

Background: Transforming growth factor β (TGFβ) induces renal hypertrophy and fibrosis.
Results: TGFβ-induced deptor down-regulation is necessary for prolonged activation of TORC1/2 and mesangial cell hypertrophy.
Conclusion: TGFβ-stimulated Smad 3 contributes to deptor suppression and mammalian target of rapamycin activation.
Significance: Sustained deptor expression may alleviate renal glomerular hypertrophy and fibrosis.
Keywords: Akt, Cell Signaling, Fibrosis, Kidney, Serine/Threonine Protein Kinase
Abstract
In many renal diseases, transforming growth factor β (TGFβ)-stimulated canonical Smad 3 and noncanonical mechanistic target of rapamycin (mTOR) promote increased protein synthesis and mesangial cell hypertrophy. The cellular underpinnings involving these signaling molecules to regulate mesangial cell hypertrophy are not fully understood. Deptor has recently been identified as an mTOR interacting protein and functions as an endogenous inhibitor of the kinase activity for both TORC1 and TORC2. Prolonged incubation of mesangial cells with TGFβ reduced the levels of deptor concomitant with an increase in TORC1 and TORC2 activity. Sustained TGFβ activation was required to inhibit association of deptor with mTOR, whereas rapid activation had no effect. Using the mTOR inhibitor PP242, we found that TGFβ-induced both early and sustained activation of TORC1 and TORC2 was necessary for deptor suppression. PP242-induced reversal of deptor suppression by TGFβ was associated with a significant inhibition of TGFβ-stimulated protein synthesis and hypertrophy. Interestingly, expression of siRNA against Smad 3 or Smad 7, which blocks TGFβ receptor-specific Smad 3 signaling, prevented TGFβ-induced suppression of deptor abundance and TORC1/2 activities. Furthermore, overexpression of Smad 3 decreased deptor expression similar to TGFβ stimulation concomitant with increased TORC1 and TORC2 activities. Finally, knockdown of deptor reversed Smad 7-mediated inhibition of protein synthesis and mesangial cell hypertrophy induced by TGFβ. These data reveal the requirement of both early and late activation of mTOR for TGFβ-induced protein synthesis. Our results support that TGFβ-stimulated Smad 3 acts as a key node to instill a feedback loop between deptor down-regulation and TORC1/2 activation in driving mesangial cell hypertrophy.
Introduction
Chronic renal diseases that eventuate in glomerulosclerosis including diabetic nephropathy are characterized by increased local production of transforming growth factor β (TGFβ). Functional consequences of TGFβ action consist of altered glomerular hemodynamics, whole kidney hypertrophy, and glomerular hypertrophy (1–3). TGFβ mediates these effects by binding to its high affinity type II receptor to form a tetrameric complex with the type I receptor (4). In the absence of TGFβ, the type I receptor binds to the negative regulatory protein FKBP12. Binding of TGFβ induces phosphorylation of TGFβ receptor I by the type II receptor in the GS domain. This phosphorylation leads to activation of the type I receptor, releasing FKBP12 to recruit receptor-specific transcription factors Smad 2 and Smad 3 onto the receptor, which phosphorylates them at the C terminus (4, 5). Phosphorylated Smads dissociate from the type I receptor and the SARA (Smad anchor for receptor activation), a Smad-recruiting protein to the plasma membrane (6). Subsequently, phosphorylated Smad 2 and Smad 3 bind to the co-Smad, Smad 4, and the heterodimer translocates to the nucleus to recruit coactivators or corepressors for target gene regulation (7–9).
Apart from the canonical signal transduction pathway described above, the TGFβ receptor activates downstream signaling kinases including ERK1/2, JNK1/2, p38 kinase, and c-Src (8, 10, 11). We have previously reported that activation of PI 3-kinase signaling in response to TGFβ in renal glomerular mesangial cells contributes to hypertrophy of these cells prior to expansion of matrix proteins that precedes fibrosis (12, 13). We also showed involvement of mechanistic target of rapamycin (mammalian target of rapamycin; mTOR)5 kinase in TGFβ-induced mesangial cell hypertrophy (14).
Although TOR exists as two independent kinases in yeast, mammalian TOR is coded by a single gene (15). Mice with homozygous deletion of mTOR die after implantation (16, 17). mTOR null embryos show deficiency in macromolecular synthesis, including protein synthesis necessary for cellular hypertrophy (16–18). The catalytic mTOR kinase forms two distinct complexes known as mTOR complex 1 (TORC1) and TORC2, which contain common, mLST8, and distinct proteins that confer functional specificity to these complexes (19, 20). Raptor and PRAS40 are exclusive components of TORC1. Similarly, rictor (protor 1), protor 2, and mSin1 distinguish TORC2 from TORC1. Raptor is essential for TORC1 activation and serves as the docking site for the substrates, whereas PRAS40 acts as a negative regulator of TORC1 activity (15, 21, 22). Similarly, rictor and mSin1 along with mLST8 maintain the integrity of the TORC2 (15, 23). Deficiency of any of these components abolishes TORC2 activity (23–25). But lack of mLST8 does not affect TORC1 activity (24).
Recently, deptor has been identified as a partner of both TORC1 and TORC2 and acts as a negative regulator of mTOR kinase activity (26). mTOR regulates protein synthesis necessary for diabetic renal hypertrophy including mesangial cell hypertrophy (27–30). TGFβ-induced mesangial cell hypertrophy is mediated by mTOR (14). Although TGFβ rapidly activates mTOR in glomerular mesangial cells, the precise mechanism by which TGFβ induces hypertrophy is not known. In the present study, we show that TGFβ induces down-regulation of deptor and prolonged activation of mTOR in mesangial cells. Both early and sustained activation of mTOR kinase by TGFβ is required for deptor down-regulation. Moreover, our results demonstrate a contribution of TGFβ-specific Smad 3 to the reduced deptor abundance necessary for mesangial cell protein synthesis and hypertrophy. These data provide evidence for the presence of a novel cross-talk between Smad 3 and mTOR to induce deptor down-regulation, resulting in mesangial cell hypertrophy.
EXPERIMENTAL PROCEDURES
Materials
Recombinant TGFβ1 was purchased from R & D Systems, Minneapolis, MN. Phenylmethylsulfonyl fluoride, Na3VO4, Nonidet P-40, cycloheximide, actin, phosphoserine, FLAG antibodies, and TRI Reagent were obtained from Sigma. First strand cDNA synthesis kit was obtained from Invitrogen. SB431542 was purchased from Calbiochem, San Diego, CA. PP242 was obtained from Chemdea Pharmaceuticals, Ridgewood, NJ. FuGENE HD was obtained from Roche Applied Science. Phospho-Akt (Ser-473), phospho-Akt (Thr-308), phospho-S6 kinase (Thr-389), phospho-C-terminal Smad 3, phospho-4EBP-1 (Thr-37/46), phospho-4EBP-1 (Ser-65), S6 kinase, mTOR, and Akt antibodies were purchased from Cell Signaling, Boston, MA. Smad 3 antibody was obtained from Zymed Laboratories Inc., San Francisco, CA. Deptor antibody and a pool of three siRNAs against Smad 3 were obtained from Santa Cruz Biotechnology. Anti-HA antibody was purchased from Covance, Princeton, NJ. Adenovirus vectors expressing HA-tagged Smad 3 (Ad Smad 3) and FLAG-tagged Smad 7 (Ad Smad 7) have been described previously (9). shRNAs targeting two different regions of deptor (deptor sh1 and deptor sh2), FLAG-deptor, and FLAG-deptor 13X were obtained from Addgene and originally constructed in the laboratory of Dr. David M. Sabatini, Whitehead Institute for Biomedical Research, Boston, MA. Vector containing scrambled RNA was kindly provided by Dr. David M. Sabatini.
Cell Culture and Adenovirus Infection
Normal human kidney glomerular mesangial cells were grown in DMEM in the presence of 10% fetal bovine serum as described previously (31–33). For the present experiments, the cells were used between passage 7 and 12. The cells were infected with adenovirus vectors at a multiplicity of infection of 50 for 24 h, essentially as described previously (9, 14). An adenovirus expressing green fluorescence protein (Ad GFP) was used as control. Before TGFβ (2 ng/ml) incubation, the cells were incubated in serum-free medium for 24 h to make them quiescent.
Cell Lysis, Immunoblotting, and Immunoprecipitation
Cells were washed with PBS and harvested in radioimmune precipitation assay buffer (20 mm Tris-HCl, pH 7.5, 5 mm EDTA, 150 mm NaCl, 1 mm Na3VO4, 1 mm PMSF, 0.1% protease inhibitor mixture, and 1% Nonidet P-40) and incubated at 4 °C for 30 min. The crude extracts were centrifuged at 12,000 × g for 30 min at 4 °C. The protein concentration was determined in the cleared cell lysate. Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis and transferred to membrane. Immunoblotting was performed using the indicated antibodies. The protein bands were developed with HRP-conjugated secondary antibody using ECL reagent as described previously (9, 14, 28, 29, 34). Equal amounts of cleared cell lysates were immunoprecipitated with the indicated antibodies as described (9, 14, 35). The immune beads were resuspended in sample buffer, proteins were separated by SDS-polyacrylamide gel electrophoresis and immunoblotted as described above.
RNA Isolation and RT-PCR
Total RNA was prepared from mesangial cells using TRI Reagent. Expression of deptor was determined by quantitative real-time RT-PCR. cDNAs were prepared by reverse transcription from 0.5 μg of total RNA using TaqMan reverse transcription reagents (number N808-0234). The cDNA was amplified and quantified in 96-well plates using TaqMan deptor primers (Applied Biosystem) in a 7500 real time PCR machine (Applied Biosystem). The PCR condition was 95 °C for 10 min followed by 45 cycles at 95 °C for 15 s and 60 °C for 1 min. The relative mRNA levels were normalized to the reference GAPDH in the same sample. Data analyses were done by the comparative Ct method as described previously (36).
Transfection
Mesangial cells were transfected with deptor shRNA plasmids using FuGENE HD as described previously (9, 14, 28, 29, 34, 35). A vector expressing scrambled RNA was used as control.
Protein Synthesis
After incubation, the mesangial cells were treated with 35S-labeled methionine and protein synthesis was determined as [35S]methionine incorporation as described (13, 14, 28, 29).
Measurement of Cellular Hypertrophy
At the end of the incubation, the cells were trypsinized and counted using a hemocytometer. Cells were then pelleted at 4000 × g at 4 °C, washed with PBS, and lysed in RIPA buffer as described above. The protein content in the total number of cells was determined. Hypertrophy was expressed as an increase in the ratio of cellular protein content to cell number as described previously (28, 29).
Statistics
The significance of the results was assessed by analysis of variance followed by Student-Newman-Keuls analysis as described previously (9, 14, 28, 29, 34, 35). The mean ± S.E. of the indicated measurements are shown. A p value of less than 0.05 was considered significant.
RESULTS
TGFβ Induces Down-regulation of Deptor for Prolonged Activation of TORC1 and TORC2
We and others have recently shown that TGFβ rapidly increases mTOR kinase activity (14, 37). One mechanism involves activation of Akt followed by phosphorylation and inactivation of the negative regulator TSC2, leading to activation of TORC1 (14). More recently, deptor was identified as a component of the mTOR kinase complex that negatively controls the activity of both TORC1 and TORC2 (26). We examined the effect of TGFβ on the expression of deptor in mesangial cells. Short-term incubation of these cells with TGFβ did not show any effect on abundance of deptor (Fig. 1A and supplemental Fig. S1A). However, prolonged treatment with TGFβ significantly decreased the levels of deptor (Fig. 1B). The reduction was evident at 6 h of TGFβ treatment and sustained for 24 h (Fig. 1B). To determine the half-life of deptor, we incubated mesangial cells with TGFβ in the presence of cycloheximide. As shown (supplemental Fig. S1B), the apparent protein half-life of deptor was 18 h. TGFβ acts through activation of its type I receptor serine/threonine kinase. SB431542, an inhibitor of TGFβ receptor I, prevented the TGFβ-induced down-regulation of deptor (Fig. 1C and supplemental Fig. S1C). The reduction in deptor protein level was associated with a significant decrease in its mRNA in response to TGFβ (Fig. 1D). SB431542 reversed the TGFβ-induced decrease in deptor mRNA (Fig. 1D). These results indicate TGFβ receptor serine/threonine kinase activity is required for the decrease in deptor abundance in response to TGFβ.
FIGURE 1.

TGFβ induces deptor down-regulation in mesangial cells. A and B, quiescent human mesangial cells were incubated with 2 ng/ml of TGFβ for the indicated time periods. Cleared cell lysates were immunoblotted with deptor and actin antibodies. Panel B, right, shows quantification of protein bands shown in the left panel. Mean ± S.E. of 4 experiments is shown. *, p < 0.05 versus control. C and D, inhibition of TGFβ receptor I prevents deptor suppression induced by TGFβ. Quiescent human mesangial cells were incubated with 5 μm SB 431542 for 1 h prior to treatment with 2 ng/ml of TGFβ for 24 h. The cell lysates were immunoblotted with deptor and actin antibodies (C). In panel D, total RNA was used to detect deptor mRNA as described under “Experimental Procedures.” Mean ± S.E. of 6 measurements is shown. *, p < 0.05 versus control; **, p < 0.01 versus TGFβ-stimulated. Quantification of A and C is shown in supplemental Fig. S1, A and C.
As deptor is a suppressor of mTOR activity and because deptor was not decreased at early time points of TGFβ stimulation, we tested the activation of this kinase. Phosphorylation of S6 kinase at Thr-389 was used as a surrogate for TORC1 activation. Incubation of mesangial cells with TGFβ increased phosphorylation of S6 kinase within 2.5 min, which was sensitive to the TGFβ receptor inhibitor SB431542 (Fig. 2A and supplemental Fig. S2, A and B). Similarly, prolonged incubation of mesangial cells with TGFβ increased phosphorylation of S6 kinase (Fig. 2B and supplemental Fig. S2C). SB431542 inhibited TGFβ-stimulated phosphorylation of S6 kinase at 24 h (supplemental Fig. S2D). Moreover, suppression of deptor using two independent shRNAs increased phosphorylation of S6 kinase supporting the hypothesis that late activation of TORC1 is mediated by a decrease in deptor levels (Fig. 2, C and D, and supplemental Fig. S2, E and F). This activation of TORC1 was confirmed by observing increased phosphorylation of another substrate, 4EBP-1, as a result of deptor down-regulation (Fig. 2, E and F, and supplemental Fig. S2, G and H). Note that early activation of TORC1 (Fig. 2A) does not coincide with deptor down-regulation (Fig. 1B). These results indicate that down-regulation of deptor is not required for early activation of TORC1. Similar to the activation of TORC1, TGFβ rapidly increased phosphorylation of Akt at the hydrophobic site, Ser-473, suggesting activation of TORC2 (Fig. 2G and supplemental Fig. S2I). Also, prolonged incubation of mesangial cells with TGFβ increased TORC2 activity (Fig. 2H and supplemental Fig. S2J). Both early and prolonged activation was inhibited by SB431542 (supplemental Fig. S2, K and L). shRNA-mediated inhibition of deptor expression also resulted in activation of TORC2 (Fig. 2, I and J, and supplemental Fig. S2, M and N). These results suggest the presence of an inverse correlation between deptor abundance and sustained TORC1/TORC2 activation in response to TGFβ. However, early activation of these kinases does not involve deptor.
FIGURE 2.

TGFβ increases TORC1 and TORC2 activities in mesangial cells. A, B, G, and H, lysates from quiescent mesangial cells treated with TGFβ for the indicated times were used for immunoblotting with phospho-S6 kinase (Thr-389) for TORC1 activity (panels A and B) and phospho-Akt (Ser-473) for TORC2 activity (panels G and H). The same cell lysates were immunoblotted with phospho-Akt (Thr-308), S6 kinase, and Akt antibodies as indicated. C–F, I, and J, human mesangial cells were transfected with deptor sh1 (panels C, E, and I) or deptor sh2 (panels D, F, and J), two independent shRNA vectors targeting different regions of deptor mRNA. Vector expressing a scrambled RNA was used as control. The cell lysates were immunoblotted with phospho-S6 kinase, S6 kinase, deptor, actin antibodies (panels C and D); phospho-4EBP-1 (Thr-37/46), phospho-4EBP-1 (Ser-65), 4EBP-1, Deptor, and actin antibodies (panels E and F); phospho-Akt (Ser-473), phospho-Akt (Thr-308), deptor, Akt, and actin antibodies (panels I and J). Quantifications of these panels are shown in supplemental Fig. S2, B, C, E–J, M, and N.
mTOR Regulates TGFβ-induced Phosphorylation of Deptor
In previous studies deptor was shown to be phosphorylated at 13 Ser/Thr residues located between the C-terminal DEP domain and PDZ domain (26). Because both TORC1 and TORC2 were activated early in response to TGFβ (Fig. 2, A and G), we considered a role of both kinases in regulating phosphorylations of deptor. We used an ATP competitive inhibitor of mTOR, PP242, which blocks both kinase complexes (38). Mesangial cells were incubated with PP242 followed by TGFβ. Deptor immunoprecipitates were immunoblotted with antibody that recognizes serine-phosphorylated proteins. TGFβ increased phosphorylation of deptor (Fig. 3A). PP242 significantly inhibited this phosphorylation (Fig. 3A and supplemental Fig. S3A). To confirm this observation, we inhibited both TORC1 and TORC2 simultaneously by down-regulating raptor and rictor, using two independent shRNAs. Inhibition of TORC1 and TORC2 blocked TGFβ-induced serine phosphorylation of deptor (Fig. 3, B and C, and supplemental Fig. S3, B andC). Moreover, we used a mutant of FLAG-tagged deptor in which all 13 phosphorylation sites were mutated to alanine (13X). This mutant and wild type deptor were transfected into mesangial cells followed by incubation with TGFβ. As shown in Fig. 3D, TGFβ did not have any effect on mutant deptor, whereas wild deptor was significantly down-regulated (supplemental Fig. S3D).
FIGURE 3.

mTORC1/2 regulate TGFβ-induced phosphorylation of deptor. A, human mesangial cells were treated with 1 μm PP242 for 1 h prior to incubation with 2 ng/ml of TGFβ for 4 h. This incubation time with TGFβ was chosen because beginning at 6 h a significant deptor down-regulation was detected (Fig. 1B). The cell lysates were immunoprecipitated (IP) with control antibody against actin or deptor antibody as indicated. The immunoprecipitates were immunoblotted with phosphoserine antibody. B and C, human mesangial cells were transfected with two shRNAs against raptor and rictor (sh1, panel B; sh2, panel C). Transfected cells were treated with TGFβ as described in panel A. The cell lysates were immunoprecipitated with actin or deptor antibody followed by immunoblotting (IB) with phosphoserine antibody as described in panel A. D, phosphorylation of deptor is required for its down-regulation by TGFβ. Human mesangial cells were transfected with FLAG-tagged phospho-deficient mutant deptor 13X (lanes 1 and 2) or wild type deptor (lanes 3 and 4). Transfected cells were incubated with 2 ng/ml of TGFβ for 24 h. The cell lysates were immunoblotted with FLAG and actin antibodies as indicated. Quantifications of these panels are shown in supplemental Fig. S3, A–D.
Deptor interacts with mTOR in both TORC1 and TORC2 (26). Because deptor is an endogenous inhibitor of mTOR activity and we found very rapid activation of both TORC1 and TORC2 (Fig. 2, A and G), we assessed the complex formation between mTOR and deptor in the presence of TGFβ at early time points. Lysates of mesangial cells incubated with TGFβ for 5 min were immunoprecipitated with mTOR antibody. This immunopurified mTOR was immunoblotted with deptor antibody. As predicted, mTOR was complexed with deptor in the absence of TGFβ (Fig. 4A). No change in complex formation between mTOR and deptor in response to TGFβ was observed at this early time point (Fig. 4A and supplemental Fig. S4A). The reciprocal experiment showed identical results (Fig. 4B and supplemental Fig. S4B). Complex formation between deptor and mTOR was inhibited in mesangial cells incubated with TGFβ for 24 h (Fig. 4, C and D, and supplemental Figs. S4, C and D). This decreased association may be due to the reduced abundance of deptor at this time of TGFβ stimulation (Fig. 1B).
FIGURE 4.

TGFβ induces dissociation of deptor from mTOR in a time-dependent manner. Quiescent mesangial cells were incubated with TGFβ (2 ng/ml) for 5 min (panels A and B) or 24 h (panels C and D). Cell lysates were immunoprecipitated (IP) with control actin antibody as indicated. Also, cell lysates were immunoprecipitated with mTOR antibody (A and C) or deptor antibody (B and D). The immunoprecipitates were immunoblotted (IB) with deptor and mTOR antibodies (panels A and C). In panels B and D, immunoprecipitates were immunoblotted with mTOR and deptor antibodies. IgG is shown in each panel. Quantifications of these panels are shown in supplemental Fig. S4, A-D.
Both mTOR Kinases Regulate TGFβ-induced Expression of Deptor
Our results above demonstrate a role of deptor down-regulation in the prolonged activation of mTOR. Furthermore, we show that mTOR regulates phosphorylation of deptor. Previously it was shown that mTOR kinase activity is required for the decrease in deptor abundance (26). But the requirement of mTOR for TGFβ-induced suppression of deptor has not been investigated. Preincubation of mesangial cells with PP242, which inhibits both TORC1 and TORC2 at an early time point, reversed the TGFβ-induced down-regulation of deptor (Fig. 5A and supplemental Fig. S5, A–C). Interestingly, PP242 also prevented the TGFβ-suppressed deptor levels when mesangial cells were treated with the drug after 15 min of incubation with TGFβ (Fig. 5A, compare lane 2 with lane 4, and supplemental Fig. S5A). This reversal of deptor abundance by PP242 was associated with inhibition of TORC1 and TORC2 (Fig. 5, B and C, and supplemental Fig. S5, D and E). These results conclusively indicate that prolonged activation of both TORC1 and TORC2 is necessary for decreased deptor abundance in response to TGFβ.
FIGURE 5.

Both TORC1 and TORC2 participate in TGFβ-induced deptor suppression. Quiescent human mesangial cells were treated with 1 μm PP242 for 1 h prior to incubation with TGFβ for 24 h (Pre-PP242). Similarly, quiescent mesangial cells were incubated with TGFβ for 15 min followed by incubation with 1 μm PP242 for 24 h in the presence of TGFβ. Cleared cell lysates were immunoblotted with deptor and actin (panel A), phospho-S6 kinase (Thr-389) and S6 kinase (panel B), phospho-Akt (Ser-473), phospho-Akt (Thr-308), and Akt antibodies as indicated. Quantifications of these panels are shown in supplemental Fig. S5, A, D, and E, respectively.
TGFβ-stimulated Prolonged Inactivation of 4EBP-1 Regulates Mesangial Cell Protein Synthesis Necessary for Hypertrophy
Phosphorylation of 4EBP-1 at Thr-37/46 and Ser-65 results in its inactivation (39). Recent reports including our own (20, 34) show involvement of both TORC1 and TORC2 in phosphorylation and inactivation of 4EBP-1. Therefore, we used PP242, which inhibits these kinase complexes (38). Mesangial cells were either treated with PP242 prior to incubation with TGFβ or first incubated with TGFβ for 15 min followed by treatment with PP242. In both cases cells were exposed to TGFβ for 24 h. TGFβ increased phosphorylation of 4EBP-1 at Thr-37/46 and Ser-65 (Fig. 6A). Both pre- and post-treatment of mesangial cells with PP242 inhibited TGFβ-induced phosphorylation of 4EBP-1 at these sites (Fig. 6A and supplemental Fig. S6). Inactivation of 4EBP-1 by TORC1-mediated phosphorylation at these specific serine/threonine sites is necessary for increased protein synthesis (39). TGFβ-induced protein synthesis was significantly inhibited by pre- as well as post-treatment with PP242 (Fig. 6B). Similarly, PP242 markedly blocked hypertrophy of mesangial cells in response to TGFβ under both conditions (Fig. 6C). These results suggest that prolonged activation of both TORC1 and TORC2 by TGFβ contributes to phosphorylation and inactivation of 4EBP-1, resulting in increased protein synthesis and hypertrophy of mesangial cells.
FIGURE 6.

TORC1 and TORC2 regulate TGFβ-stimulated 4EBP-1 phosphorylation, protein synthesis, and hypertrophy in mesangial cells. Quiescent mesangial cells were incubated with PP242 and TGFβ essentially as described in the legend of Fig. 5. Panel A, cell lysates were immunoblotted with phospho-4EBP-1 (Thr-37/46), phospho-4EBP-1 (Ser-65), and 4EBP-1 antibodies. Quantification of these data are shown in supplemental Fig. S6. B, TGFβ-incubated cells were labeled with [35S]methionine. Protein synthesis was determined essentially as described (13, 14, 28, 29). C, TGFβ-incubated cells were trypsinized and counted, and protein content was determined. The cell hypertrophy was expressed as an increase in ratio of total protein to cell number as described under ”Experimental Procedures“ (13, 28). For panels B and C, mean ± S.E. of 6 measurements are shown. *, p < 0.001 versus control; **, p < 0.001 versus TGFβ-stimulated; #, p < 0.001 versus TGFβ stimulated.
TGFβ-stimulated Smad 3 Regulates Abundance of Deptor
Activated TGFβ receptor I initiates signal transduction by direct phosphorylation of Smad 3, which regulates TGFβ-induced biological activities (4, 40). Incubation of mesangial cells with TGFβ increased phosphorylation of Smad 3 (supplemental Fig. S7A). To examine the involvement of Smad 3 phosphorylation in deptor down-regulation, we used adenovirus-mediated expression of Smad 7, an endogenous inhibitor of TGFβ-stimulated Smad 3 phosphorylation/signaling. Expression of Smad 7 in mesangial cells reversed the TGFβ-induced down-regulation of deptor concomitant with inhibition of phosphorylation of Smad 3 (Fig. 7A and supplemental Fig. S7, A and B). This reversal of TGFβ-induced deptor suppression by Smad 7 was associated with inhibition of TORC1 as well as TORC2 activities (Fig. 7, B and C, and supplemental Fig. S7, C and D). To directly test the role of Smad 3, we employed Smad 3 siRNA. A pool of three siRNAs directed against Smad 3 mRNA significantly reversed TGFβ-suppressed deptor levels (Fig. 7D and supplemental Fig. S7E). Expression of Smad siRNAs inhibited both TORC1 and TORC2 activities induced by TGFβ (Fig. 7, E and F, and supplemental Fig. S7, F and G). To further confirm the role of Smad 3, we directly expressed Smad 3. As expected, TGFβ repressed the levels of deptor (Fig. 7G). Similarly, overexpression of Smad 3 decreased the expression of deptor (Fig. 7G, compare lane 1 with lane 3 and supplemental Fig. S7H). Furthermore, expression of Smad 3 in the presence of TGFβ maintained deptor abundance analogous to the levels observed with TGFβ or Smad 3 alone (Fig. 7G). Concomitantly, the decrease in deptor expression by Smad 3 was accompanied by increased TORC1 and TORC2 activities similar to those found with TGFβ stimulation (Fig. 7, H and I, and supplemental Fig. S7, I a n dJ). These results conclusively demonstrate that TGFβ-stimulated Smad 3 signal transduction regulates deptor down-regulation.
FIGURE 7.

TGFβ-stimulated Smad 3 signaling regulates deptor down-regulation and, TORC1 and TORC2 activation. Mesangial cells were infected with Ad Smad 7 (panels A–C) and Ad Smad 3 (panels G--I) in serum-free medium for 24 h prior to incubation with 2 ng/ml of TGFβ for 24 h as described under ”Experimental Procedures.“ As control, Ad GFP was used. D–F, mesangial cells were transfected with a pool of three siRNAs against Smad 3 or scramble RNA. Transfected cells were incubated with TGFβ as described above. Cell lysates were immunoblotted with deptor and actin antibodies (panels A, D, and G); phospho-S6 kinase (Thr-389) and S6 kinase antibodies (panels B, E, and H); phospho-Akt (Ser-473), phospho-Akt (Thr-308), and Akt antibodies (panels C, F, and I). Expression of Smad 7 is shown by immunoblotting with FLAG (panels A–C). Expression of Smad 3 is shown by immunoblotting with Smad 3 (panels D–F) and anti-HA (panels G–I) antibodies, respectively. Quantification of these data are shown in supplemental Fig. S7, B–J.
TGFβ-stimulated Smad 3 Signaling Regulates Deptor-mediated Mesangial Cell Protein Synthesis and Hypertrophy
We have shown above that TGFβ-induced protein synthesis is sensitive to mTOR inhibition (Fig. 6B). We also showed that deptor down-regulation activates both TORC1 and TORC2 (Fig. 2, C-F, I, and J). We examined whether deptor regulates mesangial cell protein synthesis. Down-regulation of deptor using two independent shRNAs significantly increased protein synthesis similar to TGFβ treatment (supplemental Fig. S8, A and B). Next, we tested if the integration between the TGFβ-induced canonical Smad 3 signaling and non-canonical mTOR activation by deptor down-regulation contributes to protein synthesis. Expression of Smad 7, which inhibits TGFβ-induced activation of Smad 3 (supplemental Fig. S7A), blocked TGFβ-stimulated protein synthesis (Fig. 8, A and B, and supplemental Fig. S9, A and B). Interestingly, inhibition of deptor expression by two independent shRNAs significantly reversed the inhibitory effect of Smad 7 on TGFβ-stimulated protein synthesis (Fig. 8, A and B, and supplemental Fig. S9, A and B). Similarly, expression of Smad 7 abrogated TGFβ-stimulated mesangial cell hypertrophy, which was significantly prevented by suppression of deptor expression (Fig. 8, C and D, and supplemental Fig. S9, C and D). These results demonstrate that TGFβ-induced Smad 3 signaling regulates deptor-mediated mesangial cell hypertrophy.
FIGURE 8.

TGFβ-stimulated Smad regulates mesangial cell protein synthesis and hypertrophy via deptor down-regulation. Human mesangial cells were transfected with two independent shRNAs (250 ng/well) targeting deptor (deptor sh1 and deptor sh2) along with Ad Smad 7 infection with 50 multiplicity of infection as described under ”Experimental Procedures.“ The cells were incubated with TGFβ for 24 h. A and B, the cells were labeled with [35S]methionine and its incorporation was determined as described previously (13, 28). C and D, cellular hypertrophy was determined as ratio of protein content to cell number as described in legend of Fig. 5C (13, 28). Mean ± S.E. of triplicate measurements are shown. In panel A, *, p < 0.01 versus control; **, p < 0.05 versus TGFβ; @, p < 0.05 control; #, p < 0.01 versus TGFβ + Smad 7. In panel B, *, p < 0.01 versus control; **, p < 0.05 versus TGFβ; @, p < 0.01 control; #, p < 0.01 versus TGFβ + Smad 7. In panels C and D, *, p < 0.001 versus control; **, p < 0.001 versus TGFβ; @, p < 0.001 control; #, p < 0.001 versus TGFβ + Smad 7. Expression of Smad 7 and deptor is shown in supplemental Fig. S9.
DISCUSSION
In the present study we identified deptor, an endogenous inhibitor of mTOR activity, as a pathological target of TGFβ receptor serine/threonine kinase. We present evidence demonstrating Smad 3-dependent activation of mTOR kinase through down-regulation of its endogenous inhibitor deptor. Our results also show that this cross-talk between Smad 3 and deptor abundance regulates mesangial cell hypertrophy in response to TGFβ (Fig. 9).
FIGURE 9.
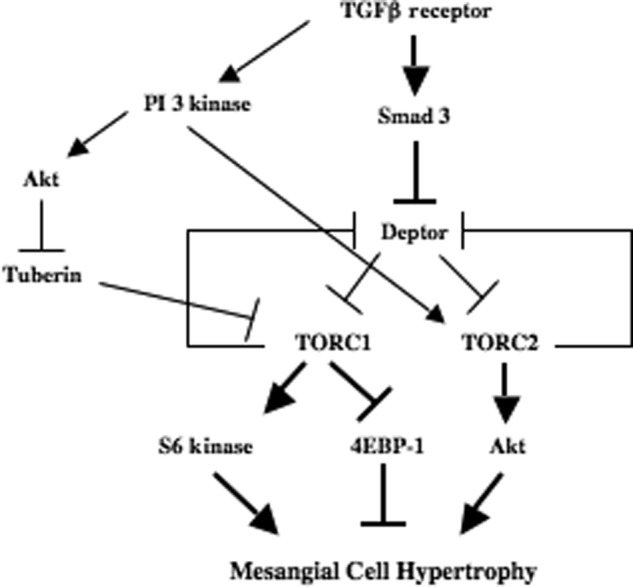
Schematic summarizing the results. Thick arrows show the results described here.
In physiologic kidney hypertrophy such as compensatory renal hypertrophy following uninephrectomy and in disease states such as diabetes, a role for TGFβ has been recognized (41–43). For example, TGFβ1 null renal cells display reduced hypertrophy in response to high glucose (44). Furthermore, type 1 diabetic mice heterozygous for the type II TGFβ receptor exhibit significantly decreased glomerular and mesangial cell hypertrophy, thus reinforcing the requirement of TGFβ-induced signal transduction in this process (45). We and others have shown a pivotal role of mTOR in pathologic renal hypertrophy in vitro and in vivo (14, 30, 42, 46). Mechanistically, we established phosphorylation of the tumor suppressor protein tuberin by the TGFβ-activated PI 3-kinase/Akt signaling pathway resulting in activation of TORC1, which lead to mesangial cell hypertrophy (14).
By definition, hypertrophy results from an increase in protein synthesis in the absence of DNA synthesis. A role of TORC1 in protein synthesis is well established (39). It is executed mainly by TORC1 substrates 4EBP-1 along with S6 kinase. Phosphorylated and activated S6 kinase binds or phosphorylates multiple proteins acting at the rate-limiting steps in phases of mRNA translation initiation and elongation (20). Moreover, studies have postulated a role of TORC2 via the Akt hydrophobic motif phosphorylation in protein synthesis, which may involve phosphorylation of 4EBP-1 (13, 34, 47). Although mTOR activity in both TORC1 and TORC2 has been shown to be positively regulated by PI 3-kinase, recent studies have identified negative regulators of mTOR, such as FBXW7 and deptor (26, 48, 49).
Deptor was originally discovered as a protein that contains two tandem DEP (dishevelled, egl-10, and pleckstrin) domains in the N terminus and a PDZ (postsynaptic density 95, discs large, zonular occludens-1) domain in the C terminus (26). It binds directly through its PDZ domain to the FAT domain of mTOR and blocks the kinase activity of both TORC1 and TORC2 (26). Moreover, deptor is an extremely unstable protein, which undergoes rapid degradation via a proteasomal pathway, resulting in activation of both TORC1 and TORC2 (26, 50). In the present study, we demonstrate that activation of the TGFβ receptor serine/threonine kinase decreased the abundance of deptor (Fig. 1).
Although deptor contains phosphorylation sites for multiple kinases including ERK1/2, PKC, and GSK3β, they do not regulate its stability (51). Recent mechanistic studies identified CK1α, S6 kinase 1, and RSK1 as other kinases that contribute to the degradation of deptor (51–53). Furthermore, deptor contains multiple phosphorylation sites for mTOR (TORC1 and TORC2) in a serine-rich domain between the second DEP and C-terminal PDZ domains (26, 51). In fact, phosphorylation of deptor by mTOR (either TORC1 or TORC2 or both) primes its phosphorylation by other kinases, which confer its ability to bind to the F box protein βTRCP1 (51). In line with this observation, we found enhanced TGFβ-induced phosphorylation of deptor, which was sensitive to both TORC1 and TORC2 inhibition (Fig. 3). Furthermore, the TGFβ-induced decrease in deptor expression was dependent on both TORC1 and TORC2 activities (Fig. 5). In fact, our results demonstrate that both rapid and prolonged activation of both kinase complexes are necessary for reduction in deptor levels, resulting in phosphorylation/activation of S6 kinase, phosphorylation/inactivation of 4EBP-1, and activation of Akt as judged by the deptor suppression-sensitive phosphorylation at Ser-473 by TORC2 (Figs. 5 and 6A). Moreover, both early activation of TORC1 and TORC2, and late activation of mTOR, which is controlled by diminished deptor levels, are necessary for TGFβ-induced protein synthesis and hypertrophy of mesangial cells (Figs. 5 and 6).
Full activation of Akt requires phosphorylation at both Thr-308 and Ser-473 sites (20, 47, 54). It has been shown previously that TORC2-mediated phosphorylation may be necessary for phosphorylation of Akt at Thr-308 by PDK-1 (20, 24). In line with this, we find that TGFβ-induced deptor down-regulation resulted in an increase in TORC2 activity, which phosphorylates Akt at Ser-473, and is concomitant with an increase in its phosphorylation at the PDK1 site Thr-308 (Fig. 2, H and J). As inhibition of TORC1 results in increased PI 3-kinase-mediated phosphorylation of Akt at Thr-308, one would expect increased phosphorylation at this site by TGFβ in the presence of PP242, which is an ATP-competitive inhibitor of mTOR (26, 38, 55, 56). In contrast to this notion, we found inhibition of Akt phosphorylation at Thr-308 in TGFβ-treated mesangial cells in the presence of PP242 (Fig. 5C). These results suggest that TGFβ forced inhibition of deptor expression and consequently TORC2 activation contributes to the full activation of Akt in mesangial cells.
The role of canonical TGFβ receptor-specific Smad 3 in mediating renal pathology, including hypertrophy, has been reported (57–59). Our results demonstrate that TGFβ-induced Smad 3 activation is necessary for deptor down-regulation (Fig. 7, A, D, and G). To our knowledge, this is the first evidence for Smad 3 regulation of deptor. TGFβ receptor-specific Smads including Smad 3 contain N-terminal MH-1 and C-terminal MH-2 domains separated by a proline-rich linker region (4). The MH-1 domain confers nuclear import of proteins and takes part in DNA binding, whereas the MH-2 domain binds to the type I receptor and forms a complex with Smad 4 (4, 60). Although Smad 3 acts as a transcriptional activator for many genes induced by TGFβ, it has also been shown to repress certain genes (4, 61). Deptor has been shown to be regulated partly at the level of mRNA expression (26). We found that TGFβ decreased the expression of deptor mRNA (Fig. 1D). In fact using a reporter plasmid, deptor promoter activity was significantly inhibited by TGFβ (data not shown). Therefore, our results demonstrating down-regulation of deptor in response to TGFβ or by Smad 3 may result from transcriptional repression (Fig. 7, A, D, and G).
The MH-2 domain of Smad 3 is known to interact with the phospholipid-binding FYVE domain containing the protein SARA, which targets Smads to the endosomes where active TGFβ signaling occurs (4, 6, 40). Recent evidence supports intracellular localization of mTOR at the endosomes (62–65). Whether Smad 3 binds mTOR in TGFβ-stimulated cells is not known. However, our results provide strong evidence for the requirement of Smad 3 for activation of TORC1/2 and mesangial cell hypertrophy (Figs. 7, B, C, E, F, H, and I, and 8).
Abundance of deptor is significantly low in many cancers (26, 50). A key biological consequence of deptor suppression is regulation of cancer cell survival (26, 50). In fact, deptor inhibition protects tumor cells from apoptosis induced by serum withdrawal. This effect occurs in the absence of phosphorylation of Akt, suggesting a lack of involvement of this kinase in the biological activity elicited by the deptor-repressed state (26). In contrast to these results, in mesangial cells we found that reduced deptor levels in response to TGFβ increased phosphorylation of Akt at both sites Ser-473 and Thr-308, leading to increased protein synthesis (Figs. 1, 2, 5, and 6). Thus, to induce mesangial cell hypertrophy, the contribution of deptor is to regulate Akt activation. In summary, the results presented here are consistent with a model in which TGFβ-stimulated Smad 3 induces deptor down-regulation, which contributes to sustained activation of TORC1 and TORC2 to maintain mesangial cell hypertrophy. Thus, deptor may be amenable for rational drug designing to increase its expression that may ameliorate renal disease such as diabetic nephropathy where mesangial cell hypertrophy plays a pathologic role.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 DK50190 and Veterans Affairs Merit Review grants (to G. G. C.).

This article contains supplemental Figs. S1–S9.
- mTOR
- mechanistic target of rapamycin
- EBP-1
- E-binding protein-1.
REFERENCES
- 1. Lehmann R., Schleicher E. D. (2000) Molecular mechanism of diabetic nephropathy. Clin. Chim. Acta 297, 135–144 [DOI] [PubMed] [Google Scholar]
- 2. Wolf G., Ziyadeh F. N. (1999) Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 56, 393–405 [DOI] [PubMed] [Google Scholar]
- 3. Mason R. M., Wahab N. A. (2003) Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 14, 1358–1373 [DOI] [PubMed] [Google Scholar]
- 4. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 5. Massagué J. (2000) How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 1, 169–178 [DOI] [PubMed] [Google Scholar]
- 6. Tsukazaki T., Chiang T. A., Davison A. F., Attisano L., Wrana J. L. (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 95, 779–791 [DOI] [PubMed] [Google Scholar]
- 7. Miyazono K. (2009) Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 85, 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moustakas A., Heldin C. H. (2009) The regulation of TGFβ signal transduction. Development 136, 3699–3714 [DOI] [PubMed] [Google Scholar]
- 9. Das F., Ghosh-Choudhury N., Venkatesan B., Li X., Mahimainathan L., Choudhury G. G. (2008) Akt kinase targets association of CBP with SMAD 3 to regulate TGFβ-induced expression of plasminogen activator inhibitor-1. J. Cell Physiol. 214, 513–527 [DOI] [PubMed] [Google Scholar]
- 10. Samarakoon R., Higgins C. E., Higgins S. P., Kutz S. M., Higgins P. J. (2005) Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp60(c-src)/MEK-dependent. J. Cell Physiol. 204, 236–246 [DOI] [PubMed] [Google Scholar]
- 11. Samarakoon R., Higgins S. P., Higgins C. E., Higgins P. J. (2008) TGF-β1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J. Mol. Cell Cardiol. 44, 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghosh Choudhury G., Abboud H. E. (2004) Tyrosine phosphorylation-dependent PI 3-kinase/Akt signal transduction regulates TGFβ-induced fibronectin expression in mesangial cells. Cell Signal. 16, 31–41 [DOI] [PubMed] [Google Scholar]
- 13. Mahimainathan L., Das F., Venkatesan B., Choudhury G. G. (2006) Mesangial cell hypertrophy by high glucose is mediated by down-regulation of the tumor suppressor PTEN. Diabetes 55, 2115–2125 [DOI] [PubMed] [Google Scholar]
- 14. Das F., Ghosh-Choudhury N., Mahimainathan L., Venkatesan B., Feliers D., Riley D. J., Kasinath B. S., Choudhury G. G. (2008) Raptor-rictor axis in TGFβ-induced protein synthesis. Cell Signal. 20, 409–423 [DOI] [PubMed] [Google Scholar]
- 15. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 16. Gangloff Y. G., Mueller M., Dann S. G., Svoboda P., Sticker M., Spetz J. F., Um S. H., Brown E. J., Cereghini S., Thomas G., Kozma S. C. (2004) Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell Biol. 24, 9508–9516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin P. M., Sutherland A. E. (2001) Exogenous amino acids regulate trophectoderm differentiation in the mouse blastocyst through an mTOR-dependent pathway. Dev. Biol. 240, 182–193 [DOI] [PubMed] [Google Scholar]
- 18. Murakami M., Ichisaka T., Maeda M., Oshiro N., Hara K., Edenhofer F., Kiyama H., Yonezawa K., Yamanaka S. (2004) mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 24, 6710–6718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guertin D. A., Sabatini D. M. (2007) Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 [DOI] [PubMed] [Google Scholar]
- 20. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR. From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fingar D. C., Blenis J. (2004) Target of rapamycin (TOR). An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151–3171 [DOI] [PubMed] [Google Scholar]
- 22. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 23. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 [DOI] [PubMed] [Google Scholar]
- 24. Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871 [DOI] [PubMed] [Google Scholar]
- 25. Sarbassov D. D., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 [DOI] [PubMed] [Google Scholar]
- 26. Peterson T. R., Laplante M., Thoreen C. C., Sancak Y., Kang S. A., Kuehl W. M., Gray N. S., Sabatini D. M. (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sataranatarajan K., Mariappan M. M., Lee M. J., Feliers D., Choudhury G. G., Barnes J. L., Kasinath B. S. (2007) Regulation of elongation phase of mRNA translation in diabetic nephropathy. Amelioration by rapamycin. Am. J. Pathol. 171, 1733–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dey N., Das F., Mariappan M. M., Mandal C. C., Ghosh-Choudhury N., Kasinath B. S., Choudhury G. G. (2011) MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J. Biol. Chem. 286, 25586–25603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dey N., Ghosh-Choudhury N., Das F., Li X., Venkatesan B., Barnes J. L., Kasinath B. S., Ghosh Choudhury G. (2010) PRAS40 acts as a nodal regulator of high glucose-induced TORC1 activation in glomerular mesangial cell hypertrophy. J. Cell Physiol. 225, 27–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mariappan M. M., Shetty M., Sataranatarajan K., Choudhury G. G., Kasinath B. S. (2008) Glycogen synthase kinase 3β is a novel regulator of high glucose- and high insulin-induced extracellular matrix protein synthesis in renal proximal tubular epithelial cells. J. Biol. Chem. 283, 30566–30575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choudhury G. G., Biswas P., Grandaliano G., Abboud H. E. (1993) Involvement of PKC-α in PDGF-mediated mitogenic signaling in human mesangial cells. Am. J. Physiol. 265, F634–642 [DOI] [PubMed] [Google Scholar]
- 32. Choudhury G. G., Biswas P., Grandaliano G., Fouqueray B., Harvey S. A., Abboud H. E. (1994) PDGF-mediated activation of phosphatidylinositol 3-kinase in human mesangial cells. Kidney Int. 46, 37–47 [DOI] [PubMed] [Google Scholar]
- 33. Choudhury G. G., Ghosh-Choudhury N., Abboud H. E. (1998) Association and direct activation of signal transducer and activator of transcription 1α by platelet-derived growth factor receptor. J. Clin. Invest. 101, 2751–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das F., Ghosh-Choudhury N., Dey N., Mandal C. C., Mahimainathan L., Kasinath B. S., Abboud H. E., Choudhury G. G. (2012) Unrestrained mammalian target of rapamycin complexes 1 and 2 increase expression of phosphatase and tensin homolog deleted on chromosome 10 to regulate phosphorylation of Akt kinase. J. Biol. Chem. 287, 3808–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Das F., Mahimainathan L., Ghosh-Choudhury N., Venkatesan B., Kasinath B. S., Abboud H. E., Ghosh Choudhury G. (2007) TGFβ intercepts nuclear glycogen synthase kinase 3β to inhibit PDGF-induced DNA synthesis in mesangial cells. FEBS Lett. 581, 5259–5267 [DOI] [PubMed] [Google Scholar]
- 36. Ghosh-Choudhury T., Mandal C. C., Woodruff K., St Clair P., Fernandes G., Choudhury G. G., Ghosh-Choudhury N. (2009) Fish oil targets PTEN to regulate NFκB for down-regulation of anti-apoptotic genes in breast tumor growth. Breast Cancer Res. Treat. 118, 213–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lamouille S., Derynck R. (2007) Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feldman M. E., Apsel B., Uotila A., Loewith R., Knight Z. A., Ruggero D., Shokat K. M. (2009) Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hay N., Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 [DOI] [PubMed] [Google Scholar]
- 40. Izzi L., Attisano L. (2004) Regulation of the TGFβ signalling pathway by ubiquitin-mediated degradation. Oncogene. 23, 2071–2078 [DOI] [PubMed] [Google Scholar]
- 41. Chen J. K., Chen J., Neilson E. G., Harris R. C. (2005) Role of mammalian target of rapamycin signaling in compensatory renal hypertrophy. J. Am. Soc. Nephrol. 16, 1384–1391 [DOI] [PubMed] [Google Scholar]
- 42. Lee C. H., Inoki K., Guan K. L. (2007) mTOR pathway as a target in tissue hypertrophy. Annu. Rev. Pharmacol. Toxicol. 47, 443–467 [DOI] [PubMed] [Google Scholar]
- 43. Kasinath B. S., Feliers D., Sataranatarajan K., Ghosh Choudhury G., Lee M. J., Mariappan M. M. (2009) Regulation of mRNA translation in renal physiology and disease. Am. J. Physiol. Renal Physiol. 297, F1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen S., Hoffman B. B., Lee J. S., Kasama Y., Jim B., Kopp J. B., Ziyadeh F. N. (2004) Cultured tubule cells from TGF-β1 null mice exhibit impaired hypertrophy and fibronectin expression in high glucose. Kidney Int. 65, 1191–1204 [DOI] [PubMed] [Google Scholar]
- 45. Kim H. W., Kim B. C., Song C. Y., Kim J. H., Hong H. K., Lee H. S. (2004) Heterozygous mice for TGF-βIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int. 66, 1859–1865 [DOI] [PubMed] [Google Scholar]
- 46. Mariappan M. M., Feliers D., Mummidi S., Choudhury G. G., Kasinath B. S. (2007) High glucose, high insulin, and their combination rapidly induce laminin-β1 synthesis by regulation of mRNA translation in renal epithelial cells. Diabetes 56, 476–485 [DOI] [PubMed] [Google Scholar]
- 47. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 48. Mao J. H., Kim I. J., Wu D., Climent J., Kang H. C., DelRosario R., Balmain A. (2008) FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321, 1499–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zinzalla V., Stracka D., Oppliger W., Hall M. N. (2011) Activation of mTORC2 by association with the ribosome. Cell 144, 757–768 [DOI] [PubMed] [Google Scholar]
- 50. Proud C. G. (2009) Dynamic balancing. DEPTOR tips the scales. J. Mol. Cell Biol. 1, 61–63 [DOI] [PubMed] [Google Scholar]
- 51. Gao D., Inuzuka H., Tan M. K., Fukushima H., Locasale J. W., Liu P., Wan L., Zhai B., Chin Y. R., Shaik S., Lyssiotis C. A., Gygi S. P., Toker A., Cantley L. C., Asara J. M., Harper J. W., Wei W. (2011) mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao Y., Xiong X., Sun Y. (2011) DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Duan S., Skaar J. R., Kuchay S., Toschi A., Kanarek N., Ben-Neriah Y., Pagano M. (2011) mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol. Cell 44, 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cantley L. C., Neel B. G. (1999) New insights into tumor suppression. PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., Gout I., Downes C. P., Lamb R. F. (2004) The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shah O. J., Hunter T. (2005) Tuberous sclerosis and insulin resistance. Unlikely bedfellows reveal a TORrid affair. Cell Cycle 4, 46–51 [DOI] [PubMed] [Google Scholar]
- 57. Wang A., Ziyadeh F. N., Lee E. Y., Pyagay P. E., Sung S. H., Sheardown S. A., Laping N. J., Chen S. (2007) Interference with TGF-β signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am. J. Physiol. Renal Physiol. 293, F1657–1665 [DOI] [PubMed] [Google Scholar]
- 58. Lan H. Y. (2011) Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 7, 1056–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meng X. M., Huang X. R., Chung A. C., Qin W., Shao X., Igarashi P., Ju W., Bottinger E. P., Lan H. Y. (2010) Smad2 protects against TGF-β/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Attisano L., Wrana J. L. (2002) Signal transduction by the TGF-β superfamily. Science 296, 1646–1647 [DOI] [PubMed] [Google Scholar]
- 61. Grocott T., Frost V., Maillard M., Johansen T., Wheeler G. N., Dawes L. J., Wormstone I. M., Chantry A. (2007) The MH1 domain of Smad3 interacts with Pax6 and represses autoregulation of the Pax6 P1 promoter. Nucleic Acids Res. 35, 890–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., Sabatini D. M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mavrakis M., Lippincott-Schwartz J., Stratakis C. A., Bossis I. (2007) mTOR kinase and the regulatory subunit of protein kinase A (PRKAR1A) spatially and functionally interact during autophagosome maturation. Autophagy 3, 151–153 [DOI] [PubMed] [Google Scholar]
- 64. Saito K., Araki Y., Kontani K., Nishina H., Katada T. (2005) Novel role of the small GTPase Rheb. Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 137, 423–430 [DOI] [PubMed] [Google Scholar]
- 65. Flinn R. J., Yan Y., Goswami S., Parker P. J., Backer J. M. (2010) The late endosome is essential for mTORC1 signaling. Mol. Biol. Cell 21, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]