

Abstract
S-Nitrosoglutathione reductase (GSNOR) is a key regulator of protein S-nitrosylation, the covalent modification of cysteine residues by nitric oxide that can affect activities of many proteins. We recently discovered that excessive S-nitrosylation from GSNOR deficiency in mice under inflammation inactivates the key DNA repair protein O6-alkylguanine-DNA alkyltransferase and promotes both spontaneous and carcinogen-induced hepatocellular carcinoma. To explore further the mechanism of tumorigenesis due to GSNOR deficiency, we compared the protein expression profiles in the livers of wild-type and GSNOR-deficient (GSNOR−/−) mice that were challenged with lipopolysaccharide to induce inflammation and expression of inducible nitric oxide synthase (iNOS). Two-dimensional difference gel electrophoresis analysis identified 38 protein spots of significantly increased intensity and 31 protein spots of significantly decreased intensity in the GSNOR−/− mice compared to those in the wild-type mice. We subsequently identified 19 upregulated and 19 downregulated proteins in GSNOR−/− mice using mass spectrometry. Immunoblot analysis confirmed in GSNOR−/− mice a large increase in the expression of the pro-inflammatory mediator S100A9, a protein previously implicated in human liver carcinogenesis. We also found a decrease in the expression of multiple members of the protein disulfide-isomerase (PDI) family and an alteration in the expression pattern of the endoplasmic reticulum (ER) chaperones in GSNOR−/− mice. Furthermore, altered expression of these proteins from GSNOR deficiency was prevented in mice lacking both GSNOR and iNOS. In addition, we detected S-nitrosylation of two members of the PDI protein family. These results suggest that S-nitrosylation resulting from GSNOR deficiency may promote carcinogenesis under inflammatory conditions in part through the disruption of inflammatory and ER stress responses.
Keywords: Animal proteomics, Cancer, Endoplasmic reticulum, Liver proteins, Oxidative stress, Two-dimensional differential in-gel electrophoresis
1 Introduction
Chronic inflammation has long been recognized as a risk factor for many human diseases including cancer [1]. One mechanistic link between inflammation and cancer may involve generation of nitric oxide (NO), superoxide, and other reactive oxygen species by macrophages and neutrophils that infiltrate the site of inflammation [2, 3]. During chronic inflammation, chemical reactions mediated by these reactive chemical species, including nitrosative deamination, oxidation and halogenation, are believed to cause DNA damage, which is fundamental to carcinogenesis [2, 3]. Whereas high levels of these reactive species formed under pathological conditions may cause damage to nucleic acids, at low levels, they can play important roles in cell signaling under physiological and pathophysiological conditions.
S-nitrosylation, the PTM of the thiol group of a cysteine residue by NO, has lately been receiving attention as a mechanism by which NO ubiquitously influences cellular signal transduction, including apoptosis and G protein-coupled receptor signaling [4]. S-nitrosoglutathione reductase (GSNOR; also known as alcohol dehydrogenase class III), is a key negative regulator of the levels of protein S-nitrosylation in vivo [5, 6]. Recently we showed that deletion of the GSNOR gene in mice during inflammatory responses significantly increased S-nitrosylation, ubiquitination, and proteasomal degradation of the key DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT), leading to rapid loss of AGT [7]. As a result of AGT depletion, repair of carcinogenic O6-ethylguanines in the livers of diethylnitrosamine-challenged GSNOR−/− mice was significantly impaired. Importantly, GSNOR−/− mice were found to be very susceptible to both spontaneous and diethylnitrosamine-induced hepatocellular carcinoma (HCC) [7]. Predisposition to HCC, S-nitrosylation, and depletion of AGT, and accumulation of O6-ethylguanines due to GSNOR deficiency, strikingly, was all abolished by concurrent deletion of inducible nitric oxide synthase (iNOS) in GSNOR−/−iNOS−/− mice, demonstrating a critical role of S-nitrosylation originated from iNOS in AGT inactivation and liver carcinogenesis in GSNOR−/− mice [7]. It has been further demonstrated most recently that hepatocyte-specific deletion of GSNOR caused nitrosative inactivation of liver AGT and increased mortality from diethylnitrosamine [8], underscoring the importance of the control of S-nitrosylation by GSNOR in liver parenchymal cells.
We showed that the expression and activity of GSNOR decreased significantly in about 50% of human HCC, the cancer mostly caused by chronic viral hepatitis with induction of iNOS [7]. Interestingly, gene-expression profiling showed that both GSNOR deficiency and iNOS overexpression in liver are closely associated with HCC development and a poor prognosis in HCC patients [9]. It has thus been hypothesized that excessive S-nitrosylation from GSNOR deficiency and concurrent iNOS overexpression in liver contributes critically to human HCC [7]. More recently, decrease in expression and activity of GSNOR has been reported for human lung adenocarcinoma [10], implicating broader involvement of GSNOR deficiency in human cancers.
The endoplasmic reticulum (ER) stress has been linked to several inflammatory response pathways in many cellular models and disease states [11–13]. ER stress is defined as an imbalance between the protein folding capacity of the ER and the client protein load, resulting in the accumulation of misfolded proteins [13]. The loss of homeostasis in the ER activates the ER stress response, known as the unfolded protein response (UPR) [12]. The UPR decreases protein translation and induces transcription of components of the ER machinery involved in folding, quality control, ER-associated degradation, and N-glycosylation. The sensing of stress in the ER lumen and transduction of signals from the ER to the cytoplasm or nucleus are mediated by three canonical ER stress transducers, inositol-requiring 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [12]. When activated, IRE1 causes splicing of X-box binding protein 1 (Xbp-1) mRNA, leading to synthesis of the spliced form of Xbp1, a potent transcription factor that induces expression of ER chaperones and folding catalysts [12]. The IRE1 branch of the UPR is also activated by nuclear factor-κB NFκB and JNK [13]. XBP1 signaling is also shown to be involved in the immune system [14, 15]. Thus, several different avenues of crosstalk exist between the UPR mediators and inflammatory responses in different types of cells under different conditions.
In this study, we have analyzed the expression of proteins in liver tissues of lipopolysaccharide (LPS)-challenged wild-type and GSNOR−/− mice. HCC develops in the context of chronic inflammation with substantial induction of iNOS. To clarify the function(s) of GSNOR in a pathophysiological condition relevant to HCC development, we challenged mice with LPS to induce inflammation and expression of iNOS. We used the 2D DIGE technique to generate quantitative protein expression profiles [16], and then used data-mining methods and MS to determine the identities of proteins associated with expression of GSNO. We found a number of protein spots whose intensities differed between the GSNOR−/− and wild-type mice, and the proteins corresponding to these spots were subsequently identified. We then evaluated expressions of several proteins that were identified by 2D DIGE and MS analyses, including the pro-inflammatory mediators, and the ER chaperones and folding catalysts, by immunoblot and confirmed that the expression of these proteins indeed differed between the GSNOR−/− and wild-type mice.
2 Materials and methods
2.1 Materials
Urea, DTT, Pharmalyte™, glycerol, SDS, tris(hydroxymethyl)aminomethane (Tris), and CyDye DIGE Fluors were purchased from GE healthcare Bioscience (Fair-field, CT, USA). 2-Iodoacetamide and lysine were purchased from Sigma-Aldrich (St. Louis, MO, USA). Thiourea, acetic acid, magnesium acetate, formic acid, and methanol were purchased form Wako Pure Chemical Industries (Osako, Japan). 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was from Doujin Molecular Technologies (Rockville, MD, USA), trypsin was from Promega (Madison, WI, USA), SYPRO® Ruby was from Invitrogen (Carlsbad, CA, USA), and the protease inhibitor Pefabloc SC PLUS was from Roche Applied Science (Basel, Switzerland). Anti-GRP78 rabbit polyclonal, anti-GRP94 rabbit polyclonal, anti- protein disulfide-isomerase (PDI) rabbit polyclonal, anti-ERp72 rabbit polyclonal, anti-ERp57 rabbit polyclonal antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA); anti-KDEL mouse monoclonal (10C3), anti-ORP150 mouse monoclonal (38), anti-ERp5 mouse monoclonal (G-5), anti-CHOP (C/EBP homologous protein) rabbit polyclonal, anti-arginase-1 rabbit polyclonal, and anti-Xbp-1 rabbit polyclonal antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); and anti-mouse S100A9 rat monoclonal antibody (MAB2065) was from R&D Systems (Minneapolis, MN, USA).
2.2 Animals
GSNOR−/− mice were reported previously [5]. GSNOR−/− mice were bred with iNOS−/− mice (The Jackson Laboratory) to obtain GSNOR−/−iNOS−/− mice [7]. All mice were maintained on normal mouse chow (5058 PicoLab Mouse Diet 20; LabDiet, Brentwood, MO, USA) in a specific pathogen-free facility at the UCSF. Polymerase chain reaction analysis of samples prepared from the liver of three GSNOR−/− mice showed that none of them were infected with Helicobater (UC Davis Comparative Pathology Laboratory). Two- to 3-month-old female wild-type, GSNOR−/− and GSNOR−/−iNOS−/− mice were injected intraperitoneally with 10 μg/g LPS (Escherichia coli, serotype 026:B6; Sigma, St. Louis, MO, USA) and sacrificed to collect tissues 48 h after LPS challenge. The LPS used (lot number 119K4044) contains three million endotoxin units per milligram. The experimental protocol was approved by the Institutional Animal Care and Use Committee of UCSF.
2.3 Protein extraction
After measuring the weight of the liver samples from LPS-challenged mice, we added ten volumes of lysis buffer (10 mM Tris, pH 8.0, 7 M urea, 2 M thiourea, 5 mM magnesium acetate, 4%(w/v) CHAPS, 4 mM protease inhibitor), and then homogenized and sonicated them. After centrifugation at 8000 × g for 10 min, protein concentration in the supernatant was measured by Bradford method using an absorption spectrophotometer (Shimadzu, Kyoto, Japan).
2.4 Fluorescent labeling
Fluorescent labeling of proteins was carried out as described previously [16] with some modification. In brief, aliquots of all samples pooled together were used as an internal control as described in Table 1. Proteins (100 μg) in the internal control sample and individual experimental samples were labeled with 200 nmol of 1-(5-carboxypentyl)-1′-propylindocarbocyamine halide (Cy2), 200 nmol of 1-(5-carboxypentyl)-1′-propylindocarbocyamine halide (Cy3), and 200 nmol of 1-(5-carboxypentyl)-1′-methylindocarbocyamine halide (Cy5), respectively. Samples were incubated on ice for 30 min, and then the reaction was terminated by incubating with 1 μL of 10 mM lysine on ice for 10 min. The labeled samples were then incubated with an equal amount of 2× sample buffer (8 M urea, 4% (w/v) CHAPS, 20 mg/mL DTT, and 2% (v/v) Pharmalyte™) on ice for 10 min.
Table 1.
Summary of 2D DIGE analysis of liver extracts. Protein extracts were prepared from the livers of LPS-treated wild-type (WT) and GSNOR −/− knockout (KO) mice
| Fluorescent dye | No. of spots | Genotype/group | |
|---|---|---|---|
| 1 | Cy3 | 4111 | WT-1 |
| Cy5 | 4111 | KO-1 | |
| Cy2 | 4111 | Standard | |
| 2 | Cy3 | 4513 | WT-2 |
| Cy5 | 4513 | KO-2 | |
| Cy2 | 4513 | Standard | |
| 3 | Cy3 | 3920 | WT-3 |
| Cy5 | 3920 | KO-3 | |
| Cy2 | 3920 | Standard | |
| 4 | Cy3 | 4264 | KO-1 |
| Cy5 | 4264 | WT-2 | |
| Cy2 | 4264 | Standard | |
| 5 | Cy3 | 3979 | KO-2 |
| Cy5 | 3979 | WT-3 | |
| Cy2 | 3979 | Standard | |
| 6 | Cy3 | 4019 | KO-3 |
| Cy5 | 4019 | WT-1 | |
| Cy2 | 4019 | Standard | |
| 7 | Cy3 | 4047 | WT-1 |
| Cy5 | 4047 | KO-2 | |
| Cy2 | 4047 | Standard | |
| 8 | Cy3 | 3874 | WT-2 |
| Cy5 | 3874 | KO-3 | |
| Cy2 | 3874 | Standard | |
| 9 | Cy3 | 4378 | WT-3 |
| Cy5 | 4378 | KO-1 | |
| Cy2 | 4378 | Standard |
Internal standard (Standard) sample was prepared by pooling equal amounts of proteins from each extract. As shown, samples were labeled with the indicated fluorescent dyes and subjected to 2D DIGE as described in Section 2. This table also includes information on the number of protein spots detected on each 2D DIGE gel and genotype/group of the sample used for the 2D DIGE analysis.
2.5 Separation by 2D PAGE
2D DIGE was carried out as reported previously [16]. In brief, the first separation was achieved using IPG DryStrip gels (24 cm long with a isoelectric point (pI) range between 3.0 and 10.0, GE Healthcare Biosciences) and Multiphore II (GE Healthcare Biosciences). After the first separation, the strips were equilibrated in the equilibration buffer A (0.25% DTT, 50 mM Tris pH 8.8, 6 M urea, 30% glycerol, and 2% SDS) and then in the equilibration buffer B (4.5% 2-iodoacetamide, 50 mM Tris pH 8.8, 6 M urea, 30% glycerol, and 2% SDS) for 10 min. The second-dimension separation was achieved using Ettan Dalt II and 12% polyacrylamide gels. After electrophoresis, gels were scanned with Typhoon9400 (GE Healthcare Biosciences) and analyzed by Decyder Ver7.0 (GE Healthcare Biosciences) to detect, subtract background of, normalize, and quantify the spots in images from a single 2D DIGE gel (Fig. 1).
Figure 1.
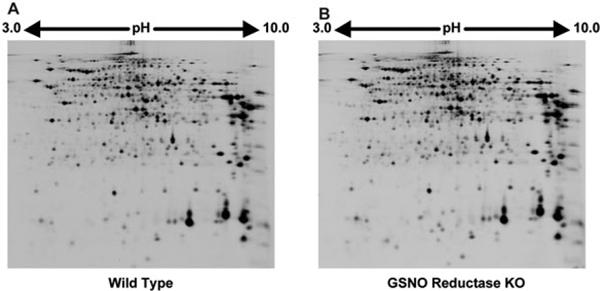
Representative 2D images of proteins in the liver extracts of wild-type (A) and GSNOR−/− mice (B) following LPS-treatment. Approximately 4000 protein spots were visualized by laser scanning.
2.6 Mass spectrometric identification of proteins
For identifying proteins by MS, equal amount of tissue extracts prepared from the wild-type and GSNOR knockout (KO) mice were mixed and labeled with 200 nmol of Cy5 on ice for 30 min. The reaction was terminated, samples were incubated with 2× sample buffer and then two-dimensional gel electrophoresis was carried out as described above. After electrophoresis, gels were scanned with Typhoon9400 and the proteins were fixed by incubating the gels in a solution containing 10% methanol and 7% acetic acid for 3 h at RT. Gels were then stained with SYPRO® Ruby and then destained by incubating in 10% methanol and 7% acetic acid. Finally, gels were scanned with Typhoon9400 and analyzed by Decyder Ver7.0. Protein spots were picked up using Ettan™ Spot Picker (GE Healthcare Biosciences).
Each picked gel plug was destained, protein in each plug was reduced with DTT, alkylated with iodoacetamide, and then digested overnight with trypsin. Samples for MS analysis were prepared by mixing separately 0.5 μL of each trypsin-digested protein sample with 0.5 μL of matrix solution containing 5 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) in 50% MeCN/0.1% trifluoroacetic acid (TFA).
MALDI-TOF MS and MS/MS analyses were performed using the AXIMA-Performance mass spectrometer (Shimadzu/Kratos, Kyoto, Japan). The operating conditions used were as follows: nitrogen laser (337 nm); reflectron mode; detection of positive ions; and acceleration voltage, 20 kV. Proteins were identified by using the MASCOT peptide mass fingerprint (PMF) and/or MS/MS ion search (MIS) of the SWISS-PROT database. The MASCOT search parameters used were as follows: taxonomy, house mouse; enzyme, trypsin; fixed modifications, carbamidomethyl (C); variable modifications, oxidation (M); peptide tolerance, 0.3 Da; MS/MS tolerance, 1.2 Da; mass values, [M+H]+ and monoisotopic; and missed cleavages, 1.
Molecular functional categories, biological processes, and cellular localizations of proteins identified by MIS were categorized using Scaffold version 3.0 (Proteome Software, Portland, OR, USA).
2.7 Western blotting
Cell lysates were prepared using Qproteome Mammalian Protein Prep Kit (Qiagen, Valencia, CA, USA). Samples were analyzed by Western blotting. After transfer to PVDF membranes, immunoreactive bands were visualized using the enhanced chemiluminescence detection system and indicated antibodies as previously described [17].
2.8 Detection of S-nitrosylated proteins
S-Nitrosothiols using resin-assisted capture (SNO-RAC) as described in [18] with some modifications. Briefly, SNO-RAC resins were prepared as described. 250 μL of cell lysates were diluted with 750 μL of HEN buffer (250 mM HEPES, 1 mM EDTA, 0.1 mM neocuproine, pH 8.0) and incubated with 1% SDS (final concentration) and 0.1% methyl methanethio-sulfonate (Sigma-Aldrich) at 50°C for 25 min. Proteins were precipitated with acetone, washed three times with 70% acetone, and resuspended in 200 μL HENS buffer (HEN containing 1% SDS). This was added to 50 μL resin slurry in the presence of sodium ascorbate (final 20 mM unless indicated otherwise), mixed by rotation in the dark for 3 h, following which the resin was washed with 4 × 1 mL HENS buffer. Captured proteins were eluted with 30 μL HENS buffer containing 100 mM 2-mercaptoethanol for 20 min at RT, and 20 μL of each eluent was used for SDS-PAGE analysis.
2.9 Statistical analyses
Values are presented as mean ± SEM. Differences between groups were examined for statistical significance using Student's t test. p values of <0.05 were considered statistically significant.
3 Results
3.1 2D DIGE analysis of protein expression in liver of LPS-treated GSNOR−/− mice
To study the role of GSNOR in liver carcinogenesis, we performed 2D DIGE to identify differentially expressed proteins in the liver tissues of wild-type and GSNOR−/− mice following LPS treatment. For our experiments, we typically prepared protein samples from three individual wild-type and GSNOR−/− mice. These samples were randomly labeled with Cy3 or Cy5 dye as listed in Table 1 to avoid any dye biases. The pooled sample prepared by mixing equal amount of protein from each individual sample was labeled with Cy2 and used as an internal standard (Table 1). After 2D DIGE separation, images of proteins expressed in the liver of wild-type and GSNOR−/− mice were acquired from the same gel under different wavelengths (Fig. 1). DeCyder 7.0 analysis identified 3874–4513 protein spots (Fig. 2). Statistical analysis of the 2D DIGE data using DeCyder 7.0 revealed that the signal intensities of 38 spots were consistently ≥1.5-fold higher (i.e. increase in protein expression) and signal intensities of 31 spots were consistently lower (i.e. decrease in protein expression) in the GSNOR−/− sample than in the wild-type control (p < 0.05, t-test with false discovery rate [FDR] correction).
Figure 2.

Location of selected spots on the 2D images. Sixty-nine protein spots whose intensities increased (A) or decreased (B) in liver tissues of LPS-treated GSNOR−/− mice are labeled on images of 2D gels. Number represents ID of each spot as assigned by DeCyder 7.0.
3.2 Identification of differentially expressed proteins by MS
To identify the differentially expressed proteins, a pooled sample containing equal amounts of proteins from each of the three wild-type and GSNOR−/− mice was labeled with Cy5, resolved on a 2D gel, and stained with SYPRO® Ruby. Spots were picked using the Ettan™ Spot Picker (GE Healthcare Biosciences) as described in section 2. Proteins in picked gel-plugs were digested in-gel with trypsin and prepared for MS analysis as described in section 2.
Nineteen proteins were identified from 38 spots whose signals increased in the GSNOR−/− mice, whereas another set of 19 proteins were identified from 31 spots whose signals decreased in the GSNOR−/− mice. These upregulated and downregulated proteins are listed in Tables 2 and 3, respectively, together with their spot number, p-value (t-test with FDR correction), averaged expression ratio (GSNOR−/− to wild-type), ID, name, calculated molecular weight, calculated pI, score, number of matched peaks, and sequence coverage (%). The spots contained one or more proteins. In the upregulated spot number 1713, we detected a single protein, 78 kDa glucose-regulated protein (GRP78): eight signals corresponding to tryptic peptides of GRP78 were identified by PMF and five of the eight (m/z 1461, 1567, 1589, 1816, and 1888) were confirmed by MIS (Fig. 3A; upper panel). In the downregulated spot number 903, we detected two proteins, PDI A4 (ERp72) and GRP78 (Fig. 3A; lower panel). Either one or both of the proteins in spot number 903 might be downregulated in GSNOR−/− samples. Interestingly, GRP78 in spot 903 had higher molecular mass and lower pI than that in spot 1713 (Fig. 2), indicating possibly two distinct forms of GRP78 from different PTM in the two spots. Thus, GSNOR deficiency increased expression of one form of GRP78, but it might also decrease the level of a differently modified GRP78 protein. We also analyzed the results of MS using Scaffold version 3.0 (Proteome Software) and found that expressions of proteins belonging to a wide range of functional categories were altered in samples from GSNOR-deficient mice (Figs. 3B, C and D).
Table 2.
Proteins with increased expression in liver of LPS-treated GSNOR −/− mice compared to wild-type control
| Spot no. | t-test | Av. ratio | ID | Protein name | MW (kDa) | P/ | Scorea) | Matchesb) | Coverage (%)c) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1357 | 0 | 1.73 | ATTY_MOUSE | Tyrosine aminotransferase | 51 | 5.4 | MIS | 82 | 2 | 4 |
| 1471 | 0 | 1.6 | FIBG_MOUSE | Fibrinogen gamma chain | 50 | 5.5 | MIS | 252 | 6 | 17 |
| PMF | 94 | 11 | 28 | |||||||
| RPAB3_MOUSE | DNA-directed RNA polymerases RPABC3 | 17 | 4.5 | PMF | 55 | 6 | 47 | |||
| ARP3_MOUSE | Actin-related protein 3 | 48 | 5.6 | MIS | 47 | 1 | 3 | |||
| 1479 | 0.01 | 2.12 | PLIN2_MOUSE | Perilipin-2 | 47 | 6.4 | MIS | 129 | 3 | 9 |
| PMF | 50 | 6 | 18 | |||||||
| 1683 | 3.20E-10 | 2.46 | ADH4_MOUSE | Alcohol dehydrogenase 4 | 41 | 7.7 | MIS | 83 | 2 | 3 |
| PMF | 57 | 5 | 14 | |||||||
| 1704 | 0.01 | 1.55 | CPSM_MOUSE | Carbamoyl-phosphate synthase | 166 | 6.5 | MIS | 57 | 2 | 1 |
| 1710 | 3.20E-10 | 1.87 | ALDOB_MOUSE | Fructose-bisphosphate aldolase B | 40 | 8.5 | MIS | 157 | 2 | 9 |
| PMF | 69 | 9 | 27 | |||||||
| 1713 | 6.40E-09 | 1.48 | GRP78_MOUSE | 78 kDa glucose-regulated protein | 73 | 5.1 | MIS | 176 | 5 | 9 |
| PMF | 56 | 8 | 15 | |||||||
| 1714 | 0 | 1.42 | ALDOB_MOUSE | Fructose-bisphosphate aldolase B | 40 | 8.5 | MIS | 238 | 5 | 15 |
| PMF | 126 | 13 | 33 | |||||||
| 1785 | 5.60E-05 | 2.58 | ARGI1_MOUSE | Arginase-1 | 35 | 6.5 | PMF | 85 | 8 | 29 |
| 1825 | 0 | 1.68 | FIBG_MOUSE | Fibrinogen gamma chain | 50 | 5.5 | MIS | 32 | 1 | 2 |
| 1963 | 0 | 1.59 | A2M_MOUSE | Alpha-2-macroglobulin | 167 | 6.2 | MIS | 37 | 2 | 0 |
| 2631 | 0 | 2.01 | GPKOW_MOUSE | G patch domain and KOW motifs-containing portein | 54 | 8.2 | PMF | 56 | 6 | 18 |
| 2704 | 2.50E-05 | 1.64 | URAD_MOUSE | 2-Oxo-4-hydroxy-4-carboxy -5-ureidoimidazoline decarboxylase | 20 | 5.7 | MIS | 40 | 1 | 7 |
| PMF | 58 | 5 | 27 | |||||||
| 3644 | 1.30E-09 | 3.57 | S10A9_MOUSE | Protein S100-A9 | 13 | 6.6 | MIS | 129 | 2 | 11 |
| 3659 | 8.90E-05 | 2.72 | S10A9_MOUSE | Protein S100-A9 | 13 | 6.6 | MIS | 154 | 2 | 11 |
| 3679 | 9.60E-05 | 1.51 | CYTB_MOUSE | Cystatin-B | 11 | 6.8 | MIS | 58 | 1 | 9 |
| UK114_MOUSE | Ribonuclease UK114 | 14 | 8.7 | MIS | 42 | 1 | 11 | |||
| 3730 | 1.60E-08 | 7.37 | SAA1_MOUSE | Serum amyloid A-1 protein | 14 | 6.5 | MIS | 25 | 1 | 7 |
| 3732 | 7.70E-06 | 3.19 | SAA1_MOUSE | Serum amyloid A-1 protein | 14 | 6.5 | MIS | 34 | 1 | 7 |
| 3735 | 8.30E-07 | 11.85 | SAA2_MOUSE | Serum amyloid A-2 protein | 14 | 6.4 | MIS | 30 | 1 | 7 |
| 3736 | 3.80E-07 | 4.1 | SAA2_MOUSE | Serum amyloid A-2 protein | 14 | 6.4 | MIS | 37 | 1 | 7 |
| 2930 | 5.10E-06 | 2.94 | APC_MOUSE | Adenomatous polyposis coli protein | 313 | 7.4 | PMF | 58 | 13 | 6 |
Mascot score for proteins identified by MALDI-TOF.
Peptides matched by Mascot search.
Percentage sequence coverage.
Table 3.
Proteins with decreased expression in liver of LPS-treated GSNOR−/− mice compared to wild-type control
| Spot no. | t-test | Av. ratio | ID | Protein name | MW (kDa) | p/ | Scorea) | Matchesb) | Coverage (%)c) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 470 | 0 | 0.53 | CPSM_MOUSE | Carbamoyl-phosphate synthase | 166 | 6.5 | MIS | 228 | 4 | 5 |
| PMF | 95 | 15 | 16 | |||||||
| 526 | 1.20E-05 | 0.59 | HYOU1_MOUSE | Hypoxia upregulated protein 1/ORP150 | 111 | 5.1 | PMF | 97 | 11 | 12 |
| 720 | 4.70E-05 | 0.6 | RRBP1_MOUSE | Ribosome-binding protein 1 | 173 | 9.4 | MIS | 53 | 2 | 1 |
| PMF | 81 | 12 | 11 | |||||||
| 903 | 3.30E-07 | 0.65 | PDIA4_MOUSE | PDI A4/ERp72 | 72 | 5.2 | MIS | 180 | 5 | 7 |
| PMF | 129 | 16 | 25 | |||||||
| GRP78_MOUSE | 78 kDa glucose-regulated protein/GRP78 | 73 | 5.1 | MIS | 52 | 2 | 4 | |||
| PMF | 56 | 9 | 16 | |||||||
| 1160 | 3.30E-07 | 0.56 | NLTP_MOUSE | Non-specific lipid-transfer protein | 60 | 7.2 | MIS | 212 | 5 | 11 |
| PMF | 94 | 12 | 32 | |||||||
| 1223 | 0 | 0.65 | CBR1_MOUSE | Carbonyl reductase [NADPH] 1 | 31 | 8.5 | PMF | 58 | 9 | 38 |
| 1306 | 2.00E-05 | 0.65 | SBP1_MOUSE | Selenium-binding protein 1 | 53 | 5.9 | MIS | 370 | 6 | 15 |
| PMF | 72 | 9 | 23 | |||||||
| 1483 | 7.80E-07 | 0.67 | PDIA6_MOUSE | PDI A6/ERp5 | 48 | 5.0 | MIS | 322 | 6 | 18 |
| PMF | 68 | 8 | 21 | |||||||
| 1573 | 2.70E-05 | 0.64 | BHMT1_MOUSE | Betaine-homocysteine S-methyltransferase 1 | 45 | 8.0 | MIS | 247 | 7 | 19 |
| PMF | 200 | 20 | 66 | |||||||
| 1717 | 9.60E-13 | 0.17 | SPYA_MOUSE | Serine-pyruvate aminotransferase | 46 | 8.6 | MIS | 97 | 3 | 8 |
| PMF | 76 | 8 | 23 | |||||||
| ADHX_MOUSE | Alcohol dehydrogenase class-3/GSNOR | 40 | 7.0 | MIS | 57 | 1 | 4 | |||
| 1950 | 7.60E-06 | 0.63 | PAHX_MOUSE | Phytanoyl-CoA dioxygenase, peroxisomal | 39 | 7.2 | MIS | 98 | 3 | 9 |
| 2197 | 0 | 0.62 | CY1_MOUSE | Cytochrome d, heme protein, mitochondrial | 36 | 9.2 | MIS | 26 | 1 | 4 |
| FLOT2_MOUSE | Flotillin-2 | 47 | 5.1 | PMF | 65 | 7 | 18 | |||
| 2648 | 7.10E-08 | 0.6 | GSTP1_MOUSE | Glutathione S-transferase P 1 | 24 | 7.7 | MIS | 269 | 4 | 29 |
| PMF | 73 | 7 | 39 | |||||||
| 3575 | 0 | 0.46 | HBA_MOUSE | Hemoglobin subunit alpha | 15 | 8.0 | MIS | 76 | 2 | 16 |
| G3ST2_MOUSE | Galactose-3-O-sulfotransferase 2 | 47 | 8.2 | PMF | 57 | 5 | 17 | |||
| 3643 | 0 | 0.56 | HBB1_MOUSE | Hemoglobin subunit beta-1 | 16 | 7.1 | MIS | 136 | 2 | 21 |
| PMF | 60 | 5 | 38 | |||||||
| 4510 | 3.30E-06 | 0.44 | BHMT1_MOUSE | Betaine-homocysteine S-methyltransferase 1 | 45 | 8.0 | MIS | 46 | 2 | 4 |
| PMF | 64 | 8 | 17 | |||||||
| 4512 | 0 | 0.52 | HBB1_MOUSE | Hemoglobin subunit beta-1 | 16 | 7.1 | MIS | 169 | 3 | 23 |
| PMF | 62 | 7 | 48 |
Mascot score for proteins identified by MALDI-TOF.
Peptides matched by Mascot search.
Percentage sequence coverage.
Figure 3.
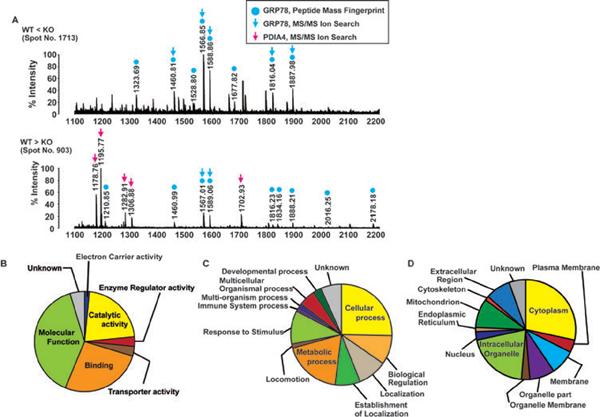
Identification of proteins by MS and categorization of the proteins. (A) Mass spectra of an upregulated protein (spot number 1713, upper panel) and a downregulated protein (spot number 903, lower panel) in liver of GSNOR−/− mice. Gel plugs corresponding to the protein spots were picked, proteins were digested in-gel with trypsin and further processed for MS analysis as described in Section 2. Blue and red arrows indicate the peptides identified by MS/MS ion search (MIS) as GRP78 and PDIA4, respectively. Blue circles indicate the peptides identified by PMF as GRP78. The differentially expressed proteins that are identified by MIS, a total of 28, can be classified into seven molecular functional categories (B), 12 biological processes (C), and 12 cellular localization sites (D).
3.3 Western blot analysis of differentially expressed proteins
First, consistent with our previous report that expression of GSNOR is deficient in GSNOR−/− mice [5], GSNOR, with serine–pyruvate aminotransferase, was identified in the spot 1717 whose intensity was decreased in the GSNOR−/− mice compared to wild-type control (Table 3), suggesting that our 2D DIGE experiment worked well. AGT, which is expressed at a lower level in GSNOR−/− mice [7], was nevertheless not among the proteins detected, possibly because of its low expression level.
Second, we detected a large increase in the expression of pro-inflammatory mediators, including S100A9 and serum amyloid A-1 and A-2 proteins (Table 2). S100A9, also known as myeloid-related protein-14, is upregulated in HCC cells and is implicated in liver carcinogenesis [19]. Immunoblot analysis confirmed that the expression of S100A9 was significantly elevated in the livers of the LPS-challenged GSNOR−/− mice as compared to the wild-type control (Figure 4). Furthermore, the increase in S100A9 expression was abolished in the LPS-challenged mice that were deficient in both GSNOR and iNOS. These data suggest that iNOS-derived S-nitrosylation in GSNOR−/− mice may cause increase in the expression of pro-inflammatory proteins.
Figure 4.
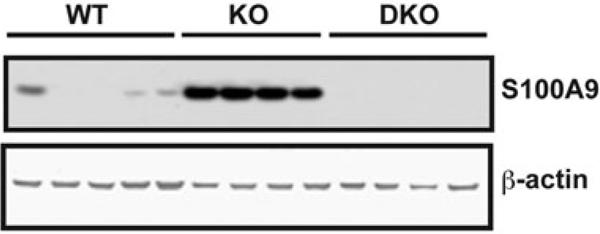
S100A9 expression was elevated in livers of GSNOR−/− mice after LPS challenge. Immunoblot of S100A9 and β-actin (control protein) in livers of wild-type (WT, n = 5), GSNOR−/− (KO, n = 4), and GSNOR−/−iNOS−/− mice (DKO, n = 4) 2 days after LPS injection.
Third, arginase-1 was detected as among the upregulated proteins in GSNOR−/− samples (Table 2). Arginase-1 may limit the activity of NOS by competing for the common substrate, L-arginine [20]. Western blot analysis showed that the expression level of arginase-1 was higher in the GSNOR−/− mice than in the wild-type mice and the level was highest in GSNOR−/−iNOS−/− mice (Figs. 5A and 5D).
Figure 5.
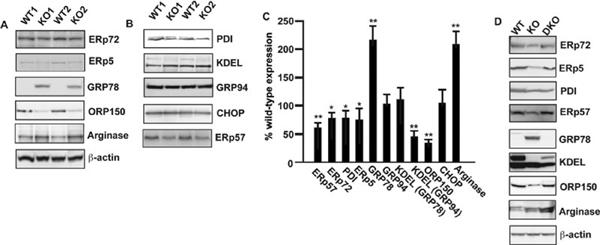
Validation of differentially expressed proteins by immunoblot. Lysates prepared from LPS-treated liver tissues of wild-type and GSNOR−/− mice were subjected to immunoblot analysis using anti-ERp72, anti-ERp5, anti-GRP78, anti-ORP150, and anti-arginase-1 antibodies (A) and using anti-PDI, anti-KDEL, anti-GRP94 anti-CHOP, and anti-ERp57 antibodies (B). Anti-β-actin antibody was used as a control to ensure that equal amount protein was loaded in each lane. (C) Semiquantitative analysis of protein-expression in GSNOR−/− mice, as measured by scanning densitometry, and is expressed as a percentage of wild-type, normalized with respect to the expression of beta-actin. Data shown are mean ± SE (n = 3); * p < 0.05 and **p < 0.01 versus control. (D) Lysates prepared from LPS-treated liver tissues of wild-type, GSNOR−/− mice and GSNOR−/−iNOS−/− mice were subjected to immunoblot analysis using anti-ERp72, anti-ERp5, anti-PDI, anti-ERp57, anti-GRP78, anti-KDEL anti-ORP150, and anti-arginase-1 antibodies. Anti-β-actin antibody was used as a control to ensure that equal amount protein was loaded in each lane.
Fourth, among the proteins with altered expression in GSNOR−/− mice are a number of proteins that are inducible via the UPR pathway and are associated with ER stress (Tables 2 and 3). We performed Western blot analysis and confirmed that hypoxia upregulated protein 1 (ORP150), PDI A4 (ERp72), and protein disulfide-protein A6 (ERp5) were all downregulated whereas GRP78 was upregulated in GSNOR−/− mice compared to wild-type mice (Figs. 5A and 5D). Thus, deficiency of GSNOR attenuated the expressions of several ER stress-related proteins, except that of GRP78. To further evaluate the effect of GSNOR deficiency on the expression of other ER stress-related proteins, Western blot analysis was performed using antibodies against PDI family A3 (ERp57), PDI, 94 kDa GRP (GRP94), C/EBP homologous protein (CHOP), and KDEL (the C-terminal tetra-peptide that is present in GRP78/Bip and GRP94 and responsible for their retention in ER [21]; Figs. 5B and 5D). Interestingly expressions of ERp57, PDI, and KDEL-positive GRP94 (using anti-KDEL antibody) were lower in the GSNOR−/− mice than in the wild-type mice, whereas the expressions of total GRP94 (using anti-GRP94 antibody), KDEL-positive GRP78 (using anti-KDEL antibody), and CHOP were comparable between wild-type and GSNOR−/− mice. The anti-KDEL antibody in Western blot specifically recognized induced expression of GRP78 and GRP94 in HepG2 cells that were treated with thapsigargin to activate UPR (results not shown). Furthermore, altered expressions of ERp72, ERp5, PDI, ERp57, total GRP78, KDEL-positive GRP94, and ORP150 in the liver of LPS-challenged GSNOR−/− mice were reversed by further genetic deletion of iNOS (Fig. 5D). These results suggest that multiple members of the PDI family are downregulated by iNOS-derived S-nitrosylation in the liver of GSNOR−/− mice. Our results also suggest that GRP78 and GRP94 might have isoforms that are differently expressed in GSNOR−/− and wild-type mice.
3.4 Detection of S-Nitrosylated ERp72 and ERp5 and evaluation of ER stress
The thioredoxin-like domains of PDI were shown to be modified by S-nitrosylation [22], suggesting that other members of the PDI protein family might also be modified by S-nitrosylation. To identify the S-nitrosylated proteins belonging to the PDI family, we analyzed cell extracts prepared from thapsigargin-treated (12 h) HepG2 cells by SNO-RAC assay. Results of this assay revealed S-nitrosylation of ERp72 and ERp5 as well as that of GADPH (Fig. 6A, 6B, 6C), which is a typical S-nitrosylated protein [23].
Figure 6.
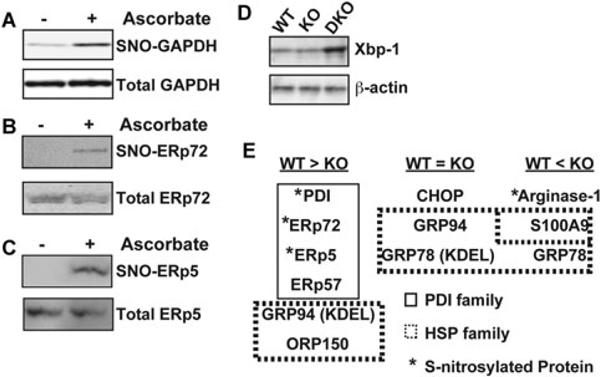
Detection of S-nitrosylated GAPDH, ERp72 and ERp5 and evaluation of ER stress. HepG2 cells were treated with thapsigargin (5 μM) for 12 h, and lysates prepared from the treated cells were analyzed by SNO-RAC assay, in which ascorbate-dependent capture of proteins by resin indicates the presence of S-nitrosylated Cys residues. Elutants were subjected to immunoblot analysis using anti-GAPDH (A), anti-ERp72 (B) and anti-ERp5 antibodies (C). (D) Lysates prepared from LPS-treated liver tissues of wild-type and GSNOR−/− mice were subjected to immunoblot analysis using anti-Xbp-1 antibodies. Anti-β-actin antibody was used as a control to ensure that equal amount protein was loaded in each lane. (E) Summary of proteins whose expression levels were evaluated by immunoblot analysis. Proteins enclosed within the solid line are members of the PDI family, whereas proteins enclosed within the dotted lines are members of the heat shock protein (HSP) family. *S-nitrosylated protein.
To evaluate ER stress in livers of LPS-challenged wild-type, GSNOR−/−, and GSNOR−/−iNOS−/− mice, immunoblot analysis were performed using anti-Xbp-1 antibody [24]. Immunoblot analysis showed the expression of Xbp-1 was not altered in the livers of the LPS-challenged GSNOR−/− mice as compared to the wild-type control, whereas the Xbp-1 expression was increased in the LPS-challenged GSNOR−/−iNOS−/− mice (Figure 6D).
4 Discussion
We have identified a set of 19 proteins whose expressions are significantly increased and another set of 19 proteins whose expressions are significantly decreased in the liver of GSNOR−/− mouse following LPS challenge. Many of these identified proteins were reported to be associated with carcinogenesis, and they were also known to be involved in a wide variety of biological processes, suggesting that GSNOR may affect various cellular functions in liver. For example, it was reported that the downregulation of the selenium binding protein 1, a sensor of reactive xenobiotics, was involved in ovarian and colon cancer [25,26]. Upregulation of glutathione S-transferase P1 was reported to be associated with acquired resistance to cancer drugs [27].
We have shown in the current study that following inflammatory stimulation by LPS challenge, GSNOR deficiency results in elevated expression of pro-inflammatory mediators, including S100A9 and serum amyloid A-1 and A-2 proteins. This effect of GSNOR deficiency is apparently dependent on iNOS activity, underscoring the causative role of S-nitrosylation. The present finding on inflammation is consistent with our previous report that following LPS challenge, GSNOR deficiency prolonged activation of pro-inflammatory iNOS in mice [5]. Inflammation has been recognized as an important enabling characteristic in tumorigenesis [28]. S100A9 in particular has been shown to be significantly upregulated in human and mouse HCC cells [19]. Furthermore, ectopic expression of S100A9 together with S100A8 protected HCC cell line Hep3B from TNF-γ-induced apoptosis [19]. Thus, inflammation promoted by S-nitrosylation from GSNOR deficiency may provide an additional oncogenic mechanism in S-nitrosylation-induced HCC.
We have also observed altered expression levels of several ER chaperones and folding catalysts in the livers of GSNOR-deficient mice. The ER chaperones and folding catalysts are categorized into four groups: chaperones of heat shock protein family, chaperone lectins, thiol oxidoreductases of the PDI family, and peptidyl-prolyl cis–trans isomerases (PPIs) [12, 29]. GRP78 and GRP94 are members of the heat shock protein family and are known to be involved in many physiological and pathophysiological processes including carcino-genesis [11]. Our results revealed that GSNOR deficiency leads to increased expression of the isoforms that were recognized by the anti-GRP78 antibody but not by the anti-KDEL antibody; conversely, GSNOR deficiency leads to decreased expression of the isoform(s) recognized by the anti-KDEL antibody but not by the anti-GRP94 antibody. These results suggest that GSNOR deficiency may alter the expression patterns of the GRP78 and GRP94 isoforms. It is noteworthy that several isoforms of GRP78 [30] and GRP94 [31] were found primarily in pathological tissues including cancer. In addition, several isoforms of GRP78 were found to be localized in the plasma membrane, but not in the ER, of cancer cells [32], suggesting that PTM, possibly glycosylation, might account for their altered cellular localization and/or atypical role(s). Our results thus suggest that GSNOR deficiency might affect cellular process by altering the expression levels of isoforms of ER chaperones.
The ER chaperones and folding catalysts are induced as a consequence of accumulation of unfolded proteins in the ER via the UPR pathway [12]. NO is thought to activate the UPR pathway by perturbing the Ca2+ homeostasis in cells. NO could perturb the Ca2+ homeostasis of ER by inhibiting the Ca2+-ATPase or by activating RyR1 and RyR2, and thereby induce expression of the ER chaperones [33]. In addition, Moncada's group showed that NO may elevate cytosolic Ca2+ and activate the UPR pathway by disrupting the respiratory chain in the mitochondria [34]. Our current study indicates that the expression of PDI, ERp57, ERp72, and ERp5 was downregulated by iNOS-derived S-nitrosylation in liver of GSNOR−/− mice, and that ERp72 and ERp5 were readily S-nitrosylated. Interestingly, expressions of CHOP and Xbp-1 were not significantly different between the wild-type and GSNOR−/− mice, suggesting that mechanism(s) independent of either CHOP or Xbp-1 are important for regulating the expression of ER chaperones in GSNOR−/− mice. Thus, decrease in the levels of the PDI family proteins in GSNOR−/− mice might result from destabilization of the proteins by iNOS-derived S-nitrosylation. S-nitrosylation has been reported to inhibit the enzymatic activity of PDI in cell culture [22], and it would be interesting to determine whether S-nitrosylation in GSNOR−/− mice can affect the activities of PDI or other members of the PDI family.
Activation of UPR and induction of ER chaperones and folding catalysts are two-sided events for cancer cells [35]. The UPR promotes adaptation to hypoxia, thus could induce expression of the ER chaperones and folding catalysts and promote survival of cancer cells under hypoxic conditions [19, 32]. The ER chaperones are also necessary to secrete angiogenic factors to promote vasculature growth [36]. On the other hand, prolonged activation of this response can terminate dormancy and lead to apoptosis of a cancer cell [35]. Activation of p38 mitogen activated protein kinase is associated with PERK-eIF2α mediated translational arrest, leading to growth arrest and dormancy, and promotes resistance to conventional chemotherapy [37]. Therefore, there are two possible ways to promote tumorigenesis as a consequence of GSNOR deficiency by manipulation of ER-stress proteins. The first possibility is that deficiency of GSNOR exacerbates ER stress, which induces cytoprotective pathways in cancer cells leading to tumorigenesis. The other possibility is that the deficiency of GSNOR ameliorates ER stress, thereby inhibits dormancy and/or apoptosis of cancer cells.
We recently demonstrated that GSNOR deficiency destabilizes the key DNA repair protein AGT and promotes carcinogenesis in the liver, suggesting that NO enhances carcinogenesis likely as a consequence of inactivation of the DNA repair system by S-nitrosylation [7]. Through analysis of protein expression profiles in the liver of LPS-challenged GSNOR−/− mice and wild-type and GSNOR−/−iNOS−/− control, we found in the present study that S-nitrosylation from GSNOR deficiency causes altered expression of a number of proteins that are involved in a wide variety of biological processes including pro-inflammatory regulation, protection from oxidative stress, urea cycle, and protein folding in ER. S-nitrosylation has been reported to directly affect protein stability, increasing the stability of hypoxia-inducible factor (HIF)-1α [38] but destabilizing AGT [7]. It has also been reported that S-nitrosylation can alter the activities of transcription factors, increasing HIF-1 activity but inhibiting the NFκB pathway [38]. It is thus conceivable that S-nitrosylation from GSNOR deficiency may influence hepatic levels of various proteins through altering protein stability and protein synthesis via gene transcription. Taken together, our results suggest that in addition to inactivation of the DNA repair system, S-nitrosylation might contribute to carcinogenesis through regulation of a wide range of other pathways including ER stress.
Acknowledgments
This research is supported by the Japan Society for the Promotion of Science (JSPS) through its Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program), by the Japan Ministry of Education, Culture, Sports and Technology (MEXT) Grants-in-Aid for Scientific Research 08107960 and 09153695, and by the National Institutes of Health of the United States (R01CA55578, R01CA122359, and P01CA123328 to L.L.).
Abbreviations
- ER
endoplasmic reticulum
- GSNOR
S-nitrosoglutathione reductase
- HCC
heptacellular carcinoma
- iNOS
inducible nitric oxide synthase
- JNK
c-jun N-terminal kinase
- NF-κB
nuclear factor-κB
- PDI
protein disulfide-isomerase
- SNO-RAC
S-nitrosothiols using resin-assisted capture
Footnotes
The authors have declared no conflict of interest.
5 References
- [1].Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- [2].Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother. Pharmacol. 2011;67:1211–1224. doi: 10.1007/s00280-011-1654-4. [DOI] [PubMed] [Google Scholar]
- [3].Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int. J. Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hess DT, Matsumoto A, Kim SO, Marshall HE, et al. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- [5].Liu L, Yan Y, Zeng M, Zhang J, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- [6].Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- [7].Wei W, Li B, Hanes MA, Kakar S, et al. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci. Transl. Med. 2010;2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wei W, Yang Z, Tang CH, Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis. 2011;32:973–977. doi: 10.1093/carcin/bgr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hoshida Y, Villanueva A, Kobayashi M, Peix J, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Marozkina NV, Yemen S, Wei C, Wallrabe H, et al. S-Nitrosoglutathione reductase in human lung cancer. Am. J. Respir. Cell Mol. Biol. 2011;46:63–70. doi: 10.1165/rcmb.2011-0147OC. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- [13].Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- [14].Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- [15].Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kondo T, Hirohashi S. Application of highly sensitive fluorescent dyes (CyDye DIGE Fluor saturation dyes) to laser microdissection and two-dimensional difference gel electrophoresis (2D-DIGE) for cancer proteomics. Nat. Protoc. 2006;1:2940–2956. doi: 10.1038/nprot.2006.421. [DOI] [PubMed] [Google Scholar]
- [17].Kimura I, Inoue D, Maeda T, Hara T, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41) Proc. Natl. Acad. Sci. USA. 2011;108:8030–8035. doi: 10.1073/pnas.1016088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Forrester MT, Thompson JW, Foster MW, Nogueira L, et al. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Németh J, Stein I, Haag D, Riehl A, et al. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology. 2009;50:1251–1262. doi: 10.1002/hep.23099. [DOI] [PubMed] [Google Scholar]
- [20].Santhanam L, Lim HK, Lim HK, Miriel V, et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ. Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- [21].Ozawa K, Kuwabara K, Tamatani M, Takatsuji K, et al. 150-kDa oxygen-regulated protein (ORP150) suppresses hypoxia-induced apoptotic cell death. J. Biol. Chem. 1999;274:6397–6404. doi: 10.1074/jbc.274.10.6397. [DOI] [PubMed] [Google Scholar]
- [22].Uehara T, Nakamura T, Yao D, Shi ZQ, et al. S-nitrosylated protein-disulphide isomerase links protein mis-folding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- [23].Hara MR, Agrawal N, Kim SF, Cascio MB, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- [24].Calfon M, Zeng H, Urano F, Till JH, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- [25].Huang KC, Park DC, Ng SK, Lee JY, et al. Selenium binding protein 1 in ovarian cancer. Int. J. Cancer. 2006;118:2433–2440. doi: 10.1002/ijc.21671. [DOI] [PubMed] [Google Scholar]
- [26].Kim H, Kang HJ, You KT, Kim SH, et al. Suppression of human selenium-binding protein 1 is a late event in colorectal carcinogenesis and is associated with poor survival. Proteomics. 2006;6:3466–3476. doi: 10.1002/pmic.200500629. [DOI] [PubMed] [Google Scholar]
- [27].Singh S, Okamura T, Ali-Osman F. Serine phosphorylation of glutathione S-transferase P1 (GSTP1) by PKCα enhances GSTP1-dependent cisplatin metabolism and resistance in human glioma cells. Biochem. Pharmacol. 2010;80:1343–1355. doi: 10.1016/j.bcp.2010.07.019. [DOI] [PubMed] [Google Scholar]
- [28].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [29].Kozlov G, Määttänen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. FEBS J. 2010;277:3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- [30].Pearce A, Farrell WE, Jenkins HA. Characterisation of the isoforms of GRP78 using two-dimensional electrophoresis. Biochem. Soc. Trans. 1995;23:79S. doi: 10.1042/bst023079s. [DOI] [PubMed] [Google Scholar]
- [31].Nciri R, Allagui MS, Vincent C, Murat JC, et al. Chronic lithium administration triggers an over-expression of GRP94 stress protein isoforms in mouse liver. Food Chem. Toxicol. 2010;48:1638–1643. doi: 10.1016/j.fct.2010.03.038. [DOI] [PubMed] [Google Scholar]
- [32].Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011;434:181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gotoh T, Mori M. Nitric oxide and endoplasmic reticulum stress. Arterioscler. Thromb. Vasc. Biol. 2006;26:1439–1446. doi: 10.1161/01.ATV.0000223900.67024.15. [DOI] [PubMed] [Google Scholar]
- [34].Xu W, Liu L, Charles IG, Moncada S. Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat. Cell Biol. 2004;6:1129–1134. doi: 10.1038/ncb1188. [DOI] [PubMed] [Google Scholar]
- [35].Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer. 2004;4:966–977. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- [36].Ozawa K, Tsukamoto Y, Hori O, Kitao Y, et al. Regulation of tumor angiogenesis by oxygen-regulated protein 150, an inducible endoplasmic reticulum chaperone. Cancer Res. 2001;61:4206–4213. [PubMed] [Google Scholar]
- [37].Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]