

SUMMARY
Recent studies indicated that sortilin-related receptor 1 (SORL1) is a risk gene for late-onset Alzheimer's disease (AD), although its role in the aetiology and/or progression of this disorder is not fully understood. Here, we report the finding of a non-coding (nc) RNA (hereafter referred to as 51A) that maps in antisense configuration to intron 1 of the SORL1 gene. 51A expression drives a splicing shift of SORL1 from the synthesis of the canonical long protein variant A to an alternatively spliced protein form. This process, resulting in a decreased synthesis of SORL1 variant A, is associated with impaired processing of amyloid precursor protein (APP), leading to increased Aβ formation. Interestingly, we found that 51A is expressed in human brains, being frequently upregulated in cerebral cortices from individuals with Alzheimer's disease. Altogether, these findings document a novel ncRNA-dependent regulatory pathway that might have relevant implications in neurodegeneration.
INTRODUCTION
Sortilin-related receptor 1 (SORL1; also known as sorting protein-related receptor 1 and LR11) is a 250-kDa type-1 membrane protein of unknown function that is expressed in neurons of the central and peripheral nervous system (Jacobsen et al., 1996; Yamazaki et al., 1996; Hermans-Borgmeyer et al., 1998; Motoi et al., 1999). Although its function is still poorly understood, SORL1 is a member of a family of neuronal receptors that share structural similarity with yeast vacuolar protein sorting 10 protein (Vps10p), a sorting-specific polypeptide that transports carboxypeptidase Y from the Golgi to vacuoles (Marcusson et al., 1994). Interestingly besides its potential role in certain cancer pathways (Akil et al., 2011; Demont et al., 2012), SORL1 expression is reduced in brain tissues from individuals with Alzheimer's disease (AD) (Ma et al., 2009), suggesting a potential role in AD pathogenesis (Rogaeva et al., 2007; Reitz et al., 2011). The link between SORL1 and AD has been further strengthened by the recent demonstration that reduction of SORL1 expression promotes an increase of neurotoxic β-amyloid peptide (Aβ) formation by a mechanism that has been only partially elucidated (Andersen et al., 2005; Small et al., 2005;
Offe et al., 2006; Wang et al., 2007). Indeed, the initial processing of amyloid precursor protein (APP) by α- and β-secretases is intimately associated with post-Golgi compartments and requires efficient transition of the precursor through these organelles (Haass et al., 1993; Yamazaki et al., 1995). In this context, SORL1 interacts with APP and affects its trafficking and proteolytic processing in the brain, acting as a sorting receptor for APP holoprotein. By contrast, the absence or downregulation of SORL1 expression shifts APP holoprotein from the retromer recycling pathway to the β-secretase cleavage pathway, increasing secreted APPα (sAPPβ) production and, subsequently, Aβ formation (Peraus et al., 1997; Khvotchev and Südhof, 2004).
The link between SORL1 and AD was also supported by the identification of AD-associated allelic variants in distinct regions of the SORL1 gene in different populations. These results also suggested that these variants might map in still-unknown intronic regulatory regions that might govern cell-type- or tissue-specific expression of SORL1 (Bruni et al., 2007; Hinerfeld et al., 2007; Klein et al., 2007; Lee et al., 2007; Matsui et al., 2007; Rogaeva et al., 2007; Shibata et al., 2007; Lee et al., 2008; Xiao et al., 2008; Massone et al., 2012). Thus, expression of these variants might affect AD risk by altering the physiological role of SORL1 in the processing of APP holoprotein (Schmidt et al., 2012).
In recent works, we have documented pivotal roles of pol-III-transcribed non-coding (nc) RNAs in gene expression regulation and, in particular, in the regulation of alternative splicing (Dieci et al., 2007; Pagano et al., 2007; Castelnuovo et al., 2010; Massone et al., 2011a; Massone et al., 2011b; Vella et al., 2012). A still uncharacterized transcription unit of our collection (hereafter referred to as 51A) maps to intron 1 of the SORL1 gene (a genomic portion subjected to alternative splicing events) in antisense configuration. Thus, we hypothesised a possible control of SORL1 pre-mRNA maturation mediated by 51A ncRNA expression. According to this working hypothesis, the synthesis of this ncRNA and its possible RNA:RNA pairing with SORL1 pre-mRNA would mask canonical splicing sites, leading to alternative splicing events. By addressing such a hypothesis, in this work we demonstrate that: (i) 51A is a newly identified ncRNA whose synthesis promotes the expression of SORL1 alternatively spliced protein variants to the detriment of the canonical SORL1 splice variant A; (ii) this event triggers an altered processing of APP that leads to its impaired internalisation; (iii) this process ultimately leads to increased amyloid secretion; and (iv) 51A is upregulated in post-mortem cerebral cortices from individuals with AD.
TRANSLATIONAL IMPACT.
Clinical issue
The mechanisms that cause Alzheimer's disease (AD) are still unclear. Rare familial forms of AD are clearly linked to mutations in a few genes (mainly involved in amyloidosis, such as those encoding APP and presenilins), but the genesis of sporadic forms is still obscure. There is therefore great interest in obtaining a deeper comprehension of disease mechanisms, and in particular the tightly regulated molecular events whose dysfunction is associated with neuronal death in AD. Recent data indicate that newly identified ncRNAs are overexpressed in the brains of AD subjects, where they modulate alternative splicing events that ultimately regulate amyloid formation. In this study, the authors investigated a previously uncharacterised ncRNA that maps within SORL1, a gene that encodes a receptor for apolipoprotein E (a well-known risk factor for late-onset AD) and that has been associated with AD. Allelic variants of SORL1 have recently been associated with AD, and preliminary results suggest that the function of SORL1 is reduced in the disease.
Results
Here, the authors describe a new ncRNA (named 51A) that maps in an antisense orientation in intron 1 of the SORL1 gene and whose synthesis promotes the expression of alternatively spliced SORL1 variants. The expression of 51A was detected in normal human brain samples, and was significantly overexpressed in AD brain. In vitro, 51A reduced the expression of SORL1 variant A while increasing the amounts of alternative isoforms. The expression of 51A also induced a significant increase in amyloid formation in cultured cells. Finally, the authors report a modest increase in the expression of 51A in post-mortem AD brains.
Implications and future directions
Based on their results, the authors hypothesise that abnormal regulation of SORL1 by the ncRNA 51A might increase susceptibility to AD by increasing amyloid formation via downregulating SORL1 variant A. This work reveals a new mechanism by which an AD-associated gene might be regulated, and highlights the importance of ncRNAs in regulating amyloid formation and potentially neurodegeneration. Knowledge about ncRNA is limited, so further studies of the role of different ncRNAs in AD susceptibility are needed. Pursuing this issue could elaborate more details of the complex mechanisms involved in this disease and could lead to the identification of new therapeutic targets.
RESULTS
51A is a newly identified PSE/DSE-dependent transcriptional unit
Alternative splicing variants of SORL1 with distinct biochemical properties have been reported (see supplementary material Fig. S1), although the regulation of this phenomenon is still obscure. We have recently identified in silico 51A, a predicted transcriptional unit that maps to intron 1 of the SORL1 gene in antisense configuration (Dieci et al., 2007; Pagano et al., 2007). Because intronic RNAs transcribed by RNA polymerase III (pol III) might regulate the splicing of protein-coding genes that are involved in AD (Massone et al., 2011a; Massone et al., 2011b), we hypothesised that the transcription of 51A might interfere with the maturation of SORL1 pre-mRNA, leading to the occurrence of alternative splicing events that are potentially interesting for AD studies.
To test this hypothesis we first established the transcriptional activity of the 51A promoter by co-transfecting SKNBE2 neuroblastoma cells with a plasmid construct expressing a luciferase-silencing hairpin driven by the 51A promoter (hereafter referred to as pSHAG-51A) together with a plasmid expressing luciferase (pGL3). In this condition, if the 51A promoter is active, the transcription of the hairpin drives the post-transcriptional silencing of a co-transfected luciferase cDNA and the decrease of its signal; by contrast, an unaltered luminescent signal indicates that luciferase is not silenced because the 51A promoter is not active.
A reduced luminescence signal was detected in pSHAG-51A-transfected cells 48 hours after transfection (0.4±0.008 as normalised to cells transfected with pGL3 alone) as a consequence of efficient transcription of the silencing hairpin driven by the 51A promoter. In the same experiment, two well-assessed pol III type 3 promoters (U6 and H1 snRNA promoters) were used as positive controls driving the active transcription of the silencing hairpin, whereas a promoterless construct [named No Promoter (NP)] was referred to as the negative control because, as expected, it did not influence luciferase expression (Fig. 1A,B). Altogether, these experiments demonstrate the transcriptional activity of the 51A pol III type 3 promoter.
Fig. 1.

The 51A transcription unit is actively transcribed in SKNBE2 cell nuclear extracts. (A) Promoter activity transfection assay in human SKNBE2 cells. A specific luciferase-silencing hairpin is transcribed by the 51A PSE/DSE-dependent promoter. The promoter region encompasses the putative pol-III type 3 regulatory regions (TATA box, PSE and DSE). pGL3/pRL, negative control; pShag-NP, pShag-No promoter negative control; pShag-U6, positive control; pShag-H1, positive control; pShag-51A, sample of interest. (B) Sequence of the transcription unit. The PSE is in bold red and underlined, as is the putative T4 termination signal. A TATA-like element is also underlined. (C) In vitro transcription of plasmid-borne 51A (lane 3) or empty pNEB193 plasmid (lane 2) was carried out in a nuclear extract from SKNBE2 cells. The main 51A-specific transcripts are indicated by arrowheads on the right. The migration position of a 300-nt RNA size marker is indicated on the left. Lane 1, no DNA. (D) Primer extension analysis was conducted on unlabelled RNA products of in vitro transcription reactions programmed with no DNA (lane 1), empty pNEB193 plasmid (lane 2) or pNEB193-51A (lane 3). Shown in lanes 4-7 are the results of sequencing reactions conducted with the same 5′-labelled oligonucleotide utilised for primer extension. The sequence of the non-transcribed DNA strand around the TSS (+1) is indicated on the right. The position of the main, 51A-specific primer extension product is indicated by an arrowhead on the left.
Next, to more directly test 51A RNA synthesis, we performed in vitro transcription of the 51A template, using a nuclear extract from SKNBE2 cells. As shown in Fig. 1C, two main 51A-specific transcripts were produced. The shorter transcript approximately co-migrated with a 300-nucleotide (nt) RNA size marker. Such a transcript size was compatible with transcription initiating downstream of the proximal sequence element (PSE) (but upstream of the putative TATA-like element) and terminating at a run of four Ts ( T4) located ~300 bp downstream (evidenced in Fig. 1B). The predominant 51A transcription product, however, was a longer transcript, whose size could be explained by pol III read-through at the T4 signal followed by termination at a downstream-located termination signal (Fig. 1B). The transcription start site (TSS) of 51A was also investigated by primer extension analysis conducted on in vitro-produced, unlabelled transcripts. In both SKNBE2 (Fig. 1D) and HeLa (not shown) cell nuclear extracts, a single predominant extension product, corresponding to transcription initiation at an A residue located 33 bp downstream of the start of the PSE, was observed. Such a short distance between PSE and the TSS is unusual, suggesting the possibility that other, still-uncharacterized cis-acting elements might influence 51A TSS selection.
51A transcription drives SORL1 alternative splicing
In order to test the possible influence of 51A synthesis on SORL1 pre-mRNA splicing, we measured by real-time RT-PCR the expression of 51A RNA and SORL1 splicing variants in different cell lines in order to identify an in vitro cellular model expressing both 51A RNA and SORL1 variants. Results show that, with the exception of HeLa cells and NCTC-2544 (human skin keratinocyte), all examined cells express 51A RNA (Fig. 2A). Similarly, SORL1 variant A showed a rather ubiquitous expression profile, whereas the two alternatively spliced forms of SORL1 [variants B and F (see supplementary material Fig. S1 for details)] are specifically expressed in SKNBE2 cells and are undetectable in all the other cell lines (Fig. 2B-D). Therefore, because only the SKNBE2 cell line simultaneously expresses A, B and F protein variants, we used this cell line as a model to test possible variations of the relative amounts of these variants (by the means of protein variants ratio) induced by the overexpression of 51A ncRNA.
Fig. 2.

The overexpression of 51A ncRNA leads to SORL1 alternative maturation. (A) 51A, (B) SORL1 variant A, (C) SORL1 variant B and (D) SORL1 variant F RT-PCR expression profile in different cell lines. (E) RT-PCR expression analysis of 51A ncRNA and (F) SORL1 variant A in 51A-transfected SKNBE2 cells and/or pMock controls; ** indicates statistical significance at P-values ≤0.01. (G) Western blot expression analysis of SORL1 variant A and B (or F) in 51A-transfected SKNBE2 cells. (H,I) Immunofluorescence detection of SORL1 alternatively spliced protein products. (H) Antibodies raised against the N-terminus of SORL1 protein form A show a clearly detectable signal in GFP-expressing cells only in the absence of a concomitant 51A overexpression, demonstrating the synthesis in this condition of the canonical protein form (1, GFP; 2, DAPI, 3, Tritch; 4, merge); three independent microscope fields are reported. (I) SORL1 variant A signal is absent or very weak in cells transfected with a construct co-expressing 51A and GFP: in this case, the transient overexpression of the ncRNA leads to the synthesis of the alternative forms of SORL1 endowed with a peculiar N-terminal portion that is not recognised by the same IgGs (1, GFP; 2, DAPI, 3, Tritch; 4, merge); three independent microscope fields are reported. Untransfected cells, which do not express GFP or 51A, are positive for SORL1, in accordance with a 51A-dependent change in the protein variant.
Next, we tested whether the synthesis of 51A RNA might drive the splicing shift of SORL1. To this aim we transiently transfected SKNBE2 cells with a plasmid construct harbouring the whole 51A transcriptional unit (p51A-EGFPN1) and measured by real-time RT-PCR the amount of the individual SORL1 splicing products (variants A, B and F). We found that, 48 hours after transfection, the increased synthesis of 51A RNA is accompanied by a strong decrease of the SORL1 splice variant A mRNA content [up to 5% of the original level (P=0.0002)] (Fig. 2E,F).
Next, taking advantage of a SORL1-specific antiserum raised against the C-terminal portion of the protein (that potentially recognises all the protein variants), we tested by western blotting whether the decrease of SORL1 variant A transcription corresponds to a reduced amount of SORL1 protein. Results showed that an immunoreactive band of about 270 kDa (a size corresponding to the long canonical variant A) is significantly reduced (up to 14% of the original level) in 51A-overexpressing cells, confirming, at protein level, that the expression of 51A ncRNA favours the downregulation of the synthesis of SORL1 protein form A. Interestingly, in the same protein extract, a SORL1 immunoreactive band at about 110 kDa was upregulated in 51A-overexpressing cells, although at present it is not possible to ascribe its signal specifically to either variant B or variant F (Fig. 2G).
Next, to further prove that the downregulation of SORL1 variant A is specifically induced by the synthesis of 51A RNA, we took advantage of a different SORL1-specific polyclonal antiserum raised against the N-terminal portion of splice variant A, thus ineffective to recognise the alternative protein forms B and F. By immunofluorescence microscopy we detected a marked signal of SORL1 in pMock-transfected cells; in this condition, 51A RNA is not overexpressed and the SORL1 variant A is predominantly synthesised and recognised by this antiserum (Fig. 2H). By contrast, a strongly decreased signal was detected in cells overexpressing 51A RNA (Fig. 2I). In this case, although we were unable to detect the increased synthesis of the alternative protein forms B and F (they harbour a different N-terminal portion and are thus not recognised by the antiserum), we found that overexpression of 51A RNA leads to a decreased amount of SORL1 variant A, which, as a consequence, is only barely detectable. In the same microscope fields, untransfected cells, which do not express GFP and 51A, were positive for SORL1, in accordance with a 51A-dependent change in the protein biochemical variant.
Altogether, the above results demonstrate that the synthesis of 51A RNA leads to the splicing shift of SORL1.
The 51A-dependent alternative splicing of SORL1 leads to the impairment of Aβ secretion
Because it has been demonstrated that the downregulation of SORL1 leads to the increase of Aβ formation, we hypothesised that the ability of 51A to specifically limit the synthesis of SORL1 variant A might also represent an upstream control of Aβ secretion. To address this hypothesis without the technical limitations imposed by the transient transfection procedure, we generated a transgenic SKNBE2 cell line permanently transfected with 51A expression plasmid (hereafter referred to as SKNBE2-51A). As a negative control we generated a SKNBE2 cell line permanently transfected with the empty vector (SKNBE2-Mock). In order to preliminarily characterise this experimental model, we measured, by real-time RT-PCR, the expression of 51A RNA and, by western blotting, the extent of SORL1 protein variant A downregulation. SKNBE-51A cells expressed 51A RNA at a 6.3-fold higher level than did SKNBE2-Mock cells and showed a parallel significant decrease in SORL1 protein content (P=0.0027) (Fig. 3A,B). To evaluate the effects of 51A expression on the different SORL1 isoforms that cannot be discriminated by the antibody used in the western blot experiments, we analysed the amount of different SORL1 mRNA variants in 51A-overexpressing cells by real-time RT-PCR. We found a decrease of SORL1 variant A mRNA (P=0.0017) and a concomitant increase of variants B (P=0.003) and F (P=0.001) mRNAs in SKNBE2-51A cells as compared with SKNBE2-Mock cells, thus demonstrating that a prolonged overexpression of 51A RNA leads to the stable shift of SORL1 splicing from the long form A to the alternative forms B and F (Fig. 3C).
Fig. 3.
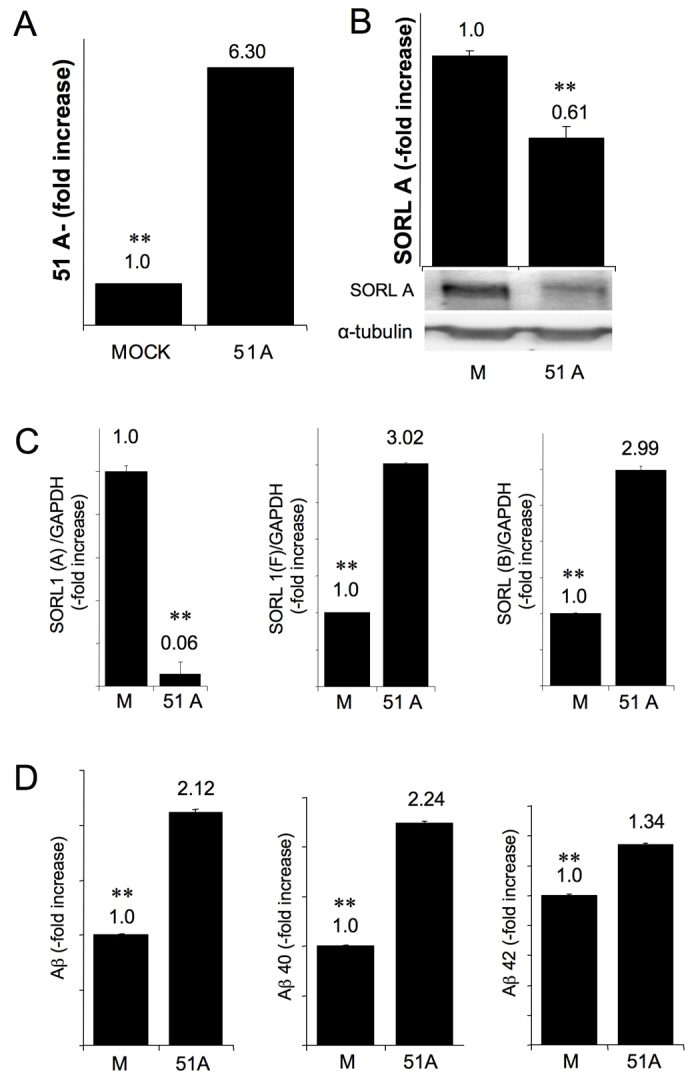
51A ncRNA overexpression and the consequent alternative splicing of SORL1 drive an impairment of amyloid release. (A) RT-PCR analysis of 51A ncRNA expression in 51A permanently transfected SKNBE cells. (B) SDS-PAGE expression analysis of SORL1 variant A in 51A permanently transfected SKNBE cells. (C) RT-PCR analysis of SORL1 variant A, B and F in 51A permanently transfected SKNBE cells. (D) Increased secretion of total Aβ, Aβ x-40 and Aβ x-42 in 51A-overexpressing SKNBE2 cells. x-axis: transfected plasmids. y-axis: quantitative determination of Aβ (pg/ml) secreted in the medium 48 hours after medium replacement as determined by sandwich ELISA (results were normalised to the pMock-transfected cell line).
Considering that proteolytic processing of APP plays a central role in AD aetiology through the formation of Aβ (Johnson et al., 1990; Selkoe, 1990) and that this processing is influenced by the downregulation of SORL1, we investigated whether the stable overexpression of 51A in SKNBE2 cells would affect the formation of Aβ. Hence, we measured by ELISA the relative amount of Aβ peptides ending at amino acid position 42 (Aβ x-42) and at position 40 (Aβ x-40) in the culture medium conditioned for 48 hours by SKNBE2-Mock or SKNBE2-51A cells. We found that the amount of both Aβ x-42 and Aβ x-40 is increased in SKNBE2-51A cells as compared with mock-transfected cells (1.35-fold, P=0.004 and 2.24- fold increase, P=0.001, respectively; Fig. 3D). Therefore, these results demonstrate that, as expected, the overexpression of 51A and the consequent downregulation of SORL1 protein isoform cause a significant overproduction of Aβ.
51A RNA is overexpressed in AD post-mortem samples
In consideration of the role of 51A RNA in the regulation of amyloid formation in the in vitro model, we tested whether its expression might contribute to AD generation in ex vivo human brain samples. To this aim, we measured by real-time RT-PCR the amount of 51A RNA in cerebral cortices from 23 AD patients and 23 non-demented control samples. Interestingly, we found that 51A is actually expressed in human brains and that, although with significant individual variations, the average 51A expression is upregulated in AD samples (mean values: control=1.13; AD=2.14; P=0.049; Fig. 4A,B). To verify whether the increase in 51A expression was specific for AD, we measured its expression in five samples of Parkinson's disease. The average level of expression (using Top1 to normalise) was slightly higher than controls (1.13 versus 1.42) but much lower than observed in AD patients (1.42 versus 2.14). Although these results suggests a possible specificity of 51A overexpression in AD in comparison to other neurodegenerative diseases, owing to the small number of samples available they do provide any definitive conclusion, and larger studies will be required to assess this issue. Importantly, the differences in 51A expression observed in control versus AD patients were not related to the time of the sampling: we observed the lack of any relationship between the post-mortem delay of brain sampling and 51A expression (data not shown).
Fig. 4.

The expression of 51A is significantly increased in AD brains. (A) 51A expression in AD cases (black columns) and non-AD control individuals (white columns) as determined by realtime RT-PCR of post-mortem cerebral cortex samples. (B) Box plot and median values are reported. Statistical analysis by Mann-Whitney U test demonstrated a significant difference between groups (P=0.049).
The findings that SORL1 variant A is downregulated in AD and that there is a functional correlation between the expression of 51A and SORL1 splicing (that leads to a decreased amount of the canonical variant A) clearly suggest that 51A RNA plays an active role in altering SORL1 expression in AD patients, leading to increased amyloid production that could ultimately induce neurodegeneration.
DISCUSSION
In previous papers, we documented the relevant role of pol-III-transcribed ncRNAs in the regulation of alternative splicing in particular (Dieci et al., 2007; Pagano et al., 2007; Massone et al., 2011a; Massone et al., 2011b), and in the molecular events leading to neuron differentiation (Dieci et al., 2007; Pagano et al., 2007; Castelnuovo et al., 2010; Gavazzo et al., 2011).
In the current work, we report the characterisation of a newly identified transcriptional unit, named 51A, that maps to intron 1 of the SORL1 gene, and from which a newly identified regulatory ncRNA is synthesised. The expression of 51A leads to a splicing shift of SORL1 mRNA that leads to the maturation of alternative protein forms instead of the canonical protein variant A. This event ultimately causes a significant downregulation of the canonical form of SORL1. Reduction of SORL1 expression has been shown to represent a condition that is associated with detrimental pathological consequences. In particular, SORL1 downregulation was reported to induce increased Aβ production (Andersen et al., 2005; Small et al., 2005; Offe et al., 2006; Wang et al., 2007). Thus, owing to its influence on SORL1 function, this ncRNA might be of particular relevance as a determinant of induction of Aβ-dependent neurodegeneration. We propose that triggering 51A RNA overexpression could represent one of the upstream regulatory events of Aβ generation.
Therefore, the results reported here support previous studies demonstrating that the downregulation of SORL1 drives the over-formation of Aβ and disclose a possible role of 51A as a newly identified regulatory element, acting upstream to Aβ formation via SORL1 variant A downregulation. In this context, the observation that a significant Aβ increase is observed in 51A-overexpressing cells whereas Aβ changes are more modest in SORL1 knockout (KO) mice (Andersen et al., 2005) might plausibly be due to the different intensity of SORL1 downregulation observed in the two experimental systems, with the activation of compensatory pathways in KO animals that are not present in our transiently transfected cells.
It is also important to underline that 51A is expressed in non-AD human brains although at low levels, indicating a potential as-yet-unidentified physiological role for this ncRNA. Interestingly, the expression of 51A is significantly increased in AD brains (although with individual variations), suggesting that it might be involved in the Aβ generation in these individuals, through the inhibition of SORL1 expression. Currently, further experiments are needed to define the cause of the individual variation in 51A expression in post-mortem AD brains and possibly the correlation of this variation with clinical-pathological conditions such as, for example, brain inflammatory conditions. In any case, the reduction of Aβ production caused by 51A overexpression is associated with a reduction of SORL1 variant A content that could, in turn, affect Aβ production. Further studies are required to show a causative involvement of the 51A-dependent SORL1 modulation in the regulation of APP processing.
In conclusion, our results identify an active role played by a pol-III-transcribed ncRNA, acting upstream of Aβ formation via SORL1 variant A downregulation, in the control of amyloid processing, providing new insight into AD-related processes.
MATERIALS AND METHODS
Genomic and cDNA clones
The genomic clone herein analysed was generated following molecular biology procedures previously reported (Pagano et al., 2003). The oligos used to generate the insert were 5′-ATGCATTAATTTAAGAGCAAGGACCTTGAT-3', and 5′-ATGCATTAATTAGTGTATCATCAGTTGGCA-3' and span a region containing the PSE/TATA pol III type 3 promoter together with the transcribed portion of 51A.
Human brain samples
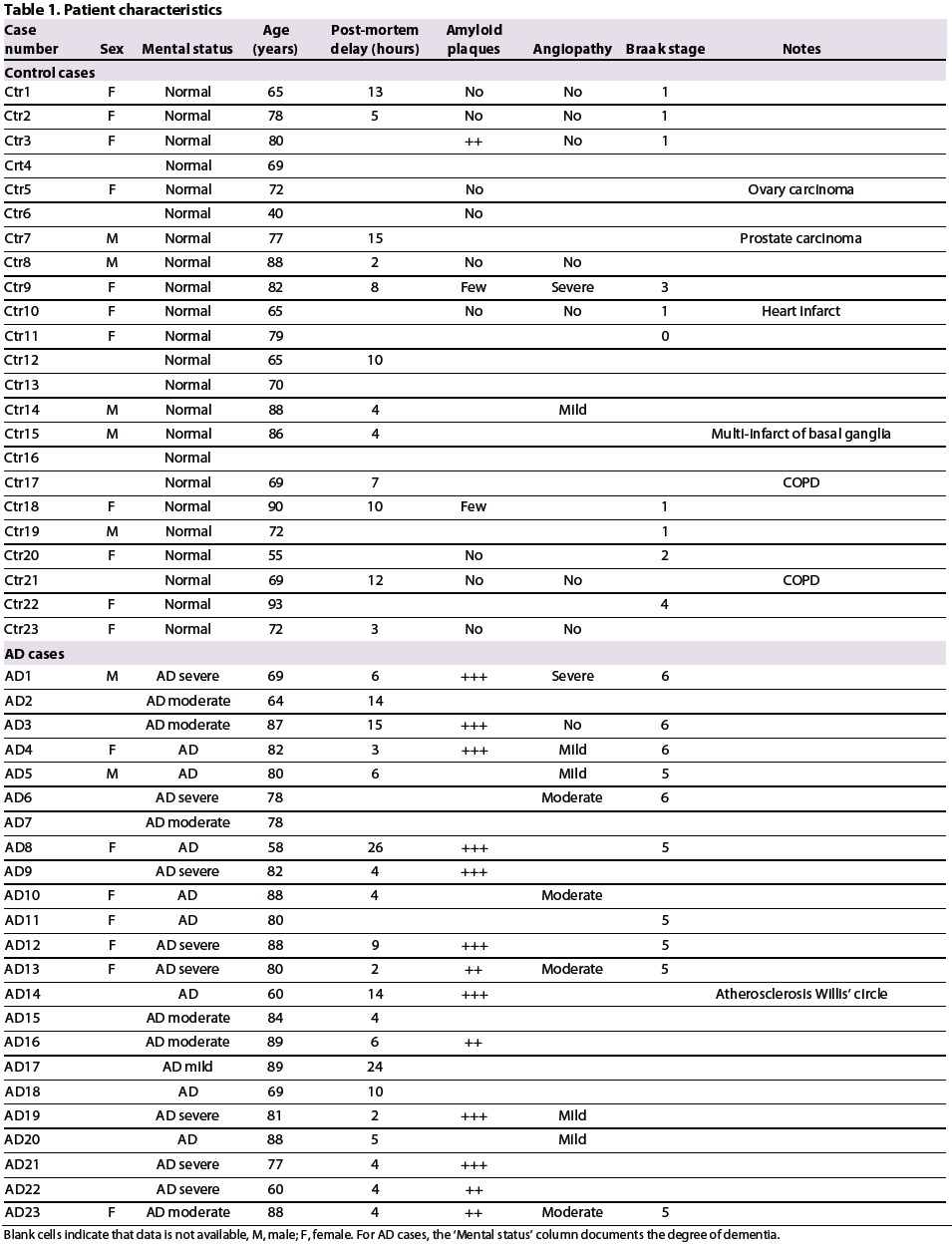
Frontal and temporal cortices from AD [clinical history of disease; pathological diagnosis according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria] and control cases (AD excluded by clinical neurological assessment and immunohistochemical analysis), as described in Table 1, derive from two different sources: the Brain Bank at Case Western Reserve University, Cleveland, OH, and the Joseph and Kathleen Bryan Alzheimer's Disease Research Center (Bryan ADRC) at Duke University Medical Center, Durham, NC. A total of 23 controls and 23 AD samples were analysed. Average age was 71 years for controls and 78 for AD brains.
Table 1.
Patient characteristics
Cell culture, transfection and luciferase assay
Different cells lines were used: HEK-293, NCTC-2544, HepG2 and HeLa cells were maintained on Dulbecco's modified Eagle's medium (DMEM ECB7501L, EuroClone, Milan, Italy), 10% FBS (LONZA DE14-801F), L-glutamine (2 mM; EuroClone, Milan, Italy) and penicillin-streptomycin (100 U/ml/ 100 (ig/ml; EuroClone, Milan, Italy); Neuroblastoma cell lines (SKNBE2 and SHSY5Y) were maintained on RPMI 1640 medium (ECB9006L EuroClone, Milan, Italy), 10% FBS (LONZA DE14-801F), L-glutamine (2 mM; EuroClone, Milan, Italy) and penicillin-streptomycin (100 U/ml/ 100 (ig/ml; EuroClone, Milan, Italy; standard medium); LoVo cells were maintained on F-12K medium (LONZA BE12-615F), 10% FBS (LONZA DE14-801F), L-glutamine (2 mM; EuroClone, Milan, Italy) and penicillin-streptomycin (100 U/ml/ 100 (ig/ml; EuroClone, Milan, Italy). SKNBE cells were transfected using polyethylenimine (PEI; P3143, Sigma, St Louis, MO; 1 μg DNA2.5 μl PEI 10 mM) with pEGFP-N1 as control (referred in the text to as Mock) or pEGFP-N1-51A (referred to as 51A). G418 (Geneticin) was used in culture medium as a means of selection up to 1000 μg/ml, until resistant clones were identified. After selection the clones were preserved in 200 μg/ml G-418 in standard culture conditions. Luciferase-based promoter activity assay was performed 48 hours after transfection by firefly luciferase activity determination with the dual-luciferase reporter-assay system (Promega) according to the manufacturer's protocol.
Real-time quantitative RT-PCR analysis
Total RNAs from samples were extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol and subjected to reverse transcription by Omniscript RT Kit (Qiagen, Valencia, CA, USA) as previously described elsewhere (Pagano et al., 2004). The total RNA from samples was measured by real-time quantitative RT-PCR using PE ABI PRISM® 7700 Sequence Detection System (Perkin Elmer, Wellesley, MA, USA) and Sybr Green method following manufacturer's instructions. The sequences of 51A forward and reverse primers were 5′-TGGGA-GAGTCAGCATCTTGAAG-3' and 5′-TGTACAGTCAGACAA-GAGGTGTGTGTAT-3', respectively. The sequences of SORL1 (Var A) forward and reverse primers were 5′-AGCCCGAGCC-CATCAAG-3' and 5′-AATCAGATGGTGGTGCACTGGG-3', respectively. The sequences of SORL1 (Var B) forward and reverse primers were 5′-TTGGTTCTCGGCAGGTGAA-3' and 5′-ATCT-GACAGCTCATACATCCTATGAGATT-3', respectively. The sequences of SORL1 (Var F) forward and reverse primers were 5′-TCCTAGCATTTATTATTACTTTTCTCTCTTAA-3' and 5′-GTAGCTAATCCAGATGGCGACTT-3', respectively. For endogenous control, expression of the human glyceraldehyde 3 phosphate dehydrogenase (G3PDH) gene was examined. The sequences for human G3PDH primers were 5′-GAAGGTGAAG-GTCGGAGTC-3' and 5′-GAAGATGGTGATGGGATTTC-3'. Relative transcript levels were determined from the relative standard curve constructed from stock cDNA dilutions, and divided by the target quantity of the calibrator following the manufacturer's instructions. For the determination of 51A RNA expression in human post-mortem brain samples, we selected the appropriate housekeeping gene [topoisomerase (DNA) I, NM003286.2], taking advantage of the geNorm Housekeeping Gene Selection Kit as described elsewhere (Penna et al., 2011).
In vitro transcription analysis
In vitro transcription reactions and primer extension experiments were carried out as previously described (Pagano et al., 2003; Pagano et al., 2007). To construct in vitro templates, a 51A-containing fragment was amplified from human genomic DNA and cloned into pGEM-Teasy vector (Promega) by using the primers 5′-ACAAACTCCATCTGCAATTCCTCG-3' and 5′-CAGGTATAGGAGGGGTGCAGC-3'.
Immunofluorescence detection
SKNBE2 cells were grown overnight on culture slides and then transfected with pEGFPN1-51A (or pEGFPN1 as control) using PEI (Sigma). At 48 hours after transfection the cells were washed in PBS, fixed for 10 minutes with 10% buffered formalin and blocked for 15 minutes with 3% BSA in PBS. Cells were subsequently incubated with primary antibody overnight at 4°C in 0.5% BSA in PBS. The next day, the cells were labelled with secondary antibodies for 45 minutes in 0.5% BSA/PBS solution. Cells were then incubated with DAPI for 5 minutes and mounted with Mowiol (Invitrogen) as described elsewhere (Thellung et al., 2011). Immunostained cells were observed with the appropriate filters on an Axiovert 200 M (Zeiss, Jena, Germany) microscope and captured at the same adjustments of laser intensity and photomultiplier sensitivity using AxioVision software.
Primary antibody was anti-SorLA (H-300), sc-33822 rabbit polyclonal antibody raised against amino acids 86-385 mapping within an N-terminal extracellular domain of SorLA of human origin (Santa Cruz). Secondary antibody was anti-rabbit Rhodamine-TRITC (1:200; Jackson ImmunoResearch).
Aβ ELISA
The amount of secreted Aβ x-40 and Aβ x-42 were evaluated by sandwich ELISA (IBL, Gumna, Japan) following the procedure here described. Media of 51A permanently transfected SKNBE2 cells were diluted in EIA buffer and processed using a kit specific for both Aβ species, following the indications of the manufacturer. The kits are solid-phase sandwich ELISA using plates pre-coated with the specific polyclonal anti-human Aβ antibody (raised against residues 38-42 or 35-40). An HRP-conjugated monoclonal human anti-Aβ antibody (11-28) was also supplied. Both assays show a linear reactivity within the range of concentration 7-1000 pg/ml for both Aβ species. Aβ concentration was determined using a Benchmark Microplate Reader and evaluated by Microplate Manager Version 5.1 software (Bio-Rad, Hercules, CA).
Western blot
The proteins were quantified using a commercial protein quantification kit (Protein Assay, Bio-Rad) following the manufacturer's instructions. The samples were subsequently analysed by 10% SDS PAGE (SDS-PAGE) and transferred to a nitrocellulose membrane (Whatman, Inc., GE Healthcare, New York, NY) as previously described (Zerega et al., 2004). In detail, the membranes were initially blocked by an incubation of 2 hours in Tris-buffered saline Tween 20 (TBST; 50 mM Tris-HCl, 150 mM NaCl, pH 7.5, 0.05% Tween 20) containing 5% non-fat dried milk. The blots were incubated for 1 hour with the appropriate primary antibodies: SorLA N-terminal (H-300, sc-33822, Santa Cruz) and anti-SORL1 C-terminal (Sigma-Aldrich, S9200). In order to normalise the protein levels of SORL1, western blot membranes were stripped with the stripping reagent ‘Restore’ (Pierce), then probed with a monoclonal antibody against α-tubulin (clone B-5-1-2; T 5168, Sigma; 1:2000). All primary antibodies were then diluted in TBS containing 0.1% NaN3 and 1% BSA. After washing with TBST, membranes were incubated with peroxidase-conjugated secondary antibodies [anti-mouse IgGs (A 0168, Sigma; 1:12,000; anti-rabbit IgGs (A 0545, Sigma; 1:20,000)] for 1 hour at room temperature. After washing, the reactive bands were revealed by ECL (Amersham Biosciences, GE Healthcare). Densitometric analysis of protein bands was performed using the ImageJ software system.
Statistics
Experiments were performed in triplicate and repeated three times. Data are reported as mean value ± standard deviation, and statistical significance was examined using the unpaired Student's t-test. For analysis of 51A expression in control and AD brains, the non-parametric Mann-Whitney U test was used. P-values less than 0.05 were considered statistically significant.
In the figures, * and ** indicate statistical significance at P-values of <0.05 and <0.01, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Prof. P. Gambetti, Dept of Neuropathology at Case Western Reserve University, Cleveland, OH, USA and Prof. C. Hulette and the Joseph and Kathleen Bryan Alzheimer's Disease Research Center, DUMC, Durham, NC, USA (NIA grant# 5P50 AG05128) for providing brain tissues.
FUNDING
G.D. was supported by Fondazione Cariparma (2010 grant program) and by the Italian Ministry of Education, University and Research (MIUR, PRIN Program). A.P. was supported by MIUR (2007 PRIN Program prot. 2007945BZN), by the Associazione Italiana Ricerca sul Cancro (2009 AIRC Program no. IG9378) and by the Associazione Italiana per la Lotta al Neuroblastoma (Genoa, Italy). T.F. was supported by Italian Ministry of Education, University and Research (MIUR-FIRB Accordi di Programma 2011 project no. RBAP11HSZS).
Footnotes
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS: S.M. and E.C. performed most of the experiments. I.P. and A.G. performed RT-PCR analysis. M.N. performed the fluorescence microscopy analysis. C.R. performed the analysis of APP C-terminal fragments. T.F., G.D. and R.C. participated in the interpretation of results and critically read the manuscript. A.P. led the project, participated in the experiments and wrote the paper.
SUPPLEMENTARY MATERIAL: Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009761/-/DC1
References
- Akil H., Perraud A., Mélin C., Jauberteau M. O., Mathonnet M. (2011). Fine-tuning roles of endogenous brain-derived neurotrophic factor, TrkB and sortilin in colorectal cancer cell survival. PLoS ONE 6, e25097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen O. M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., von Arnim C. A., Breiderhoff T., Jansen P., Wu X., et al. (2005). Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 102, 13461-13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni A. C., Momeni P., Bernardi L., Tomaino C., Frangipane F., Elder J., Kawarai T., Sato C., Pradella S., Wakutani Y., et al. (2007). Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology 69, 140-147 [DOI] [PubMed] [Google Scholar]
- Castelnuovo M., Massone S., Tasso R., Fiorino G., Gatti M., Robello M., Gatta E., Berger A., Strub K., Florio T., et al. (2010). An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J. 24, 4033-4046 [DOI] [PubMed] [Google Scholar]
- Demont Y., Corbet C., Page A., Ataman-Önal Y., Choquet-Kastylevsky G., Fliniaux I., Le Bourhis X., Toillon R. A., Bradshaw R. A., Hondermarck H. (2012). Pro-nerve growth factor induces autocrine stimulation of breast cancer cell invasion through tropomyosin-related kinase A (TrkA) and sortilin protein. J. Biol. Chem. 287, 1923-1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G., Fiorino G., Castelnuovo M., Teichmann M., Pagano A. (2007). The expanding RNA polymerase III transcriptome. Trends Genet. 23, 614-622 [DOI] [PubMed] [Google Scholar]
- Gavazzo P., Vella S., Marchetti C., Nizzari M., Cancedda R., Pagano A. (2011). Acquisition of neuron-like electrophysiological properties in neuroblastoma cells by controlled expression of NDM29 ncRNA. J. Neurochem. 119, 989-1001 [DOI] [PubMed] [Google Scholar]
- Haass C., Hung A. Y., Schlossmacher M. G., Teplow D. B., Selkoe D. J. (1993). beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 268, 3021-3024 [PubMed] [Google Scholar]
- Hermans-Borgmeyer I., Hampe W., Schinke B., Methner A., Nykjaer A., Süsens U., Fenger U., Herbarth B., Schaller H. C. (1998). Unique expression pattern of a novel mosaic receptor in the developing cerebral cortex. Mech. Dev. 70, 65-76 [DOI] [PubMed] [Google Scholar]
- Hinerfeld D. A., Moonis M., Swearer J. M., Baker S. P., Caselli R. J., Rogaeva E., St George-Hyslop P., Pollen D. A. (2007). Statins differentially affect amyloid precursor protein metabolism in presymptomatic PS1 and non-PS1 subjects. Arch. Neurol. 64, 1672-1673 [DOI] [PubMed] [Google Scholar]
- Jacobsen L., Madsen P., Moestrup S. K., Lund A. H., Tommerup N., Nykjaer A., Sottrup-Jensen L., Gliemann J., Petersen C. M. (1996). Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J. Biol. Chem. 271, 31379-31383 [DOI] [PubMed] [Google Scholar]
- Johnson S. A., McNeill T., Cordell B., Finch C. E. (1990). Relation of neuronal APP-751/APP-695 mRNA ratio and neuritic plaque density in Alzheimer's disease. Science 248, 854-857 [DOI] [PubMed] [Google Scholar]
- Khvotchev M., Südhof T. C. (2004). Proteolytic processing of amyloid-beta precursor protein by secretases does not require cell surface transport. J. Biol. Chem. 279, 47101-47108 [DOI] [PubMed] [Google Scholar]
- Klein C., Lohmann-Hedrich K., Rogaeva E., Schlossmacher M. G., Lang A. E. (2007). Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 6, 652-662 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Cheng R., Schupf N., Manly J., Lantigua R., Stern Y., Rogaeva E., Wakutani Y., Farrer L., St George-Hyslop P., et al. (2007). The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Arch. Neurol. 64, 501-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H., Barral S., Cheng R., Chacon I., Santana V., Williamson J., Lantigua R., Medrano M., Jimenez-Velazquez I. Z., Stern Y., et al. (2008). Age-at-onset linkage analysis in Caribbean Hispanics with familial late-onset Alzheimer's disease. Neurogenetics 9, 51-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. L., Galasko D. R., Ringman J. M., Vinters H. V., Edland S. D., Pomakian J., Ubeda O. J., Rosario E. R., Teter B., Frautschy S. A., et al. (2009). Reduction of SorLA/LR11, a sorting protein limiting beta-amyloid production, in Alzheimer disease cerebrospinal fluid. Arch. Neurol. 66, 448-457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusson E. G., Horazdovsky B. F., Cereghino J. L., Gharakhanian E., Emr S. D. (1994). The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 77, 579-586 [DOI] [PubMed] [Google Scholar]
- Massone S., Vassallo I., Fiorino G., Castelnuovo M., Barbieri F., Borghi R., Tabaton M., Robello M., Gatta E., Russo C., et al. (2011a). 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 41, 308-317 [DOI] [PubMed] [Google Scholar]
- Massone S., Vassallo I., Castelnuovo M., Fiorino G., Gatta E., Robello M., Borghi R., Tabaton M., Russo C., Dieci G., et al. (2011b). RNA polymerase III drives alternative splicing of the potassium channel-interacting protein contributing to brain complexity and neurodegeneration. J. Cell Biol. 193, 851-866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massone S., Ciarlo E., Vella S., Nizzari M., Florio T., Russo C., Cancedda R., Pagano A. (2012). NDM29, a RNA polymerase III-dependent non coding RNA, promotes amyloidogenic processing of APP and amyloid β secretion. Biochim. Biophys. Acta 1823, 1170-1177 [DOI] [PubMed] [Google Scholar]
- Matsui M., Kawarai T., Hase Y., Tomimoto H., Iseki K., Rogaeva E., Orlacchio A., Bernardi G., St George-Hyslop P., Takahashi R., et al. (2007). A novel mutation in the SPG3A gene (atlastin) in hereditary spastic paraplegia. J. Neurol. 254, 972-974 [DOI] [PubMed] [Google Scholar]
- Motoi Y., Aizawa T., Haga S., Nakamura S., Namba Y., Ikeda K. (1999). Neuronal localization of a novel mosaic apolipoprotein E receptor, LR11, in rat and human brain. Brain Res. 833, 209-215 [DOI] [PubMed] [Google Scholar]
- Offe K., Dodson S. E., Shoemaker J. T., Fritz J. J., Gearing M., Levey A. I., Lah J. J. (2006). The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J. Neurosci. 26, 1596-1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A., Crooijmans R., Groenen M., Randazzo N., Zerega B., Cancedda R., Dozin B. (2003). A chondrogenesis-related lipocalin cluster includes a third new gene, CALgamma. Gene 305, 185-194 [DOI] [PubMed] [Google Scholar]
- Pagano A., Giannoni P., Zambotti A., Sánchez D., Ganfornina M. D., Gutiérrez G., Randazzo N., Cancedda R., Dozin B. (2004). Phylogeny and regulation of four lipocalin genes clustered in the chicken genome: evidence of a functional diversification after gene duplication. Gene 331, 95-106 [DOI] [PubMed] [Google Scholar]
- Pagano A., Castelnuovo M., Tortelli F., Ferrari R., Dieci G., Cancedda R. (2007). New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. PLoS Genet. 3, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna I., Vella S., Gigoni A., Russo C., Cancedda R., Pagano A. (2011). Selection of candidate housekeeping genes for normalization in human postmortem brain samples. Int. J. Mol. Sci. 12, 5461-5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraus G. C., Masters C. L., Beyreuther K. (1997). Late compartments of amyloid precursor protein transport in SY5Y cells are involved in beta-amyloid secretion. J. Neurosci. 17, 7714-7724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C., Cheng R., Rogaeva E., Lee J. H., Tokuhiro S., Zou F., Bettens K., Sleegers K., Tan E. K., Kimura R., et al. (2011). Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch. Neurol. 68, 99-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E., Meng Y., Lee J. H., Gu Y., Kawarai T., Zou F., Katayama T., Baldwin C. T., Cheng R., Hasegawa H., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V., Baum K., Lao A., Rateitschak K., Schmitz Y., Teichmann A., Wiesner B., Petersen C. M., Nykjaer A., Wolf J., et al. (2012). Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer's disease. EMBO J. 31, 187-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (1990). Deciphering Alzheimer's disease: the amyloid precursor protein yields new clues. Science 248, 1058-1060 [DOI] [PubMed] [Google Scholar]
- Shibata N., Kawarai T., Meng Y., Lee J. H., Lee H. S., Wakutani Y., Shibata E., Pathan N., Bi A., Sato C., et al. (2007). Association studies between the plasmin genes and late-onset Alzheimer's disease. Neurobiol. Aging 28, 1041-1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S. A., Kent K., Pierce A., Leung C., Kang M. S., Okada H., Honig L., Vonsattel J. P., Kim T. W. (2005). Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann. Neurol. 58, 909-919 [DOI] [PubMed] [Google Scholar]
- Thellung S., Corsaro A., Villa V., Simi A., Vella S., Pagano A., Florio T. (2011). Human PrP90-231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death Dis. 2, e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella S., Conti M., Tasso R., Cancedda R., Pagano A. (2012). Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int. J. Cancer 130, 1484-1493 [DOI] [PubMed] [Google Scholar]
- Wang L., Beg F., Ratnanather T., Ceritoglu C., Younes L., Morris J.C., Csernansky J. G., Miller M. I. (2007). Large deformation diffeomorphism and momentum based hippocampal shape discrimination in dementia of the Alzheimer type. IEEE Trans. Med. Imaging 26, 462-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S., Sato C., Kawarai T., Goodall E. F., Pall H. S., Zinman L. H., Robertson J., Morrison K., Rogaeva E. (2008). Genetic studies of GRN and IFT74 in amyotrophic lateral sclerosis. Neurobiol. Aging 29, 1279-1282 [DOI] [PubMed] [Google Scholar]
- Yamazaki T., Selkoe D. J., Koo E. H. (1995). Trafficking of cell surface beta-amyloid precursor protein: retrograde and transcytotic transport in cultured neurons. J. Cell Biol. 129, 431-442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H., Bujo H., Kusunoki J., Seimiya K., Kanaki T., Morisaki N., Schneider W. J., Saito Y. (1996). Elements of neural adhesion molecules and a yeast vacuolar protein sorting receptor are present in a novel mammalian low density lipoprotein receptor family member. J. Biol. Chem. 271, 24761-24768 [DOI] [PubMed] [Google Scholar]
- Zerega B., Pagano A., Pianezzi A., Ulivi V., Camardella L., Cancedda R., Cancedda F. D. (2004). Expression of serum amyloid A in chondrocytes and myoblasts differentiation and inflammation: possible role in cholesterol homeostasis. Matrix Biol. 23, 35-46 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.