

Background: G-protein-coupled receptor (GPCR) signaling through the GNA12/13-Rho axis has been linked to cancer cell invasion and metastasis.
Results: MicroRNAs regulate GNA13 expression and cancer cell invasion.
Conclusion: Loss of miR-182 and miR-200a in prostate cancer cells induces GNA13 expression and SDF-1-mediated invasion.
Significance: Suppressing GNA13 expression using microRNAs could inhibit GPCR-mediated cancer cell invasion and metastasis in prostate cancers.
Keywords: G protein-coupled receptors (GPCR), G proteins, Invasion, MicroRNA, Oncogene
Abstract
G protein-coupled receptors (GPCRs) and their ligands have been implicated in progression and metastasis of several cancers. GPCRs signal through heterotrimeric G proteins, and among the different types of G proteins, GNA12/13 have been most closely linked to tumor progression. In this study, we explored the role of GNA13 in prostate cancer cell invasion and the mechanism of up-regulation of GNA13 in these cells. An initial screen for GNA13 protein expression showed that GNA13 is highly expressed in the most aggressive cancer cell lines. Knockdown of GNA13 in highly invasive PC3 cells revealed that these cells depend on GNA13 expression for their invasion, migration, and Rho activation. As mRNA levels in these cells did not correlate with protein levels, we assessed the potential involvement of micro-RNAs (miRNAs) in post-transcriptional control of GNA13 expression. Expression analysis of miRNAs predicted to bind the 3′-UTR of GNA13 revealed that miR-182 and miR-141/200a showed an inverse correlation to the protein expression in LnCAP and PC3 cells. Ectopic expression of miR-182 and miR-141/200a in PC3 cells significantly reduced protein levels, GNA13–3′-UTR reporter activity and in vitro invasion of these cells. This effect was blocked by restoration of GNA13 expression in these cells. Importantly, inhibition of miR-182 and miR-141/200a in LnCAP cells using specific miRNA inhibitors elevated the expression of GNA13 and enhanced invasion of these cells. These data provide strong evidence that GNA13 is an important mediator of prostate cancer cell invasion, and that miR-182 and miR-200 family members regulate its expression post-transcriptionally.
Introduction
Despite substantial progress in therapies, prostate cancer is still the second largest killer among cancers in males; in large part due to its propensity to metastasize (1). Efforts to identify new genes and signaling pathways that promote tumor invasion and metastasis have revealed that several G-protein-coupled receptors (GPCRs)2, and their respective ligands function as metastatic drivers in prostate as well other cancer types (2–5). One of the best-studied GPCRs is CXCR4, which is known to be up-regulated in prostate and other cancer types. Increased activity of CXCR4 leads to increased migration of cancer cells toward its ligand SDF-1 and enhanced metastasis (6, 7). Similarly, Thrombin-PAR1 (8) and LPA-LPAR3 (9) are the other ligand-GPCR combinations that are known to be highly expressed in metastatic prostate cancers (2).
GPCRs signal primarily through heterotrimeric G-proteins, consisting of Gα, Gβ, and Gγ subunits. GPCRs trigger binding of GTP to Gα, which are then rendered capable of activating downstream signaling pathways, which can lead to cell proliferation, migration, invasion, and even metastasis of cancer cells (7, 10, 11). Among the different types of Gα proteins that exist in the cells, Gα12/13 (GNA12/GNA13) are among the most important in the context of cancer as these are known to be up-regulated in aggressive cancer cells as well as advanced cancer tissues (11–14). The metastatic potential of GNA12/13 proteins appears to involve activation of its major downstream signaling intermediate, the Rho GTPase (13, 15, 16). Blocking GNA12/13 signaling using the specific inhibitor p115-RGS inhibits invasion, migration, and prevents distant metastasis in mice (12, 13, 17, 18). Conversely, expression of Gα12-QL (a dominant active mutant of GNA12) in breast and prostate cancer cells has been shown to induce in vitro invasion and metastatic spread in mice (12, 13, 19).
Most studies of the roles of GNA12/13 in cancer biology to date have focused on GNA12, and little is known about the specific role(s) of GNA13 in cancers. What is known, however, is that GNA13 expression is markedly increased during prostate cancer progression (13). Thus, one of the primary objectives of this study was to investigate the mechanism of regulation of expression of GNA13 in prostate cancer cells, and the impact of this regulation on cancer cell invasion and migration.
MicroRNAs (miRNAs) are endogenously expressed noncoding RNAs that regulate gene expression post-transcriptionally. Binding of the miRNAs to the 3′-UTR or coding sequence of the target gene can either lead to translational repression or mRNA degradation, eventually suppressing the protein production from the target gene (20). Recently, large-scale variations in miRNA expression have been implicated in development and progression of cancers. miRNAs could either act as OncomiRs by inhibiting tumor suppressor genes or as tumor suppressor miRs by targeting potential oncogenes in the cells (21). For example, miR-21 is a well known OncomiR that is up-regulated in different types of cancers and induces tumor formation, invasion, and metastasis by targeting multiple tumor suppressor genes such as PDCD4, PTEN, etc. (22). The miR-200 family is an example of tumor suppressor miRs, which are lost during cancer progression. MiR200 members target ZEB1 and ZEB2 transcriptional factors and prevent invasion and distant metastasis by preventing epithelial-to-mesenchymal (EMT) transition (23, 24).
In the current study, we found that GNA13 expression is essential for prostate cancer cell invasion and migration and that miR-182 and miR-200 family members post-transcriptionally regulate GNA13. Identifying GNA13 as an important contributor to cancer cell invasion, and a regulatory mechanism for its expression could facilitate development of novel strategies to inhibit cancer invasion and metastasis.
EXPERIMENTAL PROCEDURES
Cell Lines, Reagents, and Plasmids
HEK293, HEK293T, MDA-MB-231, MCF-10a, and PC3 were purchased from American Type Culture Collection (ATCC). LnCAP cells were a kind gift from Dr. Marie-Veronique Clement (Department of Biochemistry, National University of Singapore, Singapore). Cells were maintained in RPMI or DMEM complete media with 10% FBS and 1% penicillin/streptomycin (GIBCO). Matrigel inserts, plates, and growth factor reduced Matrigel were purchased from BD Biosciences. Monoclonal antibody against Gα13 (ST1629) was from Calbiochem, Germany and monoclonal antibodies against α-tubulin (#010M4813) were purchased from Sigma. cDNAs expressing Gα13 cloned in pCDNA3.1 were obtained from University of Missouri cDNA Resource, Rolla, MO. MicroRNA mimics (PremiRs) for miR-182 (MSY0000259), miR-141 (MSY0000432), miR-200a (MSY0000682), PremiR-control, and antimiR-182 (MIN0000259), antimiR-141 (MIN0000432), antimiR-200a (MIN0000682), and antimiR-control were purchased from Qiagen GmBH, Germany. Recombinant SDF-1, thrombin, and EGF were purchased from Sigma.
Construction of GNA13-3′-UTR-Luciferase Plasmid
The full-length GNA13 3′-UTR (4941 base pairs) was cloned by performing a nested PCR using genomic DNA from PC3 cells (Fwd.Primer: 5′-TCTGCATGACAACCTCAAGC-3′, Rev.Primer: 5′-TTGAATTGTTTACAAATGTTTATTAAATGTC-3′). The amplified product was purified using PCR purification kit (Qiagen GmBH, Germany), and the 1:10 diluted PCR product from the external PCR was used to amplify a 4941 base pair product (Fwd.XhoI: ATCGCTCGAGTGTACAAAAGACTTGCTGTTTTAATATCTT, Rev.NotI: ATCATATGCGGCCGCAAATGTCAGTAATTTTTACAAAGCAAA) that was then cloned downstream to Renilla luciferase gene (reporter) driven by 5′-LTR promoter in miR-Sens retroviral reporter vector (25) using XhoI and NotI sites. The miR-Sens-Vector also carries a firefly luciferase gene driven by thymidine kinase promoter, which is used as a control to normalize the Renilla (reporter) activity in the reporter assays as detailed previously (25).
Site-directed Mutagenesis of miR-Sens-GNA13–3′-UTR
The seed sequences for miR-182 at 1838–1844 within the GNA13–3′-UTR “UUGCCAAA” was mutated to “UUGAACCC,” and miR-141/200a at “CAGUGUUA” to “CAGUCGGC” as shown here in underlined letters using specific primers carrying the mutant sequence, using the QuickChangeTM site-directed mutagenesis kit as per the manufacturer's instruction (Stratagene). Briefly, the wild type miR-Sens-GNA13–3′-UTR was used as a template to amplify the mutant-182 or mutant-141/200a 3′-UTR using specific primers (carrying the mutations) as shown in the supplemental Table S1. The parental plasmid was digested using DPN-1, and the mutant plasmid was amplified by bacterial transformation. The double mutant was created using MUT-182 as a template and mutational primers of mutant 200a to amplify the double mutant. The wild type and all the mutants were verified by sequencing.
Retro Viral Packaging and Luciferase Assays
Virus for the miR-Sens-Vector, miR-Sens -GNA13–3′UTR wt., miR-Sens-MUT-182, miR-Sens-MUT-200a, and miR-Sens-DD (double mutant), was produced using (pCL-Ampho), amphotropic virus packaging plasmid in HEK293T cells as described previously (25). Briefly, retroviruses were prepared by transfection of HEK293T cells with DNA precipitated using lipofectamine (Invitrogen) according to standard procedures. Retroviruses were pseudotyped with amphotropic (pCL-Ampho) envelope. DNA of the miR-Sens vector constructs was cotransfected along with the packaging plasmid. Retroviral supernatants were harvested at 48 and 72 h after transfection, pooled on ice, aliquoted, and flash frozen. For basal reporter assays, cell transduction (2–5 × 104 cells) was carried out with 150 μl of viral supernatant and 350 μl of media in the presence of 8 μg/ml of polybrene (Sigma) in triplicates in 24-well plates. For reporter assays with premiRs and antimiRs, 100 nm PremiRs in PC3, and 100 nm antimiRs in LnCAP cells as indicated or respective controls were transfected using Lipofectamine 24 h after viral transduction. Luciferase assay was performed after 48 h of viral transduction using Dual Luciferase assay kit (Promega) as described previously (22, 25). The relative light units (RLU) were calculated using the ratio of Renilla luciferase/firefly luciferase, and the final values were normalized against miR-Sens-Vector control and the normalized values were plotted in the graph, respectively.
Preparation of Cell Lysates and Western Blots
Cells were seeded in a 6-well plates and transfected with premiRs, antimiRs, or control miRs (100 nm) as indicated or 1 μg of pCDNA3.1-GNA13 or shRNA-containing plasmid, respectively. After 48 h of transfection, the cells were washed with phosphate-buffered saline and lysed in protein extraction buffer (50 m HEPES pH 7.5, 1 mm EDTA, 3 mm dithiothreitol, 10 mm MgSO4, 1% polyoxyethylene-10-lauryl ether) with protease inhibitors. Protein concentration was determined by BCA (Pierce). Aliquots (20 μg) were separated on a 10% SDS-PAGE and transferred to nitrocellulose membrane. The membrane was incubated with the specific antibody (GNA13 or tubulin) followed by horseradish peroxidase-linked immunoglobulin G (Millipore), and visualized by chemiluminescence (ECL, Thermo Scientific).
Real-time PCR-based Detection of MicroRNAs and GNA13-mRNA
Total RNA from cells was extracted using Trizol (Qiazol, Qiagen). cDNA synthesis was performed using 1 μg of total RNA using miScript-RT-II kit (Qiagen). Expression of mature miRNAs was determined using the miScript primer assays for specific microRNAs (Qiagen GmBH, Germany), and normalized using the 2−ΔΔCTmethod, using Syber Green real-time PCR (26) relative to small nucleolar RNA25 (snoRD25). GNA13-mRNA was quantified using Quantitect primer assays specific for GNA13 (QT000079968) (Qiagen) using the same cDNA. All Syber Green PCRs were performed in triplicates using Quantitect Syber Green PCR mix (Qiagen). A melting curve analysis was performed for each of the primer sets used, and each showed a single peak indicating the specificity of each of the primers tested.
Matrigel Invasion Assays
Cells were transfected with 100 nm control-anti-miR, or anti-miRs, control-PremiR, or pre-miRs as indicated, (1 μg) pCDNA3.1-GNA13 and or shRNA-600, respectively. Lipofectamine was used to transfect LnCAP cells while PC3 cells were transfected with Jetprime (Polyplus). After 24 h (LnCAP) or 48 h (PC3), cells were trypsinized, and 1 × 105 cells were seeded on transwell chambers precoated with 20 μg of Matrigel for PC3 cells and 10 μg of Matrigel for LnCAP cells. Media with or without 100 ng/ml SDF-1 in the lower chamber served as chemo attractant. After an additional 24 h, non-invading cells were removed with cotton swabs. Invading cells were trypsinized and counted using the Cell-Titer Glo (ATP-luminescence-based cell proliferation assay kit) as described previously (Promega) (27). Remaining cells were plated into a 6-well plate for protein and RNA isolation after 24 h. The same method was applied to analyze PC3 cells stably expressing shRNA.
Rho Activity Assays
PC3-shRNA stable cells (1 × 106) were seeded in a 10-cm dish. After 24 h, the cells were placed in starvation media for the next 24 h. SDF-1 (100 ng/ml) or thrombin (2 units/ml) was used to stimulate the cells for 10–15 min. The cells were lysed with 200 μl of G-LISA™ lysis buffer with protease inhibitors provided with G-LISA™ Rho activity assay kit (Cytoskeleton). The RhoA G-LISA™ kit contains a Rho-GTP-binding protein linked to the wells of a 96-well plate. Active, GTP-bound Rho in cell lysates binds to the wells while inactive GDP-bound Rho is removed during washing steps. The bound active RhoA is detected with a RhoA specific antibody and chemiluminescence. The degree of RhoA activation is determined by comparing readings from activated cell lysates versus non-activated cell lysates; 30 μl of the cell lysate was added in each well, which was then incubated at 4 °C for 30 min followed by washes and addition of primary Rho specific antibody (1:1000, 45 min at 4 °C) and secondary antibody (1:2000, 45 min at room temperature). Chemiluminescence was measured using the ECL reagents provided by the manufacturer. The light units obtained using TECAN reader were then normalized to the total protein concentration of each sample and plotted in the graph relative to starved cells.
Cloning of shRNAs and Production of Stable Cell Lines
Oligos targeting GNA13 were generated using iRNAi software for siRNA and shRNA designing. The plasmid expressing shRNA targeting GNA13 were constructed by ligating annealed oligonucleotides encoding for the shRNA with 5′-BglII and 3′-HindIII overhangs (supplemental Fig. S1) to a modified pRetro-Super vector at the BglII/HindIII sites. Prior to ligation, 500 pmol of each complementary sequence of the shRNA encoding sequences were annealed in a total volume of 11 μl of hybridization buffer (0.1 m NaCl, 10 mm MgCl2, 20 mm Tris-HCl, pH 7.4) by heating at 96 °C for 5 min and gradually cooled to room temperature in 2 h. A 5× molar excess of the annealed oligonucleotides over the linearized plasmid was utilized in the ligation performed using 1 unit of T4 DNA ligase (Fermentas) at 22 °C for 1 h, followed by heat inactivation at 65 °C for 10 min. The sequence of the plasmid was verified by restriction mapping and sequencing. Retroviral packaging was done as described above for miR-sens retroviral constructs. 1 ml of retroviral supernatant was used to transduce 5 × 105 of PC3 cells in a 10-cm dish in the presence of 8 μg/ml of polybrene (Sigma). A second transduction was performed the following day. The PC3 cells were selected using 10 μg/ml of blasticidine for 7–14 days after the second transduction. Western blots were performed to assess the efficiency of GNA13 knockdown in the selected cells.
Statistics and Online miRNA Binding Prediction Tool
Experiments were performed at least three times, generally in triplicate. The significance was calculated using Student's t test (for all the biological replicates), and a p value less then 0.05 was considered as significant. TargetScan was used to predict the microRNA binding to the GNA13–3′-UTR and the same was validated using miRANDA, PicTar, and miRwalk.
RESULTS
GNA13 Protein Expression Correlates to the Aggressiveness of Cancer Cells
We showed previously that GNA12 and GNA13 are highly expressed in the most aggressive cancer cells (12, 13). Further, blockade of GNA12/GNA13 signaling with a common inhibitor, p115RGS, inhibited invasion and migration of cancer cells in vitro and distant metastasis in vivo (12). Since most of the studies to date have focused on GNA12 and little is known specifically about GNA13 in this context, we wanted to study the effect of GNA13 on invasion and migration of cancer cells. To initiate these studies, we first examined protein expression of GNA13 in four different cell lines of various origins. As shown in Fig. 1A, the more aggressive prostate cancer cell line PC3 had the highest level of GNA13, while the non-aggressive prostate cancer cell line LnCAP had much lower expression as did cell lines such as HEK-293 (embryonic kidney cells) and IMR90 (lung fibroblasts). A similar observation was made with breast cancer cells (supplemental Fig. S2). These data show that GNA13 expression correlates to the aggressiveness of the cancer cells, which is consistent with previous observations (12, 13).
FIGURE 1.

GNA13 is required for PC3 cells invasion and Rho activation. A, expression of GNA13 in different cell lines. Immunoblot analysis of GNA13 and tubulin (loading control) in the indicated cell lines. HEK-G13 is HEK293 cells overexpressing GNA13 (i.e. positive control). B, knockdown of GNA13 protein expression in PC3 cell stably expressing different shRNAs. Shown in the immunoblot GNA13 and tubulin (loading control) in PC3 cells stably expressing the indicated shRNA. C, impact of GNA13 knockdown on SDF-1 induced invasion of PC3 cells. Cells were transduced with shRNA-vector control, shRNA-600, or shRNA-UTR were used to perform in vitro matrigel invasion experiment. The invasion relative to unstimulated sh-UTR (100%) is shown. Each experiment was performed three times with triplicates). White bars, no chemoattractant; black bars, invasion in the presence of SDF-1 (*, p < 0.05). D, impact of GNA13 knockdown on migration of PC3 cells. Cells stably expressing shRNA-600, shRNA-UTR, or shRNA-vector (control) was subjected to wound healing assay. The relative distance migrated was measured at 4 points in the wound. The y axis represents the migrated distance (initial wound width minus wound width after 12 h) in millimeters. The wound width at 0 h is sh-Control = 3.5 mm, sh-600 = 3.84 mm, and sh-UTR = 3.98 mm, respectively. The results are quantified in the lower panel (n = 8; *, p < 0.05). E, impact of GNA13 knockdown on Rho activity in PC3 cells. Assays were performed using G-LISA Rho activity assay kit as per the protocol. RLU corresponds to GTP-bound Rho in the samples. RLU is normalized to the respective total protein concentration measured separately and plotted relative to the unstimulated control (n = 8; *, p < 0.5).
Knockdown of GNA13 Expression Inhibits SDF-1-induced Invasion, Migration, and Rho-A Activity in PC3 Cells
As PC3 cells exhibited high expression of GNA13, they were chosen to examine the functional impact of silencing of this protein expression. To this end, four different specific shRNAs targeting either the coding regions or the 3′-UTR of GNA13 were expressed in PC3 cells using a retroviral vector. All four shRNAs were able to reduce protein expression of GNA13 with various efficacies (Fig. 1B). The stable lines sh-600 (60% knockdown) and sh-UTR (80% knockdown) were selected for further studies.
To determine whether GNA13 was required for PC3 cell invasion and migration, in vitro matrigel invasion assays were performed with SDF-1 as a chemoattractant. Knockdown of the endogenous expression of GNA13 was sufficient to significantly inhibit basal (sh-600 p = 0.01; sh-UTR p = 0.01) as well as SDF-1 induced invasion of these cells (sh-600 p = 0.04; sh-UTR p = 0.02) (Fig. 1C). In a wound-healing assay of cell migration, knockdown of GNA13 similarly inhibited the migration of the cells (p < 0.01) (Fig. 1D). However, GNA13 knockdown did not significantly impact proliferation in PC3 cells (supplemental Fig. S3). Since Rho activation has been implicated in the ability of GNA13 to induce invasion and migration, Rho activation in response to SDF-1 and thrombin stimulation was assessed in the cells. Consistent with the inhibition of migration and invasion, loss of GNA13 dramatically impaired Rho-A activation in these cells (SDF-1, p values Sh-600 = 0.009, Sh-UTR = 0.002; thrombin, p values Sh-600 = 0.008, Sh-UTR = 0.0002 respectively in relation to respective controls) (Fig. 1E). These data indicate that GNA13 is required for the basal as well specific GPCR ligand (e.g. SDF-1)-induced invasion, migration, and Rho activation in PC3 cells.
GNA13 Protein Expression Is Controlled Post-transcriptionally
Because our screen for protein expression showed substantial differential expression of GNA13 in aggressive versus non-aggressive cells, and that silencing of endogenous GNA13 impacted cell invasion and migration, we felt it was important to study the molecular mechanisms controlling expression of GNA13 in cancer cells. Expression of a protein can be regulated at various stages, i.e. transcription, post-transcription, translation, or post-translation. To determine whether there were changes at the transcriptional level that correlate with protein levels, we quantitated mRNA levels for GNA13 using real-time PCR. Interestingly, mRNA expression of GNA13 (Fig. 2A) did not correlate with protein expression (see Fig. 1A) in the four cell lines examined. For example, IMR90 had the highest mRNA expression in the panel while it had very little protein expression in comparison to PC3 cells, which had high protein expression, but relatively less mRNA abundance (Figs. 1A and 2A). These data indicated that GNA13 expression is most likely regulated at the post-transcriptional level.
FIGURE 2.
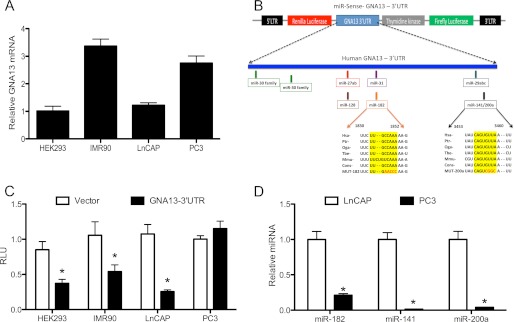
microRNAs -182 and -141/200a regulate GNA13 expression post-transcriptionally. A, GNA13 mRNA expression in 4 different cell lines. The relative GNA13 mRNA, quantified by real-time PCR expression, is shown relative to that in HEK-293 cells. B, schematic representation of GNA13–3′-UTR in the miR-Sens-Luciferase reporter vector, and the predicted miRNA binding sites within the UTR sequence. The miR-182 and miR-141/200a binding sites that are conserved across mammalian species are indicated, and mutations experimentally introduced into the sequences are shown in red. C, GNA13–3′-UTR activity in different cell types. The indicated cells were transduced with miR-Sens-Vector control or miR-Sens-GNA13–3′-UTR, and reporter activity was calculated using Renilla luciferase values normalized to firefly luciferase values (Renilla Luc/Firefly Luc = RLU). Data and statistics were calculated using 9 points (*, p < 0.05). D, expression of specific microRNAs in LnCAP and PC3 cells. Real-time PCR was performed using Qiagen primers specific for microRNAs -182 and -141/200a, and the relative microRNA concentration calculated using the 2−ΔΔCT method relative to the miR-182 level in LnCAP cells (n = 9; *, p < 0.05).
One of the key mechanisms of post-transcriptional regulation in the cells is through small, 22-nucleotide long, non-coding RNAs called microRNAs (miRNA). MiRNAs bind to complementary sequences at the 3′-UTR sequence of specific mRNA and lead to either translational blockage or degradation of mRNA (20). To determine whether GNA13 might be subject to such form of post-transcriptional regulation, we cloned the full-length 3′-UTR (4,941 bp) of GNA13 gene into a retroviral vector (miR-Sens) downstream to a Renilla luciferase gene (Fig. 2B). miR-Sens also carries a firefly luciferase gene driven by thymidine kinase promoter, which was used as a normalizing control. Using this construct, basal GNA13–3′-UTR-Reporter activity of the same four cell lines noted above (Fig. 2C) was found to correlate well with the basal GNA13 protein expression of these cells (see Fig. 1A). HEK293 (p = 0.02), IMR90 (p = 0.02), and LnCAP (p < 0.01) cells with low GNA13 protein expression showed significantly reduced basal GNA13–3′-UTR reporter activity in comparison to vector control (Fig. 2C), while PC3 cells, which had the highest GNA13 protein expression, showed essentially no suppression in the 3′-UTR reporter activities. These data suggested the presence of repressive elements in the GNA13–3′-UTR that might be responsible for the inhibition of expression of GNA13 in the low expressing cells, i.e. HEK293, IMR90, and LnCAP, but that the elements are not active in the high-expressing PC3 cells. Taken together these data indicate that the mechanism of regulation of GNA13 in these cells is post-transcriptional, and point to the possibility of microRNA binding to the GNA13–3′-UTR as being responsible for this control.
GNA13 Expression Is Regulated by MicroRNAs 182 and 141/200a
Because we observed a significant inhibition of GNA13–3′-UTR reporter activity in those cells in which GNA13 protein expression is low, we examined the GNA13–3′-UTR sequence for microRNA binding sites using TargetScan, an online microRNA binding prediction tool. An initial assessment predicted hundreds of microRNA binding sites within the GNA13–3′-UTR (data not shown). However, when we narrowed the list to those binding sites that were highly conserved across mammalian species, seven distinct sites where different microRNAs were predicted to bind were identified (Fig. 2B). The miR-30 family had 2 binding sites, one for miR-27ab/miR-128, and one for miR-31, one for miR-182, one for the miR-29 family, and one for miR-141/200a. These same sites were validated using additional online microRNA binding prediction tools.
To identify which, if any, of the identified microRNAs might be relevant to the regulation of GNA13 expression, we determined the basal expression of the microRNAs in LnCAP (low GNA13) and PC3 (high GNA13) cells and compared this to GNA13–3′-UTR-reporter activity and to basal GNA13 protein expression in these cells. Expression of microRNAs-182, -141, and -200a showed an exact inverse correlation to the basal GNA13–3′-UTR reporter activity as well as to the basal protein expression (Fig. 2, C and D; see also Fig. 1A). LnCAP cells, which showed the lowest GNA13 protein expression (Fig. 1A) and 3′-reporter activity (Fig. 2C), had severalfold higher expression of mir-182, -141, and -200a (Fig. 2D) while PC3 cells had very little or no expression of these microRNAs (Fig. 2D) but high protein expression (Fig. 1A) and reporter activity (Fig. 2C). Other microRNAs did not show significant correlation neither to GNA13–3′-UTR reporter activity nor to the GNA13 basal protein expression (supplemental Fig. S4). These data indicated that microRNAs -182, -141, and -200a were likely candidates for regulating GNA13 expression in LnCAP and PC3 cells.
MiR-182 and miR-141/200a Inhibit GNA13 Expression by Binding to the GNA13–3′-UTR
To determine whether miR-182 and miR-141/200a indeed bind to the GNA13–3′-UTR, we introduced either the miR-Sens-vector or miR-Sens-GNA13–3′-UTR reporters into PC3 cells, and 12 h later these cells were transfected with either the individual PremiRs -182, -141, or -200a, or a combination of PremiRs-182 + 141 or PremiRs-182 + 200a. The reporter assay performed 48 h later indicated that the GNA13–3′-UTR-reporter activity was inhibited 30–35% by miR-182 (p = 0.1), miR-141 (p = 0.01), and miR-200a (p = 0.02), when compared with control microRNA-transfected cells (Fig. 3A). More importantly, the cells transfected with a combination of PremiRs-182 + 141 (p < 0.01) or 182 + 200a (p = 0.02) showed >50% inhibition of the basal GNA13–3′-UTR activity (Fig. 3A). Hence, miR-182 and miR-141/200a inhibit GNA13–3′-UTR reporter activity synergistically.
FIGURE 3.

Enforced expression of miR-182 and miR-141/200a inhibits GNA13 expression in PC3 cells. A, impact of microRNA expression on GNA13-UTR reporter activity in PC3 cells. Cells were transfected with PremiRs or PremiR-control as shown. Reporter assays were performed, and activity was normalized against each respective vector control. Experiments were performed in triplicates and repeated three times (n = 9; *, p < 0.05). B, mutation of seeds sequences alleviates microRNA repression of GNA13–3′-UTR reporter activity. The GNA13–3′-UTR construct was subjected to site-directed mutagenesis at microRNA binding sties as described under “Experimental Procedures.” Reporter assays were performed either in the presence (black bars) or in the absence (white bars) of premirs-182 + 200a (50 nm each). Average reporter activity was normalized against respective vector controls in three independent experiments in triplicates (n = 9; *, p < 0.05). C, impact of microRNA expression on GNA13 protein levels in PC3 cells. Cells were transfected with individual premiRs (100 nm) alone or in combination (50 nm each) as shown. Immunoblot analysis was performed after 48 h; tubulin was probed as a loading control. D, impact of microRNA expression on SDF-1-induced invasion of PC3 cells. Cells were transfected with PremiR-control or a mixture of or premiRs-182 and -141/200a, and the pcDNA-GNA13 as indicated. Invasion assays were performed invasion plotted relative to that of the control (n = 9; *, p < 0.05). E, cell lysates were prepared from the conditions in D and immunoblot analysis of GNA13 and tubulin (loading control) performed.
To confirm that the identified microRNAs directly bind to the GNA13–3′-UTR and that their impact on GNA13–3′-reporter activity was not an indirect effect, the 3′-UTR sequences where these microRNAs are predicted to bind, the so-called “seed” sites (see Fig. 2B), were mutated to abolish binding. Reporter assays performed in PC3 cells with the mutant reporters showed that mutating the individual sites of either miR-182 (p = 0.01) or miR-200a (p = 0.01) rescued the reporter activity from suppression induced by the PremiRs- 182 + 200a combination (Fig. 3B). Moreover, a synergistic rescue of the GNA13–3′-UTR reporter activity from PremiRs-182 + 200a induced suppression was observed when we mutated both miR-182 and miR-141/200a binding sites (p < 0.01) (Fig. 3B). These data provide conclusive evidence that both microRNAs 182 and 141/200a are able to bind to the 3′-UTR of GNA13 and down-regulate the 3′-UTR reporter activity in PC3 cells.
MiR-182 and miR-141/200a Synergistically Inhibit GNA13 Protein Expression and Impair SDF-1 Induced Invasion in PC3 Cells
The data detailed above revealed that miR-182 and miR-141/200a bind to the GNA13–3′-UTR and synergistically inhibit activity of a reporter element carrying the UTR. To test the prediction that these microRNAs would therefore impact expression of the endogenous GNA13 gene, we transfected PC3 cells (that show a high GNA13 protein expression) with PremiRs miR-182, -141, and -200a. Real-time PCR performed 48 h following transfection of PremiRs showed that these microRNAs were expressed in the cells (supplemental Fig. S5). At the same time point, GNA13 protein expression was analyzed in cell lysates by immunoblot. As shown in Fig. 3C, microRNAs-182, -141, and -200a individually suppressed GNA13 protein expression ≈40% when compared with control microRNA. Moreover, a synergistic inhibition of GNA13 expression was observed in those cells in which combinations of these PremiRs were transfected. The combination of PremiR-182 + 141 showed ≈ 70%, and that of PremiR-182 + 200a showed ≈80%, inhibition of GNA13 protein expression, respectively (Fig. 3C). These data indicate that enforced expression of mir-182 and miR-141/200a in PC3 can inhibit the expression of GNA13. Taken together these data indicate that GNA13 is an important target of miR-182 and -141/200a, which bind to the 3′-UTR of GNA13 and down-regulate its protein expression in PC3 cells.
The finding that knockdown of GNA13 in PC3 cells impaired SDF-1 induced invasion (Fig. 1) and that that microRNAs -182 and -200a were able to inhibit the expression of GNA13 (Fig. 3C), prompted us to study the effect of these two microRNAs on PC3 cell invasion. We used miR-200a in these experiments because both miR-141 and miR-200a have the same target sequence and a single binding site at the GNA13–3′-UTR. Cells were transfected with PremiRs-182 + 200a, and 24 h later an in vitro invasion assay was performed using SDF-1 as a chemo-attractant. The combination of microRNAs was indeed able to significantly inhibit invasion of these cells (p = 0.01) (Fig. 3D). Importantly, ectopic expression of GNA13 in the cells effected a partial rescue of invasion from the suppression induced by the PremiRs (Fig. 3D). The invasion of these cells is consistent to GNA13 protein expression in these cells (Fig. 3E). Taken together these data point to GNA13 being an important regulator of SDF-1-induced invasion in PC3 cells and microRNAs -182 and -200a down-regulate invasion partially through down-regulating GNA13 protein expression.
Inhibiting MicroRNA Activity Rescues GNA13 Expression in LnCAP Cells and Confers Responsiveness to SDF-1
In our cell line screen, LnCAP cells had the lowest GNA13 protein expression (Fig. 1A) and GNA13–3′-UTR reporter activity (Fig. 2C), which correlated with high miR-182 and miR-141/200a activity in these cells (Fig. 2D). These data suggest that suppression by these microRNAs might be the reason for the low GNA13 expression in LnCAP cells. To determine whether this was indeed the case, we examined whether blocking miR-182 and miR-141/200a activity in the cells through use of miRNA inhibitors (anti-miRs) could rescue expression of GNA13. LnCAP cells expressing the GNA13–3′-UTR reporter were transfected with Anti-miRs -182, -141, -200a individually or in combination. We found that blocking microRNA activity in these cells significantly increased GNA13–3′-UTR reporter activity (Fig. 4B). The individual AntimiRs-182, -141, and 200a showed a ≈2-fold induction of GNA13–3′-UTR reporter activity (AntimiR-182 p = 0.03, AntimiR-200a p < 0.01) when compared with control. Moreover, an increase in reporter activity of nearly 10-fold was observed with combination of anti-miRs-182 + 141 (p < 0.01) or antimiRs-182 + 200a (p < 0.01) (Fig. 4A). To confirm that binding of these microRNAs to the GNA-13–3′-UTR was required for this activity; we mutated the miRNA binding sites individually or in combination. Mutating either miR-182 (p < 0.01) seed sequence or miR-141/200a (p < 0.01) seed sequence was sufficient to rescue the GNA13–3′-UTR reporter activity from miR-182- and miR-141/200a-induced suppression, and mutation of both sites provided essentially complete rescue (p < 0.01) (Fig. 4B). Taken together, these data clearly indicate that, both mir-182 and miR-141/200a inhibit GNA13 expression in LnCAP cells by directly binding to the GNA13–3′-UTR, and suggested a molecular mechanism for the very low level of GNA13 protein expression in this cell line.
FIGURE 4.

Blockade of miR-182 and miR-141/200a activity rescues GNA13 expression and SDF-1 induced invasion in LnCAP cells. A, LnCAP cells harboring the GNA13–3′-UTR reporter construct were transfected with AntimiRs as shown, and reporter assays performed. GNA13–3′-UTR reporter activity was normalized against the respective vector control. Each experiment was performed in triplicates and repeated twice (n = 6; *, p < 0.05). B, mutation of microRNA binding sites enhances expression of the GNA13–3′-UTR reporter in LnCAP cells. The GNA13–3′-UTR was subjected to site-directed mutagenesis as described under “Experimental Procedures.” Reporter assays were performed 48 h after transduction with either the vector, wild type -3′-UTR, or the mutant UTRs. Reporter activity was normalized against respective vector controls from two independent experiments performed in triplicates (n = 6; *, p < 0.05). C, blockade of miR-182 and miR-141/200a activity rescues GNA13 expression in LnCAP cells. Cells were transfected with the indicated antimiRs alone (100 nm) or in combination (50 nm each) as indicated, and immunoblot was performed after 48 h. Levels of GNA13 and tubulin (loading control) were determined by immunoblot analysis. D, blockade of miR-182 and miR-141/200a confers SDF-1 responsiveness to LnCAP cells. Cells were transfected with a mixture of antimiRs -182 and -141/200a, shRNA-600 targeting GNA13, or the pcDNA3.1-GNA13 construct as indicated. Invasion assays were performed in the presence and absence of SDF-1 as the chemoattractant. Data were normalized to control vector in the absence of SDF-1; (n = 9; *, p < 0.05). E, cell lysates were prepared from the conditions in D and immunoblot analysis of GNA13 and tubulin (loading control) performed.
To directly determine whether mir-182 and miR-141/200a were responsible for the low GNA13 protein expression observed in LnCAP cells, we transfected the cells with antimiRs -182, -141, and -200a, individually and in combination. A robust increase of GNA13 protein expression was observed, particularly with the combination of anti-miRs-182 + 141 or antimiRs-182 + 200a (Fig. 4C). These data are consistent to the increase of GNA13–3′-UTR reporter activity observed by treatment of these cells with the antimiRs (Fig. 4A), and provide compelling evidence that miR-182 and miR-141/200a control protein expression of GNA13 in LnCAP cells.
LnCAP cells are relatively non-responsive to SDF-1 stimulation (Fig. 4D). Since the studies with the PC3 cells (Fig. 1) indicated that the presence of GNA13 in prostate cancer cells was required for the ability of SDF1 to promote invasion, we reasoned that restoring GNA13 expression in LnCAP cells via blockade of miR-182, -141, and -200a activity might confer responsiveness to SDF1. To test this hypothesis LnCAP cells were transfected with, antimiRs-182 + 200a, and the ability of SDF1 to stimulate invasion was assessed. Blocking activity of the microRNAs using specific antimiRs could indeed confer responsiveness to SDF-1 in terms of inducing invasion of these cells (p = 0.02) (Fig. 4D). Importantly, shRNA-mediated knockdown GNA13 blocked the ability of the antimiRs to confer SDF-1-stimulated invasion onto the cells (p = 0.01) (Fig. 4D), and expression of GNA13 alone showed a robust increase in SDF1-induced invasion (Fig. 4D). The invasion of these cells is consistent to GNA13 protein expression in these cells (Fig. 4E).
DISCUSSION
GPCRs are one of the most important classes of cell surface receptors and play pleotropic roles in cell physiology. Recently, many of these receptors and their ligands, e.g. SDF-1, thrombin, LPA, S1P, and endothelin, have been implicated in tumor formation and organ-specific metastasis in prostate, breast, and other cancers (2). All these GPCRs signal through heterotrimeric G proteins, and in particular the G12 subfamily has been implicated in the impact of these signaling pathways on tumor progression. This subfamily consists of two members Gα12 (GNA12) and Gα13 (GNA13), both of which are highly expressed in aggressive breast and prostate cancers (12, 13) and, in their dominant-active forms, can induce invasion and metastatic behavior of many cell types (19, 28, 29).
We report here that one of the most aggressive prostate cancer cell lines, PC3, is highly dependent on GNA13 protein expression for basal as well as SDF-1-induced Rho activity, invasion, and migration. This finding is consistent with observations made in other cell types that a CXCR4-GNA13-Rho axis is important in SDF-1-induced invasion (30–32). Importantly, we also observed thrombin, which is a ligand for PAR-1, induced Rho activity in these cells that was dependent on GNA13 expression. While these data collectively indicate that GNA13 overexpression might be a specific marker for invasiveness of prostate cancer cells, little has been known about the mechanism of deregulation of GNA13 in these cells.
Our finding that there was no clear correlation between GNA13 mRNA and protein expression in prostate cancer cells indicated that regulation of GNA13 might be a post-transcriptional event. Since it has recently become clear that one of the important post-transcriptional mechanisms of gene expression is microRNAs (21, 33), and deregulation of microRNAs has been closely associated with tumor formation, invasion, and metastasis in several cancer types (21, 33), we focused our attention on this possibility. These studies led to the finding that miR-182 and miR-141/200a directly bind to the 3′-UTR of GNA13 and inhibit its protein expression in prostate cancer cells. Indeed, simply by suppressing the activities of these miRNAs it was possible to restore GNA13 expression, and response to SDF-1 induced invasion, in LnCAP cells.
We also observed a synergistic ability of ectopically expressed mir-182 and miR-141/200a to inhibit the expression of GNA13 and impair SDF-1 induced invasion of PC3 cells. Restoring GNA13 expression in PremiR-treated cells was able to partially, but not fully, rescue the SDF-1 induced invasion, indicating that these microRNAs likely (and not unexpectedly) have additional targets important for cell invasion apart from GNA13. In this regard, miR-182 has been reported to impact invasion by inhibiting the PI3K-AKT-mTOR pathway and also by regulating the expression of FOXO1 in medulloblastoma and Hodgkin's Lymphoma (34, 35). However, miR-182 has been reported to induce invasion in melanoma cell lines, indicating that impact of miR-182 on invasion is context dependent (36). Similarly, independent studies have shown that miR-200 family, which includes (miR-141/miR-200a and miR-200c) impact invasion by inhibiting the EMT-inducing transcriptional factors ZEB1 and SIP1/ZEB2 in breast and colorectal cancers (23, 24, 37). One of the important mechanisms by which miR-200a inhibits invasion in meningioma is by inducing E-cadherin and subsequently induction of WNT signaling pathways (38). In our experiments with PremiR 200a induced suppression of invasion in PC3 cells; however, we did not see an up-regulation of E-cadherin (supplemental Fig. S6). Additionally, several reports have shown that both miR-182 and mir-141/200a are lost during progression of several cancer types (23, 24, 37, 39). These findings underscore an emerging role for both miR-182 and miR-141/200a in inhibiting cancer cell invasion by targeting multiple proteins in cells.
In summary, we have found that GNA13 is required for basal and SDF-1-induced invasion of prostate cancer cells, and identified two microRNAs, miR-182 and miR-141/200a, which synergistically inhibit GNA13 protein expression and cancer cell invasion. Since GNA13 could act as a common mediator for multiple oncogenic GPCR signals in cancer cells, identifying mechanisms of its deregulation could lead to novel microRNA based, strategies to inhibit cancer invasion and metastasis in the future. Further studies should reveal whether loss of miR-182 and miR-200 family together, with the gain of GNA13 in the same patient tissues, could be used as a prognostic marker in prostate cancers.

This article contains supplemental Table S1 and Figs. S1—S6.
- GPCR
- G-protein-coupled receptor
- miRNA
- microRNA
- EMT
- epithelial-to-mesenchymal
- RLU
- relative light units.
REFERENCES
- 1. Siegel R., Naishadham D., Jemal A. (2012) Cancer Statistics. Cancer J. Clin. 62, 10–29 [DOI] [PubMed] [Google Scholar]
- 2. Lappano R., Maggiolini M. (2012) GPCRs and cancer. Acta Pharmacologica Sinica 33, 351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spiegelberg B. D., Hamm H. E. (2007) Roles of G-protein-coupled receptor signaling in cancer biology and gene transcription. Curr. Opin. Gen. Dev. 17, 40–44 [DOI] [PubMed] [Google Scholar]
- 4. Pi M., Quarles L. D. (2012) GPRC6A regulates prostate cancer progression. The Prostate 72, 399–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kasina S., Macoska J. A. (2012) The CXCL12/CXCR4 axis promotes ligand-independent activation of the androgen receptor. Mol. Cell Endocrin. 351, 249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Darash-Yahana M., Pikarsky E., Abramovitch R., Zeira E., Pal B., Karplus R., Beider K., Avniel S., Kasem S., Galun E., Peled A. (2004) Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 18, 1240–1242 [DOI] [PubMed] [Google Scholar]
- 7. Li S., Huang S., Peng S. B. (2005) Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int. J. Oncol. 27, 1329–1339 [PubMed] [Google Scholar]
- 8. Hains M. D., Wing M. R., Maddileti S., Siderovski D. P., Harden T. K. (2006) Galpha12/13- and rho-dependent activation of phospholipase Cϵ by lysophosphatidic acid and thrombin receptors. Mol. Pharm. 69, 2068–2075 [DOI] [PubMed] [Google Scholar]
- 9. Zeng Y., Kakehi Y., Nouh M. A., Tsunemori H., Sugimoto M., Wu X. X. (2009) Gene expression profiles of lysophosphatidic acid-related molecules in the prostate: relevance to prostate cancer and benign hyperplasia. The Prostate 69, 283–292 [DOI] [PubMed] [Google Scholar]
- 10. Wettschureck N., Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 [DOI] [PubMed] [Google Scholar]
- 11. Kelly P., Casey P. J., Meigs T. E. (2007) Biologic functions of the G12 subfamily of heterotrimeric g proteins: growth, migration, and metastasis. Biochemistry 46, 6677–6687 [DOI] [PubMed] [Google Scholar]
- 12. Kelly P., Moeller B. J., Juneja J., Booden M. A., Der C. J., Daaka Y., Dewhirst M. W., Fields T. A., Casey P. J. (2006) The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U.S.A. 103, 8173–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelly P., Stemmle L. N., Madden J. F., Fields T. A., Daaka Y., Casey P. J. (2006) A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J. Biol. Chem. 281, 26483–26490 [DOI] [PubMed] [Google Scholar]
- 14. Cheong S. C., Chandramouli G. V., Saleh A., Zain R. B., Lau S. H., Sivakumaren S., Pathmanathan R., Prime S. S., Teo S. H., Patel V., Gutkind J. S. (2009) Gene expression in human oral squamous cell carcinoma is influenced by risk factor exposure. Oral. Oncology 45, 712–719 [DOI] [PubMed] [Google Scholar]
- 15. Kozasa T., Hajicek N., Chow C. R., Suzuki N. (2011) Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J. Biochem. 150, 357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Z., Guo L., Hadas J., Gutowski S., Sprang S. R., Sternweis P. C. (2012) Activation of p115-RhoGEF Requires Direct Association of Gα13 and the Dbl Homology Domain. J. Biol. Chem. 287, 25490–25500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malchinkhuu E., Sato K., Maehama T., Mogi C., Tomura H., Ishiuchi S., Yoshimoto Y., Kurose H., Okajima F. (2008) S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Comm. 366, 963–968 [DOI] [PubMed] [Google Scholar]
- 18. Shumay E., Tao J., Wang H. Y., Malbon C. C. (2007) Lysophosphatidic acid regulates trafficking of β2-adrenergic receptors: the Gα13/p115RhoGEF/JNK pathway stimulates receptor internalization. J. Biol. Chem. 282, 21529–21541 [DOI] [PubMed] [Google Scholar]
- 19. Kim E. S., Lee K. M., Noh D. Y., Moon A. (2011) Regulation of matrix metalloproteinases and invasion by G(α12/13) proteins in NIH3T3 mouse fibroblast cells. Oncology Res. 19, 297–301 [DOI] [PubMed] [Google Scholar]
- 20. Huang Y., Shen X. J., Zou Q., Wang S. P., Tang S. M., Zhang G. Z. (2011) Biological functions of microRNAs: a review. J. Physiol. Biochem. 67, 129–139 [DOI] [PubMed] [Google Scholar]
- 21. Rutnam Z. J., Yang B. B. (2012) The involvement of microRNAs in malignant transformation. Histology Histopathol. 27, 1263–1270 [DOI] [PubMed] [Google Scholar]
- 22. Asangani I. A., Rasheed S. A., Nikolova D. A., Leupold J. H., Colburn N. H., Post S., Allgayer H. (2008) MicroRNA-21 (miR-21) post-transcriptionally down-regulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27, 2128–2136 [DOI] [PubMed] [Google Scholar]
- 23. Gregory P. A., Bert A. G., Paterson E. L., Barry S. C., Tsykin A., Farshid G., Vadas M. A., Khew-Goodall Y., Goodall G. J. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601 [DOI] [PubMed] [Google Scholar]
- 24. Burk U., Schubert J., Wellner U., Schmalhofer O., Vincan E., Spaderna S., Brabletz T. (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beillard E., Ong S. C., Giannakakis A., Guccione E., Vardy L. A., Voorhoeve P. M. (2012) miR-Sens–a retroviral dual-luciferase reporter to detect microRNA activity in primary cells. RNA 18, 1091–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rasheed S. A., Efferth T., Asangani I. A., Allgayer H. (2010) First evidence that the antimalarial drug artesunate inhibits invasion and in vivo metastasis in lung cancer by targeting essential extracellular proteases. Int. J. Cancer 127, 1475–1485 [DOI] [PubMed] [Google Scholar]
- 28. Sayas C. L., Avila J., Wandosell F. (2002) Glycogen synthase kinase-3 is activated in neuronal cells by Gα12 and Gα13 by Rho-independent and Rho-dependent mechanisms. J. Neuro. 22, 6863–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Juneja J., Cushman I., Casey P. J. (2011) G12 signaling through c-Jun NH2-terminal kinase promotes breast cancer cell invasion. PloS One 6, e26085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan W., Martin D., Gutkind J. S. (2006) The Gα13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J. Biol. Chem. 281, 39542–39549 [DOI] [PubMed] [Google Scholar]
- 31. Shan D., Chen L., Wang D., Tan Y. C., Gu J. L., Huang X. Y. (2006) The G protein Gα(13) is required for growth factor-induced cell migration. Dev. cell 10, 707–718 [DOI] [PubMed] [Google Scholar]
- 32. Yagi H., Tan W., Dillenburg-Pilla P., Armando S., Amornphimoltham P., Simaan M., Weigert R., Molinolo A. A., Bouvier M., Gutkind J. S. (2011) A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Science Signal. 4, ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z. J., Ma S. L. (2012) miRNAs in breast cancer tumorigenesis (Review). Oncology Rep. 27, 903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weeraratne S. D., Amani V., Teider N., Pierre-Francois J., Winter D., Kye M. J., Sengupta S., Archer T., Remke M., Bai A. H., Warren P., Pfister S. M., Steen J. A., Pomeroy S. L., Cho Y. J. (2012) Pleiotropic effects of miR-183∼96∼182 converge to regulate cell survival, proliferation and migration in medulloblastoma. Acta Neuropathol. 123, 539–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie L., Ushmorov A., Leithäuser F., Guan H., Steidl C., Färbinger J., Pelzer C., Vogel M. J., Maier H. J., Gascoyne R. D., Möller P., Wirth T. (2012) FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 119, 3503–3511 [DOI] [PubMed] [Google Scholar]
- 36. Segura M. F., Hanniford D., Menendez S., Reavie L., Zou X., Alvarez-Diaz S., Zakrzewski J., Blochin E., Rose A., Bogunovic D., Polsky D., Wei J., Lee P., Belitskaya-Levy I., Bhardwaj N., Osman I., Hernando E. (2009) Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc. Natl. Acad. Sci. U.S.A. 106, 1814–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu M., Xia M., Chen X., Lin Z., Xu Y., Ma Y., Su L. (2010) MicroRNA-141 regulates Smad-interacting protein 1 (SIP1) and inhibits migration and invasion of colorectal cancer cells. Dig. Dis. Sci. 55, 2365–2372 [DOI] [PubMed] [Google Scholar]
- 38. Saydam O., Shen Y., Würdinger T., Senol O., Boke E., James M. F., Tannous B. A., Stemmer-Rachamimov A. O., Yi M., Stephens R. M., Fraefel C., Gusella J. F., Krichevsky A. M., Breakefield X. O. (2009) Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol. Cell Biol. 29, 5923–5940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barshack I., Lithwick-Yanai G., Afek A., Rosenblatt K., Tabibian-Keissar H., Zepeniuk M., Cohen L., Dan H., Zion O., Strenov Y., Polak-Charcon S., Perelman M. (2010) MicroRNA expression differentiates between primary lung tumors and metastases to the lung. Pathol. Res. Prac. 206, 578–584 [DOI] [PubMed] [Google Scholar]