

Abstract
Introduction
α7-nicotinic acetylcholine receptor (α7-nAChR) is one of the major neuronal nAChR subtypes. α7-nAChR is involved in variety of neuronal processes and disorders including schizophrenia and Alzheimer’s disease. A number of α7-nAChR PET radioligands have been developed, but a quality radiotracer remains to be discovered.
Methods
High binding affinity α7-nAChR ligands A-833834 and A-752274 were radiolabeled with 11C. Baseline and blockade biodistribution studies in the mouse brain of [11C]A-833834 (5-(6-(5-[11C]methylhexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)pyridazin-3-yl)-1H-indole) and [11C]A-752274 (2-(6-[11C]methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-7-(6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9H-fluoren-9-one) were performed. [11C]A-752274 was evaluated in a baseline baboon PET study.
Results
[11C]A-833834 and [11C]A-752274 were synthesized by radiomethylation of corresponding des-methyl precursors. The radioligands were prepared with radiochemical yield of 12–32%, high specific radioactivity (330–403 GBq/μmol) and radiochemical purity > 95%. Dissection studies with [11C]A-833834 demonstrated low specific α7-nAChR binding in the mouse brain. [11C]A-752274 specifically (~50%) labeled α7-nAChR in the mouse thalamus. However, [11C]A-752274 exhibited low brain uptake in baboon (%SUV<100).
Conclusion
Two novel α7-nAChR ligands radioligands were synthesized and studied in animals. Specific binding of [11C]A-833834 in the mouse brain is low due to the insufficient binding affinity of the radioligand. The very high binding affinity [11C]A-752274 exhibited good specific binding in the α7-nAChR – rich mouse brain regions. The low uptake of [11C]A-752274 in the baboon brain is due to its high hydrophilicity, rapid metabolism or other properties. Future development of α7-nAChR PET radioligands will be based on compounds with high binding affinities and good blood-brain barrier permeability.
Keywords: positron emission tomography, PET, radioligand, nicotinic acetylcholine receptor, α7-nAChR
1. Introduction
Nicotinic acetylcholine receptors (nAChR) are members of the ligand-gated cation channel superfamily and involved in many physiological functions.[1, 2] Neuronal central nAChR populations are mostly comprised of the homomeric α7-nAChR and heteromeric α4β2-nAChR.[3, 4] These acetylcholine receptor subtypes are involved in great variety of neuronal processes and of interest as targets for therapeutic intervention in a number of conditions and disorders.[5, 6]
The α7-nAChR subtype is highly expressed in the hippocampus and cortex, the brain regions that are involved in cognition and memory.[7, 8] Recent research suggests that this receptor subtype is associated with several neurodegenerative and psychiatric disorders.[9–13] Post-mortem studies of human brains have demonstrated reduced expression of α7-nAChR in schizophrenics versus control brains.[14, 15] Successful experiments with the α7-nAChR agonist DMXB that improves the auditory gating deficit in animal models of schizophrenia [16, 17] have led to the first clinical study.[18]
Reduced expression of α7-nAChR protein has also been observed in the hippocampus of patients suffering from Alzheimer’s disease (AD).[11, 13, 19] The neuroprotective effects of nicotine against β-amyloid toxicity can be blocked by selective α7-nAChR antagonists,[20] and inhibition of α7-nAChR mRNA and protein expression aggravate the toxicity of β-amyloids in vitro.[21] These findings suggest a therapeutic role for α7-nAChR agonists in the treatment of AD.[20, 21]
Recent progress in the pharmacology of α7-nAChR agonists has stimulated the interest of pharmaceutical companies in these compounds as potential medications for treatment of schizophrenia and AD and several new drug candidates that are now undergoing clinical trials.[22]
Successful imaging of α7-nAChR with positron-emission tomography (PET) would open an opportunity to evaluate novel nicotinic drugs and, perhaps, lead to an understanding of the relationship of these receptors to brain function and the progression of various diseases. While PET imaging of α 4β2-nAChR in human subjects is a current reality (see for review [23–25]), a lack of PET radioligands for α7-nAChR in human subjects impedes progress in the neuroscience of this receptor subtype. A number of α7-nAChR PET radioligands have been developed, but a quality radiotracer remains to be synthesized [26–28] (see also recent reviews [29, 30]).
Abbott Laboratories has recently developed two α7-nAChR ligands (A-833834 and A-752274) with very high binding affinities (Fig 1) [31, 32]. This paper describes our endeavor to explore radiolabeled [11C]A-833834 and [11C]A-752274 as potential PET tracers for imaging the α7-nAChR.
Figure 1.

Structures of the high affinity α7-nAChR ligands A-833834 [31] and A-752274 [32]. In the publication [31] the inhibition binding constant value (Ki = 0.24 nM) of A-833834 has been determined under the assay conditions that do not match those of [32]. In order to uniform the binding affinity data the α7-nAChR inhibition constant of A-833834 (Ki = 1.53 nM) has been determined here under the same conditions as those of A-752274 (Ki = 0.092 nM [32]) using [3H]MLA as a radioligand.
1. Methods
1.1. General
1H NMR spectra were recorded on a GE QE-300 (300 MHz) spectrometer, and chemical shifts are reported in parts per million (ppm) downfield of tetramethylsilane (TMS) as internal standard. Mass spectra were acquired on a JEOL JMS-SX-102 spectrometer. Unless otherwise specified, all reagents and solvents were obtained from commercial sources and used without further purification. Preparative high performance liquid chromatography (HPLC) for cold chemistry was performed on an automated system consisting of Gilson 333 pumps, a Gilson 155 UV/VIS detector, a Gilson 8452 injector module, and Gilson 215 liquid handling system. A Waters 30x100 mm XBridge® column, eluted with a binary gradient of 0.1 M ammonium carbonate buffer (pH~10) and methanol (80:20 to 5:95 over 15 min), was used for all non-radioactive chemistry work.
The high performance liquid chromatography (HPLC) system used in the radiochemistry experiments consisted of a Waters model 610 pump, a Valco injector, a Varian Prostar 325 LC detector set to 254 nm, a Bioscan Flow-Count PMT radioactivity detector. The analytical HPLC was performed using a Varian Prostar 210 pump with a Prostar 410 Autosampler, a Varian Prostar 325 LC detector set to 254 nm, and a Bioscan Flow-Count PMT radioactivity detector. All HPLC data were recorded and analyzed with Varian Galaxie Chromatography Data System software (version 1.9.302.952). A dose calibrator (Capintec 15R) was used for all radioactivity measurements.
The HPLC conditions used in this work for radiochemistry are described below:
Method A: Waters XBridge C18 column (150x10 mm); mobile phase is acetonitrile : 0.1M aq. ammonium formate : 25% ammonia (25:75:0.1); flow rate 8 ml/min
Method B: Phenomenex Luna C18, 10 micron, (250x4.6 mm); mobile phase is acetonitrile : 0.1M aq. ammonium formate : triethylamine (32:86:0.2); flow rate 3 ml/min
Method C: Waters XBridge C18 column (250x10 mm); mobile phase is acetonitrile : water : triethylamine (36:64:0.2); flow rate 12 ml/min
Method D: Waters XBridge C18 column, 3.5 micron (100x4.6 mm); mobile phase is acetonitrile : water : triethylamine (36:64:0.2); flow rate 2 ml/min
[11C]Methyl iodide was prepared from 11CO2 using a Tracerlab FX MeI module (General Electric) and a PETtrace biomedical cyclotron (General Electric). [11C]Methyl triflate was prepared by reaction of [11C]methyl iodide with silver triflate.
1.2. Chemistry
t-Butyl (cis)-5-(6-(1H-indol-5-yl)pyridazin-3-yl)hexahydropyrrolo[3,4-c]pyrrole-2-carboxylate (3)
Pd2dba3 (75.3 mg, 0.082 mmol) and 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (104 mg, 0.245 mmol) were added at room temperature to a mixture of t-butyl cis- 5-(6-chloropyridazin-3-yl)hexahydropyrrolo[3,4-c]pyrrole-2-carboxylate (1) (271 mg, 0.834 mmol) [31] and 1H-indol-5-ylboronic acid (2) (161 mg, 1.000 mmol) in dioxane (20 mL). The reaction flask was evacuated and purged with nitrogen (4 cycles). Aqueous 20% Na2CO3 (2 mL, 4 mmol) was added, and the flask was again evacuated and purged with nitrogen (4 cycles). The mixture was heated under nitrogen at 85 °C for 19 h, then cooled to room temperature. The dark mixture was diluted with EtOAc (40 mL) and filtered through diatomaceous earth. The filtrate was concentrated and the residue purified by flash chromatography (SiO2, eluted with hexanes-EtOAc, 50:50 - 0:100) to provide the title compound as a friable, amber foam (196 mg, 58%): 1H NMR (300 MHz, CD3OD) 1.46 (s, 9 H), 3.05–3.16 (m, 2 H), 3.30–3.36 (m, 2 H), 3.49 (dd, J=10.5, 3.7 Hz, 2 H), 3.62–3.73 (m, J=9.5, 7.5 Hz, 2 H), 3.82 (dd, J=10.7, 7.3 Hz, 2 H), 6.53 (d, J=3.1 Hz, 1 H), 7.03 (d, J=9.5 Hz, 1 H), 7.27 (d, J=3.1 Hz, 1 H), 7.47 (d, J=8.8 Hz, 1 H), 7.71 (dd, J=8.6, 1.5 Hz, 1 H), 7.87 (d, J=9.5 Hz, 1 H), 8.07 ppm (d, J=1.7 Hz, 1 H); MS (DCI/NH3) m/z 406 (M+H)+.
cis-5-(6-(Hexahydropyrrolo[3,4-c]pyrrol-2-yl)pyridazin-3-yl)-1H-indole (4)
p-Toluenesulfonic acid monohydrate (238 mg, 1.251 mmol) was added to a suspension of 3 (243 mg, 0.599 mmol) in ethylacetate (55 mL). The mixture was heated under nitrogen at 75 °C for 5 h, then cooled to room temperature overnight. The solid precipitate was collected by filtration, washed with ethylacetate (20 mL) and dried under vacuum to provide the title compound as its bis(tosylate) salt (337 mg, 87%). This material was purified by HPLC (30x100 mm Waters Xbridge® column eluted with ammonium carbonate buffer-MeOH (95:5 – 5 :95 over 20 min)) to provide the free base 4 as a white solid (117 mg, 64%): 1H NMR (300 MHz, CD3OD) δ 2.83 (dd, J=11.1, 3.6 Hz, 2 H), 2.99–3.09 (m, 2 H), 3.18 (dd, J=11.3, 7.3 Hz, 2 H), 3.51 (dd, J=11.1, 3.2 Hz, 2 H), 3.74 (dd, J=10.9, 7.7 Hz, 2 H), 6.53 (d, J=3.2 Hz, 1 H), 7.07 (d, J=9.5 Hz, 1 H), 7.27 (d, J=3.2 Hz, 1 H), 7.47 (d, J=8.3 Hz, 1 H), 7.71 (dd, J=8.5, 1.8 Hz, 1 H), 7.87 (d, J=9.5 Hz, 1 H), 8.07 (d, J=1.6 Hz, 1 H); MS (DCI/NH3) m/z 306 (M+H)+.
(1R,5S)-t-Butyl 3-(7-bromo-9-oxo-9H-fluoren-2-yl)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (7)
(rac)-2,2′-Bis(biphenylphosphino)-1,1′-binaphthyl (19.9 mg, 0.032 mmol) and tris(dibenzylidine)dipalladium(0) (13.9 mg, 0.015 mmol) were combined in a test tube with toluene (2 mL) and warmed to 80 °C for 1 min, then cooled to room temperature. This catalyst solution was added to a mixture of 2,7-dibromo-9H-fluoren-9-one (1.06 g, 3.15 mmol), sodium tert-butoxide (116 mg, 1.21 mmol) and t-butyl (1R,5S)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (6) [33] (156 mg, 0.787 mmol) in toluene (50 mL). The reaction flask was evacuated and purged with nitrogen (3 cycles), and heated at 90–95 °C under nitrogen for 24 h. The reaction mixture was cooled to room temperature and concentrated under vacuum. The residue was purified by flash chromatography (silica gel, eluted with hexanes-EtOAc, 80:20 – 50:50) to provide the title compound as a red-purple foam (235 mg, 66%): 1H NMR (300 MHz, CD3OD) δ1.45 (s, 9 H), 2.96 (dd, J=11.0, 4.6 Hz, 1 H), 3.01–3.12 (m, 1 H), 3.17 – 3.28 (m, 2 H), 3.55–3.66 (m, 1 H), 3.82 (d, J=10.2 Hz, 1 H), 3.91–4.02 (m, 1 H), 4.04–4.18 (m, 1 H), 6.92 (dd, J=8.5, 2.4 Hz, 1 H), 7.07 (d, J=2.4 Hz, 1 H), 7.38 (d, J=8.5 Hz, 1 H), 7.48 (d, J=8.1 Hz, 1 H), 7.53 – 7.63 ppm (m, 2 H); MS (ESI) m/z 455/457 (M+H)+.
2-Bromo-7-((1S,5S)-6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9H-fluoren-9-one (8)
Aqueous formalin (0.3 mL, 36%, 3.92 mmol) was added to a solution of (1R,5S)-tert-butyl 3-(7-bromo-9-oxo-9H-fluoren-2-yl)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (7) (165 mg, 0.362 mmol) in formic acid (6 mL). The mixture was heated at 90–95 °C for 2.5 h, then the purple solution was cooled to room temperature and concentrated under vacuum. The residue was purified by flash chromatography (silica gel eluted with CH2Cl2-MeOH-NH4OH, 95:5:0.5) to provide a dark purple solid (70 mg, 52%): 1H NMR (300 MHz, CD3OD) δ 2.39 (s, 3 H), 3.05 (dd, J=11.1, 4.8 Hz, 1 H), 3.20–3.28 (m, 2 H), 3.29–3.40 (m, 2 H), 3.70–3.83 (m, 2 H), 4.02 (dd, J=6.3, 4.4 Hz, 1 H), 6.86 (dd, J=8.3, 2.4 Hz, 1 H), 7.02 (d, J=2.4 Hz, 1 H), 7.36 (d, J=8.3 Hz, 1 H), 7.46 (d, J=7.9 Hz, 1 H), 7.54 – 7.61 ppm (m, 2 H); MS (DCI/NH3) m/z 369/371 (M+H)+.
(1R,5S)-t-Butyl 3-(7-((1S,5S)-6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9-oxo-9H-fluoren-2-yl)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (9)
(rac)-2,2′-Bis(biphenylphosphino)-1,1′-binaphthyl (19.9 mg, 0.032 mmol) and tris(dibenzylidine)dipalladium(0) (13.9 mg, 0.015 mmo) were combined in a test tube with toluene (2 mL) and warmed to 80 °C for 5 min, then cooled to room temperature. The catalyst solution was added to a mixture of 2-bromo-7-((1S,5S)-6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9H-fluoren-9-one (8) (71 mg, 0.192 mmol), sodium tert-butoxide (30 mg, 0.312 mmol) and (1R,5S)-tert-butyl 3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (6) [33] (45 mg, 0.227 mmol) in toluene (8 mL). The reaction flask was evacuated and purged with nitrogen (3 cycles) and heated at 90–95 °C under nitrogen for 5 h. The mixture was cooled to room temperature and concentrated under vacuum. The residue was purified by HPLC (30 x 100 mm Waters XBridge® column eluted with aqueous (NH4)2CO3 - MeOH, 80:20 - 0:100 over 15 min) to provide the product as a deep blue powder (50.5 mg, 54%): 1H NMR (300 MHz, CD3OD) δ1.46 (s, 9 H), 2.39 (s, 3 H), 2.87 (dd, J=11.0, 4.2 Hz, 1 H), 2.92 – 3.02 (m, 2 H), 3.12–3.26 (m, 3 H), 3.32–3.41 (m, 1 H), 3.56–3.68 (m, 1 H), 3.69–3.82 (m, 3 H), 3.90–4.14 (m, 3 H), 6.80 (dd, J=8.3, 2.5 Hz, 1 H), 6.84 (dd, J=8.1, 2.4 Hz, 1 H), 6.98 (dd, J=9.2, 2.4 Hz, 2 H), 6.97 (d, J=2.4 Hz, 1 H), 7.00 (d, J=2.4 Hz, 1 H), 7.27 ppm (d, J=8.1 Hz, 2 H); MS (ESI) m/z 487 (M+H)+.
(1R,5S)-3-(7-((1S,5S)-6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9-oxo-9H-fluoren-2-yl)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (10)
Trifluoroacetic acid (2 mL, 26.0 mmol) was added to an ice-cooled solution of (1R,5S)-tert-butyl 3-(7-((1S,5S)-6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9-oxo-9H-fluoren-2-yl)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (9) (49 mg, 0.101 mmol) in CH2Cl2 (4 mL). The solution was stirred at 0 °C for 1 h, then allowed to warm to room temperature for 3 h. The purple solution was concentrated under vacuum, and the residue was purified by HPLC (30 x 100 mm Waters XBridge® column eluted with aqueous (NH4)2CO3 - MeOH, 80:20 - 0:100 over 15 min) to provide the product as a blue solid (32 mg, 82%): 1H NMR (300 MHz, CD3OD) δ2.38 (s, 3 H), 2.93–3.06 (m, 3 H), 3.12–3.26 (m, 3 H), 3.33–3.45 (m, 3 H), 3.69–3.93 (m, 5 H), 3.96–4.04 (m, 1 H), 4.54–4.61 (m, 1 H), 6.80 (dd, J=8.3, 2.4 Hz, 1 H), 6.88 (dd, J=8.1, 2.2 Hz, 1 H), 6.97 (d, J=2.4 Hz, 1 H), 7.04 (d, J=2.0 Hz, 1 H), 7.26 (s, 1 H), 7.29 ppm (s, 1 H); MS (ESI) m/z 387 (M+H)+.
1.3. Radiochemistry
1.3.1. Radiosynthesis of [11C]A833834
[11C]Methyl iodide was swept by argon flow into the vial containing a solution of 1 mg of 5-(6-(hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)pyridazin-3-yl)-1H-indole (4) in 0.2 mL anhydrous DMF. After the accumulated radioactivity reached a plateau, the vial was assayed in the dose calibrator, and then heated at 80 °C for 5 min. HPLC mobile phase (200 μL) was added and the solution was injected onto the semi-preparative HPLC column (method A). The retention time of the normethyl precursor was 5.6 min. The product peak having retention time of 10.8 min was collected in 50 mL of HPLC water. The water solution was transferred through an activated Waters C-18 Sep-Pak Plus cartridge. The product was eluted with 1 mL ethanol into a vial and diluted with 10 mL of 0.9% saline through the same Sep-Pak and sterile microfilter assembly. An aliquot was removed to determine specific radioactivity and radiochemical purity of the final product. [11C]A833834 was assayed by analytical HPLC (method B) and its retention time was 2.9 min.
1.3.2. Radiosynthesis of [11C]A-752274
[11C]Methyl triflate was swept by argon flow into the vial containing a solution of 0.5 mg of 2-(3,6-diazabicyclo[3.2.0]heptan-3-yl)-7-(6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9H-fluoren-9-one (10) in 0.1 mL anhydrous DMF. After the accumulated radioactivity reached a plateau, the vial was assayed in the dose calibrator, and then heated at 60 °C for 5 min. A mixture of water (200 μL) and 1 μL triethylamine was added and the solution was injected onto the semi-preparative HPLC column (method C). The retention time of the normethyl precursor was 6.7 min. The product peak had a retention time of 11.5 min and was collected in 50 mL of water and 0.25 g sodium ascorbate. The aqueous solution was passed through an activated Waters Oasis HLB Plus LP purification cartridge (30 mg). The product was eluted with 1 mL ethanol into a vial and diluted with 10 mL of 0.9% saline containing 0.05 g sodium ascorbate through a sterile filter unit. An aliquot was removed to determine specific radioactivity and radiochemical purity of the final product [11C]A752274 by analytical HPLC (method D), and its retention time was 6 min.
1.4. Animal studies
All animal protocols were approved by the Animal Care and Use Committee of the Johns Hopkins University.
1.4.1. Biodistribution dissection studies in mice
CD1 mice (all males, 23–28 g) from the Charles River Laboratories (Wilmington, MA) were used in the animal experiments. The animals were sacrificed by cervical dislocation at various times following injection of [11C]-radioligands (0.1 mCi in 0.2 mL saline, specific radioactivity ~5000 mCi/μmol) into a lateral tail vein. The brains were rapidly removed and dissected on ice. The brain regions of interest were weighed and their radioactivity content was determined in a γ-counter with a counting error below 3%. Aliquots of the injectate were used as standards and their radioactivity content was counted along with the tissue samples. The percent of injected dose per gram of tissue (%ID/g tissue) was calculated.
1.4.2. Blocking dissection studies in mice
In vivo blocking studies were performed by intravenous co-injection of radiotracers [11C]A-833834 or [11C]A-752274 (0.1 mCi in 0.2 mL saline; specific radioactivity ~5000 mCi/μmol) with unlabeled ligands A-833834 (3 mg/kg) or A-752274 (0.1 mg/kg), respectively. Sixty or ninety minutes after administration of the radiotracers, brain tissues were harvested, and the radioactivity content was determined. There were three animals per time-point in both the baseline and blockade groups.
3. Results and discussions
3.1. Chemistry
The nor-methyl precursors 3 and 7 for radiolabeling [11C]A-833834 and [11C]A-752274 were synthesized as shown in Schemes 1 and 2. Since both precursors are highly polar compounds, their purification with silica-gel flash chromatography was expected to be problematic. Therefore, the final purification of free base 3 and 7 were performed with preparative reverse-phase HPLC.
Scheme 1.
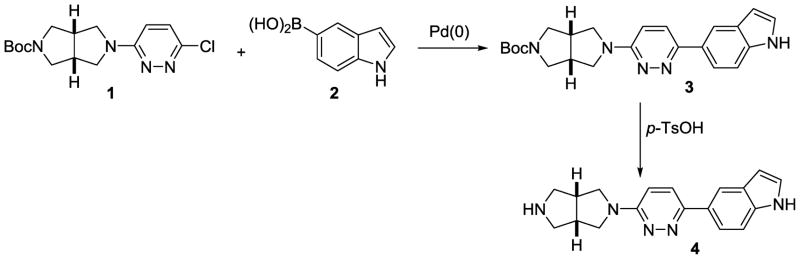
Preparation of cis-5-(6-(hexahydropyrrolo[3,4-c]pyrrol-2-yl)pyridazin-3-yl)-1H-indole (4), a precursor for radiosynthesis of [11C]A-833834
Scheme 2.

Preparation of (1R,5S)-3-(7-((1S,5S)-6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9-oxo-9H-fluoren-2-yl)-3,6-diazabicyclo[3.2.0]heptane-6-carboxylate (10), a precursor for radiosynthesis of [11C]A-752274.
3.2. Radiochemistry
The carbon-11 radiosynthesis of 5-(6-(5-[11C]methylhexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)pyridazin-3-yl)-1H-indole ([11C]A-833834) was achieved by reaction of the corresponding nor-methyl precursor 4 with [11C]methyl iodide in DMF solution (Scheme 3) followed by semi-preparative HPLC separation and formulation as a solution in sterile saline with 7% alcohol. The precursor molecule 4 has two potential nucleophilic centers (secondary amine and indole nitrogen), but methylation of indole takes place only in the presence of a strong base and, thus, none was detected under the methylation conditions that we used here.
Scheme 3.

Radiosynthesis of [11C]A-833834.
The total synthesis time including production of [11C]methyl iodide and quality control was 37 min. The average non-decay corrected radiochemical yield (32 ± 3%, n = 6) was determined with respect to starting radioactivity of [11C]methyl iodide. The average specific radioactivity of the final [11C]A-833834 was 403 ± 70 GBq/μmol (14900 ± 2580 mCi/μmol) determined at end of synthesis, and radiochemical purity was greater than 99% by HPLC analysis. Repeat HPLC analysis of formulated [11C]A-833834 at 60 min after the end-of-synthesis demonstrated satisfactory radiochemical purity (96%) of the radiotracer.
The radiosynthesis of 2-(6-[11C]methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-7-(6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9H-fluoren-9-one ([11C]A-752274) by radiomethylation of 2-(3,6-diazabicyclo[3.2.0]heptan-3-yl)-7-(6-methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)-9H-fluoren-9-one (10) utilized a similar approach, but used [11C]methyl triflate as a radiomethylating agent (Scheme 4); the same reaction with [11C]methyl iodide produced some radioproduct in low yield (data not presented). The final [11C]A-752274 was formulated as a sterile solution in saline with 7% alcohol and 0.5% sodium ascorbate to prevent radiolysis. Without ascorbate [11C]A-752274 undergoes rapid radiolysis in saline solution.
Scheme 4.

Radiosynthesis of [11C]A-752274
The total synthesis time of [11C]A-752274 was 39 min. The average non-decay corrected radiochemical yield (12 ± 4%, n = 10) of [11C]A-752274 with respect to the starting radioactivity of [11C]methyl triflate was relatively low due to the presence of unidentified radiolabeled by-product in the reaction mixture. The average specific radioactivity of [11C]A-752274 was 332 ± 51 GBq/μmol (12300 ± 1890 mCi/μmol) as determined at end of synthesis, and the radiochemical purity was 99% by HPLC analysis. Repeat HPLC analysis of the formulated [11C]A-752274 at 60 min after the end-of-synthesis demonstrated satisfactory radiochemical purity (95%) of the radiotracer.
The identities of the radiotracers [11C]A-833834 and [11C]A-752274 were confirmed by co-injection with corresponding non-radioactive reference compounds A-833834[31] and A-752274[32] onto an analytical HPLC system.
3.3. Mice studies
The radioactivity accumulation for [11C]A-833834 and [11C]A-752274 was determined as %ID/g tissue in several regions of the CD1 mouse brain after intravenous injection of the radioligands.
[11C]A-833834. After intravenous injection, [11C]A-833834 entered the mouse brain with a peak concentration of 1.4 ± 0.2 % I.D. (mean of 3; ± SD) at 30 min after injection. The highest accumulation of [11C]A-833834 radioactivity was seen in three brain regions: the frontal cortex, thalamus, and hippocampus with a peak at 30–45 min post-injection with some decline thereafter (Fig. 2-left). The washout of radioactivity from the mouse cerebellum, a region with the lowest concentration of radioactivity, was more rapid.
Figure 2.

Biodistribution studies with [11C]A-833834 in CD1 mice. Left panel: Regional time-uptake curves in the mouse brains. Right panel: Comparison of baseline (open bars) and blockade (black bars), 60 min post injection. The difference between baseline and blockade are not significant. Blocker = unlabeled A833834 (3 mg/kg, co-injected intravenously with radiotracer). Abbreviators: FrCtx = frontal cortex; Th = thalamus; Hipp = hippocampus; CB = cerebellum; Rest = rest of the brain. Data = Mean ± SD.
The regional radioactivity concentrations of [11C]A-833834 in the mouse brain matches in vitro and in vivo data on the distribution of α7-nAChRs (see for review [30]). The tissue/cerebellum ratios increased steadily over the observation period, reaching values of 1.5 in the frontal cortex at 60 min after injection (Figure 2).
Unlabeled A-833834 (3 mg/kg) was injected with [11C]A-833834 to study saturation of the tracer binding. Animals were sacrificed 60 min after the administration. The blockade showed some reduction of the radioactivity uptake in all brain regions studied (Fig. 2-right), but the difference versus the baseline study was not significant. This experiment demonstrates that the specific binding of [11C]A-833834 in the mouse brain is low and the radiotracer does not display sufficient properties for further PET experiments.
[11C]A-752274. Similar mouse experiments with [11C]A-752274 demonstrated that the radiotracer slowly enters the animal brain (Fig. 3). The uptake of radioactivity did not reach its peak during the 90 min experiment. The greatest accumulation of [11C]A-752274 radioactivity was seen in the α7-nAChR - rich thalamus (0.9%), superior colliculus (0.8%) and hippocampus (0.6%) and the lowest uptake in the α7-nAChR - poor cerebellum (0.4%). The tissue/cerebellum ratios continued to climb throughout the duration of experiment reaching the values of 2.1, 1.8 and 1.4 at 90 min post injection for the thalamus, superior colliculus and hippocampus, respectively. It is noteworthy that in the same experiment [11C]A-752274 exhibited strikingly high accumulation (~ 20%ID/g tissue) in the adrenal gland, an organ with high density of α7-nAChR (graph not shown).
Figure 3.

Right: Regional brain Time-uptake curves of [11C]A-752274 in CD1 mouse. Data = mean ± SD. Left: Tissue cerebellum ratio from the same study. This experiment demonstrates a very low rate of cerebral uptake by [11C]A-752274.
The shape of the cerebral time-uptake curves (Fig 3) clearly suggests that an extended duration of imaging might reveal increased signal-to-noise ratios. However, this is not possible with [11C]A-752274 due to the short half-life of the 11C (~20 min).
An experiment using low specific radioactivity radiotracer was performed to study the receptor saturability by coinjection of [11C]A-752274 with unlabeled A-752274. The blocking dose of A-752274 significantly inhibited [11C]A-752274 binding in the thalamus (Figure 4) suggesting that the binding in this region is specific, whereas little displacement was observed in the α7-nAChR-poor cerebellum [30].
Figure 4.

Comparison of regional brain uptake of [11C]A-752274 in mice at the 90 min time-point in control (white bars) and blocking experiments with unlabeled A-752274 (0.1 mg/kg, iv) (black bars). Th = thalamus; CB = cerebellum. Data = mean ± SD. *P< 0.05, significantly different from controls; **P> 0.05, insignificantly different from controls (ANOVA single-factor analysis). There was a significant blockade in the thalamus and no blockade in the cerebellum. This result demonstrates that [11C]A-752274 specifically labels α7-nAChR in the mouse thalamus.
3.4. Baboon study
The typical approach of the bench-to-bed side development of new PET radioligands involves animal experiments that often start with rodents. The candidate radioligands that show best results in rodents are selected for further pre-clinical experiments in non-human primates (NHP) as a final preclinical step. In general, the in vitro regional distribution of the α7-nAChR in the mouse brain[7] is similar to that in non-human primates with the greatest density of α7-nAChR receptor binding in the monkey hippocampus and thalamus, moderate density in cortical regions and basal ganglia and lowest density in cerebellum[34–36]. However, a detailed mapping of α7-nAChR in the non-human primate brain remains to be studied. With [11C]A-752274 exhibiting some specific binding in the mouse experiments, a single baboon baseline PET study with this radiotracer was performed. The 90 min PET experiment showed a low uptake (%SUV < 30) of [11C]A-752274 in the baboon brain (image not shown) that was consistent with low uptake rate of the radiotracer in the mouse brain. This SUV value was lower than the conventional range for a successful cerebral radioligand (%SUV>100) and insufficient for further PET experiments. HPLC analysis of the baboon plasma demonstrated that [11C]A-752274 underwent a rapid metabolism and formed several highly hydrophilic radiometabolites. At 90 min post injection the fraction of the parent [11C]A-752274 in the plasma was 7%.
The largely negative imaging results of this study are methodologically important for further development of PET radioligands for imaging α7-nAChR. We can hypothesize that the low specific binding of [11C]A-833834 in the mouse brain is likely to be due to the insufficient binding affinity of the radioligand. Previous studies with PET radioligands for imaging another nicotinic receptor subtype α4β2-nAChR suggest that a successful compound of this class should exhibit very high binding affinity Ki in the picomolar range [25]. Because the density of the α7-nAChR subtype is lower than that of α4β2-nAChR it is obvious that picomolar binding affinity values are essential for a quality α7-nAChR PET radioligands. The better specific binding of [11C]A-752274 in the mouse brain is in accord with the nearly 16-fold greater binding affinity of this compound than that of [11C]A-833834 (Ki = 0.092 nM vs. 1.53 nM, respectively).
The low uptake of the high affinity [11C]A-752274 in the mouse and baboon brain is likely because of the high hydrophilicity of this compound. Both aliphatic amino-groups of [11C]A-752274 are protonated at the physiological pH and, consequentially, the compound exhibits very low lipophilicity logD7.4 = −2.7 (calculated with ACD/logD suite software). It is well understood that CNS active drugs and radiotracers should display lipophilicity values in the approximate range of 0 – 4 (see for review [37]). Yet, there are a number of successful cerebral PET radiotracers with very low lipophilicities including [11C]MP4a (logD7.4 = − 0.7), an acetylcholinesterase radiotracer, and 2-[18F]fluoro-A85380 (logD7.4 = − 1.4), a radioligand for imaging cerebral α4β2-nAChR (see for review [25, 38]) and a cut off value of logD for cerebral radiotracers is probably lower than that for CNS active drugs. Competing systemic clearance of [11C]A-752274 and/or P-glycoprotein efflux from the brain could also contribute to low brain uptake of this radioligand. Prediction of P-glycoprotein substrate specificity is still in a state of development [39–41] and fully reliable structure-activity relationships are not available. Roughly, compounds with (N + O) > 8, MW > 400 and acid pKa < 4 are likely to be P-gp substrates, whereas compounds with (N + O) < 4, MW < 400 and base pKa > 8 are likely to be non-substrates. Compound A-752274 ((N+O) = 5, MW = 401, and strong base) does not fall definitely in either group and could potentially exhibit some P-gp substrate activity.
4. Conclusion
Radiolabeling of α7-nAChR ligands A-833834 and A-752274 with [11C] have been achieved with high radiochemical yields and specific radioactivities by 11C-methylation of corresponding nor-methyl precursors that were synthesized in this study.
Regional brain distribution studies demonstrated that [11C]A-833834 enters the mouse brain but its specific binding in the α7-nAChR - rich brain regions was low due to insufficient binding affinity.
The very high binding affinity radioligand [11C]A-752274 specifically labeled α7-nAChR in the mouse thalamus, a region with a high density of α7-nAChR. However, [11C]A-752274 exhibited a slow rate of brain uptake in mice and baboon because the radioligand lipophilicity value was too low for passive blood-brain barrier transport.
Future research and development of α7-nAChR PET radioligands will be based on compounds with very high binding affinities and good blood-brain barrier permeability.
Acknowledgments
We are grateful to Robert C. Smoot for radiochemistry assistance, and David J. Clough and Karen Edmonds for PET scanner operation and Paige Finley for help with animal experiments. The authors thank Mrs. Judy Buchanan for her kind editorial assistance. This research was supported in part by the Division of Nuclear Medicine of Johns Hopkins University School of Medicine and by NIH Grants DA020777 and MH079017 (AGH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kellar KJ, Xiao Y. Neuronal nicotinic receptors: one hundred years of progress. In: Sibley DR, editor. Handbook of contemporary neuropharmacology. Wiley-Interscience; 2007. pp. 108–31. [Google Scholar]
- 2.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–96. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- 4.Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–91. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Lippiello PM, Bencherif M, Hauser TA, Jordan KG, Letchworth SR, Mazurov AA. Nicotinic receptors as targets for therapeutic discovery. Expert Opinion on Drug Discovery. 2007;2:1185–203. doi: 10.1517/17460441.2.9.1185. [DOI] [PubMed] [Google Scholar]
- 6.Jones HE, Garrett BE, Griffiths RR. Subjective and physiological effects of intravenous nicotine and cocaine in cigarette smoking cocaine abusers. J Pharmacol Exp Ther. 1999;288:188–97. [PubMed] [Google Scholar]
- 7.Whiteaker P, Davies AR, Marks MJ, Blagbrough IS, Potter BV, Wolstenholme AJ, et al. An autoradiographic study of the distribution of binding sites for the novel alpha7-selective nicotinic radioligand [3H]-methyllycaconitine in the mouse brain. Eur J Neurosci. 1999;11:2689–96. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- 8.Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38:679–90. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 9.Philip NS, Carpenter LL, Tyrka AR, Price LH. Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology (Berl) 2010;212:1–12. doi: 10.1007/s00213-010-1932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishikawa M, Hashimoto K. alpha7 nicotinic acetylcholine receptor as a potential therapeutic target for schizophrenia. Curr Pharm Des. 2011;17:121–9. doi: 10.2174/138161211795049561. [DOI] [PubMed] [Google Scholar]
- 11.Parri HR, Hernandez CM, Dineley KT. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem Pharmacol. 2011;82:931–42. doi: 10.1016/j.bcp.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 12.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodruff-Pak DS, Gould TJ. Neuronal nicotinic acetylcholine receptors: involvement in Alzheimer’s disease and schizophrenia. Behav Cogn Neurosci Rev. 2002;1:5–20. doi: 10.1177/1534582302001001002. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Ruiz CM, Haroutunian VH, Long P, Young AH, Davis KL, Perry EK, et al. Dementia rating and nicotinic receptor expression in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2003;54:1222–33. doi: 10.1016/s0006-3223(03)00348-2. [DOI] [PubMed] [Google Scholar]
- 15.Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, et al. Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J Chem Neuroanat. 2001;22:115–26. doi: 10.1016/s0891-0618(01)00117-x. [DOI] [PubMed] [Google Scholar]
- 16.O’Neill HC, Rieger K, Kem WR, Stevens KE. DMXB, an a7 nicotinic agonist, normalizes auditory gating in isolation-reared rats. Psychopharmacology (Berlin, Germany) 2003;169:332. doi: 10.1007/s00213-003-1482-2. [DOI] [PubMed] [Google Scholar]
- 17.Stevens KE, Kem WR, Mahnir VM, Freedman R. Selective à 7 -nicotinic agonists normalize inhibition of auditory response in DBA mice. Psychopharmacology (Berl) 1998;136:320. doi: 10.1007/s002130050573. [DOI] [PubMed] [Google Scholar]
- 18.Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–8. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- 19.Guan ZZ, Zhang X, Ravid R, Nordberg A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J Neurochem. 2000;74:237–43. doi: 10.1046/j.1471-4159.2000.0740237.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275:5626–32. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 21.Qi XL, Nordberg A, Xiu J, Guan ZZ. The consequences of reducing expression of the alpha7 nicotinic receptor by RNA interference and of stimulating its activity with an alpha7 agonist in SH-SY5Y cells indicate that this receptor plays a neuroprotective role in connection with the pathogenesis of Alzheimer’s disease. Neurochem Int. 2007;51:377–83. doi: 10.1016/j.neuint.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 22.D’Hoedt D, Bertrand D. Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin Ther Targets. 2009;13:395–411. doi: 10.1517/14728220902841045. [DOI] [PubMed] [Google Scholar]
- 23.Horti AG, Wong DF. Clinical Perspective and Recent Development of PET Radioligands for Imaging Cerebral Nicotinic Acetylcholine Receptors. PET Clin. 2009;4:89–100. doi: 10.1016/j.cpet.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horti AG, Gao Y, Kuwabara H, Dannals RF. Development of radioligands with optimized imaging properties for quantification of nicotinic acetylcholine receptors by positron emission tomography. Life Sci. 2010;86:575–84. doi: 10.1016/j.lfs.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horti AG, Villemagne VL. The quest for Eldorado: development of radioligands for in vivo imaging of nicotinic acetylcholine receptors in human brain. Curr Pharm Des. 2006;12:3877–900. doi: 10.2174/138161206778559605. [DOI] [PubMed] [Google Scholar]
- 26.Ettrup A, Mikkelsen JD, Lehel S, Madsen J, Nielsen EO, Palner M, et al. 11C-NS14492 as a novel PET radioligand for imaging cerebral alpha7 nicotinic acetylcholine receptors: in vivo evaluation and drug occupancy measurements. J Nucl Med. 2011;52:1449–56. doi: 10.2967/jnumed.111.088815. [DOI] [PubMed] [Google Scholar]
- 27.Ogawa M, Nishiyama S, Tsukada H, Hatano K, Fuchigami T, Yamaguchi H, et al. Synthesis and evaluation of new imaging agent for central nicotinic acetylcholine receptor alpha7 subtype. Nucl Med Biol. 2010;37:347–55. doi: 10.1016/j.nucmedbio.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Deuther-Conrad W, Fischer S, Hiller A, Becker G, Cumming P, Xiong G, et al. Assessment of alpha7 nicotinic acetylcholine receptor availability in juvenile pig brain with [(1)F]NS10743. Eur J Nucl Med Mol Imaging. 2009;38:1541–9. doi: 10.1007/s00259-011-1808-y. [DOI] [PubMed] [Google Scholar]
- 29.Horti AG, Villemagne VL. The quest for Eldorado: development of radioligands for in vivo imaging of nicotinic acetylcholine receptors in human brain. Curr Pharm Des. 2006;12:3877–900. doi: 10.2174/138161206778559605. [DOI] [PubMed] [Google Scholar]
- 30.Toyohara J, Wu J, Hashimoto K. Recent development of radioligands for imaging alpha7 nicotinic acetylcholine receptors in the brain. Curr Top Med Chem. 2010;10:1544–57. doi: 10.2174/156802610793176828. [DOI] [PubMed] [Google Scholar]
- 31.Bunnelle WH, Tietje KR, Frost JM, Peters D, Ji J, Li T, et al. Octahydropyrrolo[3,4-c]pyrrole: a diamine scaffold for construction of either alpha4beta2 or alpha7-selective nicotinic acetylcholine receptor (nAChR) ligands. Substitutions that switch subtype selectivity. J Med Chem. 2009;52:4126–41. doi: 10.1021/jm900249k. [DOI] [PubMed] [Google Scholar]
- 32.Schrimpf MR, Sippy KB, Briggs CA, Anderson DJ, Li T, Ji J, et al. SAR of alpha7 nicotinic receptor agonists derived from tilorone: Exploration of a novel nicotinic pharmacophore. Bioorg Med Chem Lett. 2012;22:1633–8. doi: 10.1016/j.bmcl.2011.12.126. [DOI] [PubMed] [Google Scholar]
- 33.Ji J, Schrimpf MR, Sippy KB, Bunnelle WH, Li T, Anderson DJ, et al. Synthesis and structure-activity relationship studies of 3,6-diazabicyclo[3.2.0]heptanes as novel alpha4beta2 nicotinic acetylcholine receptor selective agonists. J Med Chem. 2007;50:5493–508. doi: 10.1021/jm070755h. [DOI] [PubMed] [Google Scholar]
- 34.Kulak JM, Carroll FI, Schneider JS. [125I]Iodomethyllycaconitine binds to alpha7 nicotinic acetylcholine receptors in monkey brain. Eur J Neurosci. 2006;23:2604–10. doi: 10.1111/j.1460-9568.2006.04804.x. [DOI] [PubMed] [Google Scholar]
- 35.Kulak JM, Schneider JS. Differences in alpha7 nicotinic acetylcholine receptor binding in motor symptomatic and asymptomatic MPTP-treated monkeys. Brain Res. 2004;999:193–202. doi: 10.1016/j.brainres.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 36.Han ZY, Zoli M, Cardona A, Bourgeois JP, Changeux JP, Le Novere N. Localization of [3H]nicotine, [3H]cytisine, [3H]epibatidine, and [125I]alpha-bungarotoxin binding sites in the brain of Macaca mulatta. J Comp Neurol. 2003;461:49–60. doi: 10.1002/cne.10659. [DOI] [PubMed] [Google Scholar]
- 37.Testa B, Caron G, Crivori P, Rey S, Reist M, Carrupt PA. Lipophilicity and related molecular properties as determinants of pharmacokinetic behavior. Chimia. 2000;54:672. [Google Scholar]
- 38.Ryu EK, Chen X. Development of Alzheimer’s disease imaging agents for clinical studies. Front Biosci. 2008;13:777–89. doi: 10.2741/2719. [DOI] [PubMed] [Google Scholar]
- 39.Seelig A. Toward understanding P-glycoprotein structure-activity relationships. Methods and Principles in Medicinal Chemistry. 2009;40:497–519. [Google Scholar]
- 40.Wiese M, Pajeva I. Algorithms to predict affinity for transporters. Solvay Pharmaceuticals Conference. 2006;6:187–209. [Google Scholar]
- 41.Didziapetris R, Japertas P, Avdeef A, Petrauskas A. Classification analysis of P-glycoprotein substrate specificity. J Drug Target. 2003;11:391–406. doi: 10.1080/10611860310001648248. [DOI] [PubMed] [Google Scholar]