

Methanotrophs consume methane as their major carbon source and play an essential role in the global carbon cycle by limiting escape of this greenhouse gas to the atmosphere1–3. These bacteria oxidize methane to methanol via soluble (sMMO) and particulate (pMMO) methane monooxygenases1–4. sMMO contains three protein components, a 251 kDa hydroxylase (MMOH), a 38.6 kDa reductase (MMOR), and a 15.9 kDa regulatory protein (MMOB) required to couple electron consumption with substrate hydroxylation at the catalytic diiron center of MMOH2. Until now, the role of MMOB has remained ambiguous owing to lack of atomic-level information about the MMOH-MMOB (hereafter H-B) complex. Here we remedy this deficiency by providing a crystal structure of H-B, which reveals the manner by which MMOB controls the conformation of residues in MMOH critical for substrate access to the active site. MMOB docks at the α2β2 interface of α2β2γ2 MMOH and triggers simultaneous conformational changes in the α-subunit that modulate O2 and CH4 access as well as proton delivery to the diiron center. Without such careful control by MMOB of these substrate routes to the diiron active site, the enzyme operates as an NADH oxidase rather than a monooxygenase5. Biological catalysis involving small substrates is often accomplished in nature by large proteins and protein complexes. The structure presented in this work provides an elegant example of this principle.
Bacterial multicomponent monooxygenases (BMMs) are unique among diiron proteins by virtue of their ability to hydroxylate a broad spectrum of hydrocarbon substrates1–3. Soluble methane monooxygenases (sMMOs), alkene monooxygenases (AMOs), phenol hydroxylases (PHs), and alkene/aromatic monooxygenases (TMOs) belong to the BMM superfamily1,2,4. sMMO is the only BMM capable of catalyzing the conversion of methane selectively to methanol by activation of O2 for insertion of an oxygen atom into a C–H bond (104.9 kcal/mol), as illustrated in eq 12,3. The crystal
| (1) |
structure of MMOH revealed a dimeric (α2β2γ2) architecture with a glutamate-bridged diiron active site in each α-subunit6,7. Substrate turnover in sMMO is initiated by electron transfer from MMOR to the resting state diiron(III) hydroxylase MMOHox, converting it to the reduced diiron(II) state, MMOHred. In the presence of MMOB, oxygen activation at the active site of MMOHred yields a diiron(III) peroxo intermediate that rapidly decays to form Q, the diiron(IV) species that performs methane oxidation, returning the enzyme to the resting state8,9. Comparison of oxidized and reduced hydroxylase structures revealed charge neutral active sites, reduction being accompanied by conversion of two bridging OH− ions to water (Supplementary Fig. 1)6,7,10. When MMOB binds MMOH, the (FeIII)2 → (FeII)2 reduction potential is lowered, but only in the absence of MMOR11. Binding of MMOB to MMOH increases the rate and specificity of substrate hydroxylation12–14. The nature of the internal MMOH conformational changes induced by MMOB has remained unknown owing to the absence of structural information about the complex formed between these two component proteins.
A crystal of H-B that diffracted to 2.9 Å resolution allowed us to determine the X-ray structure by molecular replacement, as outlined in Methods Summary and Supplementary Table 1. There are two H-B complexes in the asymmetric unit comprising four crystallographically independent αβγB protomers (Supplementary Fig. 2). Within individual dimers, the protomers are related by a non-crystallographic twofold symmetry axis (Fig. 1a and Supplementary Fig. 3) and have nearly identical overall structures (Supplementary Table 2). MMOB binds to the hydroxylase with its core residues (Asp 36 ~ Leu 129) located primarily in a `canyon' region7 formed at the α2β2 interface of the two MMOH protomers. Similar canyon motifs occur in the hydroxylase components of phenol hydroxylase (PH) and toluene-4-monooxygenase (T4MO) for binding their respective regulatory proteins15–17, but these proteins lack the N-terminal tail that is critical for the function of sMMO (Supplementary Fig. 4). Proof that MMOB binds in the canyon of MMOH, the archetypal and most investigated member of the BMM family, and the structure and function of the MMOB N-terminus, are provided for the first time by the present structure determination.
Figure 1. MMOB induces conformational changes that affect function.
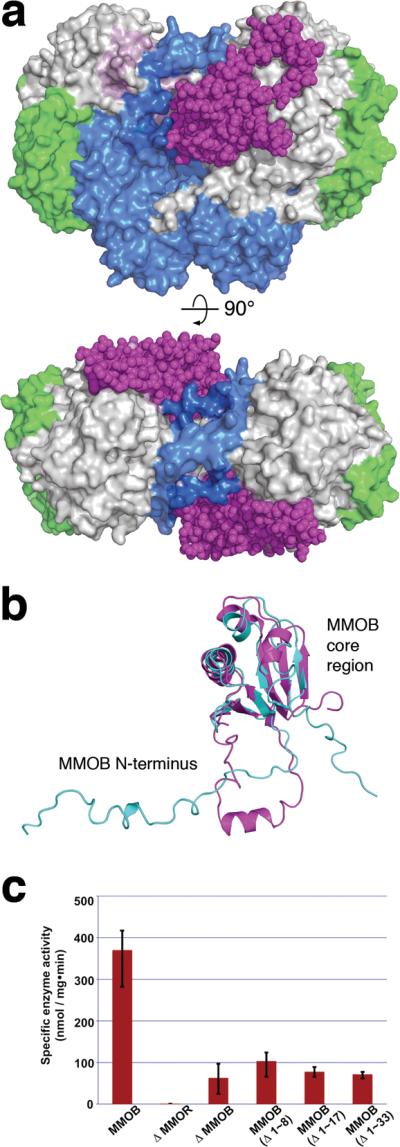
(a) Front (upper) and top (lower) views of a space-filling representation of the H-B complex. MMOB (magenta) binds to the canyon region formed by the α- (grey) and β- (blue) subunits of MMOH. The MMOH γ-subunit is depicted in green. (b) Structural alignment of the solution NMR structure of MMOB from M. capsulatus (Bath) (PDB Code: 1CKV) (cyan) with that of MMOB in the H-B complex (magenta). An α helix (Gly 17-Phe 25) forms in the MMOB N-terminus upon complexation with MMOH. (c) sMMO activity assay with wild type and truncated versions of MMOB. Propylene is converted to propylene oxide in the presence of NADH. Native MMOB is required for maximum sMMO activity. N-terminal truncated MMOB constructs tested, (Δ1–8), (Δ1–17), and (Δ1–33) show activity profiles similar to that observed in the absence of MMOB (n = 3, mean values ± deviation).
NMR spectroscopic analysis18 of unbound MMOB from M. capsulatus (Bath) revealed a compact core region (Fig. 1b) and an unstructured N-terminal tail that is ~35 amino acids longer than the corresponding region of regulatory proteins from all other BMM subclasses (Supplementary Fig. 4). In the H-B complex, the MMOB core exhibits only minor structural changes with respect to that in the unbound protein, as reflected by Cα root-mean-square-deviation values of ~2.1 Å. The N-terminus of MMOB becomes very well ordered in H-B, forming a remarkable ring-shaped structure on the α-subunit of MMOH (Figs. 1a and 1b). The extended N-terminus in MMOB was previously noted to be critical for sMMO catalysis19,20, and those results are confirmed in the present study, wherein N-terminal truncates (Δ 1–8, Δ 1–17, and Δ 1–33) displayed substantially reduced activity with respect to full length MMOB (Fig. 1c).
The MMOB N-terminus binds to helices H and 4 of MMOH in the complex through hydrogen-bonding as well as hydrophobic interactions (Supplementary Fig. 5). A small α helix (Gly 17-Phe 25) in the MMOB tail facilitates formation of its ring-shaped structure on the MMOH surface (Fig. 1a and Supplementary Fig. 5). Within this ring structure, Phe residues 20, 24, and 25 of MMOB generate hydrophobic interactions with Lys 303 (helix H), Val 302, and Tyr 340 (helix 4 of MMOH), respectively, as shown in Supplementary Fig. 5. These features of the N-terminus may help anchor MMOB on the MMOH surface, making it difficult for MMOR to displace it from a preformed H-B complex. Such a consequence would account for the diminished rate of intermolecular electron transfer observed between MMOR and MMOH in preformed H-B21. In addition, hydrophilic residues including Lys 18, Asp 19, Asp 22, and Gln 23 of MMOB, located opposite the H-B binding interface, contribute to the solubility of the H-B complex.
Additionally, when MMOB docks onto the α-subunit of MMOH, it imparts important conformational changes in the hydroxylase. These structural changes are largely confined to the α-subunits and involve particularly helices E, F, H, and 4 (Supplementary Figs. 6 and 7). In the H-B complex, Tyr 8 and Ser 111 of MMOB allosterically induce significant amino acid side chain movements near the diiron active site in MMOH helix E. Tyr 8 forms hydrogen bonds with Arg 307 and Glu 299 in MMOH helix H reorienting Trp 308 (Figs. 2a and 2b). This reorientation of Trp 308 is stabilized by π-interactions with Tyr 76 and Trp 78 (β3 strand) of MMOB. In addition, Ser 111 of MMOB forms a hydrogen bond with Asn 214 in MMOH helix E, which triggers a side chain reorientation in Thr 213, an active site, second coordination sphere residue of importance for the formation of oxygenated intermediates in the catalytic cycle2,3,9 and possibly proton-coupled electron transfer. In H–B, the conformational change of Thr 213 generates hydrogen bonds with Glu 240. This event closes a pore in the MMOH structure, the shortest access route between the diiron active site and the protein surface defined by residues Glu 240, Thr 213 and Asn 214 (Figs 2c, d and 3). This pore was previously proposed to be involved in proton transfer22.
Figure 2. Conformational changes near the diiron center and pore residues in MMOH upon MMOB binding.

In both (a) and (b), protein backbones are shown as ribbons in black (MMOHox), grey (MMOH from the H-B complex), and magenta (MMOB from the H-B complex). Iron atoms are depicted as black (MMOHox) or grey (H-B) van der Waals spheres. Interactions between key residues at the protein-protein interface are depicted as sticks; carbon atoms are colored to match to the protein backbone from which they stem, nitrogen atoms are shown in blue, and oxygen atoms in red; hydrogen bonds are represented as green dashes. Conformational changes in the MMOH pore residues upon MMOB binding are illustrated in MMOHox (c) and H-B (d). Iron-ligating helices B, C, E, and F and iron atoms are shown as black (c) and grey (d) ribbons and MMOB as magenta ribbons. Active site side chain ligands are shown as sticks in grey. Residues Thr 213 (green), Asn 214 (cyan), and Glu 240 (yellow) in MMOH, and Ser 111 in MMOB (magenta), are rendered as sticks. Nitrogen and oxygen atoms are shown in blue and red, respectively. The blue spheres in (c) are water molecules or hydronium ion. 2Fo-Fc electron density at 1.0 sigma from the H-B structure is drawn as a light blue mesh about key residues. The Glu 240 carboxylate side chain may function to deliver protons from solvent to the diiron site while closing the pore (see text).
Figure 3. Pore closure and cavity opening upon MMOH-MMOB complex formation.

Views of cavities 1 (green), 2 (cyan), and 3 (magenta), and the pore region (orange) are shown as translucent van der Waals surfaces in the hydroxylase interior. Protein residues that contribute to the interior surfaces are shown as sticks and colored to match the surfaces to which they primarily contribute. Active site iron atoms are depicted as grey spheres. MMOH from the MMOHox (PDB Code: 1MTY) is shown in (a), and the H-B complex is depicted in (b). The pore region in the hydroxylase interior becomes completely occluded by structural rearrangements that occur in the iron-ligating helices E and F, and in residues Asn 214 and Glu 240 in particular, upon complex formation with the regulatory protein. Cavity-gating residue Phe 188 also changes orientation as a consequence of the regulatory protein binding-induced rearrangements in helices E and F, and as a result cavities 1 and 2 (cyan/green interface) merge. Hydrophobic cavities 2 and 3 in MMOH have sufficient space to accommodate the binding and translocation of these two gaseous substrates.
Protons are an important substrate in BMM catalytic cycles9,22–24, and the H-B structure provides insight into the role that MMOB may have in facilitating proton access to the catalytic diiron center in soluble MMO. In the H-B complex, the conformational change of Thr 213 is accompanied by formation of a bifurcated hydrogen bond between the hydroxyl group of this residue and the carboxylate side chain of Glu 240. Glu 243 simultaneously undergoes a `carboxylate shift'25 (vide infra). In the absence of MMOB, the Glu 240 and Asn 214 side chains in MMOH are solvent accessible and linked through hydrogen bonding to a water molecule or hydronium ion (Fig. 2c). Upon H-B complex formation (Fig. 2d), Glu 240 shifts toward the protein interior possibly delivering a proton in the process. One possible scenario is that, during the O2 activation steps by MMOH to form the peroxo and Q intermediates, both of which require a proton transfer,9,23 MMOB core binding, release, and rebinding to the hydroxylase might facilitate delivery of the requisite two solvent-derived protons through the pore. The anchoring of MMOB by its N-terminus may allow the core to function in this manner without complete dissociation of the regulatory protein from the hydroxylase. The presence of these protons in the active site would also facilitate product and hydroxide ion release during reduction of the diiron(III) center in MMOHox to form MMOHred. Delivering protons through the pore may be one of the primary functions of the regulatory proteins in the BMM family.
Yet another important feature of MMOB binding to the α-subunit of MMOH is to control methane and O2 access to the active site. Previous structural analyses of MMOH crystals soaked in solutions of halogenated substrate analogs or pressurized with Xe identified a putative access route for these substrates26,27. In the MMOHox structure, cavities 2 and 3 are connected, but there is a discontinuity between cavities 1 and 2. This break in freely diffusible space blocks access of methane and oxygen to the active site (Fig. 3a and Supplementary Fig. 8). Molecular access to the diiron site via the cavities is gated by residues Phe 188 and Leu 1107,22,26. In the H-B complex, cavities 1 and 2 become connected as a consequence of a change in conformation of the Phe 188 side chain, and a structural comparison of MMOHox with that of H-B highlights the difference (Fig. 3 and Supplementary Figs. 8 and 9). A major function of MMOB binding to MMOH is, therefore, to facilitate methane and O2 access to the diiron active site by opening the gate. It is noteworthy that this structural alteration occurs concomitantly with closure of the pore (Fig. 3 and Supplementary Figs. 8 and 9). Opening the pore upon MMOB dissociation also supports its previously proposed role as a hydrophilic route for methanol release26.
Changes also occur in the geometry of the diiron center upon MMOB binding, in accord with spectroscopic studies that revealed conformational rearrangements of coordinated amino acid side chains in the H-B complex2,3,9,14. The coordination environments of the iron atoms in H-B (Fig. 4a and Supplementary Fig. 10) exhibit many similarities to, as well as some key differences from, those observed in MMOHox and MMOHred (Figs. 4b and 4c and Supplementary Fig. 10)7,10,28. As in the other structures of MMOH, Fe1 and Fe2 in H-B are positioned within the four-helix bundle formed by helices B, C, E, and F. The positions of helices E and F shift upon MMOB binding, moving Fe2 ~1.1 Å from its location in MMOHox (Supplementary Fig. 11). The coordination of Glu 243 resembles that in Hred, but the Fe⋯Fe distance in H-B is closer to that in MMOHox.
Figure 4. Coordination geometry at the diiron active site of MMOH.
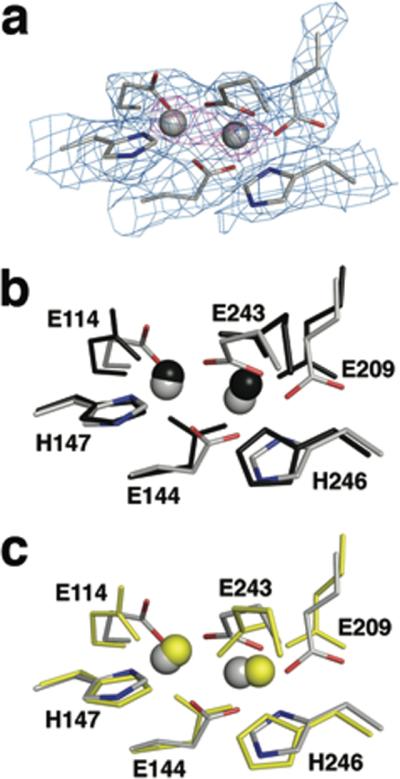
(a) Electron density at the diiron site in H-B. Side chain ligands are shown as sticks in grey (carbon), blue (nitrogen), and red (oxygen). 2Fo-Fc electron density at 1.0 sigma and 5.0 sigma is drawn as a mesh in light blue and magenta, respectively. Views comparing the diiron site in H-B (grey) with that in MMOHox (b, PDB code: 1MTY, black) and in MMOHred (c, PDB code: 1FYZ, yellow) are also presented in (b) and (c), respectively. Upon MMOB binding, Glu 243 undergoes a substantial conformational change, involving simultaneous chelation of Fe2 and bridging to Fe1. Such bidentate coordination of the Glu 243 side chain resembles that in MMOHred, but distances between the carboxylate oxygen atoms and Fe2 (OE1-Fe2 and OE2-Fe2) are shorter in H-B (1.9 and 2.0 Å) than in MMOHred (2.4 and 2.4 Å). This result is more consistent with an Fe(III) than an Fe(II) oxidation state for the iron atoms in H-B. Solvent-derived ligands, such as hydroxide ion and water, are not observed in the H-B complex owing to the 2.9 Å resolution.
Individual refinement of crystallographically independent diiron active sites within the four protomers revealed the same coordinated ligands, although with slightly different geometries (Supplementary Fig. 12). The Fe1 and Fe2 ions bond to the δ-N atoms of His 147 and His 246, respectively, Glu 144 bridges the two metals, and Glu 209 binds in a monodentate fashion to Fe2, all as in MMOHox structures (Fig. 4 and Supplementary Figs. 10 and 12). The most notable change occurs in the Glu 243 side chain carboxylate, which chelates Fe2 in a bidentate manner while being singly bonded to Fe1. In MMOHox, the carboxylate of Glu 243 forms a single bond with Fe2, and the dangling oxygen atom hydrogen bonds to a terminal water coordinated to Fe1 and a hydroxide ion bridging Fe1 and Fe2. The MMOHox active site contains a water molecule terminally bound to Fe1 that is not observed in H-B, either because it cannot be distinguished at 2.9-Å resolution or because it is not present in H-B structure.
In conclusion, the present structure reveals how the MMOB regulatory protein controls substrate access to the diiron center in the sMMO hydroxylase. Docking of the MMOB core in the MMOH canyon is accompanied by ordering of its long N-terminal tail on the α-subunit of the hydroxylase, triggering allosteric changes that control proton, methane, and oxygen access to the active site. The timed entry of these substrates is important to assure events required for MMOR conversion of MMOHox to MMOHred and the generation of oxygenated intermediates that react with methane during the critical oxygen activation and substrate hydroxylation steps of the catalytic cycle. In this manner, MMOB can function to couple the consumption of electrons with efficient hydrocarbon hydroxylation. Finally, the present results can be used as a leading example of the use by nature of a large protein complex to delineate access pathways of some of its smallest substrates to the active site of a metalloenzyme to achieve a remarkable catalytic reaction.
Methods Summary
M. capsulatus (Bath) cultures were grown by fermentation and MMOH was purified as described previously9. Recombinant full-length and truncated MMOB proteins were expressed and purified from Escherichia coli as described29, 30. Crystallization, crystal structure determination, and enzyme activity studies with the MMOB N-terminal deletion mutants were performed as described in Methods. Data collection was performed at the Advanced Light Source beamline 8.2.2. at Lawrence Berkeley National Laboratory and the structure was determined by molecular replacement using the program “Phaser” with MMOHox (PDB Code: 1MTY) and MMOB (PDB Code: 1CKV) as search models.
Methods
sMMO fermentation and purification of MMOH
M. capsulatus (Bath) cultures were fermented and MMOH was purified via DEAE-sepharose fast-flow, S-300 size exclusion, Q sepharose, and S-200 size exclusion chromatography9,29–32. The final eluent was concentrated to form a pale yellow solution.
MMOB and truncated version of MMOB expression and purification
The wild type and truncated versions of MMOB were prepared recombinantly in Escherichia coli. From a recombinant glycerol stock of native MMOB (pkk223-3-mmoB, JM105) or truncated versions of MMOB (pET22b(+)-mmoB, BL21(DE3)), cells were grown and expressed for 3 hr at 37 °C. The native and truncated regulatory proteins were purified using Q sepharose fast-flow and S-75 size exclusion chromatography to obtain a colorless solution18,19,29–31.
Enzyme activity measurement of MMOH in the presence of the full-length or N-terminal truncated regulatory subunit (MMOB)
MMOH (1.0 μM), MMOB (2.0 μM), and MMOR (0.5 μM) were incubated with propylene in 25.0 mM phosphate buffer at pH 7.0. Steady-state kinetic data were recorded by using an HP8452 diode array spectrophotometer19,31. The temperature was controlled at 45 °C with a circulating water bath. The reaction was initiated by addition of NADH (167.0 μM) in the presence of propylene (approximately 1.0 mM). The consumption of NADH was monitored spectrophotometrically at 340 nm and quantified by using an extinction coefficient of 6,220 M−1cm−1.
Crystallization, data collection, and structure determination
Purified MMOH (α2β2γ2) and MMOB, which was stored in 30 mM HEPES [pH7.5], 100 mM NaCl, and 1 mM TCEP, were mixed with at a 1:2.2 molar ratio and the final concentration was adjusted to ~10 mg/ml. Crystals were grown for one month at 18 °C by the sitting drop vapor diffusion method in 0.1 M MES [pH 6.5] and 15% PEG 20,000 (w/v). Crystals were flash frozen in liquid nitrogen after transferring to a cryo-protectant solution containing the precipitant and 20% glycerol. The crystal in space group P212121, a=183.6, b=249.0, c=122.3, gave recordable diffracted to a minimum Bragg spacing of 2.9 Å at the Advanced Light Source (ALS) beamline 8.2.2. at Lawrence Berkeley National Laboratory. Data were processed using XDS33 and scaled with SCALA34. Molecular replacement computations with MMOHox (PDB code: 1MTY) and MMOB (PDB code: 1CKV) were performed using the program “Phaser”35. Model building and refinement were accomplished using Coot36 and PHENIX.refine37. We generated restraints for iron atoms and ligands using the program “PHENIX” (PHENIX.metal_coordination)37 and applied them during the refinement of the diiron center; no NCS restraints were applied in any stage of the refinement. The final refined model contains MMOH α-subunit (residues 15–526), β-subunit (residues 2–389) and γ-subunit (residues 3–168), and MMOB (residues 2–133) with Rfactor and Rfree values of 20.6 and 25.8 respectively.
Supplementary Material
Acknowledgements
This work was supported by grant GM 32114 from the National Institute of General Medical Sciences to S. J. Lippard. We thank the staff at the Advanced Light Source (ALS) beamline 8.2.2. in Lawrence Berkeley National Laboratory for the data collection, Dr. Stephen C. Harrison for resources and helpful comments on the manuscript, and Mr. T. C. Johnstone, Ms. A. D. Liang, and Dr. T.-T. Lu for helpful discussions.
Footnotes
Author Contributions
S. J. Lee designed experiments, purified proteins, measured the enzyme activity, analyzed data, and wrote the manuscript; M.S.M. analyzed the enzyme cavity, analyzed data, prepared figures, and wrote the manuscript; S. J. Lippard directed the project, designed experiments, analyzed data, and wrote the manuscript; and U-S.C. obtained crystals, solved, and refined the structures, analyzed data, and wrote the manuscript. All authors discussed the results and commented on the manuscripts.
Author Information
Coordinates and structure factors for the crystal structure of the H-B complex have been deposited with the Protein Data Bank under the accession code 4GAM. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiol Rev. 1996;60:439–471. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merkx M, et al. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins. Angew Chem Int Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 3.Wallar BJ, Lipscomb JD. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 4.Leahy JG, Batchelor PJ, Morcomb SM. Evolution of the soluble diiron monooxygenases. FEMS Microbiol Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 5.Gassner GT, Lippard SJ. Component interactions in the soluble methane monooxygenase system from Methylococcus capsulatus (Bath) Biochemistry. 1999;38:12768–12785. doi: 10.1021/bi990841m. [DOI] [PubMed] [Google Scholar]
- 6.Elango N, et al. Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyzes the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 8.Shu L, et al. An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 9.Tinberg CE, Lippard SJ. Revisiting the mechanism of dioxygen activation in soluble methane monooxygenase from M. capsulatus (Bath): Evidence for a multi-step, proton-dependent reaction pathway. Biochemistry. 2009;48:12145–12158. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whittington DA, Lippard SJ. Crystal structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) demonstrating geometrical variability at the dinuclear iron active site. J Am Chem Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 11.Paulsen KE, et al. Oxidation-reduction potentials of the methane monooxygenase hydroxylase component from Methylosinus trichosporium OB3b. Biochemistry. 1994;33:713–722. doi: 10.1021/bi00169a013. [DOI] [PubMed] [Google Scholar]
- 12.Froland WA, Andersson KK, Lee SK, Liu Y, Lipscomb JD. Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions. J Biol Chem. 1992;267:17588–17597. [PubMed] [Google Scholar]
- 13.Liu KE, et al. Kinetic and spectroscopic characterization of Intermediates and component interactions in reactions of methane monooxygenase from Methylococcus capsulatus (Bath) J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 14.Liu Y, Nesheim JC, Lee SK, Lipscomb JD. Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J Biol Chem. 1995;270:24662–24665. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- 15.Bailey LJ, Mccoy JG, Phillips GN, Fox BG. Structural consequences of effector protein complex formation in a diiron hydroxylase. Proc Natl Acad Sci USA. 2008;105:19194–19198. doi: 10.1073/pnas.0807948105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sazinsky MH, Lippard SJ. Correlating structure with function in bacterial multicomponent monooxygenases and related diiron proteins. Acc Chem Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 17.Sazinsky MH, Dunten PW, McCormick MS, DiDonato A, Lippard SJ. X-ray structure of a hydroxylase-regulatory protein complex from a hydrocarbonoxidizing multicomponent monooxygenase, Pseudomonas sp. OX1 phenol hydroxylase. Biochemistry. 2006;45:15392–15404. doi: 10.1021/bi0618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walters KJ, Gassner GT, Lippard SJ, Wagner G. Structure of the soluble methane monooxygenase regulatory protein B. Proc Natl Acad Sci USA. 1999;96:7877–7882. doi: 10.1073/pnas.96.14.7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandstetter H, Whittington DA, Lippard SJ, Frederick CA. Mutational and structural analyses of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) Chem Biol. 1999;6:441–449. doi: 10.1016/s1074-5521(99)80062-3. [DOI] [PubMed] [Google Scholar]
- 20.Chang SL, Wallar BJ, Lipscomb JD, Mayo KH. Residues in Methylosinus trichosporium OB3b methane monooxygenase component B involved in molecular interactions with reduced- and oxidized-hydroxylase component: a role for the N-terminus. Biochemistry. 2001;40:9539–9551. doi: 10.1021/bi0103462. [DOI] [PubMed] [Google Scholar]
- 21.Blazyk JL, Gassner GT, Lippard SJ. Intermolecular electron-transfer reactions in soluble methane monooxygenase: A role for hysteresis in protein function. J Am Chem Soc. 2005;127:17364–17376. doi: 10.1021/ja0554054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCormick MS, Lippard SJ. Analysis of substrate access to active sites in bacterial multicomponent monooxygenase hydroxylases: X-ray crystal structure of xenon-pressurized phenol hydroxylase from Pseudomonas sp OX1. Biochemistry. 2011;50:11058–11069. doi: 10.1021/bi201248b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SK, Lipscomb JD. Oxygen activation catalyzed by methane monooxygenase hydroxylase component: proton delivery during the O-O bond cleavage steps. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 24.Song WJ, et al. Active site threonine facilitates proton transfer during dioxygen activation at the diiron center of toluene/o-xylene monooxygenase hydroxylase. J Am Chem Soc. 2010;132:13583–13585. doi: 10.1021/ja1063795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rardin RL, Tolman WB, Lippard SJ. Monodentate carboxylate complexes and the carboxylate shift: Implications for polymetalloprotein structure and function. New J Chem. 1991;15:417–430. [Google Scholar]
- 26.Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- 27.Whittington DA, Sazinsky MH, Lippard SJ. X-ray crystal structure of alcohol products bound at the active site of soluble methane monooxygenase hydroxylase. J Am Chem Soc. 2001;123:1794–1795. doi: 10.1021/ja0031725. [DOI] [PubMed] [Google Scholar]
- 28.Rosenzweig AC, et al. Crystal structures of the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): Implications for substrate gating and component interactions. Proteins. 1997;29:141–152. [PubMed] [Google Scholar]
- 29.Coufal DE, et al. Sequencing and analysis of the Methylococcus capsulatus (Bath) soluble methane monooxygenase genes. Eur J Biochem. 2000;267:2174–2185. doi: 10.1046/j.1432-1327.2000.01210.x. [DOI] [PubMed] [Google Scholar]
- 30.Valentine AM, Stahl SS, Lippard SJ. Mechanistic studies of the reaction of reduced methane monooxygenase hydroxylase with dioxygen and substrates. J Am Chem Soc. 1999;121:3876–3887. [Google Scholar]
- 31.Beauvais LG, Lippard SJ. Reactions of the peroxo intermediate of soluble methane monooxygenase hydroxylase with ethers. J Am Chem Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 32.Liu KE, Johnson CC, Newcomb M, Lippard SJ. Radical clock substrate probes and kinetic isotope effect studies of the hydroxylation of hydrocarbons by methane monooxygenase. J Am Chem Soc. 1993;115:939–947. [Google Scholar]
- 33.Kabsch W. Xds. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 35.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.