

In tumor cells that coexpress Cx43 and Cx26, the assembly of Cx43 is selectively impaired due to endocytosis. Assembly can be restored upon expressing a Cx43 sorting-motif mutant and mutants that cannot be phosphorylated on Ser-279 or Ser-282.
Abstract
The molecular mechanisms regulating the assembly of connexins (Cxs) into gap junctions are poorly understood. Using human pancreatic tumor cell lines BxPC3 and Capan-1, which express Cx26 and Cx43, we show that, upon arrival at the cell surface, the assembly of Cx43 is impaired. Connexin43 fails to assemble, because it is internalized by clathrin-mediated endocytosis. Assembly is restored upon expressing a sorting-motif mutant of Cx43, which does not interact with the AP2 complex, and by expressing mutants that cannot be phosphorylated on Ser-279 and Ser-282. The mutants restore assembly by preventing clathrin-mediated endocytosis of Cx43. Our results also document that the sorting-motif mutant is assembled into gap junctions in cells in which the expression of endogenous Cx43 has been knocked down. Remarkably, Cx43 mutants that cannot be phosphorylated on Ser-279 or Ser-282 are assembled into gap junctions only when connexons are composed of Cx43 forms that can be phosphorylated on these serines and forms in which phosphorylation on these serines is abolished. Based on the subcellular fate of Cx43 in single and contacting cells, our results document that the endocytic itinerary of Cx43 is altered upon cell–cell contact, which causes Cx43 to traffic by EEA1-negative endosomes en route to lysosomes. Our results further show that gap-junctional plaques formed of a sorting motif–deficient mutant of Cx43, which is unable to be internalized by the clathrin-mediated pathway, are predominantly endocytosed in the form of annular junctions. Thus the differential phosphorylation of Cx43 on Ser-279 and Ser-282 is fine-tuned to control Cx43’s endocytosis and assembly into gap junctions.
INTRODUCTION
Gap junctions, formed of proteins called connexins (Cxs), are ensembles of several cell–cell channels that signal by permitting the direct exchange of small molecules between the cytoplasmic interiors of contiguous cells. Evidence is mounting that this form of signaling fulfills a homeostatic role through buffering of spatial gradients of nutrients and small molecules of <1500 Da (Goodenough and Paul, 2009). Connexins, which are designated according to molecular mass, are family of 21 related proteins, some of which are expressed in a tissue-specific manner, while others are expressed redundantly (Beyer and Berthoud, 2009). A cell–cell channel is formed when newly synthesized Cxs oligomerize as a hexamer to form a connexon that, upon reaching the cell surface, docks with a connexon in an adjacent cell. A gap junction, often called a gap-junctional plaque, is formed when several such channels cluster. The half-life of most Cxs has been determined to lie between 2 and 5 h both in vivo and in vitro, revealing gap-junctional plaques to be highly dynamic macromolecular complexes (Laird, 2006; Goodenough and Paul, 2009). Because a gap junction is a bicellular structure and is assembled by the collaborative effort of two cells, it is as yet not precisely known how the formation of a nascent gap-junctional plaque is initiated at the site of cell–cell contact, how the plaque assembles and grows, and how it is endocytosed and disassembled (Musil, 2009).
With regard to assembly, current evidence supports the notion that, once a plaque has been nucleated or a nascent gap junction formed, the plaque grows either when connexons, which have been delivered to the cell surface randomly, are recruited to its periphery by diffusion (Gaietta et al., 2002; Lauf et al., 2002; Thomas et al., 2005), or when connexons are directly delivered to the plaque itself (Shaw et al., 2007). Moreover, it is thought that docking of connexons occurs either in the plaque itself or in its vicinity. Furthermore, given the fact that an ensemble of cells has gap junctions of diverse sizes, it is also not known whether larger plaques arise from the coalescence of smaller plaques or by the continuous accretion of undocked or docked connexons to the plaque periphery. With regard to disassembly, recent studies have shown that gap junctions disassemble intricately, either through the endocytosis of the plaque in its entirety or through endocytosis of senescent plaque components from the center as double-membrane vesicles (Jordan et al., 2001; Gaietta et al., 2002; Piehl et al., 2007; Nickel et al., 2008; Falk et al., 2009). Recently gap junctions have also been found to be degraded by autophagy upon internalization (Hesketh et al., 2010; Lichtenstein et al., 2011; Bejiarno et al., 2012; Fong et al., 2012).
Our previous studies with cadherin-null human squamous carcinoma cells showed that the assembly of Cx43, which is ubiquitously expressed (Laird, 2006), was facilitated by cell–cell adhesion mediated by cadherins although cadherins were not required to initiate the formation of nascent gap-junctional plaques. On the other hand, the assembly of Cx32, which is expressed in well-differentiated and polarized cells (Bosco et al., 2011), was induced only when cells acquired a partially polarized state (Chakraborty et al., 2010). Our subsequent studies with nontransformed rat epithelial cells showed that the assembly of Cx43 was differentially regulated by E-cadherin and N-cadherin at sites of cell–cell contact. When only E-cadherin was expressed, Cx43 was preponderantly assembled into large gap junctions, and when only N-cadherin was expressed, Cx43 was endocytosed prior to assembly into gap junctions (Govindarajan et al., 2010). These studies suggest that the assembly of Cxs into gap junctions upon arrival at the cell surface is subject to regulatory mechanisms at the site of cell–cell contact and may be dependent upon additional proteins that permit or impede assembly. The extent to which the assembly of a particular connexon into gap junctions upon arrival at the cell surface is determined by factors intrinsic to the connexon itself or by extrinsic factors, such as the direct or indirect interaction of a Cx with other cell surface-associated protein(s), has not yet been explored (Laird, 2010; Herve et al., 2012). Although many proteins have been shown to interact with Cxs either directly or indirectly, not much is known about the molecular nature of these interactions and how they impose local constraints to restrict or permit the assembly of a particular connexon upon arrival at the cell surface (Laird, 2010; Herve et al., 2012).
As a first step toward elucidating the molecular mechanisms by which the assembly of a particular connexon is regulated at the site of cell–cell contact, we chose a tumor cell line that expressed two or more Cxs and in which the assembly of one of the Cxs was efficient, while that of the other Cx was inefficient. Using human pancreatic tumor cell line BxPC3, which expresses an exocrine pancreas-specific Cx, Cx26, as well as a ubiquitously expressed Cx, Cx43, we show that only the assembly of Cx43 into gap junctions is impaired, because it is selectively internalized prior to assembly. Moreover, we show that the defective assembly of Cx43 is caused by the endocytosis of connexons by the clathrin-mediated pathway. Furthermore, we provide evidence that defective assembly is restored by expressing a sorting-motif mutant of Cx43 that cannot be endocytosed by the clathrin-mediated pathway. In addition, we document that assembly of Cx43 can also be restored by preventing phosphorylation of Ser-279 or Ser-282, previously shown to be phosphorylated by mitogen-activated protein (MAP) kinase (Warn-Cramer et al., 1998). Finally, our results document that defective assembly can only be restored when connexons are composed of Cx43 forms that can be phosphorylated on Ser-279 and Ser-282 and Cx43 forms in which phosphorylation of these serines is abolished. Our results suggest that differential phosphorylation of Cx43 on these serines is fine-tuned to control endocytosis, plaque growth, and size.
RESULTS
Defective assembly of Cx43 in BxPC3 and Capan-1 cells
Cx32 and Cx26 are expressed in the acinar cells of the exocrine pancreas (Bavamian et al., 2009). We screened several human pancreatic cancer cell lines by semiquantitative reverse transcriptase PCR (RT-PCR) for the expression of Cxs (unpublished data) and chose two cell lines, BxPC3 and Capan-1, which expressed Cx43 and Cx26. As was reported earlier (Lahlou et al., 2005), upon immunocytochemical and Western blot analysis, we found that Cx43 was not assembled into gap junctions in either cell line and lay predominantly as discrete intracellular vesicular puncta dispersed throughout the cytoplasm (Figure 1A). On the other hand, Cx26 formed gap junctions in BxPC3 cells but remained intracellular in Capan-1 cells (Supplemental Figure S1, left panels). To substantiate the immunocytochemical data, we determined the extent of assembly of Cx43 into gap junctions biochemically by Western blot analysis of total and Triton X-100 (TX100)-soluble and TX100-insoluble fractions (VanSlyke and Musil, 2000) and upon in situ extraction with 1% TX100. The results showed that Cx43 remained predominantly detergent-soluble in both cell lines, as assessed by the disappearance of intracellular puncta (Figure 1B) and by the majority of Cx43 appearing in the detergent-soluble fraction (Figure 1C). For example, the mean number of Cx43 intracellular puncta in both cell lines after detergent extraction was reduced from 82 ± 13 (n = 14) and 104 ± 19 (n = 17) per cell to 11 ± 4 (n = 13) and 7 ± 3 (n = 14) per cell in BxPC3 and Capan-1 cells, respectively. Because Cx26 was assembled into gap junctions, whereas Cx43 was not, we next examined the assembly of Cx32 in BxPC3 and Capan-1 cells upon retroviral transduction. We found that Cx32 assembled into gap junctions in BxPC3 cells, but not in Capan-1 cells, as assessed immunocytochemically and biochemically with TX100-solubility assays (Figure S1). Altogether the results shown in Figures 1 and S1 suggest the following: 1) In BxPC3 cells, both Cx26 and Cx32 are efficiently assembled into gap junctions, but the assembly of Cx43 is selectively impaired. 2) In Capan-1 cells, the assembly of all three Cxs is impeded.
FIGURE 1:

Cx43 fails to assemble into gap junctions in BxPC3 and Capan-1 cells. (A) Cells were immunostained for Cx43. Note that in both BxPC3 and Capan-1 cells, Cx43 (red) is seen as discrete intracellular puncta dispersed throughout the cytoplasm. (B) Loss of Cx43 intracellular puncta upon in situ extraction with 1% TX100 in BxPC3 and Capan-1 cells. Note loss of Cx43 intracellular puncta upon extraction with TX100. (C) Western blot analysis of Cx43 in total (T), TX100-soluble (S), and TX100-insoluble (I) fractions of cell lysates in BxPC3 and Capan-1 cells. Note that the majority of Cx43 is soluble in TX100 in both cell types. (D) Cx43 traffics to the cell surface in BxPC3 and Capan-1 cells. The cell surface proteins of BxPC3 and Capan-1 cells were biotinylated. Biotinylated proteins were pulled down by immobilized streptavidin and immunoblotted for Cx43. Biotinylation of E-cadherin (E-cad) was used as a positive control. For input, 10 μg total cell lysate was used. Note that Cx43 and E-cad were efficiently biotinylated in both cell lines. (E) Cells were immunostained for Cx43 (red), clathrin, and EEA1. Enlarged images of the boxed regions are shown on the right. Note that Cx43 does not colocalize discernibly with either marker.
To examine whether the failure of Cx43 to assemble into gap junctions was due to impaired trafficking or to endocytosis prior to assembly into gap junctions, we used cell surface biotinylation, as well as markers for the secretory and the endocytic compartments, to assess its subcellular fate. Using biotinylation of E-cadherin as a positive control, we found that Cx43 was biotinylated significantly in both BxPC3 and Capan-1 cells (Figure 1D). However, immunocytochemical analysis showed that Cx43 barely colocalized with clathrin (Roth, 2006), the early endocytic marker EEA1 (Mills et al., 1998), or caveolin 1 (Parton and Simons, 2007; Figures 1E and S2A). This conclusion was supported by quantitative analysis (Figure S2C). On the other hand, discernible colocalization was observed with GM130, a cis-Golgi–resident protein (Nakamura et al., 1995), and with caveolin 2 (Parton and Simons, 2007), which are markers for the secretory compartments (Figure S2A), as well as with the lysososmal marker Lamp1 (Rohrer et al., 1996; Hunziker and Geuze, 2011; Figure S2B). Taken together, these data suggest that intracellular accumulation of Cx43 in BxPC3 and Capan-1 cells is not caused by impaired trafficking to the cell surface, but by some mechanism that selectively interferes with its assembly into gap junctions, causing it to accumulate in intracellular vesicles, yet permitting the assembly of Cx32 and Cx26 into gap junctions.
Endocytosis of Cx43
Because Cx43 trafficked to the cell surface, yet failed to colocalize discernibly with endocytic markers, we used other approaches to examine whether it was endocytosed prior to assembly into gap junctions. We first used hypertonic sucrose to inhibit the clathrin-mediated pathway (Heuser and Anderson, 1989) and filipin to inhibit the lipid-mediated pathway (Schnitzer et al., 1994). We found that exposure of cells to hypertonic conditions for 1–2 h caused the disappearance of Cx43 intracellular puncta and the concomitant increase in cell surface–associated Cx43, whereas filipin had no effect (unpublished data). These findings hinted that clathrin-mediated endocytosis was involved. Earlier studies had shown that the cytoplasmic tail of Cx43 harbored a consensus protein–protein interaction proline-rich motif and an overlapping putative tyrosine-based sorting motif (278-LSPMSPPGYKLV-289) that potentially targeted it for internalization via the clathrin-mediated pathway (Thomas et al., 2003). We therefore directly tested whether Cx43 interacted with the μ2 subunit of the clathrin adapter complex AP2 by using a yeast two-hybrid analysis (Figure 2A). The results showed that, while the cytoplasmic tail of wild-type (WT) Cx43 interacted with the μ2 subunit of AP2, Cx43 mutants (Y286A, V289D, and Y286A/V289D), in which either tyrosine or valine or both amino acids of the sorting motif had been mutated, did not. The mutant Y286A/V289D hereafter will be referred to as YA/VD-Cx43. All the mutants and engineered constructs used are diagrammed in Figure 12 under Materials and Methods later in the paper.
FIGURE 2:

(A) In the yeast two-hybrid assay, Cx43 interacts with the μ2 subunit of the AP2 complex in a sorting motif–dependent manner. Yeast were cotransformed with the indicated GAL4-binding domain and GAL4 transcription activation domain fusion constructs. Measured by growth on selective media (−His), only the wild-type (WT) tail of Cx43 interacted with the μ2 subunit of the AP2 complex. PVA3 (p53) and PTD1 (large T-antigen) served as positive control, while rabinosyn-5 served as the negative control. (B) Cx43 (green) colocalizes with clathrin and EEA-1 in single cells. (C and D) Cx43 (green) colocalizes with the AP2 complex (red) in both single and contacting cells. (B–D) Outlined boxes are shown at higher power.
FIGURE 12:
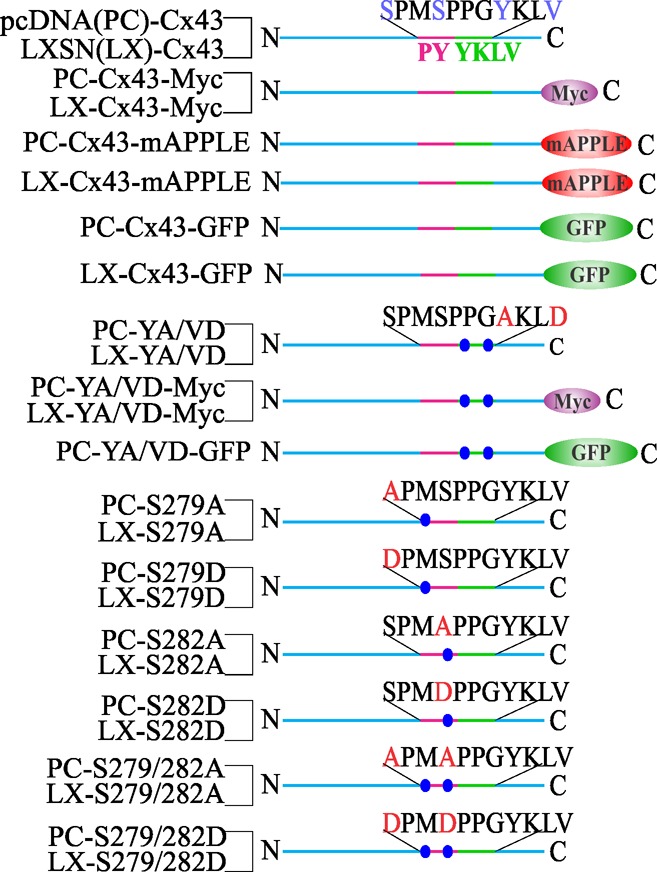
Diagram of the Cx43 constructs. Shown are the maps of wild-type and mutant Cx43 cloned into pcDNA (PC) and retroviral vector LXSN (LX), as indicated on the left. The constructs are tagged either with Myc or with red or green fluorescent proteins. The regions indicated are as follows: the proline rich region (PY) is pink, whereas the tyrosine-based sorting motif is green. The amino acid sequence of each indicated region is shown above each construct. Mutated residues are highlighted by red letters, and the relative position of each mutant is marked by blue circles. The amino and the carboxyl termini are indicated by N and C, respectively.
Our previous studies showed that Cx43 colocalized extensively with clathrin and EEA1 in single cells but not in contacting cells (Govindarajan et al., 2010). Therefore we examined the subcellular fate of Cx43 in single BxPC3 and Capan-1 cells 24–36 h after seeding to determine whether it was endocytosed by the clathrin-mediated pathway. We rationalized that, due to the short half-life of Cx43, intracellular puncta in single cells would plausibly represent endocytosed connexons that had trafficked to the cell surface. We observed a marked colocalization of Cx43 with clathrin and EEA1 (Figure 2B), as well as with Lamp1 (Figure S3A), but no colocalization with caveolin 1 (Figure S3A). The data shown in Figure 2A prompted us to examine whether Cx43 would colocalize with the AP2 complex. The results showed that Cx43 colocalized discernibly with the AP2 complex both in contacting (Figure 2C) and single cells (Figure 2D). To determine whether AP2 complex interacts with WT-Cx43, we transiently expressed the monomeric enhanced green fluorescent protein (mEGFP)-tagged μ2 subunit of the adaptor protein (AP2) complex (Ohno et al., 1995) in HEK293 cells, which express endogenous Cx43. The results showed that Cx43 could be immunoprecipitated from the total cell lysates with antibody to green fluorescent protein (GFP) green fluorescent protein (Figure S3B). We used HEK293 cells due to the low transfection efficiencies of BxPC3 and Capan-1 cells. Altogether these data indicate that Cx43 interacts with the μ2 subunit of AP2 complex either directly or indirectly.
To lend further support to the above data, and given the involvement of Rab5 in clathrin-mediated endocytosis (Bucci et al., 1992; Zerial and McBride, 2001), we also examined whether Rab5 was involved in the endocytosis of Cx43 (Figure S3). We transiently expressed wild-type Rab5 (Rab5-WT) and its dominant-active (Rab5-DA) and dominant-negative (Rab5-DN) mutants conjugated with EGFP in BxPC3 and Capan-1 cells. The results showed that expression of Rab5-DN profoundly reduced the number of EEA1-positive vesicles, suggesting that the generation of EEA1-positive vesicles was Rab5-dependent in both cell lines (unpublished data). In agreement with the effect of Rab5-DN on the generation of EEA1-positive vesicles, we found that the expression of Rab5-DN also drastically reduced the number of Cx43 intracellular puncta (Figure S3A). These results suggest that the generation of Cx43-positive intracellular vesicles depends on Rab5, which also regulates the productive endocytosis of Cx43. Altogether the above data demonstrate that Cx43 is endocytosed by the clathrin-mediated pathway in a Rab5-dependent manner in both cell types, but that Cx43 colocalizes with clathrin—and predominantly traffics via EEA1-positive endosomes—only in single cells, whereas in contacting cells, it traffics via EEA1-negative endosomes
Cells expressing YA/VD-Cx43 assemble gap junctions
Various Cx subtypes have been shown to oligomerize to form heteromeric connexons that are incorporated into gap-junctional plaques (Jiang and Goodenough, 1996; Valiunas et al., 2001; Churko et al., 2010). We hypothesized that the sorting-motif mutant YA/VD-Cx43, by forming heteromers with the endogenous Cx43, might affect junction assembly by inhibiting endocytosis. To pursue this hypothesis, we investigated whether expression of YA/VD-Cx43 would restore defective gap junction assembly in BxPC3 and Capan-1 cells. WT-Cx43 or YA/VD-Cx43 were transiently expressed in each cell type along with EGFP to mark transfected cells, and the formation of gap junctions was assessed immunocytochemically. The results showed that expressing YA/VD-Cx43 induced the formation of large gap-junctional plaques, while expressing WT-Cx43 did not (Figure 3A, top panels). To rule out that the restoration of junction assembly was an artifact of overexpression, we also expressed WT-Cx43 or YA/VD-Cx43 via recombinant retroviruses. The results showed that the retroviral expression of YA/VD-Cx43 restored gap junction assembly with a concomitant decrease in intracellular puncta, although not as robustly as upon transient expression (Figure 3A, bottom panels). Detergent-solubility assays showed the detergent-insoluble fraction was substantially increased upon expressing YA/VD-Cx43 (Figure 3C), substantiating the data obtained from the immunocytochemical analysis.
FIGURE 3:

The sorting-motif mutant YA/VD-Cx43 restores gap junction assembly. (A) WT-Cx43 (WT) and YA/VD-Cx43 (YA/VD) were expressed in cells by transient transfection (top row) or by retroviral infection (bottom row). Cells were immunostained for Cx43. For transient expression, WT-Cx43 and YA/VD-Cx43 were cotransfected with EGFP to mark the transfected cells. Note the formation of gap junctions (arrows) in cells expressing YA/VD-Cx43. (B) The percentage of transfected cells that had gap junctions was determined by examining 20 cells from two experiments (blue bars: cells expressing WT-Cx43; red bars: cells expressing YA/VD-Cx43). Note the increase in gap junction number in cells transfected with YA/VD-Cx43. (C) Total (T), TX100-soluble (S), and TX100-insoluble (I) fractions were prepared from cells expressing retrovirally introduced WT-Cx43 or YA/VD-Cx43. Note the robust increases in the detergent-insoluble fractions in cells infected with YA/VD-Cx43.
To examine whether YA/VD-Cx43 restored assembly by incorporating into gap-junctional plaques that also contained the endogenous Cx43, we transiently expressed Myc-tagged YA/VD-Cx43 in BxPC3 and Capan-1 cells and examined whether all forms of Cx43 were incorporated into gap-junctional plaques. Within the sensitivity of the light microscope and based on the pattern of colocalization, the results showed that total Cx43 colocalized nearly completely with YA/VD-Cx43-Myc (Figure 4A). If endogenous Cx43 did not assemble with YA/VD-Cx43-Myc, some of the red signal would appear alone. To determine whether YA/VD-Cx43 formed heteromers with WT-Cx43, and to substantiate the immunocytochemical data, we transiently expressed both WT-Cx43EGFP and YA/VD-Cx43-Myc (or the reciprocally labeled protein) in HeLa cells. We examined whether the two proteins could be coimmunoprecipitated from the detergent-soluble fraction, since that fraction is likely to contain only connexons that are not incorporated into gap-junctional plaques (VanSlyke and Musil, 2000). We used HeLa cells for coimmunoprecipitation, because of the low transfection efficiencies of BxPC3 and Capan-1 cells. The results showed that WT-Cx43 and YA/VD-Cx43 reciprocally coimmunoprecipitated with each other (Figure 4B).
FIGURE 4:

Sorting-motif mutant YA/VD-Cx43 restores defective gap junction assembly by incorporating into gap junctions and inhibiting clathrin-mediated endocytosis. (A) BxPC3 and Capan-1 cells expressing endogenous Cx43 were transiently transfected with YA/VD-Cx43-Myc (YA/VD-Myc) and immunostained for Myc and Cx43. Note the robust colocalization of YA/VD-Cx43-Myc with total Cx43 (WT) and the formation of gap junctions. (B) Coimmunoprecipitation of WT-Cx43 and YA/VD-Cx43. HeLa cells were transfected with YA/VD-Cx43EGFP (YA/VD-GFP), WT-Cx43-Myc (WT-Myc), WT-Cx43EGFP (WT-GFP), and YA/VD-Cx43-Myc (YA/VD-Myc) either alone or in the various combinations indicated. GFP-tagged proteins were pulled down with immobilized anti-GFP antibody. Note that YA/VD-Cx43-Myc coimmunoprecipitated with WT-Cx43EGFP and WT-Cx43-Myc coimmunoprecipitated with YA/VD-Cx43EGFP. (C) Capan-1 cells, expressing endogenous Cx43 along with retrovirally introduced WT-Cx43 or YA/VD-Cx43, were seeded as single cells and immunostained for Cx43 (red) and either clathrin, Caveolin 1 (Cav1), or Lamp1 (green). Note that puncta composed of both endogenous Cx43 and retrovirally introduced WT-Cx43 colocalize extensively with clathrin and Lamp1 but not with Cav1. In contrast, puncta composed of both endogenous Cx43 and retrovirally introduced YA/VD-Cx43 fail to colocalize with clathrin, but colocalize discernibly with Cav1 at cell borders.
To test whether the formation of heteromers between endogenous Cx43 and YA/VD-Cx43 restored gap junction assembly by inhibiting endocytosis of connexons, we examined the colocalization of Cx43 with clathrin in single Capan-1 cells expressing retrovirally introduced WT-Cx43 or YA/VD-Cx43 in addition to endogenous Cx43. The results showed that, in contrast with discernible colocalization of Cx43 with clathrin in single cells expressing retroviral WT-Cx43, no colocalization was observed in cells that expressed YA/VD-Cx43 (Figure 4C). We also found that, in contrast with WT-Cx43, heteromers of endogenous Cx43 and YA/VD-Cx43 colocalized discernibly with caveolin 1 (Figure 4C, bottom middle panel). Both WT-Cx43 and the heteromers of endogenous Cx43 and YA/VD-Cx43 were routed to lysosomes, as assessed by colocalization of Cx43 with Lamp1 in single cells expressing either WT-Cx43 or YA/VD-Cx43 (Figure 4C). Collectively the data shown in Figures 3 and 4 document that the expression of the YA/VD-Cx43 mutant restores gap junction assembly in BxPC3 and Capan-1 cells and that the restoration occurs through inhibition of clathrin-mediated endocytosis of connexons.
Phosphorylation of Ser-279 and Ser-282 of Cx43 and gap junction assembly
Phosphorylation of serine and tyrosine residues in or adjacent to sorting motifs modulates the endocytosis of many transmembrane proteins (Bonifacino and Traub, 2003; Moore et al., 2007; Traub, 2009). Thus we rationalized that the phosphorylation of Ser-279 and Ser-282 of Cx43, which lay near the sorting motif, might disrupt its assembly into gap junctions by inducing endocytosis. To test this notion, we created the following Cx43 mutants: 1) S279A and S279D, in which Ser-279 was substituted with alanine or its phosphomimetic, aspartic acid, respectively. 2) S282A and S282D, in which Ser-282 was substituted with alanine or aspartic acid, respectively. 3) S279A/S282A and S279D/S282D, in which both Ser-279 and Ser-282 were substituted with alanine or aspartic acid, respectively. WT-Cx43 and the above mutants were transiently expressed in BxPC3 and Capan-1 cells along with EGFP to mark the transfected cells. The results showed that the expression of serine-to-alanine mutants (S279A and S282A) restored gap junction assembly, whereas expression of serine-to-aspartic acid mutants (S279D and S282D) had no discernible effect (Figure 5A). Moreover, mutants S279A and S282A restored gap junction assembly as robustly as YA/VD-Cx43 (Figure 5A). Furthermore, restoration with the double mutant S279A/S282A was not more robust than with the single mutants. To test whether mutants restored assembly by incorporating into gap-junctional plaques that also contained endogenous Cx43, we transiently expressed the Myc-tagged mutant S279A-Myc in BxPC3 and Capan-1 cells. The results showed that S279A-Myc colocalized extensively with total Cx43 both in the gap-junctional plaques and in intracellular vesicles (Figure 5B). Collectively these data suggest that phosphorylation on Ser-279 and/or Ser-282 of Cx43 impairs its assembly into gap junctions in BxPC3 and Capan-1 cells and preventing phosphorylation of these residues restores assembly.
FIGURE 5:

S279A and S282A mutants restore gap junction assembly. (A) BxPC3 and Capan-1 cells were transiently cotransfected with EGFP and the indicated Cx43 constructs. Cells were immunostained for Cx43 (red). Note that the S279A and S282A mutants restored gap junction assembly as robustly as the sorting-motif mutant YA/VD-Cx43, whereas S279D and S282D did not. (B) Capan-1 and BxPC3 cells were transiently transfected with Myc-tagged S279A and were immunostained for Cx43 (red) and Myc (green) after 24 h. Note the robust colocalization of total Cx43 with Myc.
Only the sorting-motif mutant assembles into gap junctions in cells with endogenous Cx43 knocked down
BxPC3 and Capan-1 cells express abundant endogenous Cx43 (Figure 1). To test whether those mutants that restored defective gap junction assembly upon transient expression would assemble into gap junctions by themselves, we stably knocked down endogenous Cx43 in both cell types with a short hairpin RNA (shRNA) targeting the 3′ untranslated region (UTR). As assessed by Western blot analysis, the expression of endogenous Cx43 was nearly abolished in both cell lines upon knockdown (Figure S4A). Knockdown of Cx43 had no off-target effect on the expression level of seven other randomly chosen cell junction and cytoskeletal proteins (Figure S4B). These stably knocked-down cells are hereafter referred to as Bx43KD (for BxPC3) and CP43KD (for Capan-1) cells.
We used recombinant retroviruses to knock in wild-type and mutant Cx43 in Bx43KD and CP43KD cells. Western blot analysis showed that the knocked-in WT-Cx43 and Cx43 mutants were expressed robustly (Figure 6A). However, unexpectedly, we observed that only YA/VD-Cx43 assembled into gap junctions, whereas the other mutants did not (Figure 6B). Hereafter, Bx43KD and CP43KD with knocked-in WT or mutant Cx43 will be referred to as Bx43KDKI and CP43KDKI cells, followed by the name of the constructs expressed. To substantiate the immunocytochemical data, we determined the assembly of knocked-in WT-Cx43, sorting-motif mutant YA/VD-Cx43, and mutants S279A and S279D biochemically by the TX100-solubility assay and upon in situ extraction with 1% TX100. We also performed functional analysis by scrape-loading and microinjection of gap junction–permeable fluorescent tracers. The results showed that only the YA/VD-Cx43 mutant assembled into detergent-insoluble, gap-junctional plaques, as assessed immunocytochemically (Figure 7A) and biochemically by the TX100-solubility assay (Figure 7B). Moreover, as assessed by scrape-loading and microinjection of gap junction–permeable fluorescent tracers Lucifer Yellow (LY), Alexa Fluor 560, and Alexa Fluor 594, only the YA/VD-Cx43 mutant formed functional cell–cell channels (Figure S4, C and D; see also Table 1). Finally, to further substantiate the above data, we retrovirally introduced the same mutants into the human prostate cancer cell line, LNCaP, which we have shown assembles Cxs into gap junctions efficiently (Mehta et al., 1999; Mitra et al., 2006; Kelsey et al., 2012). The results showed that, compared with WT-Cx43, mutant YA/VD-Cx43 formed large gap junctions, whereas mutants S279A and S279D assembled into small gap-junctional puncta, many of which appeared to be intracellular (Figure S5A). In sum, the data shown in Figures 6 and 7 suggest that the mutants S279A and S282A are unable to assemble into gap junctions in the absence of WT-Cx43.
FIGURE 6:

Assembly of sorting-motif and serine mutants of Cx43 into gap junctions in knockdown cells. (A) WT-Cx43 or the indicated mutants were knocked in to Bx43KD and CP43KD cells, and lysates were analyzed for Cx43 expression by Western blotting. Note that, compared with parental cells expressing endogenous Cx43 (Endo-Cx43), the knocked-in WT-Cx43 and Cx43 mutants are robustly expressed. No detectable expression is seen in CP43KD and Bx43KD cells. (B) Bx43KD and CP43KD cells expressing the knocked-in WT-Cx43 or Cx43-mutants were immunostained for Cx43. Note that only the sorting motif mutant YA/VD-Cx43 (YA/VD) assembles into gap junctions.
FIGURE 7:

Detergent solubility of knocked-in WT-Cx43 and its sorting motif and serine mutants. (A) CP43KD cells expressing knocked-in WT-Cx43 and various mutants were extracted in situ with 1% TX100 and immunostained for Cx43. Note that only knocked-in YA/VD-Cx43 (YA/VD) assembles into detergent-resistant gap junction puncta. Note also that intracellular puncta of knocked-in WT-Cx43 (WT) and all other mutants are lost upon extraction with 1% TX100. (B) Western blot analysis of Cx43 in Total (T), TX100-soluble (S), and TX100-insoluble (I) fractions from CP43KD cells expressing knocked-in WT-Cx43 and the mutants. Note that only cells expressing knocked-in YA/VD-Cx43 have a discernible detergent-insoluble fraction. The blots were stripped and reprobed for β-actin to verify equal loading.
TABLE 1:
Junctional transfer of various fluorescent tracers in parental Capan-1, CP43 knockdown (CP43KD) cells and in CP43KD cells expressing WT-Cx43 and various Cx43 mutants.
| Cell type | Junctional tracer | ||
|---|---|---|---|
| LY | Alexa Fluor 488 | Alexa Fluor 594 | |
| Capan-1 (parental) | 0 (21) | 0 (24) | 15.4 ± 3.3 (12) |
| CP43KD | 0 (22) | 0 (13) | 0 (11) |
| CP43KDKI-WT-Cx43 | 0 (32) | 0 (19) | 0 (8) |
| CP43KDKI-S279A-Cx43 | 0 (24) | 0 (14) | 0 (10) |
| CP43KDKI-S279D-Cx43 | 0 (23) | 0 (24) | 0 (9) |
| CP43KDKI-YA/VD-Cx43 | 16 ± 6 (33) | 18 ± 5 (21) | 7 ± 1 (40) |
Cells were seeded in 6-cm dishes and grown to 70% confluence. Junctional transfer was measured after microinjecting the indicated fluorescent tracers. The numbers of fluorescent cell neighbors (mean ± SE) were determined 1 min (LY), 3 min (Alexa Fluor 488), or 15 min (Alexa Fluor 594) after microinjection into the test cells. The total number of injection trials is shown in parentheses.
Wild-type and mutant Cx43 are required for gap junction assembly
To determine directly whether both WT-Cx43 and the S279A mutant are required for junction assembly, we tested whether expression of WT-Cx43 would restore gap junction assembly in Bx43KDKI cells expressing knocked-in S279A. As controls, we also chose Bx43KDKI cells stably expressing either knocked-in WT-Cx43 or Cx43-S279D. WT-Cx43 was transiently expressed along with EGFP to mark the transfected cells (Figure 8A). The results showed the expression of WT-Cx43 restored junction assembly in Bx43KDKI cells expressing S279A but not in cells expressing S279D or WT-Cx43 (Figure 8A, compare middle panel with right and left panels). Similar data were obtained when WT-Cx43 was transiently transfected into CP43KDKI cells stably expressing S279A, S279D, or WT-Cx43 (unpublished data). To test whether WT-Cx43 was incorporated into gap-junctional plaques that also contained S279A, we checked for colocalization. The results showed extensive colocalization of Myc-tagged WT-Cx43 with total Cx43. Notably, junction assembly was restored only in cells expressing S279A (Figure 8B, top row). Gap junctions were also assembled when Myc-tagged S279A was introduced into Bx43KDKI and CP43KDKI cells stably expressing S279D (unpublished data). Altogether the data shown in Figure 8, combined with those shown in Figures 5–7, suggest that S279A assembled into gap junctions only in the presence of WT-Cx43.
FIGURE 8:

Mutant S279A assembles into gap junctions only in the presence of WT-Cx43. (A) Bx43KD cells stably expressing knocked-in WT-Cx43, S279A, and S279D were transfected with WT-Cx43 along with EGFP and immunostained for Cx43 (red). Note that only mutant S279A is assembled into gap junctions upon expression of WT-Cx43. (B) Bx43KD cells stably expressing knocked-in S279A-Cx43 (S279A) or S279D-Cx43 (S279D) were transiently transfected with Myc-tagged WT-Cx43 (Cx43-Myc) and immunostained for Cx43 (red) and Myc (green). Note that total Cx43 and Myc colocalize extensively, but only mutant S279A is assembled into gap junctions.
Trafficking, degradation, and subcellular fate of knocked-in WT-Cx43 and its mutants
In the next series of experiments, we examined whether the failure of knocked-in WT-Cx43 and mutants S279A, S279D, S282A, and S282D to assemble into gap junctions was due to impaired trafficking or differential degradation at the cell surface. Therefore we determined the trafficking and the kinetics of degradation of WT-Cx43 and the mutants by cell surface biotinylation. We found that WT-Cx43 and the various Cx43 mutants trafficked to the cell surface (Figure S5B). Moreover, the kinetics of degradation of all mutants, including YA/VD-Cx43, was not noticeably different from WT-Cx43 (Figure S5C). Because endogenously expressed Cx43 colocalized with clathrin in single parental BxPC3 and Capan-1 cells, we also examined the subcellular fate of knocked-in WT-Cx43, the sorting-motif mutant YA/VD-Cx43, and mutants S279A and S279D in CP43KDKI single cells. The results showed that, in single cells, the knocked-in WT-Cx43 and S279D colocalized discernibly with clathrin, whereas YA/VD-Cx43 and S279A barely colocalized (Figure S6A). Together the data shown in Figures S5 and S6 suggest that the efficient assembly of YA/VD-Cx43 and the inefficient assembly of WT-Cx43 and mutants S279A and S279D can be attributed neither to differential trafficking nor to differential degradation. These data further document that clathrin-mediated endocytosis appears to be the predominant pathway to internalize WT-Cx43 and mutant S279D connexons in single cells, whereas mutant YA/VD-Cx43 and S279A connexons are likely internalized by a non–clathrin-mediated pathway.
Internalization and degradation of mutant YA/VD-Cx43
Intracellular vesicular puncta in cells expressing knocked-in YA/VD-Cx43, the only mutant that formed gap junctions (Figure 6), were fewer, larger, and of diverse sizes that colocalized neither with caveolin 1 nor with clathrin (Figure S6B). We therefore investigated whether this mutant was internalized in the form of annular gap junctions. We examined whether these intracellular puncta were resistant to in situ extraction with 1% TX100. The results showed that the majority of intracellular puncta in YA/VD-Cx43–expressing cells was resistant to in situ extraction with TX100 (Figure 9A). To determine whether this mutant is targeted upon internalization for degradation by autophagy, we starved these cells for various times in the absence and presence of bafilomycin A1, which inhibits autophagy (Klionsky et al., 2008; Sarkar et al., 2009; Mizushima et al., 2010). We found that this mutant was degraded upon starvation (Figure 9B). Moreover, this mutant was predominantly internalized in the form of annular gap junctions upon starvation, and the degradation was prevented upon treatment with bafilomycin A1 (Figure 9, A and B). In contrast, degradation of knocked-in WT-Cx43 was minimally affected upon starvation and was not inhibited by bafilomycin A1 (unpublished data). These results suggest that the mutant YA/VD-Cx43 is predominantly internalized in the form of annular gap junctions, most likely by a clathrin-independent pathway, and is likely degraded by autophagy.
FIGURE 9:

YA/VD-Cx43 is likely internalized in the form of annular gap junctions that are detergent resistant and degraded by autophagy. (A) Bx43KD and CP43KD cells expressing YA/VD-Cx43 were immunostained for Cx43 (green) and β-catenin (red). Cells were extracted in situ with 1% TX100 (b and d) or starved in PBS without (PBS e and g) or with bafilomycin A1 (BFL PBS+BFL, f and h) for 3 h. (B) CP43KD (left) or Bx43KD (right) cells expressing YA/VD-Cx43 were starved in PBS with or without bafilomycin A1 for the indicated times, and cell lysates were immunoblotted for Cx43. Note that the level of YA/VD-Cx43 decreases upon starvation, and the decrease is prevented by BFL. (A) Gap junctions at the areas of cell–cell contact are indicated by the pink arrows, whereas annular gap junctions are indicated by the white arrows. Note that both in normal medium and in PBS, the mutant YA/VD is internalized in the form of annular gap junctions and internalization is not prevented upon treatment with BFL.
Cell–cell contact and the endocytic itinerary of Cx43
Because discernible colocalization of Cx43 with clathrin and EEA1 was observed only in single cells, and because Cx43 significantly colocalized with AP2 and Lamp1 both in single and contacting cells, we rationalized that cell–cell contact might determine the endocytic route of Cx43. To test this notion, we first determined whether intracellular vesicles in contacting BxPC3 and Capan-1 cells were comprised of docked or undocked connexons or represented internalized miniscule annular gap junctions. To determine this, we first retrovirally expressed Cx43EGFP and Cx43mAPPLE in CP43KD and Bx43KD cells. We then cocultured Bx43KD cells expressing nearly equal levels of either Cx43EGFP or Cx43mAPPLE and, after allowing cells to aggregate as doublets in suspension, seeded them on glass coverslips (see Materials and Methods). We did the same for CP43KD cells infected with the constructs. The doublets of cells were examined 24 h after plating to quantify intracellular puncta that contained either Cx43EGFP or Cx43mAPPLE alone or both. As a control, the same experiment was done with cocultures of the human prostate cancer cell line LNCaP expressing retrovirally introduced Cx43EGFP or Cx43mAPPLE. The results showed that, in both CP43KD and Bx43KD cells, the intracellular vesicular puncta contained either Cx43EGFP or Cx43mAPPLE alone, but not both (Figure 10B). In contrast, LNCaP cells formed gap junctions and intracellular vesicles composed of both Cx43EGFP and Cx43mAPPLE (Figure 10C).
FIGURE 10:

Cell–cell contact and the subcellular fate of Cx43. (A) Cell–cell contact alters the endocytic itinerary of Cx43. Bx43KD and CP43KD cells stably expressing knocked-in WT-Cx43EGFP were cocultured with Bx43KD and CP43KD cells, and the cultures were immunostained for clathrin (red). Note that Cx43EGFP does not discernibly colocalize with clathrin when the Cx43EGFP-expressing cells are in contact with Cx43KD cells. Occasional puncta that seem to colocalize with clathrin are likely to be undocked connexons that were endocytosed from the noncontacting cell surface. (B) Cx43 connexons fail to dock upon arrival at the cell surfaces of BxPC3 and Capan-1 cells. Bx43KD and CP43KD cells stably expressing knocked-in WT-Cx43EGFP or WT-Cx43mAPPLE were seeded on glass coverslips, as described in Materials and Methods. After 24 h, cells were fixed, stained for 4′,6-diamidino-2-phenylindole (DAPI), and examined for colocalization of Cx43EGFP and Cx43mAPPLE. Note the absence of colocalization at the areas of cell–cell contact, as well as in the cytoplasm. (C) Cx43 is endocytosed as annular gap junctions in assembly-efficient LNCaP cells. LNCaP cells stably expressing Cx43EGFP or Cx43mAPPLE were cocultured for 24 h, fixed, stained for DAPI, and examined for the colocalization of Cx43EGFP and Cx43mAPPLE. Note that both gap junctions (red arrows) and intracellular vesicular puncta (white arrows) are composed of both Cx43EGFP and Cx43mAPPLE.
To examine whether cell–cell contact alters the endocytic itinerary of Cx43, we cocultured CP43KD and Bx43KD cells with cells expressing knocked-in Cx43EGFP. The cocultures were immunostained for clathrin to examine whether clathrin colocalized with Cx43EGFP. We found that, in both CP43KD and Bx43KD cells expressing Cx43EGFP, no discernible colocalization of clathrin with Cx43 was observed in contacting cells (Figure 10A) but extensive colocalization was observed in single isolated cells (unpublished data). Taken together, the above results suggest that cell-to-cell contact alters the endocytic itinerary of Cx43 and that intracellular vesicular puncta in contacting BxPC3 and Capan-1 cells are made up of undocked connexons.
DISCUSSION
Molecular cues that govern the assembly of Cxs into gap junctions upon arrival at the surface are poorly understood. Through two different approaches, based on the subcellular fate of Cx43 in single and contacting cells and on the knock down and knock in of WT-Cx43 and its various mutants, we have uncovered some novel aspects of the molecular circuitry that orchestrates the assembly and the disassembly of gap junctions. First, our study shows that the assembly of Cx43 is impaired in BxPC3 and Capan-1 cells because it is endocytosed by the clathrin-mediated pathway prior to its assembly into gap junctions. Second, our findings demonstrate that assembly is restored by expressing a sorting-motif mutant of Cx43 that cannot be endocytosed. Third, our results show that the phosphorylation on Ser-279 and Ser-282 disrupts gap junction assembly by inducing endocytosis of Cx43 by the clathrin-mediated pathway. Fourth, our study shows that Cx43 is assembled into gap junctions only when connexons are composed of forms that can be phosphorylated on Ser-279 and Ser-282 and forms in which phosphorylation on these serines is abolished. Finally, our results document that the endocytic itinerary of Cx43 is altered upon cell–cell contact: Cx43 traffics by EEA1-negative endosomes in contacting cells versus EEA1-positive endosomes in single cells. Our results imply that endocytosis of connexons seems to be the major control point that determines gap junction size and growth.
Endocytosis of Cx43 and gap junction assembly
The use of human pancreatic cancer cell lines Capan-1 and BxPC3, which coexpressed Cx43 and Cx26 endogenously and in which either both Cxs or only one of the two failed to assemble into gap junctions, allowed us to address an important question: What are the intrinsic and extrinsic determinants that dictate the assembly of Cx43 into gap junctions in these cell lines, and what are their respective contributions? To explore the molecular basis of defective assembly of Cx43 in BxPC3 and Capan-1 cells, we searched for motifs in Cx43 itself that might govern its assembly into gap junctions. We first discovered that Cx43 trafficked to the cell surface in both cell types but was endocytosed prior to its assembly into gap junctions by the clathrin-mediated pathway in a Rab5-dependent manner via a tyrosine-based sorting motif in its cytoplasmic tail that likely mediated its interaction with the AP2 complex (Figures 1 and 2). Because both Cx26 and Cx32 assembled into gap junctions in BxPC3 cells, these results suggested that the defective assembly of Cx43 was not caused by spatial constraints imposed by extrinsic determinants, such as the expression of cell–cell adhesion molecules, which might have hindered the docking of connexons to form cell–cell channels and their subsequent clustering into gap junctions (Chakraborty et al., 2010; Musil, 2009; Musil et al., 1990; Wang and Mehta, 1995), but by the endocytosis of Cx43 itself.
Restoration of defective gap junction assembly
Our results showed that transient, as well as stable, expression of the sorting-motif mutant YA/VD-Cx43 restored defective gap junction assembly in BxPC3 and Capan-1 cells expressing endogenous Cx43. Moreover, assembly was also restored upon expressing mutants S279A, S282A, and S279/282A, which mimicked the nonphosphorylated state, but not upon expressing S279D, S282D, and S279/282D, which mimicked the phosphorylated state (Figure 5). Furthermore, restoration most likely occurred through the formation of heteromers with the endogenous Cx43 (Figures 4 and 5). These results raise intriguing issues with regard to the possible mechanisms that dictate the assembly of Cx43 into gap junctions in these cell types. The most likely explanation for these data are that phosphorylation of Ser-279 or Ser-282 regulates sorting motif–mediated endocytosis and that gap junction assembly is enhanced due to the inhibition of endocytosis of connexons composed of heteromers of Cx43 and either YA/VD-Cx43, S279A, or S282A, permitting them to be incorporated in the plaque. This interpretation is supported by the fact that both Ser-279 and Ser-282 lie close to the proline-rich sorting motif of Cx43, and phosphorylation and dephosphorylation of serines near sorting motifs of many G-protein–coupled receptors have been shown to modulate their endocytosis by regulating their interaction with the AP2 complex (Pitcher et al., 1999; Bonifacino and Traub, 2003; Rapacciuolo et al., 2003; Chen et al., 2004; Kittler et al., 2005; Toshima et al., 2009; Traub, 2009; Moeller et al., 2010; Shukla et al., 2011). Moreover, Ser-279 and Ser-282 have previously been shown to be phosphorylated by MAP kinase (Warn-Cramer et al., 1996, 1998), which has been thought to mediate internalization of gap junctions in response to epidermal growth factor and tumor promoters (Kanemitsu and Lau, 1993; Ruch et al., 2001; Leithe and Rivedal, 2004a, 2004b; Leithe et al., 2009; Solan and Lampe, 2008, 2009). Furthermore, our results showing lack of discernible colocalization of clathrin and EEA1 with Cx43 in single cells that expressed heteromers of Cx43 and mutant YA/VD-Cx43 (Figure 4) but a robust colocalization in single cells that expressed only WT-Cx43 (Figures 2 and 4) lend further credence to the above notion. In contrast with their lack of colocalization with clathrin and EEA1, connexons composed of either Cx43 alone or of both Cx43 and YA/VD-Cx43, or S279A colocalized extensively with Lamp1, indicating that all were degraded in the lysosome (Figure S6). Moreover, it seems highly unlikely that the restored gap junctions were formed of cell–cell channels composed of homomeric connexons containing only Cx43, YA/VD-Cx43, or S279A, because channels formed of heteromeric connexons of different Cx subtypes have been demonstrated to readily assemble into functional gap junctions in diverse cell types (Jiang and Goodenough, 1996; Valiunas et al., 2001; Churko et al., 2010). Furthermore, in contacting cells that coexpressed endogenous Cx43 and YA/VD-Cx43 (Figure 4A) or mutant YA/VD-Cx43 alone (Figure S6B), only large vesicular Cx43 puncta of diverse sizes were observed these did not colocalize with clathrin and caveolin 1 and may represent annular gap junctions.
Ser-279 and Ser-282 of Cx43 and gap junction assembly
A salient and the most striking result of our study is that gap junction assembly was induced only when connexons were composed of Cx43 forms that can be phosphorylated on Ser-279 and Ser-282 and forms in which phosphorylation was abolished (mutants S279A, S282A, and S279/282A). Gap junction assembly was impaired when connexons were predominantly composed of Cx43 forms that mimicked the phosphorylation on Ser-279 and Ser-282 and when connexons were composed of Cx43 forms in which phosphorylation on these residues was abolished (Figures 6–8). The most plausible interpretation of these data is that the phosphorylated Ser-279 and Ser-282 (either alone or together) in the Cx43 cytoplasmic tail, previously documented to be the substrates for MAP kinase (Warn-Cramer et al., 1996, 1998), regulate junction assembly not only by modulating endocytosis of connexons but also by influencing plaque growth by a mechanism(s) that remains to be defined. Several lines of evidence support this interpretation.
First, knock in of S279A, S282A, S279/282A, S279D, S282D, or S279/282D alone in Bx43KD and CP43KD cells did not induce gap junction assembly. Moreover, the assembly of S279A and S279D was also robustly attenuated in assembly-efficient LNCaP cells (Figure S5), which suggested that these mutants were able to assemble into small gap junction plaques, but the plaques either did not grow or failed to coalesce to form larger plaques. Second, transient expression of mutants S279A or S282A, but not mutants S279D or S282D, restored gap junction assembly in cells that expressed endogenous Cx43, and the effect was as robust as with YA/VD-Cx43 (Figure 5). Third, expression of Myc-tagged, WT-Cx43 induced gap junction assembly in Bx43KD cells stably expressing S279A but not in cells expressing S279D (Figure 8). Moreover, S279A and S282A, like YA/VD-Cx43, also restored gap junction assembly through the formation of heteromers with the WT-Cx43 (Figures 5 and 8). Fourth, retroviral expression of only YA/VD-Cx43 resulted in the formation of gap junctions in Bx43KD and CP43KD cells and in assembly-efficient LNCaP cells (Figure S5). Although not demonstrated directly, YA/VD-Cx43 connexons are likely to contain Cx43 forms that are both phosphorylated and not phosphorylated on Ser-279/Ser-282. Moreover, cell surface biotinylation of YA/VD-Cx43, S279A, and S279D showed that they trafficked to the cell surface and were degraded with similar kinetics (Figure S5).
NEDD4 is a ubiquitin ligase that interacts with Cx43 via its tryptophan-tryptophan (WW) domains (Leykauf et al., 2006). NEDD4-dependent ubiquitylation of Cx43 could thus play a role in the failure of BxPC3 and Capan-1 to assemble Cx43-containing gap junctions. The WW2 domain of NEDD4 interacted with peptides containing the proline-rich tyrosine-based (PY) motif of Cx43 and was shown to have a slightly higher affinity for peptides with phosphoserines 279 and 282 than for their nonphosphorylated counterparts. However, the WW3 domain was speculated to be the dominant mediator of the binding of NEDD4 to Cx43, and full-length Cx43 with phosphoserines 279 and 282 did not interact with the WW3 domain. The phosphorylation of Ser-279 and Ser-282 by MAP kinase (Warn-Cramer et al., 1996, 1998) has been thought to mediate internalization of gap junctions in response to epidermal growth factor and tumor promoters (Kanemitsu and Lau, 1993; Leithe and Rivedal, 2004a, 2004b; Leithe et al., 2009; Solan and Lampe, 2008, 2009). However, we found that S279A and S282A failed to assemble in both Bx43KD and CP43KD (Figures 6 and 7). In addition, we found YA/VD-Cx43 assembled efficiently into gap junctions in the knockdown cells (Figures 6 and 7), in spite of the fact that Cx43-Y286A could still get ubiquitylated (Leykauf et al., 2006; Girao et al., 2009; Catarino et al., 2011). Thus the role that NEDD4 plays in the cells used in this study is unclear. Finally, the fact that gap junctions composed of mutant YA/VD-Cx43 in BxPC3 and Capan-1 cells appeared to be internalized in the form of annular junctions under both basal and starved conditions (Figure 9) suggests that clathrin-mediated endocytosis by AP2 cannot account for the internalization of this mutant. The pathway by which YA/VD-Cx43 and S279A and S282A are internalized and degraded remains to be explored in future studies.
Endocytic itinerary of Cx43 and cell–cell contact
A salient aspect of our results was that a major fraction of Cx43 appeared to reside in intracellular vesicles upon endocytosis. Despite this, discernible colocalization of Cx43 with clathrin and EEA1 was observed only in single cells—not in contacting cells (Figure 2). In contrast, in both single and contacting cells, Cx43 discernibly colocalized with AP2 and with Rab5, which is involved in clathrin-mediated endocytosis and acts upstream of EEA1 (Bucci et al., 1992; Zerial and McBride, 2001; Semerdjieva et al., 2008; Figures 2 and S3). These results were intriguing, given the fact that Cx43 trafficked to the cell surface in both cell types, and nearly all the Cx43 resided in intracellular vesicles that could be extracted in situ with 1% TX100 (Figures 1 and 7). Thus the intracellular vesicular puncta observed in these cells were not internalized annular gap junctions or aggregates of Cx43 that were detergent-resistant but represented vesicles that trafficked via EEA1-negative endosomes in contacting cells en route to the lysosomes. As Cx43 also did not discernibly colocalize with caveolin 1 (Figure S2), one plausible explanation for these data is that undocked connexons, upon clathrin-mediated endocytosis in single cells, traverse via EEA1-positive endosomes, whereas docked connexons and small miniscule annular gap junctions—which may not have been resolved immunocytochemically—traverse via vesicles that are EEA1-negative. Although highly unlikely, sporadic colocalization of intracellular Cx43 with clathrin observed in contacting cells may represent clathrin-coated secretory vesicles (Bonifacino, 2004; Weisz and Rodriguez-Boulan, 2009; Gravotta et al., 2012).
When Bx43KD and CP43KD cells expressing either Cx43EGFP or Cx43mAPPLE were cocultured, we did not observe vesicles containing both Cx43EGFP and Cx43mAPPLE, suggesting that the vesicles arose predominantly from undocked connexons (Figure 10B). Moreover, in cocultures with knockdown cells, we saw no discernible colocalization of clathrin with Cx43 in contacting cells (Figure 10A), in contrast with robust colocalization seen in single cells. One interpretation of these results is that the endocytic itinerary of Cx43 is altered upon cell–cell contact, such that, after clathrin-mediated endocytosis, Cx43 traffics by EEA1-negative endosomes in contacting cells and by EEA1-positive endosomes in single cells. It is also possible that the spatiotemporal characteristics of clathrin coats of endocytic vesicles in contacting cells are different from those in single cells, such that they cannot be discerned in the captured static images of contacting cells. The existence of at least two modes of endocytic coat formation at the plasma membrane—classical clathrin-coated pits and clathrin-coated plaques with distinct spatiotemporal characteristics—has been described (Saffarian et al., 2009). It is worth considering that the connexons, unlike other well-studied transmembrane proteins, such as growth factor receptors and G-protein–coupled receptors (Moore et al., 2007; Shenoy and Lefkowitz, 2011), are hexamers of Cxs, which are known to interact with cytoplasmic proteins—that are also part of other junctional complexes—and are recruited to the site of cell–cell contact upon cell–cell adhesion (Laird, 2010; Herve et al., 2012). Thus, although there is no coherent theme for the roles of these proteins in junction assembly or disassembly, these attributes of connexons might impose unique spatial constraints on the endocytic machinery, causing Cxs to follow a noncanonical endocytic route in contacting cells but not in single cells (Boulant et al., 2011; McMahon and Boucrot, 2011; Sigismund et al., 2012).
Recent findings have shown that, upon internalization, cell surface–associated Cxs are degraded by autophagy and that inhibition of autophagy stabilizes the cell surface–associated pool of Cxs (Hesketh et al., 2010; Lichtenstein et al., 2011; Bejiarno et al., 2012; Fong et al., 2012). Given the fact that the plasma membrane has now been shown to contribute to the formation of preautophagosomal structures (Ravikumar et al., 2010), it is tempting to speculate that cell–cell contact dictates the choice of the endocytic itinerary of Cx43, such that it traffics from noncontacting surfaces by EEA1-positive endosomes, whereas it is linked from contacting surfaces to vesicles destined to fuse with autophagosomes. Recent studies have identified the existence of a new class of early endosomes that bear the membrane adaptor proteins APPL1 and APPL2 (Miaczynska et al., 2004). These endosomes have been shown to be the precursors of EEA1-positive endosomes (Zoncu et al., 2009). Many transmembrane proteins have been demonstrated to interact with APPL1 and APPL2, which have been shown to act not only as effectors of Rab5 (Zhu et al., 2007) but also as dynamic scaffolds that modulate Rab5-associated signaling (Caruso-Neves et al., 2006; Mao et al., 2006; Varsano et al., 2006; Chial et al., 2008). Earlier studies had demonstrated or assumed that Cx43, after being endocytosed by the clathrin-mediated pathway, trafficked via EEA1-positive early endosomes to lysosomes for degradation (Leithe et al., 2006, 2009; Piehl et al., 2007; Gumpert et al., 2008; Nickel et al., 2008). Although it is as yet unknown how endocytic vesicles are directed to the autophagosomes, our results provide a plausible explanation for the distinct endocytic routes of Cx43 in single versus contacting cells. The nature of the vesicles used for the trafficking of internalized Cx43 in contacting cells and how the endocytic fate of Cx43 is modulated by cell–cell contact remain to be investigated in these cell types at ultrastructural levels.
Conclusions
Although phosphorylation of the cytoplasmic tail of Cx43 on specific serines and tyrosines by protein kinases has been documented to regulate the function of cell–cell channels in both tissues and cell lines, evidence regarding its role in the assembly and disassembly of gap junctions has been correlative, and no coherent theme has yet emerged (Laird, 2005; Solan and Lampe, 2009). Our results demonstrate that phosphorylation on Ser-279 and Ser-282 not only disrupts gap junction assembly by triggering endocytosis of Cx43 prior to its assembly but also likely regulates plaque growth. Earlier pioneering studies of Musil and Goodenough demonstrated the existence of plaque-associated and detergent-resistant multiple phosphorylated forms of Cx43 and postulated these forms to be involved in the establishment and/or maintenance of gap-junctional plaques (Musil et al., 1990; Musil and Goodenough, 1991). Our results, showing that gap junction assembly is induced only when connexons are composed of Cx43 forms that can be phosphorylated and forms that cannot be phosphorylated on Ser-279 and Ser-282, provide direct evidence for this role. Our results, however, do not exclude the important role of phosphorylation and dephosphorylation of other serines in the cytoplasmic tail of Cx43, for example, Ser-365, Ser-364, and Ser-368, in determining gap junction assembly and disassembly (Solan et al., 2007; Solan and Lampe, 2008, 2009; TenBroek et al., 2001). Because Ser-279 and Ser-282 have also been found to be phosphorylated by multiple signaling pathways (Solan and Lampe, 2008), a question that remains to be answered is how the stoichiometry of heteromerization of the various phosphoforms of Cx43 and the differential phosphorylation and dephosphorylation of these sites is fine-tuned to control plaque growth and dynamics in BxPC3 and Capan-1 cells. It also remains to be determined whether this mechanism is used in normal cells in vitro and in vivo to regulate gap junction assembly. On the basis of our studies, we propose that endocytosis of connexons is the major control point that determines gap junction size and growth (as summarized in Figure 11).
FIGURE 11:
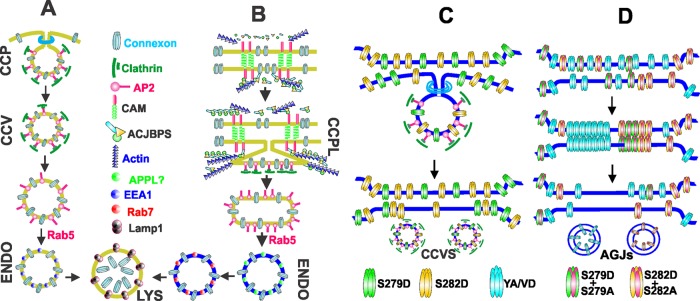
(A) In single cells, and from cell surfaces that are not in contact with other cells, connexons are endocytosed by clathrin-coated pits, which are directed toward the lysosomes for degradation by Rab5 via EEA1-positive endosomes. (B) Cell–cell contact is assumed to recruit various cell junction–associated proteins to the cell surface, where they establish a direct or an indirect link between the connexons and the actin cytoskeleton. From cell–cell contact areas, connexons are postulated to be endocytosed via clathrin-coated plaques, which are directed toward the lysosomes by Rab5 via noncanonical endosomes and intermediate vesicles yet to be characterized. (C) Phosphorylation of Cx43 on S279 and S282 induces connexon endocytosis via the clathrin-mediated pathway by enhancing Cx43’s interaction with the AP2 complex, which is postulated to attenuate gap junction assembly. (D) Gap junction assembly is robustly induced when the interaction of the AP2 complex with the sorting motif is abolished through mutation or through dephosphorylation of Ser-279 and Ser-282 of some, but not all, connexin molecules in a connexon. It is postulated that the stoichiometry of phosphorylation and dephosphorylation of Ser-279 and Ser-282 of some connexin molecules in a connexon determines gap junction size, not only by regulating connexon endocytosis, but also by facilitating connexon–connexon docking to form cell–cell channels and their subsequent incorporation into gap-junctional plaques. Inhibition of clathrin-mediated endocytosis of connexons triggers internalization via the formation of annular gap junctions. ACJBPS = actin and cell junction binding proteins AGJs = annular gap junctions CAM = cell adhesion molecule CCP = clathrin-coated pit CCPL = clathrin-coated plaque CCV and CCVS = clathrin-coated vesicles ENDO = endosome, LYS = lysosome YA/VD = sorting motif mutant Cx43.
MATERIALS AND METHODS
Cell culture
Several human pancreatic tumor cell lines were used initially to screen for the expression of Cx26, Cx32, Cx36, and Cx43 by RT-PCR (unpublished data). The cell lines were a gift from Surinder K. Batra (Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center). For subsequent studies, two human pancreatic cancer cell lines, BxPC3 (CRL-1687) and Capan-1 (HTB-79), were purchased from the American Type Culture Collection (ATCC, Manassas, VA). The characterization of these cell lines has been described (Tan et al., 1986; Fanjul and Hollande, 1993). BxPC3 and Capan-1 cells were grown, respectively, in RPMI 1640 and DMEM (GIBCO, Grand Island, NY) containing 5% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) in an atmosphere of 5% CO2 at 37°C. Stock cultures were maintained weekly by seeding 5 × 105 cells per 10-cm dish in 10 ml of complete culture medium with a medium change at day 3 or 4. New stocks were initiated every 3 mo from cells frozen in liquid nitrogen. The retroviral packaging cell lines EcoPack and PTi67 were grown as described previously (Mehta et al., 1999; Mitra et al., 2006). BxPC3 and Capan-1 cells were infected with various recombinant retroviruses, and pooled polyclonal cultures from ∼2000 colonies were grown and maintained in complete medium containing G418 (200 μg/ml), puromycin (0.5 μg/ml for BxPC3 and 1 μg/ml for Capan-1), or G418 plus puromycin (see Recombinant DNA constructs and retrovirus production and infection).
Antibodies and immunostaining
Rabbit polyclonal and mouse monoclonal antibodies against Cx43 and Cx32 and mouse anti-GM130, mouse anti-clathrin heavy chain, mouse anti–β-catenin, and rabbit anti–β-actin have been described previously (Mitra et al., 2006; Chakraborty et al., 2010; Govindarajan et al., 2010). A mouse monoclonal antibody against human Cx26 (13–8100) was purchased from Zymed Laboratories (San Francisco, CA). Mouse anti–caveolin 1 (610058), anti–caveolin 2 (610684), and anti-EEA1 (610457) were purchased from BD Transduction (San Jose, CA). We also used mouse monoclonal antibodies against Lamp1 (SC-20011; Santa Cruz Biotechnology, Santa Cruz, CA), c-Myc (MMS-150P; Covance, Princeton, NJ), and GFP (Roche Indianapolis, IN). A monoclonal antibody against human desmoglien-2 was a gift from James E. Wahl (Department of Oral Biology, University of Nebraska Medical Center).
For immunostaining, 5 × 105 BxPC3 and 4 × 105 Capan-1 cells were seeded on glass coverslips in six-well clusters and allowed to grow for 72 h, after which they were fixed with 2% paraformaldehyde and immunostained as described previously (Chakraborty et al., 2010; Govindarajan et al., 2010). Anti-rabbit and anti-mouse secondary antibodies, conjugated with Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen), were used as appropriate. Immunostained cells were mounted on glass slides in SlowFade antifade (Invitrogen, Carlsbad, CA) medium. Images of immunostained cells were acquired with a Leica DMRIE microscope (Leica Microsystems, Wetzler, Germany) equipped with a Hamamatsu ORCA-ER2 CCD camera (Hamamatsu City, Japan) using a 63× oil objective (numerical aperture 1.35). Colocalization was measured in Z-stacked images taken 0.3 μm apart using the commercial image analysis program Volocity 6.0.1 (Improvision, Lexington, MA). Saturation of the detector, which would alter background adjustment, was prevented by minimizing exposure of each fluorescently tagged antibody during acquisition. A background-corrected Pearson's r was used to determine fluorescence colocalization as previously described (Barlow et al., 2010)
Detergent extraction and Western blot analysis
BxPC3 (3 × 106) and Capan-1 (2 × 106) cells were seeded in replicate 10-cm dishes with 10 ml of complete medium per dish. Cells were grown for 72 h. Cell lysis, detergent solubility assay with 1% Triton X-100 (TX100), and Western blot analysis were performed as described previously (Mitra et al., 2006; Chakraborty et al., 2010; Govindarajan et al., 2010). For the analysis of detergent-soluble and detergent-insoluble fractions by SDS–PAGE, normalization was based on equal cell number. For the detergent extraction of live cells in situ, BxPC3 and Capan-1 cells were seeded in six-well clusters containing glass coverslips at a density of 5 × 105 and 4 × 105 cells per well, respectively. After 72 h, cells were extracted in situ with 1% TX100 in isotonic medium (30 mM HEPES, pH 7.2, 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) supplemented with protease inhibitor cocktail (Sigma-Aldrich) for 60 min at 4ºC and then fixed and immunostained essentially as described previously (Govindarajan et al., 2002, 2010; Chakraborty et al., 2010).
Cell surface biotinylation assay
BxPC3 (5 × 105) and Capan-1 (4 × 105) cells were seeded in 6-cm dishes in replicate and grown to 70–80% confluence. The biotinylation reaction was performed at 4°C using freshly prepared EZ-LinkSulfo-NHS-SS Biotin reagent (Pierce, Rockford, IL) at 0.5 mg/ml in phosphate-buffered saline (PBS) supplemented with 1 mM CaCl2 and 1 mM MgCl2 (PBS-PLUS) for 1 h. The reaction was quenched with PBS-PLUS containing 20 mM glycine, and cells were lysed as described previously (Govindarajan et al., 2002, 2010; Chakraborty et al., 2010). The affinity precipitation of biotinylated proteins was performed with 200 μg of total protein using 100 μl of streptavidin-agarose beads (Pierce) on a rotator overnight at 4°C. The streptavidin-bound biotinylated proteins were eluted and resolved by SDS–PAGE followed by Western blotting as described previously (Govindarajan et al., 2002, 2010; Chakraborty et al., 2010). The kinetics of degradation of cell surface–associated Cx43 was determined as follows: after biotinylation, washing, and quenching of biotin, biotinylated proteins were chased at 37°C for various times before affinity precipitation with streptavidin. These methods have been described in our earlier studies (Govindarajan et al., 2010). The protein concentration was determined using the BCA reagent (Pierce).
Recombinant DNA constructs and retrovirus production and infection
Wild-type rat Cx43 and its various mutants were cloned into pcDNA3.1 and the retroviral vector pLXSN using PCR cloning and standard recombinant DNA protocols. Mutants were generated by site-directed mutagenesis using a QuikChange kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. WT-Cx43 and its various mutants tagged in-frame with green (EGFP) fluorescent protein and with a Myc epitope were constructed using pEGFP-N1 and pMyc-CMV (Clontech, Mountain View, CA). All constructs were verified by DNA sequencing (ACGT, Wheeling, IL). These mutants are shown in Figure 12. For construction of an EGFP-tagged μ2 subunit of AP2 complex, a fragment of mouse μ2 corresponding to that in clone 3 μ2 (Ohno et al., 1995) was tagged at the N-terminus with mEGFP (GFPμ2). Rat Cx43 tagged in-frame with mAPPLE was a gift from Matthias Falk (Department of Biological Sciences, Lehigh University). The source of the retroviral vector pSUPER.retro.puro and the construction of the retroviral vector LXSNCx32 have been described in our earlier studies (Mitra et al., 2006; Chakraborty et al., 2010). Rab5 and their constitutively active (pRab5Q67L) and dominant-negative (pRab5S34N) mutants tagged with EGFP were a gift from Steve Caplan (Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center).
The retroviral vectors were used to produce recombinant retroviruses in EcoPack and PTi67 packaging cell lines as follows. EcoPack (Clontech) cells were seeded in duplicate at a density of 106 cells per 10-cm dish. The cells were transfected with 20 μg of retroviral vectors (plasmids) using Fugene for 6 h (Roche). The medium was replaced with 10 ml of fresh medium, and cells were incubated for additional 48 h, after which the medium containing the recombinant retrovirus was collected and filtered (0.45 μM; Millipore, Billerica, MA). For production of recombinant retrovirus for infection of target cells, amphotropic PTi67 cells were infected with the transiently produced recombinant retrovirus from EcoPack and selected in G418 (400 μg/ml), when infected with LXSN constructs, or puromycin (1 μg/ml), when infected with pSuper.retro.puro. The recombinant retrovirus produced from the pooled polyclonal cultures of PTi67 cells was assayed for the virus titer by colony-forming units as previously described (Mehta et al., 1991). PTi67 cells producing the virus were frozen. BxPC3 and Capan-1 cells were multiply (two to four times) infected with various recombinant retroviruses produced from PTi67 cells and selected in either G418 (400 μg/ml) or puromycin (1 μg/ml for BxPC3 and 2 μg/ml for Capan-1) for 2–3 wk in complete medium. Pooled cultures from ∼2000 colonies obtained from three to four dishes were expanded, frozen, and maintained in selection media containing G418 (200 μg/ml) and in G418 plus puromycin (1 μg/ml) for CP43KD and Bx43KD cells expressing the knocked-in constructs. Pooled polyclonal cultures were used within two to three passages for immunocytochemical and biochemical analyses.
Cell transfection
Twenty-four hours prior to transfection, Capan-1 (1.5 × 106) and BxPC3 (106) cells were seeded on glass coverslips in six-well clusters. Cells were transfected with various plasmids in duplicate using Fugene (Roche Diagnostics) according to the manufacturer's instructions. For transfection of single plasmids, 2 μg of plasmid DNA was used per well. For cotransfection of two plasmids, pEGFP (0.5 μg) and the plasmid DNA (1.5 μg), containing the gene whose expression was to be detected were mixed. Expression was analyzed 16–24 h posttransfection after cells were fixed and immunostained with the desired antibodies as previously described (Mitra et al., 2006).
Yeast two-hybrid analysis
To test for the direct interaction of Cx43 with the μ2 subunit of AP-2 complex, we used yeast two-hybrid analysis (BD Biosciences/Clontech, Franklin Lakes, NJ). Briefly, the cytoplasmic tail of Cx43 (residues 240–382) was cloned into the EcoRI and BamHI sites of pGBKT7 to give pGBKT743-WT. Site-directed mutagenesis was then used to generate pGBKT743-Y286A, pGBKT743-V289D, and pGBKT743-Y286A/V289D. pGADT7-T containing the μ2 subunit of AP2 complex was a gift from Steve Caplan (Biochemistry and Molecular Biology, University of Nebraska Medical Center). These plasmids were used to transform Saccharomyces cerevisiae strain AH109 (BD Biosciences/Clontech) by the lithium acetate procedure, using the MATCHMAKER two-hybrid kit (BD Biosciences/Clontech) essentially as described (Naslavsky et al., 2006).
Immunoprecipitation
For the coimmunoprecipitation experiments shown in Figure 4, HeLa cells, grown to 50% confluence in 10-cm dishes, were transfected with 10 μg of pcDNACx43EGFP, pcDNACx43-Myc, pcDNA-Y286A/V289D-Cx43-Myc (YA/VD-Cx43-Myc), and pcDNA-YA/VD-Cx43-EGFP (YA/VD-Cx43EGFP) either alone or in combination. Twenty-four hours posttransfection, cells were harvested and lysed as described above in Detergent extraction and Western blot analysis. Following lysis and protein estimation, 1 mg of total protein was precleared with the Control Agarose Resin in the Pierce Co-IP Kit (Pierce) at 4°C for 1 h with gentle end-over-end mixing. The precleared lysate was then added to AminoLink Plus Coupling Resin, containing covalently bound monoclonal-GFP antibody (Roche) and incubated at 4°C for 16 h with gentle end-over-end mixing. After incubation, the GFP resin was washed three times in immunoprecipitation/wash buffer (Pierce) for 10 min with gentle shaking. Bound proteins were eluted from the GFP resin using the Co-IP kit elution buffer, containing primary amines (Pierce). For Western blot analysis, 5X reducing lane Marker Sample Buffer (Pierce) was added to the eluted samples to a final concentration of 1X. Pull down was assessed by Western blotting with monoclonal-Myc antibody.
Connexin43 knockdown
Accell small interfering RNAs (siRNAs) with target sequences to the 3′ UTR of human Cx43 were purchased from Dharmacon/Thermo Scientific (Lafayette, CO). BxPC3 cells were seeded at a density of 8 × 104 cells per well in a 12-well cluster and, after 24 h, were transfected with the siRNAs using Oligofectamine according to the manufacturer's instructions (Invitrogen). Cells were incubated in the transfection medium mixture for 72 h, after which the medium was replaced with complete RPMI medium, and the cells were allowed to grow for an additional 24 h. Cells were harvested and lysed as described above, and knockdown was assessed by Western blot analysis.
The most potent Cx43 siRNA was selected and used to construct an shRNA. As a control, a scrambled sequence of the Cx43 siRNA was generated using siRNA Wizard software (InvivoGen, San Diego, CA). The shRNA oligos were purchased from Integrated DNA Technologies (Coralville, IA). The retroviral vector, pSUPER.retro.puro (OligoEngine, Seattle, WA), was used to produce shRNAs with the scrambled (Sh-Scr) Cx43 sequence (5′-GCUAUAGGAUACGGAUAAU-3′) and Cx43-targeting (Sh-Cx43) sequence (5′-GUAGUGGAUUCAAAGAACU-3′) according to the manufacturer's instructions. The knockdown efficiency was assessed by transient transfection of these plasmids in BxPC3 and Capan-1 cells followed by Western blot and immunocytochemical analysis, as described above. After verification of knockdown, recombinant retroviruses containing Sh-Scr and Sh-Cx43 were produced in PTi67 cells, as described above. For stable knockdown, BxPC3 and Capan-1 cells were multiply infected with the recombinant retroviruses, and pooled polyclonal cultures were established as described in Recombinant DNA Constructs and retrovirus production and infection. Stable Cx43 knockdown BxPC3 and Capan-1 cells were designated as Bx43KD and CP43KD, respectively.
Single-cell and coculture experiments
For the single-cell experiments shown in Figures 2, S3, 4, and S6, BxPC3 and Capan-1 cells were seeded in six-well clusters at a density of (2–5) × 103 cells per well in 2 ml of complete culture medium as previously described (Govindarajan et al., 2010). For the coculture experiments shown in Figure 10, Cx43EGFP or Cx43mAPPLE was introduced into Bx43KD and CP43KD cells using LXCx43EGFP and LXCx43mAPPLE, and pooled polyclonal cultures expressing each construct were established separately upon selection in G418, as described above. Bx43KD and CP43KD cells expressing either Cx43EGFP or Cx43mAPPLE were suspended in medium and then mixed together in a 1:1 ratio and allowed to aggregate in suspension for 4 h at 37°C in an atmosphere of 5% CO2 to maximize the formation of doublets, triplets, and quadruplets. After 4 h, these cells were plated into six-well clusters at a density of 105 cells per well. After 20 h, cells were fixed and mounted on slides. For coculture experiments with the human prostate cancer cell line LNCaP, cells were infected with the retroviruses containing Cx43EGFP or Cx43mAPPLE, and cells expressing them were cocultured using the approach described above for BxPC3 and Capan-1 cells. To examine whether cell–cell contact determines Cx43 colocalization with clathrin, Bx43KDKI and CP43KDKI cells expressing Cx43EGFP were cocultured with Bx43KD and CP43KD cells at a ratio of 1:4, as described above, and colocalization of Cx43 with clathrin was determined upon immunostaining.
Quantitation of gap junction assembly/restoration
For quantifying gap junction assembly, pEGFP (0.5 μg) and the wild-type or mutant cDNAs (1.5 μg) were cotransfected into BxPC3, Capan-1, Bx43KD, and CP43KD cells as described above in Cell transfection. After 16–24 h, pairs of cells expressing equal levels of EGFP (as assessed visually) were counted for the formation of gap junctions, as exemplified by Figure 3. The percentage of cells with restored gap junctions was determined by counting 15–20 EGFP-expressing cells.
Communication assays
Gap-junctional communication was assayed by microinjecting fluorescent tracers LY (Lucifer Yellow; MW 443 Da lithium salt), Alexa Fluor 488 (MW 570 Da A-10436), or Alexa Fluor 594 (MW 760 Da A-10438), and by the scrape-loading assay as previously described (Govindarajan et al., 2002, 2010; Chakraborty et al., 2010; Kelsey et al., 2012). All Alexa dyes were purchased as hydrazide sodium salts from Molecular Probes (Carlsbad, CA), and stock solutions for microinjection were prepared in water at 10 mM. Eppendorf InjectMan and FemtoJet microinjection systems (models 5271 and 5242; Brinkmann Instrument, Westbury, NY) mounted on a Leica DMIRE2 microscope were used to microinject the fluorescent tracers. Junctional transfer of fluorescent tracers was quantitated by scoring the number of fluorescent cells (excluding the injected one) from the captured TIFF images either at 1 min (LY), 3 min (Alexa 488), or 15 min (Alexa 594) after microinjection into test cells as previously described (Mehta et al., 1986, 1999; Mitra et al., 2006; Chakraborty et al., 2010). Cells were scrape-loaded in 1 ml of medium containing rhodamine-conjugated fluorescent dextrans (10 kDa, 1 mg/ml, fixable) and LY (0.5%) essentially as previously described (Govindarajan et al., 2010).
Supplementary Material
Acknowledgments
We thank Souvik Chakraborty for helpful discussion and suggestions. We thank Steve Caplan and Naava Naslavsky for help with the yeast two-hybrid analysis, as well as for helpful suggestions regarding endocytosis throughout the course of this study. We thank Surinder Batra for providing us with the various pancreatic cancer cell lines for the initial screen, Maneesh Jain for RT-PCR analysis, and Matthias Falk for providing us with Cx43mAPPLE. This research was supported by National Institutes of Health grants CA113903, DOD PCRP PC-081198, and PC-111867 and Nebraska State grants LB506 (P.P.M.) and R0-1DE12308 (K.R.J.). We gratefully acknowledge support from the Nebraska Center for Cellular Signaling and Cancer Graduate Research Training Program of Eppley Institute in the form of fellowships to K.E.J.
Abbreviations used:
- AP
adaptor protein
- Bx43KD
BxPC3 stably knocked-down cells
- Bx43KDKI
Bx43KD cells with knocked-in WT or mutant Cx43
- CP43KD
Capan-1 stably knocked-down cells
- CP43KDKI
CP43KD cells with knocked-in WT or mutant Cx43
- Cx
connexin
- DAPI
4′,6-diamidino-2-phenylindole
- GFP
green fluorescent protein
- LY
Lucifer Yellow
- MAP
mitogen-activated protein
- mEGFP
Monomeric enhanced green fluorescent protein
- PBS
phosphate-buffered saline
- Rab5-DA
dominant-active Rab5
- Rab5-DN
dominant-negative Rab5
- Rab5-WT
wild-type Rab5
- RT-PCR
reverse transcriptase polymerase chain reaction
- shRNA
short hairpin RNA
- TX100
Triton X-100
- UTR
untranslated region
- WT
wild type
- YA/VD-Cx43
mutant Y286A/V289D
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-07-0537) on January 30, 2013.
REFERENCES
- Barlow AL, MacLeod A, Noppen S, Sanderson J, Guérin CJ. Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of Pearson's correlation coefficient. Microsc Microanal. 2010;16:710–724. doi: 10.1017/S143192761009389X. [DOI] [PubMed] [Google Scholar]
- Bavamian S, Klee P, Allagnat F, Haefliger J-A, Meda P. Connexins and secretion. In: Harris A., Locke D., editors. Connexins: A Guide. New York: Springer; 2009. pp. 511–528. [Google Scholar]
- Bejiarno E, Girao H, Yuste A, Patel B, Marques C, Spray D, Pereira P, Cuervo A. Autophagy modulates dynamics of connexons at the plasma membrane in an ubiquitin-dependent manner. Mol Biol Cell. 2012;23:2156–2169. doi: 10.1091/mbc.E11-10-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer EC, Berthoud VM. The family of connexin genes. In: Harris A., Locke D., editors. Connexins: A Guide. New York: Springer; 2009. pp. 3–26. [Google Scholar]
- Bonifacino JS. The mechanism of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Bosco D, Haefliger JA, Meda P. Connexins: key mediators of endocrine function. Physiol Rev. 2011;91:1393–1445. doi: 10.1152/physrev.00027.2010. [DOI] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol. 2011;13:1124–1131. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- Caruso-Neves C, Pinheiro AA, Cai H, Souza-Menezes J, Guggino WB. PKB and megalin determine the survival or death of renal proximal tubule cells. Proc Natl Acad Sci USA. 2006;103:18810–18815. doi: 10.1073/pnas.0605029103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino S, Ramahlo J, Marques C, Pereira P, Girao H. Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem J. 2011;437:255–269. doi: 10.1042/BJ20102059. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Mitra S, Falk MM, Caplan S, Wheelock MJ, Johnson KR, Mehta PP. E-cadherin differentially regulates the assembly of connexin43 and connexin32 into gap junctions in human squamous carcinoma cells. J Biol Chem. 2010;285:10761–10776. doi: 10.1074/jbc.M109.053348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Ren XR, Nelson CD, Barak LS, Chen JK, Beachy PA, de Sauvage F, Lefkowitz RJ. Activity-dependent internalization of Smoothened mediated by β-arrestin 2 and GRK2. Science. 2004;306:2257–2260. doi: 10.1126/science.1104135. [DOI] [PubMed] [Google Scholar]
- Chial HJ, Wu R, Ustach CV, McPhail LC, Mobley WC, Chen YQ. Membrane targeting by APPL1 and APPL2: dynamic scaffolds that oligomerize and bind phosphoinositides. Traffic. 2008;9:215–229. doi: 10.1111/j.1600-0854.2007.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko JM, Langlois S, Pan X, Shao Q, Laird DW. The potency of the fs260 connexin43 mutant to impair keratinocyte differentiation is distinct from other disease-linked connexin43 mutants. Biochem J. 2010;429:473–483. doi: 10.1042/BJ20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Baker SM, Gumpert A, Segretain D, Buckheit RW. Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009;20:3342–3352. doi: 10.1091/mbc.E09-04-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanjul M, Hollande E. Morphogenesis of “duct-like” structures in three-dimensional cultures of human cancerous pancreatic duct cells (Capan-1) In Vitro Cell Dev Biol. 1993;29:574–584. doi: 10.1007/BF02634151. [DOI] [PubMed] [Google Scholar]
- Fong J, Kells R, Gumpert A, Marzillier J, Davidson M, Falk M. Internalized gap junctions are degraded by autophagy. Autophagy. 2012;8:738–755. doi: 10.4161/auto.19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta GM, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien R, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Girao H, Catarino S, Pereira P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res. 2009;315:3587–3597. doi: 10.1016/j.yexcr.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol. 1:a002576. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan R, Chakraborty S, Falk MM, Johnson KR, Wheelock MJ, Mehta PP. Assembly of connexin43 is differentially regulated by E-cadherin and N-cadherin in rat liver epithelial cells. Mol Biol Cell. 2010;21:4089–4107. doi: 10.1091/mbc.E10-05-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan R, Song X-H, Guo R-J, Wheelock MJ, Johnson KR, Mehta PP. Impaired trafficking of connexins in androgen-independent human prostate cancer cell lines and its mitigation by α-catenin. J Biol Chem. 2002;277:50087–50097. doi: 10.1074/jbc.M202652200. [DOI] [PubMed] [Google Scholar]
- Gravotta D, Carvajal-Gonzalez J, Mattera R, Deborde S, Banfelder J, Bonifacino J, Rodriguez-Boulan E. The clathrin adaptor AP-1A mediates basolateral polarity. Developmental Cell. 2012;22:811–823. doi: 10.1016/j.devcel.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 2008;582:2887–2892. doi: 10.1016/j.febslet.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herve JC, Derangeon Ml, Sarrouilhe D, Giepmans BNG, Bourmeyster N. Gap junctional channels are parts of multiprotein complexes. Biochim Biophys Acta. 2012;1818:1844–1865. doi: 10.1016/j.bbamem.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Hesketh GG, Shah MH, Halperin VL, Cooke CA, Akar FG, Yen TE, Kass DA, Machamer CE, Van Eyk JE, Tomaselli GF. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ Res. 2010;106:1153–1163. doi: 10.1161/CIRCRESAHA.108.182147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Geuze H. Intracellular trafficking of lysosomal proteins. EMBO J. 2011;18:379–389. doi: 10.1002/bies.950180508. [DOI] [PubMed] [Google Scholar]
- Jiang JX, Goodenough DA. Heteromeric connexins in lens gap junction channels. Proc Natl Acad Sci USA. 1996;93:1287–1291. doi: 10.1073/pnas.93.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K, Chodock R, Hand A, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci. 2001;114:763–773. doi: 10.1242/jcs.114.4.763. [DOI] [PubMed] [Google Scholar]
- Kanemitsu M, Lau A. Epidermal growth factor stimulates the disruption of gap junctional communication and connexin43 phosphorylation independent of 12-0-tetradecacoylphorbol 13-acetate-sensitive protein kinase C: the possible involvement of mitogen-activated protein kinase. Mol Biol Cell. 1993;4:837–848. doi: 10.1091/mbc.4.8.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey L, Katoch P, Johnson K, Batra SK, Mehta P. Retinoids regulate the formation and degradation of gap junctions in androgen-responsive human prostate cancer cells. PLoS One. 2012;7:E32846. doi: 10.1371/journal.pone.0032846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, et al. Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc Natl Acad Sci USA. 2005;102:14871–14876. doi: 10.1073/pnas.0506653102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahlou H, Fanjul M, Pradayrol L, Susini C, Pyronnet Sp. Restoration of functional gap junctions through internal ribosome entry site-dependent synthesis of endogenous connexins in density-inhibited cancer cells. Mol Cell Biol. 2005;25:4034–4045. doi: 10.1128/MCB.25.10.4034-4045.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010;20:92–101. doi: 10.1016/j.tcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Lauf U, Giepmans BNG, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci USA. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithe E, Brech A, Rivedal E. Endocytic processing of connexin43 gap junctions: a morphological study. Biochem J. 2006;393:59–67. doi: 10.1042/BJ20050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithe E, Kjenseth A, Sirnes S, Stenmark H, Brech A, Rivedal E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci. 2009;122:3883–3893. doi: 10.1242/jcs.053801. [DOI] [PubMed] [Google Scholar]
- Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004a;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem. 2004b;279:50089–50096. doi: 10.1074/jbc.M402006200. [DOI] [PubMed] [Google Scholar]
- Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, Alonso A. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J. Cell Sci. 2006;119:3634–3642. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci. 2011;124:910–920. doi: 10.1242/jcs.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Mehta P, Bertram J, Loewenstein W. Growth inhibition of transformed cells correlates with their junctional communication with normal cells. Cell. 1986;44:187–196. doi: 10.1016/0092-8674(86)90497-6. [DOI] [PubMed] [Google Scholar]
- Mehta P, Hotz-Wagenblatt A, Rose B, Shalloway D, Loewenstein WR. Incorporation of the gene for a cell-to-cell channel protein into transformed cells leads to normalization of growth. J Membr Biol. 1991;124:207–225. doi: 10.1007/BF01994355. [DOI] [PubMed] [Google Scholar]
- Mehta PP, Perez-Stable C, Nadji M, Mian M, Asotra K, Roos B. Suppression of human prostate cancer cell growth by forced expression of connexin genes. Dev Genet. 1999;24:91–110. doi: 10.1002/(SICI)1520-6408(1999)24:1/2<91::AID-DVG10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Mills IG, Jones AT, Clague MJ. Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Curr Biol. 1998;8:881–884. doi: 10.1016/s0960-9822(07)00351-x. [DOI] [PubMed] [Google Scholar]
- Mitra S, Annamalai L, Chakraborty S, Johnson K, Song X, Batra SK, Mehta PP. Androgen-regulated formation and degradation of gap junctions in androgen-responsive human prostate cancer cells. Mol Biol Cell. 2006;17:5400–5416. doi: 10.1091/mbc.E06-04-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller HB, Praetorius J, Rutzler MR, Fenton RA. Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc Natl Acad Sci USA. 2010;107:424–429. doi: 10.1073/pnas.0910683107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C, Milano S, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Musil L, Cunningham BA, Edelman G, Goodenough D. Differential phosphorylation of the gap junction protein connexin43 in junctional communicaton-competent and -deficient cell lines. J Cell Biol. 1990;111:2077–2088. doi: 10.1083/jcb.111.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS. Biogenesis and degradation of gap junctions. In: Harris A., Locke D., editors. Connexins: A Guide. New York: Springer; 2009. pp. 225–240. [Google Scholar]
- Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Rahajeng J, Sharma M, Jović M, Caplan S. Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol Biol Cell. 2006;17:163–177. doi: 10.1091/mbc.E05-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel BM, DeFranco BH, Gay VL, Murray SA. Clathrin and Cx43 gap junction plaque endoexocytosis. Biochem Biophys Res Commun. 2008;374:679–682. doi: 10.1016/j.bbrc.2008.07.108. [DOI] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 1995;269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher C, Höning S, Fingerhut A, Bowers K, Marsh M. Cluster of differentiation antigen 4 (CD4) endocytosis and adaptor complex binding require activation of the cd4 endocytosis signal by serine phosphorylation. Mol Biol Cell. 1999;10:677–691. doi: 10.1091/mbc.10.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates β-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer J, Schweizer A, Russell D, Kornfeld S. The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J Cell Biol. 1996;132:565–576. doi: 10.1083/jcb.132.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MG. Clathrin-mediated endocytosis before fluorescent proteins. Nat Rev Mol Cell Biol. 2006;7:63–68. doi: 10.1038/nrm1783. [DOI] [PubMed] [Google Scholar]
- Ruch RJ, Trosko J, Madhukar B. Inhibition of connexin43 gap junctional intercellular communication by TPA requires ERK activation. J Cell Biochem. 2001;83:163–169. doi: 10.1002/jcb.1227. [DOI] [PubMed] [Google Scholar]
- Saffarian S, Cocucci E, Kirchhausen T. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009;7:e1000191. doi: 10.1371/journal.pbio.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, Winslow AR, Rubinsztein DC. Methodological considerations for assessing autophagy modulators: a study with calcium phosphate precipitates. Autophagy. 2009;5:307–313. doi: 10.4161/auto.5.3.7664. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semerdjieva S, Shortt B, Maxwell E, Singh S, Fonarev P, Hansen J, Schiavo G, Grant BD, Smythe E. Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J Cell Biol. 2008;183:499–511. doi: 10.1083/jcb.200806016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. β-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev. 2012;92:273–366. doi: 10.1152/physrev.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 in LA-25 cells with active V-Src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes. 2008;15:75–84. doi: 10.1080/15419060802014016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Nowak N, Loor R, Ochi H, Sandberg A, Lopez C, Pkckern W, Cergian R, Douglass H, Chu T. Characterization of a new primary human pancreatic tumor line. Clin Invest. 1986;4:15–23. doi: 10.3109/07357908609039823. [DOI] [PubMed] [Google Scholar]
- TenBroek E, Lampe PD, Solan JL, Reynhout JK, Johnson R. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–1318. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Zosso N, Scerri I, Demaurex N, Chanson M, Staub O. A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J Cell Sci. 2003;116:2213–2222. doi: 10.1242/jcs.00440. [DOI] [PubMed] [Google Scholar]
- Thomas T, Jordan K, Simek J, Shao Q, Jedeszko C, Walton P, Laird DW. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J Cell Sci. 2005;118:4451–4462. doi: 10.1242/jcs.02569. [DOI] [PubMed] [Google Scholar]
- Toshima JY, Nakanishi Ji, Mizuno K, Toshima J, Drubin DG. Requirements for recruitment of a G protein-coupled receptor to clathrin-coated pits in budding yeast. Mol Biol Cell. 2009;20:5039–5050. doi: 10.1091/mbc.E09-07-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- Valiunas V, Gemel J, Brink PR, Beyer EC. Gap junction channels formed by coexpressed connexin40 and connexin43. Am J Physiol Heart Circ Physiol. 2001;281:H1675-H1689. doi: 10.1152/ajpheart.2001.281.4.H1675. [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Musil LS. Analysis of connexin intracellular transport and assembly. Methods. 2000;20:156–164. doi: 10.1006/meth.1999.0933. [DOI] [PubMed] [Google Scholar]
- Varsano T, Dong MQ, Niesman I, Gacula H, Lou X, Ma T, Testa JR, Yates JR, Farquhar MG. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol. 2006;26:8942–8952. doi: 10.1128/MCB.00305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mehta PP. Facilitation of gap junctional communication and gap junction formation by inhibition of glycosylation. Eur J Cell Biol. 1995;67:285–296. [PubMed] [Google Scholar]
- Warn-Cramer B, Cottrell GT, Burt JM, Lau AF. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem. 1998;273:9188–9196. doi: 10.1074/jbc.273.15.9188. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer B, Lampe P, Kurata W, Kanemitsu M, Loo L, Eckhart W, Lau A. characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- Weisz OA, Rodriguez-Boulan E. Apical trafficking in epithelial cells: signals, clusters and motors. J Cell Sci. 2009;122:4253–4266. doi: 10.1242/jcs.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- Zhu G, et al. Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5. EMBO J. 2007;26:3484–3493. doi: 10.1038/sj.emboj.7601771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell. 2009;136:1110–1121. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.