating that PSD-95 remained immobile within the synapse. Thus, the PSD-95 scaffold and subsynaptic AMPAR confinement are primarily intact even after depletion of spine actin filaments indicating that the PSD matrix restricting receptor mobility is actin-independent.
We then asked whether glutamate receptor activation is capable of regulating intrasynaptic AMPAR mobility. A very low concentration of glutamate (8 μm) applied to neurons for 5 min (Ackermann and Matus, 2003) did not alter fluorescence recovery in fully bleached synapses, indicating that this protocol did not affect synaptic receptor turnover (Fig. 7H; at 600 s, Control 38 ± 3%, Glutamate 36 ± 2%, p = 0.55). However, intrasynaptic mobility of AMPARs was substantially increased, as revealed by a strong increase in recovery following partial photobleaching (Fig. 7H,I; 600 s Partial 60 ± 7, p = 0.001). Despite the change in receptor mobility, exchange and intrasynaptic mobility of PSD-95 were unaffected (Fig. 7J). Together, these data suggest surprisingly that while integrity of the neuronal cytoskeleton is important for maintenance of synaptic AMPAR content over long time scales, spine actin does not anchor receptors. Instead, its remodeling acutely destabilizes PSD internal spatial organization.
Discussion
In this study, we measured the distribution and subsynaptic dynamics of AMPARs within single living synapses. Photobleaching of synapse subregions demonstrated that most synaptic AMPARs were not free to diffuse across the PSD but rather were restricted to subdomains. However, synaptic AMPAR cluster morphology was continuously dynamic, driven by the actin cytoskeleton. Within the synapse, receptor position was most closely tied only to a subset of PSD scaffold proteins, and changes in the relative distribution of receptors and scaffolds were also propelled by actin. Surprisingly, elimination of filamentous actin prompted only the gradual loss of receptors and only a quite small increase in their mobility while in the synapse. Thus, we conclude that AMPAR positioning within the synapse is not principally determined by intrasynaptic diffusion of mobile receptors, nor directly by the actin cytoskeleton, but instead by dynamic mechanisms intrinsic to the postsynaptic scaffold.
Characteristics of the PSD that determine synaptic AMPAR position
Whole-synapse AMPAR photobleaching revealed that ∼30% of receptors exchanged between synaptic and extrasynaptic membranes within 10 min, consistent with previous analyses using FRAP (Sharma et al., 2006; Heine et al., 2008; Frischknecht et al., 2009; Arendt et al., 2010) or single-particle tracking (Ehlers et al., 2007). A strikingly small fraction (<5%) of receptors underwent exchange in seconds. By precisely targeting the photobleaching laser to synaptic subdomains, we found that AMPARs rarely migrate the few hundred nanometers from one side of the synapse to the other, even within 10 min. These results confirm and greatly extend our understanding of receptor confinement (Ehlers et al., 2007; Opazo and Choquet, 2011) by making clear that synaptic AMPARs not labeled with antibodies remain segregated within small confines of the PSD not just for seconds, but for minutes. This argues strongly against a “corral” model of PSD construction and indicates instead that receptors are embedded in a matrix that determines their subsynaptic position. This in turn emphasizes the importance of determining characteristics of PSD internal structure and potential dynamic organization, because these features will determine receptor position at the synapse.
Synapses capture diffusing AMPARs via a mechanism that substantially depends on the interaction between PSD-95 and Stargazin and is triggered by phosphorylation of Stargazin by CaMKII (Bats et al., 2007; Opazo et al., 2010). Subsynaptic AMPARs placement could likewise be mediated by binding interactions with anchoring proteins in the synapse, most notably PSD-95 (Chen et al., 2000; Bats et al., 2007). Consistent with this notion, we find that the subsynaptic distribution of PSD-95 closely matches that of AMPARs. However, peptides that eliminate TARP binding to PSD-95 reduce mEPSC amplitude by only ∼25% (Sainlos et al., 2011). Further, deletion of the GluA1 C-terminal PDZ-binding ligand did not alter receptor mobility or subsynaptic pattern with respect to GKAP (Fig. 4), and deletion of the C-terminal of GluA2 binding PICK1 or ABP/GRIP also does not alter its intrasynaptic mobility (Bats et al., 2007).
Another significant possibility stems from the fact that dense packing of PSD molecules (Chen et al., 2008; Dani et al., 2010) creates an environment so crowded that diffusion of receptors is limited even if they do not remain bound to specific scaffold partners, a mechanism supported by computational modeling of AMPAR diffusion among obstacle proteins (Santamaria et al., 2010). In this case, receptor retention and positioning would thus rely not only on proteins that interact biochemically with AMPARs, but also on the space-filling characteristics of diverse transmembrane and cytosolic PSD constituents. Interestingly, variation in the stoichiometry of AMPARs and TARPs in different synapse subpopulations (Shi et al., 2010) suggests that the balance of binding and crowding mechanisms may be synapse-specific: receptors with fewer TARPs may be less likely to bind PSD-95 but equally sensitive to macromolecular crowding.
Stable PSDs are internally dynamic
Regardless of how the influence of binding and crowding are balanced within a synapse, two critical characteristics of the underlying scaffold determine subsynaptic receptor positioning: the overall morphological features of the structure in which receptors are retained, and the lateral distribution of proteins within the PSD. We explored each. Most obvious was that striking changes occur in synapse morphology over the course of just seconds to minutes. This phenomenon was quite distinct from spontaneous growth or loss of synapses measured over the scale of hours (Okabe et al., 1999) or days (Minerbi et al., 2009; Li et al., 2011) and instead reflects coordinated reorganization of existing constituents, scaffolds (Blanpied et al., 2008) and receptors. It is tempting to speculate that scaffold-receptor binding interactions at the PSD edge help capture receptors, which then distribute into the synapse as a whole over quite long time scales and are internally restrained through a combination of crowding and binding interactions.
To analyze the lateral organization of live PSDs, we developed a straightforward time-lapse fluorescence correlation method. AMPARs were most clearly associated with the distribution of PSD-95, and significantly but less strongly correlated with other scaffolds. Surprisingly, the scaffold-receptor spatial relation changes over time, indicating considerable malleability within deeper layers of the PSD. Because subsynaptic photobleaching showed receptor and PSD-95 confinement within domains smaller than the microscope point-spread function, this pliability may occur over similarly small spatial dimensions rather than stemming from wholesale free motion of scaffolds across the PSD.
Conflicting evidence has left unclear whether the pattern of AMPAR positions within a synapse is consistent across synapses or brain regions (Takumi et al., 1999; Momiyama et al., 2003; Tanaka et al., 2005; Masugi-Tokita et al., 2007; Tarusawa et al., 2009). Recent immunocytochemical evidence indicates that individual synapses even within the same brain region can display quite divergent patterns of AMPAR placement (Dani et al., 2010). Though this divergence might indicate that individual synapses fall into distinct categories of molecular organization, a simple explanation for this variety may be that, as we have seen here, the distribution of receptors in single synapses is time-variant. In addition, though small changes in activity level only mildly and transiently alter the morphological dynamics of the PSD (Blanpied et al., 2008) and do not strongly modify AMPAR dynamics within the synapse (Tardin et al., 2003), we found that direct glutamate receptor activation induces a substantial increase in subsynaptic receptor mobility. Comparable effects on synaptic AMPAR mobility were seen using single-particle tracking following exposure to 100 μm glutamate for 20 min (Tardin et al., 2003). It will be important to identify the mechanism of this influence, and to determine how it may relate to activity-driven changes in receptor numbers.
Actin drives PSD dynamics but does not directly anchor receptors
Though actin is commonly portrayed as anchoring AMPARs in the synapse, our observations suggest this is an oversimplified view at best. Latrunculin does induce a slow reduction in synaptic AMPAR number (Allison et al., 1998; Kim and Lisman, 1999), but kinetically, this effect lags far behind the loss of actin filaments (Fig. 6). Given that receptors are capable of rapid escape from the synapse (Heine et al., 2008), their continued retention for at least minutes in the absence of actin filaments clearly indicates that the spine cytoskeleton does not constrain receptors by directly binding or indirectly fencing them within the PSD. Gradual downregulation of receptor number may instead result from subtle modulation of receptor trafficking steps distant from the synapse. Quite in contrast to the anchoring notion, we conclude that at the synapse itself, the most prominent acute role for actin is in fact to promote dynamics of receptor positioning: actin treadmilling drives changes in synapse morphology and prompts continual adjustment of the relative distribution of scaffolds and receptors.
It could be that actin filaments interact directly with AMPARs to drive morphological contortion of the synapse. However, filaments are sparse within the PSD itself, as visualized by EM (Fifková and Delay, 1982; Capani et al., 2001) or deduced by single-molecule tracking (Frost et al., 2010). This suggests instead that filaments contact peripheral or distal portions of the PSD. Indeed, a variety of actin-binding proteins including Abp1 (Haeckel et al., 2008), cortactin (Naisbitt et al., 1999), and neurabin (Allen et al., 1997; Terry-Lorenzo et al., 2005) interact with Shank or other PSD scaffold proteins, linking regulation of the actin cytoskeleton to the synaptic scaffold itself.
We favor a model in which actin actively regulates the pattern of receptors across the face of the synapse, but only indirectly (Fig. 8). In this view, highly dynamic perisynaptic actin (Frost et al., 2010) powers distortion of the PSD by interaction with distal PSD components. Through a slippage or clutch-like mechanism intrinsic to the PSD milieu, this indirectly affects receptors by repositioning their binding partners and other PSD constituents. We note that this model does not exclude the additional possibility that AMPARs, which link to actin through a variety of intermediates including N-cadherin, α-actinin, and Band 4.1N (Shen et al., 2000; Cingolani and Goda, 2008; Zhou et al., 2011), conversely drive or contribute to restructuring of the PSD. The subsynaptic malleability and reorganization we have observed suggests that the relationship between release sites and receptors may not be constant, potentially regulating synaptic transmission (Raghavachari and Lisman, 2004; Newpher and Ehlers, 2008). Given this, our model suggests disease-associated perturbations of the spine actin cytoskeleton or PSD (Bonaglia et al., 2001; Durand et al., 2007; Gauthier et al., 2010) may induce pathological disruption of synapse function even without overt loss of spines, through altered cytoskeletal control of receptor positioning.
Figure 8.
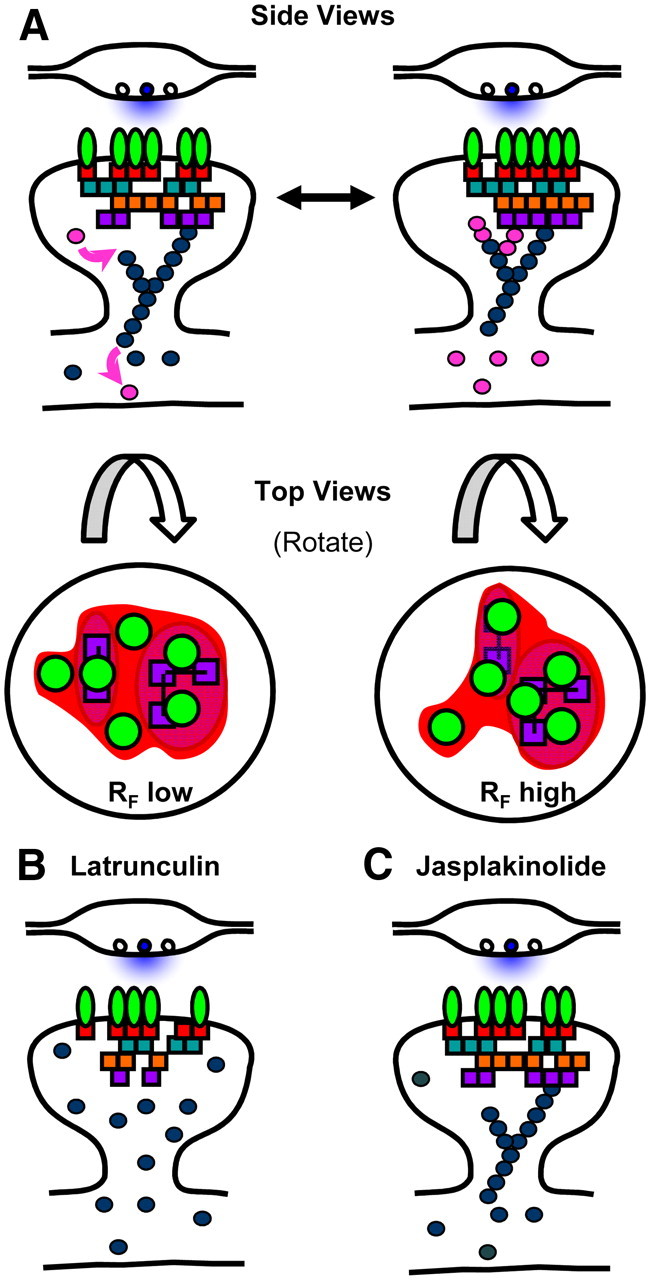
Potential mechanism of actin-regulated AMPAR subsynaptic distribution via PSD reorganization. A, A synapse viewed from the side (top) and from the top (bottom) illustrating the heterogeneous axial and lateral distribution of PSD proteins. Force generated from dynamic perisynaptic actin directly and constitutively regulates PSD lateral organization. We suspect that prolific intermolecular engagement within the PSD creates a sufficiently rigid structure to prevent mixing of PSD components over large distances. Alterations of the PSD interior in turn regulate subsynaptic AMPAR distribution, potentially by setting the position of either specific binding partners or local domains of nonspecific molecular crowding. While known binding interactions as well as the rank order of RF correlation values observed here imply a general sequence of molecular interactions that could relay information from filaments to receptors, there is not a great deal of evidence to support a specific molecular mechanism; the highly interconnected nature of PSD proteins suggests there may be many alternatives. B, Latrunculin depolymerizes spine actin filaments and at least in young neurons induces a partial loss of some PSD scaffold components. The loss of dynamic actin filaments blocks both morphology changes and subsynaptic receptor-scaffold reorganization. Receptors remain stably anchored within subsynaptic domains for long periods. While eventually the number of receptors in the synapse decreases, this is likely due to alterations in extrasynaptic receptor trafficking, not disruption of receptor stability in the synapse. C, Jasplakinolide treatment blocks synapse morphology changes and subsynaptic receptor-scaffold reorganization by preventing actin dynamics. Jasplakinolide does not strip away PSD components and does not elicit a reduction in receptor numbers, but prevents force generation and halts actin-driven reorganization of the PSD interior.
Footnotes
Support was provided by NIH NRSA F31-MH086177 and Training Grant NIGMS T32GM008181 (J.M.K.), and by NIMH Grant R01-MH080046, NARSAD, and the Katherine D. and Theodore J. Carski Foundation (T.A.B.). We thank H. D. MacGillavry, N. Frost, H. E. Lu, S. Raghavachari, and S. M. Thompson for helpful discussions, and M. Contreras and H. Kong for technical assistance. We thank Miranda Karson and the School of Medicine Confocal Core Facility for help and support.
References
- Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci. 2003;6:1194–1200. doi: 10.1038/nn1135. [DOI] [PubMed] [Google Scholar]
- Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci U S A. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Royo M, Fernández-Monreal M, Knafo S, Petrok CN, Martens JR, Esteban JA. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2010;13:36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24:5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Kerr JM, Ehlers MD. Structural plasticity with preserved topology in the postsynaptic protein network. Proc Natl Acad Sci U S A. 2008;105:12587–12592. doi: 10.1073/pnas.0711669105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001;69:261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Bressloff PC, Earnshaw BA. A dynamic corral model of receptor trafficking at a synapse. Biophys J. 2009;96:1786–1802. doi: 10.1016/j.bpj.2008.12.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capani F, Martone ME, Deerinck TJ, Ellisman MH. Selective localization of high concentrations of F-actin in subpopulations of dendritic spines in rat central nervous system: a three-dimensional electron microscopic study. J Comp Neurol. 2001;435:156–170. doi: 10.1002/cne.1199. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Chen X, Winters C, Azzam R, Li X, Galbraith JA, Leapman RD, Reese TS. Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci U S A. 2008;105:4453–4458. doi: 10.1073/pnas.0800897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, Reese TS. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci. 2011;31:6329–6338. doi: 10.1523/JNEUROSCI.5968-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D. Fast AMPAR trafficking for a high-frequency synaptic transmission. Eur J Neurosci. 2010;32:250–260. doi: 10.1111/j.1460-9568.2010.07350.x. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Fifková E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol. 1982;95:345–350. doi: 10.1083/jcb.95.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ. Independent sources of quantal variability at single glutamatergic synapses. J Neurosci. 2003;23:3186–3195. doi: 10.1523/JNEUROSCI.23-08-03186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht R, Heine M, Perrais D, Seidenbecher CI, Choquet D, Gundelfinger ED. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci. 2009;12:897–904. doi: 10.1038/nn.2338. [DOI] [PubMed] [Google Scholar]
- Frost NA, Shroff H, Kong H, Betzig E, Blanpied TA. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 2010;67:86–99. doi: 10.1016/j.neuron.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, Lapointe M, Peng H, Côté M, Noreau A, Hamdan FF, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Joober R, Fathalli F, Mouaffak F, Haghighi AP, Néri C, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeckel A, Ahuja R, Gundelfinger ED, Qualmann B, Kessels MM. The actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head and synapse formation. J Neurosci. 2008;28:10031–10044. doi: 10.1523/JNEUROSCI.0336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S. Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci. 2000;23:141–146. doi: 10.1016/s0166-2236(00)01576-9. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Sudhöf TC. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Takamiya K, Petralia RS, Sattler R, Yu S, Zhou W, Kalb R, Wenthold R, Huganir R. Persistent hippocampal CA1 LTP in mice lacking the C-terminal PDZ ligand of GluR1. Nat Neurosci. 2005;8:985–987. doi: 10.1038/nn1432. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Seeburg PH, Kennedy MB. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriu T, Inoue A, Bito H, Sobue K, Okabe S. Differential control of postsynaptic density scaffolds via actin-dependent and -independent mechanisms. J Neurosci. 2006;26:7693–7706. doi: 10.1523/JNEUROSCI.0522-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Li J, Erisir A, Cline H. In vivo time-lapse imaging and serial section electron microscopy reveal developmental synaptic rearrangements. Neuron. 2011;69:273–286. doi: 10.1016/j.neuron.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE. 2006;2006:re11. doi: 10.1126/stke.3562006re11. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Raghavachari S, Tsien RW. The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci. 2007;8:597–609. doi: 10.1038/nrn2191. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Lu J, Helton TD, Blanpied TA, Rácz B, Newpher TM, Weinberg RJ, Ehlers MD. Postsynaptic positioning of endocytic zones and AMPA receptor cycling by physical coupling of dynamin-3 to homer. Neuron. 2007;55:874–889. doi: 10.1016/j.neuron.2007.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry HD, Kerr JM, Blanpied TA. Lateral organization of the postsynaptic density. Mol Cell Neurosci. 2011;48:321–331. doi: 10.1016/j.mcn.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Martin S, Henley JM, Holman D, Zhou M, Wiegert O, van Spronsen M, Jöels M, Hoogenraad CC, Krugers HJ. Corticosterone alters AMPAR mobility and facilitates bidirectional synaptic plasticity. PLoS One. 2009;4:e4714. doi: 10.1371/journal.pone.0004714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masugi-Tokita M, Tarusawa E, Watanabe M, Molnár E, Fujimoto K, Shigemoto R. Number and density of AMPA receptors in individual synapses in the rat cerebellum as revealed by SDS-digested freeze-fracture replica labeling. J Neurosci. 2007;27:2135–2144. doi: 10.1523/JNEUROSCI.2861-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Stevens CF. Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci U S A. 2000;97:6173–6178. doi: 10.1073/pnas.100126497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Minerbi A, Kahana R, Goldfeld L, Kaufman M, Marom S, Ziv NE. Long-term relationships between synaptic tenacity, synaptic remodeling, and network activity. PLoS Biol. 2009;7:e1000136. doi: 10.1371/journal.pbio.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama A, Silver RA, Hausser M, Notomi T, Wu Y, Shigemoto R, Cull-Candy SG. The density of AMPA receptors activated by a transmitter quantum at the climbing fibre-Purkinje cell synapse in immature rats. J Physiol. 2003;549:75–92. doi: 10.1113/jphysiol.2002.033472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. 2008;58:472–497. doi: 10.1016/j.neuron.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonenko I, Boda B, Steen S, Knott G, Welker E, Muller D. PSD-95 promotes synaptogenesis and multiinnervated spine formation through nitric oxide signaling. J Cell Biol. 2008;183:1115–1127. doi: 10.1083/jcb.200805132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H. Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci. 1999;2:804–811. doi: 10.1038/12175. [DOI] [PubMed] [Google Scholar]
- Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46:1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Raghavachari S, Lisman JE. Properties of quantal transmission at CA1 synapses. J Neurophysiol. 2004;92:2456–2467. doi: 10.1152/jn.00258.2004. [DOI] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Iino R, Fujiwara T, Murase K, Kusumi A. The fence and picket structure of the plasma membrane of live cells as revealed by single molecule techniques (Review) Mol Membr Biol. 2003;20:13–18. doi: 10.1080/0968768021000055698. [DOI] [PubMed] [Google Scholar]
- Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, Sassoè-Pognetto M, Banchaabouchi MA, Giustetto M, Triller A, Choquet D, Witke W. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010;29:1889–1902. doi: 10.1038/emboj.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainlos M, Tigaret C, Poujol C, Olivier NB, Bard L, Breillat C, Thiolon K, Choquet D, Imperiali B. Biomimetic divalent ligands for the acute disruption of synaptic AMPAR stabilization. Nat Chem Biol. 2011;7:81–91. doi: 10.1038/nchembio.498. [DOI] [PubMed] [Google Scholar]
- Santamaria F, Gonzalez J, Augustine GJ, Raghavachari S. Quantifying the effects of elastic collisions and non-covalent binding on glutamate receptor trafficking in the post-synaptic density. PLoS Comput Biol. 2010;6:e1000780. doi: 10.1371/journal.pcbi.1000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Fong DK, Craig AM. Postsynaptic protein mobility in dendritic spines: Long-term regulation by synaptic NMDA receptor activation. Mol Cell Neurosci. 2006;31:702–712. doi: 10.1016/j.mcn.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi Y, Suh YH, Milstein AD, Isozaki K, Schmid SM, Roche KW, Nicoll RA. Functional comparison of the effects of TARPs and cornichons on AMPA receptor trafficking and gating. Proc Natl Acad Sci U S A. 2010;107:16315–16319. doi: 10.1073/pnas.1011706107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Renz M, Lippincott-Schwartz J, Verkhusha VV. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132:6481–6491. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swulius MT, Kubota Y, Forest A, Waxham MN. Structure and composition of the postsynaptic density during development. J Comp Neurol. 2010;518:4243–4260. doi: 10.1002/cne.22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi Y, Matsubara A, Rinvik E, Ottersen OP. The arrangement of glutamate receptors in excitatory synapses. Ann N Y Acad Sci. 1999;868:474–482. doi: 10.1111/j.1749-6632.1999.tb11316.x. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Matsuzaki M, Tarusawa E, Momiyama A, Molnar E, Kasai H, Shigemoto R. Number and density of AMPA receptors in single synapses in immature cerebellum. J Neurosci. 2005;25:799–807. doi: 10.1523/JNEUROSCI.4256-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C, Cognet L, Bats C, Lounis B, Choquet D. Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 2003;22:4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarusawa E, Matsui K, Budisantoso T, Molnár E, Watanabe M, Matsui M, Fukazawa Y, Shigemoto R. Input-specific intrasynaptic arrangements of ionotropic glutamate receptors and their impact on postsynaptic responses. J Neurosci. 2009;29:12896–12908. doi: 10.1523/JNEUROSCI.6160-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry-Lorenzo RT, Roadcap DW, Otsuka T, Blanpied TA, Zamorano PL, Garner CC, Shenolikar S, Ehlers MD. Neurabin/Protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol Biol Cell. 2005;16:2349–2362. doi: 10.1091/mbc.E04-12-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tom Dieck S, Sanmartí-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kämpf U, Fränzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142:499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron. 2008;59:359–374. doi: 10.1016/j.neuron.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Weinberg RJ. Laminar organization of the NMDA receptor complex within the postsynaptic density. J Neurosci. 2001;21:1211–1217. doi: 10.1523/JNEUROSCI.21-04-01211.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Liaw JS, Baudry M, Berger TW. Novel expression mechanism for synaptic potentiation: alignment of presynaptic release site and postsynaptic receptor. Proc Natl Acad Sci U S A. 1997;94:6983–6988. doi: 10.1073/pnas.94.13.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Kanda T, Eguchi K, Takahashi T. Vesicular glutamate filling and AMPA receptor occupancy at the calyx of Held synapse of immature rats. J Physiol. 2009;587:2327–2339. doi: 10.1113/jphysiol.2008.167759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hu J, Passafaro M, Xie W, Jia Z. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J Neurosci. 2011;31:819–833. doi: 10.1523/JNEUROSCI.3869-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]