

Abstract
Background
MicroRNAs (miRNAs) are endogenous regulators of a broad range of physiological processes and act by either degrading mRNA or blocking its translation. Oilseed rape (Brassica napus) is one of the most important crops in China, Europe and other Asian countries with publicly available expressed sequence tags (ESTs) and genomic survey sequence (GSS) databases, but little is known about its miRNAs and their targets. To date, only 46 miRNAs have been identified in B. napus.
Results
Forty-one conserved and 62 brassica-specific candidate B. napus miRNAs, including 20 miRNA* sequences, were identified using Solexa sequencing technology. Furthermore, 33 non-redundant mRNA targets of conserved brassica miRNAs and 19 new non-redundant mRNA targets of novel brassica-specific miRNAs were identified by genome-scale sequencing of mRNA degradome.
Conclusions
This study describes large scale cloning and characterization of B. napus miRNAs and their potential targets, providing the foundation for further characterization of miRNA function in the regulation of diverse physiological processes in B. napus.
Background
Endogenous small RNAs (sRNAs) are known to be important regulators of gene expression at the transcriptional and post-transcriptional levels. They fall into a number of different classes in plants: transacting siRNAs (tasiRNAs), heterochromatin-associated siRNAs, natural antisense siRNAs (nat-siRNAs) and miRNAs [1]. These classes of non-coding RNAs are distinguished by their biogenesis pathways and the types of genomic loci from which they arise [2].
miRNAs are non-coding RNAs of approximately 21 nucleotides that have been identified as important regulators of gene expression in both animals and plants [2-5]. Plant miRNAs are generated from hairpin-structured non-coding transcripts by DCL1(DICER-LIKE 1), which cleaves a short (21 bp) duplex from the stem region [6]. The duplex is incorporated into an AGO1 complex and the miRNA* strand is subsequently degraded. The mature miRNA strand guides the AGO1 complex (RNA-induced silencing complex, RISC) to protein-coding RNAs, which are cleaved by AGO1 at a specific position (opposite to the 10th and 11th nucleotides of the miRNA) [7]. Recent findings have shown that the inhibition of gene expression via translational arrest by the miRNA-guided AGO complex is more common in plants than was previously believed [8]. The mature miRNAs function within large complexes to negatively regulate specific target mRNAs. Plant miRNAs generally interact with their targets through perfect or near-perfect complementarity and direct mRNA target degradation [9,10]. Due to their evolutionary conservation, miRNAs have been found to exist in both plants [9,11] and animals [12-14]. Conserved miRNA molecules can also be found in ferns, mosses and fungi [15,16].
In plants, miRNAs not only post-transcriptionally regulate their own targets but also interact with each other in regulatory networks to affect many aspects of development, such as developmental timing [17-21], senescence [22-24], leaf morphogenesis [25-31], reproductive development [32-35], and modulation of root architecture [36-42]. miRNAs are also reported to be involved in plant responses to biotic and abiotic stresses [31]. To date an increasing number of miRNAs have been identified and deposited in miRBase V17.0 (http://www.mirbase.org/). Among these miRNAs, there are 19,724 plant miRNAs and miRNAs*, from a total of 153 species. The species with the fastest growing number of miRNAs is Brachypodium distachyon, with 120 miRNAs being recently added. Initially, miRNAs were identified by the traditional Sanger sequencing method, which used for relatively small-size cDNA libraries of plant sRNAs from Arabidopsis, rice and poplar (Populus spp.). Comparison of miRNAs from these species led to the conclusion that plant miRNAs are highly conserved [16]. This was supported by observations that even ferns shared common miRNAs with flowering plants [43]. However, it was also noticed that a small number of miRNAs were not present in the genomes of some species, suggesting that they have evolved more recently [25]. As non-conserved miRNAs are often expressed at a lower level than conserved miRNAs, many non-conserved miRNAs were not found in small-scale sequencing projects. However, high-throughput sequencing technologies have allowed the identification of many non-conserved miRNAs in several species [44-47]. To date, hundreds of miRNAs have been isolated by direct cloning or by deep sequencing in higher plants [48]. Elucidating the function of these tiny molecules requires efficient approaches to identify their targets. Originally, plant miRNA targets have been studied via computational prediction, which is based on either perfect or near -perfect sequence complementarity between miRNA and the target mRNA or sequence conservation among different species [10]. However, target prediction is very challenging, especially when a high level of mismatches exists in miRNA:target pairing [49]. Recently, a new method called degradome sequencing, which combines high-throughput RNA sequencing with bioinformatic tools, has been successfully established to screen for miRNA targets in Arabidopsis[50-52]. Using degradome sequencing, many of the previously validated and predicted targets of miRNAs and tasiRNAs were verified [50,51,53,54], indicating that it is an efficient strategy to identify sRNA targets on a large scale in plants.
Rape (Brassica napus) is one of the most important oil crops, and also is one of the major economic crops. However, unlike Arabidopsis and other plants, much less is known about its miRNA classification and miRNA targets, especially the roles of miRNAs in the developmental process of Brassica napus. Currently, miRBase lists 46 miRNAs forming 17 miRNA families in Brassica napus. The exploration of sRNA-based regulatory networks in Brassica napus is an important step towards our better understanding of sRNA-based genic regulation. Here, we describe the high-throughput sequencing analysis of sRNAs from a cultivated variety of B. napus, cv Westar, using the Illumina Solexa platform.
The sRNAs library was prepared for Solexa sequencing from greenhouse cultivated rape plants, and produced more than 2 million unique sequences. The most abundant classes were represented by 21 and 24 nt-long sRNAs. Forty-one conserved B. napus miRNAs and 62 candidate novel B. napus-specific miRNAs were firstly identified. Twelve conserved miRNAs and 10 B. napus-specific candidates were further verified by real-time RT-PCR. To identify miRNA targets, a degradome sequencing approach was used, which globally identifies the remnants of sRNA-directed target cleavage by sequencing the 5′ ends of uncapped RNAs [50,51]. We identified a total of 33 non-redundant target ESTs for 25 conserved miRNAs, and 19 non-redundant target ESTs for 17 B. napus-specific miRNAs. Approximately 70% of the identified targets for conserved miRNAs were transcriptional factors.
Results and discussion
Sequencing B. napus miRNAs using Solexa technology
We used Solexa technology to deeply sequence B. napus sRNAs. Total RNAs from different B. napus tissues were pooled and submitted for small RNA sequencing. A total of 13,020,106 reads were generated from the sequencing machine. After removing adaptor sequences, filtering out low quality tags and cleaning up sequences derived from adaptor-adaptor ligation, 2,149,116 unique sequences were obtained. Among these unique sequences, 73,931 (3.44%) were found to be similar to known miRNAs (Table 1).
Table 1.
Statistics of sRNA sequences from B. napus
| |
Redundant |
Non-redundant |
||
|---|---|---|---|---|
| Number of counts | % of total | Number of unique sequence | % of total | |
| Raw reads |
13020106 |
\ |
2149116 |
\ |
| Adaptor removed |
30673 |
0.24 |
25713 |
1.2 |
| Junk Filter a |
5182 |
0.04 |
2362 |
0.11 |
| Length filter |
1379794 |
10.6 |
646357 |
30.08 |
| Simple sequence filter b |
69175 |
0.53 |
11859 |
0.55 |
| Copy number <3 |
1219472 |
9.37 |
1130389 |
52.6 |
| Hit mRNA, RFam, Repbase |
9877816 |
75.87 |
57637 |
2.68 |
| Mappable | 437994 | 3.36 | 30400 | 1.41 |
a the sequences are filtered out if they contain 3 Ns (N is undetermined nucleotide) and only A and C without G and T, vice versa; b the simple sequences are filtered out if they contain 2 Ns (N is undetermined nucleotide), 7 consecutive As, 8 consecutive Cs, 6 consecutive Gs or 7 consecutive Ts and 10 repeats of any dimer, 6 repeats of any trimer, or 5 repeats of any tetramer. These numbers were from the statistics of miRbase ver16.
SRNAs with known function were commonly 20–24 nt in size [53]; therefore, we analyzed the size distribution patterns of the original and unique reads (Figure 1). The majority of sRNAs were 21 nt in size, followed by 24 nt and 23 nt (Figure 1a), which is consistent with the typical size distribution of sRNAs from other plants. The 21 nt class showed the highest redundancy, whereas the 24 nt class showed lower redundancy (Figure 1a and b).
Figure 1.
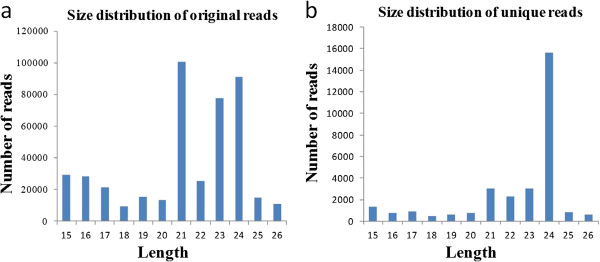
Size distribution of sequenced small RNAs.
Identification of conserved B. napus miRNAs
Conserved families of miRNAs are found in many plant species and have important functions in plant development and responses to stresses [55]. In this study, to identity the conserved miRNAs in B. napus, our dataset was mapped onto the the genome and ESTs of B. napus, B. rapa and B. oleracea, allowing one or two mismatches between sequences. all retained sequences were compared to known miRNAs from miRBase 17.0 (http://www.mirbase.org/), and secondary structures of these matched miRNAs were predicted. Based on genome mapping and the miRbase results and hairpin prediction, a total of 55conserved miRNAs derived from B. napus were identified, including 41 miRNAs and miRNAs* (22 families) were firstly identified together with 14 already in miRbase (Additional file 1: Figure S1, Additional file 2: Table S2). 41conserved miRNAs and miRNAs* were potentially generated from 26 non-redundant ESTs and 3 genomic survey sequence (GSS) loci (Table 2; Additional file 3: Table S3). The precursors of four miRNAs named Bna-miR166f, Bna-miR824*, Bna–miR1140b and Bna–miR1140b* were matched in the genome of B. rapa (Additional file 3: Table S3).
Table 2.
Conserved miRNAs in B. napus
|
miRNA |
miR sequence (5'→3') |
miR length (nt) |
Reads |
Precursor from EST |
Mature miRNA position |
Stem-loop position |
||
|---|---|---|---|---|---|---|---|---|
| miR start | miR end | Precursor start | Precursor end | |||||
| Bna--miR159* |
GGGCTCCTTATAGTTCAAACG |
21 |
79 |
EX039355 |
189 |
209 |
186 |
368 |
| Bna--miR159b |
TTTGGATTGAAGGGAGCTCTT |
21 |
66 |
EV097138 |
224 |
244 |
216 |
408 |
| Bna-miR160a |
TGCCTGGCTCCCTGTATGCCA |
21 |
4447 |
ES904429 |
153 |
173 |
149 |
237 |
| Bna-miR160a* |
GCGTACAGAGTAGTCAAGCATA |
22 |
7 |
ES904429 |
214 |
235 |
149 |
237 |
| Bna-miR161* |
GCAAGTCGACTTTGGCTCTG |
20 |
97 |
BZ512955 |
431 |
451 |
329 |
463 |
| Bna-miR162a |
TCGATAAACCTCTGCATCCAG |
21 |
193 |
DY025212 |
495 |
515 |
420 |
523 |
| Bna-miR162b |
TCGATAAACCTCTGCATCCAG |
21 |
EV208491 |
295 |
315 |
204 |
329 |
|
| Bna-miR162b* |
GGAGGCAGCGGTTCATCGATC |
21 |
32 |
EV208491 |
222 |
242 |
212 |
323 |
| Bna-miR165a |
TCGGACCAGGCTTCATCCCCC |
21 |
2205 |
FP063045 |
53 |
73 |
34 |
160 |
| Bna--miR166a* |
GGAATGTTGTTTGGCTCGAAG |
21 |
29 |
DX911364 |
806 |
826 |
697 |
831 |
| Bna-miR166e |
GGAATGTTGTCTGGCTCGAGG |
21 |
328 |
CU967744 |
48 |
68 |
39 |
185 |
| Bna-miR167d |
TGAAGCTGCCAGCATGATCTA |
21 |
6110 |
CT022223 |
721 |
741 |
635 |
758 |
| Bna-miR167e |
TGAAGCTGCCAGCATGATCTA |
21 |
AC189327 |
249 |
269 |
153 |
286 |
|
| Bna-miR167e* |
GATCATGTTCGTAGTTTCACC |
21 |
47 |
AC189327 |
169 |
189 |
153 |
286 |
| Bna-miR167f |
TGAAGCTGCCAGCATGATCTT |
21 |
160 |
ES910254 |
45 |
65 |
43 |
134 |
| Bna-miR168b |
TTCGCTTGGTGCAGGTCGGGA |
21 |
14 |
DU984956 |
357 |
377 |
258 |
396 |
| Bna-miR169n |
GCAAGTTGACTTTGGCTCTGT |
21 |
1463 |
CU944678 |
404 |
424 |
354 |
528 |
| Bna-miR169n* |
TGAGCCAAAGATGACTTGCCG |
21 |
11 |
CU944678 |
459 |
479 |
356 |
530 |
| Bna-miR171a* |
AGATATTAGTGCGGTTCAATC |
21 |
7 |
DX044654 |
128 |
148 |
119 |
219 |
| |
|
|
|
DU980843 |
223 |
243 |
159 |
246 |
| Bna--miR171f* |
TATTGGCCTGGTTCACTCAGA |
21 |
34 |
DU106747 |
666 |
686 |
647 |
756 |
| Bna-miR172a |
GGAATCTTGATGATGCTGCAT |
21 |
95 |
EV092015 |
731 |
751 |
637 |
769 |
| Bna-miR172a* |
GTGGCATCATCAAGATTCACA |
21 |
3 |
EV092015 |
654 |
674 |
629 |
777 |
| Bna-miR172b |
AGAATCTTGATGATGCTGCAT |
21 |
223 |
CU946172 |
157 |
177 |
69 |
189 |
| Bna-miR319a |
GAGCTTTCTTCGGTCCACTC |
20 |
105 |
ES908144 |
308 |
327 |
301 |
477 |
| Bna-miR319b-1 |
ATCTGCCGACTCATCCATCCA |
21 |
11 |
CN829704 |
153 |
173 |
76 |
312 |
| Bna-miR319b-2 |
GAGATTCTTTCAGTCCAGTCA |
21 |
3 |
CN829704 |
103 |
123 |
74 |
310 |
| Bna-miR390d |
AAGCTCAGGAGGGATAGCGCC |
21 |
1157 |
EE544982 |
541 |
561 |
471 |
581 |
| Bna-miR390d* |
CGCTGTCCATCCTGAGTTTCA |
21 |
348 |
EE544982 |
441 |
461 |
421 |
531 |
| Bna -miR393* |
ATCATGCGATCTCTTCGGATT |
21 |
30 |
DU101699 |
224 |
242 |
116 |
242 |
| Bna-miR396 |
AATAAAGCTGTGGGAAGATAC |
21 |
24 |
DU106522 |
54 |
74 |
35 |
216 |
| Bna-miR398 |
TGTGTTCTCAGGTCACCCCTG |
21 |
66 |
EE426846 |
89 |
109 |
1 |
123 |
| Bna-miR399d |
TGCCAAAGGAGATTTGCCCGG |
21 |
71 |
CX190537 |
106 |
126 |
9 |
146 |
| Bna-miR399f |
TGCCAAAGGAGAGTTGCCCTG |
21 |
62 |
EE556998 |
584 |
604 |
475 |
635 |
| Bna-miR400 |
TATGAGAGTATTATAAGTCAC |
21 |
25 |
CX189066 |
239 |
259 |
212 |
346 |
| Bna-miR408a |
ACAGGGAACAAGCAGAGCATG |
21 |
305 |
ES903146 |
59 |
79 |
49 |
157 |
| Bna-miR408a* |
ATGCACTGCCTCTTCCCTGGC |
21 |
141 |
ES903146 |
128 |
148 |
49 |
157 |
| Bna -miR2111c |
TAATCTGCATCCTGAGGTTTA |
21 |
35 |
BH986382 |
394 |
414 |
375 |
493 |
| DX056967 | 280 | 300 | 176 | 326 | ||||
The read number of the conserved miRNAs was highly variable, indicating different expression levels of these miRNAs. Among them, Bna-miR159, Bna-miR166a, Bna-miR164, Bna-miR171f and Bna-miR168 had relatively high number of reads, indicating that these miRNAs are likely to be expressed at a higher level, whereas Bna-miR169 family members had a low number of reads, and are, therefore, likely to be expressed at a lower level (Additional file 4: Table S1). The relative expression level of a few known miRNA families, such as miR159, miR167, miR160, miR165 and miR390, were similar to that of Arabidopsis[44] (Table 2).
Brassica-specific miRNAs
A distinct feature of miRNAs is the ability of their pre-miRNA sequences to adopt the canonical stem-loop hairpin structure. After removal of conserved miRNAs, the rest sRNA reads were predicted for each mapped locus for potential stem-loop structures. From this analysis, we identified 62 miRNA and miRNA*candidates (47 families) that were potentially generated from 62 EST or GSS loci (Additional file 5: Figure S2, Table 3).
Table 3.
Candidate new brassica-specific miRNAs
|
miRNA |
miR sequence (5'→3') |
miR length (nt) |
Reads |
Precursor from EST |
Mature miRNA position |
Stem-loop position |
||
|---|---|---|---|---|---|---|---|---|
| miR start | miR end | Precursor start | Precursor end | |||||
| Bna-miRC1 |
CCATACTAAATCTGGATCATTT |
22 |
115 |
CU943501 |
519 |
540 |
519 |
634 |
| Bna-miRC2 |
ATAAATCCCAAGCATCATCCA |
21 |
1011 |
EV202910 |
179 |
199 |
173 |
261 |
| Bna-miRC3 |
TGGGATTGGCTTTGGGCTTTTC |
22 |
12 |
CU940792 |
108 |
129 |
82 |
281 |
| Bna-miRC4 |
TTTCAGTCGTCATAGGTTAGT |
21 |
11 |
GT084890 |
55 |
75 |
51 |
158 |
| Bna-miRC5-1 |
TGTGTTGTGATGATAATCCGA |
21 |
306 |
CU971106 |
285 |
305 |
132 |
350 |
| Bna-miRC5-1* |
AATCGGATTATCATCACAACA |
21 |
7 |
CU971106 |
93 |
113 |
29 |
291 |
| Bna-miRC5-2 |
TCAACCAAATACACATTGTGG |
21 |
4 |
CU971106 |
52 |
72 |
35 |
306 |
| Bna-miRC5-3 |
TTATCATCACAACACTAGATC |
21 |
536 |
CU971106 |
76 |
96 |
19 |
281 |
| Bna-miRC5-3* |
TCTTGTGTTGTGATGATAATC |
21 |
216 |
CU971106 |
288 |
308 |
136 |
349 |
| Bna-miRC5-4 |
TGATAATCCGACTTCTATGAC |
21 |
29 |
CU971106 |
272 |
292 |
122 |
356 |
| Bna-miRC5-5 |
TTGGTTTGGATCTTGGAAATC |
21 |
8 |
CU971106 |
123 |
143 |
42 |
304 |
| Bna-miRC5-6 |
TCGGATTATCATCACAACACT |
21 |
182 |
CU971106 |
89 |
109 |
27 |
289 |
| Bna-miRC6 |
ATAGATCCTTCTGATGACGCA |
21 |
16 |
DU099814 |
306 |
326 |
254 |
327 |
| Bna-miRC7 |
CAAATCCTGTCATCCCTACCA |
21 |
89 |
GT079632 |
102 |
122 |
102 |
229 |
| Bna-miRC8 |
CAGGAGAGATTGTTGGATCCA |
21 |
3 |
CU931338 |
337 |
357 |
337 |
443 |
| Bna-miRC9 |
TGCCTGGCTCCCTGTATACCA |
21 |
118 |
EV193539 |
387 |
407 |
380 |
474 |
| Bna-miRC10 |
TCAATGTTGGCTCAATTATGT |
21 |
12 |
CU934632 |
731 |
751 |
666 |
751 |
| Bna-miRC11 |
GGCGAGTCACCGGTGTCGGTC |
21 |
6 |
FP328714 |
415 |
435 |
406 |
534 |
| Bna-miRC12 |
GGGTCGATATGAGAACACATG |
21 |
15 |
EE426846 |
15 |
35 |
1 |
123 |
| Bna-miRC13 |
ACCCTGTTGAGCTTGTCTCTA |
21 |
3 |
CU980942 |
490 |
510 |
449 |
524 |
| Bna-miRC14 |
CAGCTGGACGACTTAGTAGAC |
21 |
7 |
CU943399 |
123 |
143 |
103 |
229 |
| Bna-miRC15-1 |
ACATTGGACTACATATATTAC |
21 |
8 |
ES901619 |
392 |
412 |
299 |
430 |
| Bna-miRC15-2 |
TCAATACATTGGACTACATAT |
21 |
9 |
ES901619 |
387 |
407 |
299 |
430 |
| Bna-miRC16 |
GTTTTGAGAGATTGGGAAGCT |
21 |
3 |
EV146378 |
77 |
97 |
58 |
216 |
| Bna-miRC17a-1 |
TTTCCAAATGTAGACAAAGCA |
21 |
7132 |
ES913560 |
96 |
116 |
31 |
137 |
| Bna-miRC17a-1* |
CTTTGTCTATCGTTTGGAAAAG |
22 |
782 |
ES913560 |
53 |
74 |
31 |
137 |
| Bna-miRC18 |
TCGCGATCTTAGATCCTCTAA |
21 |
41 |
EV179238 |
441 |
461 |
288 |
564 |
| Bna-miRC19 |
CGAGTTGGTCGGGAAAGACGG |
21 |
12 |
DU102764 |
104 |
124 |
35 |
128 |
| Bna-miRC20 |
CTCTCGTGGAGCGTCTCGAGG |
21 |
3 |
EV192419 |
700 |
720 |
567 |
746 |
| Bna-miRC21 |
GGAGGCAGCGGTTGATCGATC |
21 |
7 |
DY025212 |
429 |
449 |
420 |
523 |
| Bna-miRC22a-1 |
CAAGTAGACGACTTTCCAGAC |
21 |
10 |
CU945922 |
359 |
379 |
298 |
403 |
| Bna-miRC22a-2 |
CGTGGTCGTCCAAGTAGACGA |
21 |
13 |
CU945922 |
363 |
383 |
292 |
397 |
| Bna-miRC22a-3 |
TTGGACGACTTTGTAGACGAC |
21 |
9 |
CU945922 |
303 |
323 |
297 |
402 |
| Bna-miRC23a-1 |
TCAGAACCAAACCCAGAACAAG |
22 |
54 |
CU958057 |
25 |
46 |
3 |
241 |
| Bna-miRC23a-2 |
TTACAGAACAGCAACAAGCTGT |
22 |
150 |
CU958057 |
47 |
68 |
7 |
238 |
| Bna-miRC23a-3 |
TATCTACTGCTTATGCCACCA |
21 |
65 |
CU958057 |
54 |
74 |
1 |
215 |
| Bna-miRC23a-3* |
GATGCATAACCACTAGATACG |
21 |
8 |
CU958057 |
140 |
160 |
1 |
215 |
| Bna-miRC24 |
TTAGGATTGAGATCTTAGCGA |
21 |
7 |
EV176533 |
225 |
245 |
214 |
395 |
| Bna-miRC25 |
TTGGACTGAAGGGAACTCCCT |
21 |
23719 |
FP023833 |
319 |
339 |
168 |
343 |
| Bna-miRC25* |
AGAGTTTCCTTAAGTCCATTC |
21 |
34 |
FP023833 |
173 |
193 |
167 |
343 |
| Bna-miRC26 |
TGAGCCAAAGATGACTTGTCG |
21 |
45 |
BZ021311 |
68 |
88 |
66 |
323 |
| Bna-miRC27 |
TAAGATGATGGAACACTGGCC |
21 |
25 |
EE438385 |
18 |
38 |
14 |
279 |
| Bna-miRC28 |
ATGGATCCGCCGGATAAGGAT |
21 |
6 |
CU965419 |
466 |
486 |
350 |
511 |
| Bna-miRC29 |
TTGAGGTTTTGAGGACTGGCC |
21 |
6 |
EV093069 |
644 |
664 |
564 |
668 |
| Bna-miRC30 |
TCCTGGACGACTTTCAAGTAAG |
22 |
9 |
CZ888137 |
161 |
182 |
20 |
250 |
| Bna-miRC31 |
AGATCATCCTGCGGCTTCATT |
21 |
26 |
EV134163 |
290 |
310 |
233 |
335 |
| Bna-miRC32 |
GCAAGTTGACTTTGGCTCCGT |
21 |
51 |
BZ021311 |
52 |
72 |
13 |
184 |
| Bna-miRC33 |
TTTTGCCTACTCCTCCCATACC |
22 |
268 |
CU981257 |
103 |
124 |
95 |
223 |
| Bna-miRC34 |
ATCCTCGGGACACAGATTACC |
21 |
113 |
EV076017 |
357 |
377 |
339 |
459 |
| Bna-miRC35 |
ATGGTGTAGGTACTGAGCAGA |
21 |
13 |
EV194620 |
298 |
318 |
294 |
400 |
| Bna-miRC36 |
CGTCCGGGGAAAGCAAAGTCG |
21 |
11 |
EV088144 |
141 |
161 |
64 |
186 |
| Bna-miRC37 |
TGATTTATCCAAGGGTTCAGG |
21 |
31 |
DU101557 |
509 |
529 |
367 |
608 |
| Bna-miRC38 |
CAAGTAGACTACTTTCCAGACG |
22 |
9 |
GT084321 |
52 |
73 |
1 |
92 |
| Bna-miRC39 |
TAAGATGATGGGACGTTGGATC |
22 |
11 |
DY002174 |
42 |
63 |
40 |
306 |
| Bna-miRC40 |
CGCTCACAGCATCTGAACTCT |
21 |
21 |
CD842549 |
99 |
119 |
78 |
243 |
| Bna-miRC41 |
TTTTGGAGAAGGCTGTAGGCA |
21 |
13 |
DU109430 |
791 |
811 |
780 |
890 |
| Bna-miRC42 |
TTCCCCGGACGACTTTAAATT |
21 |
15 |
EV088144 |
90 |
110 |
3 |
125 |
| Bna-miRC43 |
TGTGAATGATGCGGGAGATGT |
21 |
15 |
CN829704 |
219 |
239 |
69 |
315 |
| Bna-miRC44 |
TTGGCCACAACGGATTTAACA |
21 |
9 |
EV006438 |
66 |
86 |
66 |
141 |
| Bna-miRC45 |
TTTCATCTTAGAGAATGTTGTC |
22 |
42 |
EV178795 |
578 |
599 |
478 |
617 |
| Bna-miRC46 |
ACTTGTCTCACTCATCAGTT |
20 |
7 |
EV063926 |
5 |
24 |
3 |
215 |
| Bna-miRC47 | CAAATGTAGACAAAGCAAAAC | 21 | 4 | ES913560 | 100 | 120 | 31 | 137 |
Generally, new species-specific miRNAs are considered to be young miRNAs that have evolved recently, and are often expressed at a lower level than conserved miRNAs, as was reported for Arabidopsis and wheat [44,46,56]. This observation is also true for many of the new B. napus miRNAs identified here. However, few new miRNAs were expressed at a high level, which was opposite with this observation (Figure 2). In some cases we observed considerable inconsistency between the level of miRNAs identified by Solexa sequencing and quantitative RT-PCR (qRT-PCR) analysis, however, though we do not know the explanation for these differences. It is possible that the primers used for stem-loop real-time reactions can bind miRNA species with a few mismatches that were not considered by the bioinformatic analysis.
Figure 2.
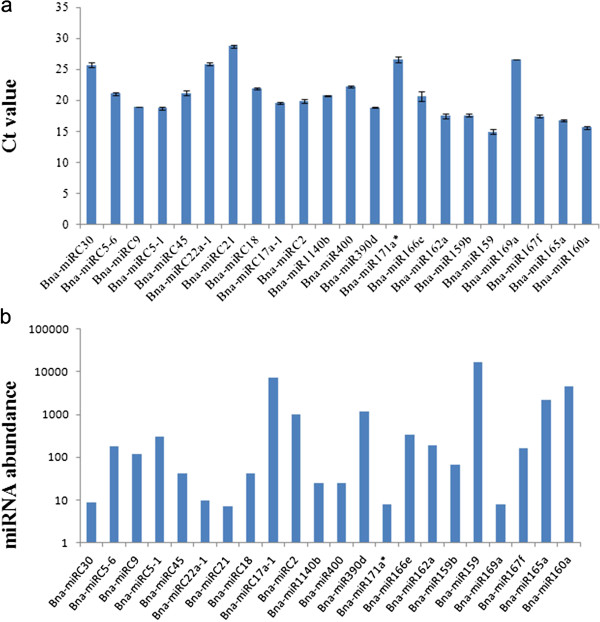
Expression levels of Bna-miRNAs by two methods. (a) Profile of qRT-PCR Ct values for Bna-miRNAs; (b) Profile of sequencing frequencies for Bna-miRNAs.
Stem-loop qRT-PCR validation and measurement of B. napus miRNAs
To verify the existence of the newly identified rape miRNA candidates, the same RNA preparation used in the Solexa sequencing was subjected to stem-loop qRT-PCR [57]. Finally, Twelve conserved miRNAs and 10 brassica-specific candidates, which were randomly selected, could be readily detected by qRT-PCR (Figure 2a), suggesting that miRNAs are bona fide miRNAs. Most results of qRT-PCR analysis agreed with the sequencing data, as in the cases of Bna-miR159, Bna-miR159b, Bna-miR160a, Bna-miR165a, Bna-miR166e, Bna-miR167f, Bna-miR169a, Bna-miR171a*, Bna-miR390d, Bna-miR400, Bna-miR1140b, Bna-miRC2, Bna-miRC5-1, Bna-miRC5-6, Bna-miRC17a-1, Bna-miRC18, Bna-miRC21, Bna-miRC22a-1, Bna-miRC30 and Bna-miRC45. In some cases, however, a discrepancy was also observed between the qRT-PCR and sequencing data (Bna-miR162a and Bna-miRC9; Figure 2a, b; Table 2, 3). The results suggested that Solexa sequencing was capable of successfully discovering candidate novel miRNAs from this species with high accuracy and efficiency.
Targets of known B. napus miRNAs
In B. napus, many conserved miRNA targets have been predicted previously [58,59], but few miRNA targets were identified experimentally. We therefore employed the recently developed high-throughput experimental approach [50,51,60] allowed us to identify target genes for known miRNAs and candidate new miRNAs identified in this work. The poly-A fraction of a balanced total RNA mix from leaf, petiole, stalk and root tissue was analyzed for the identification of target transcripts of known and new miRNAs. We obtained a total number of 8, 356, 060 reads with an average size of ~20 nt, representing the 5′ ends of uncapped, polyadenylated RNAs. After initial processing, 6,999,869 reads were obtained, and could be mapped to mRNAs. Previous studies established that the 5′ terminal nucleotide of miRNA-cleaved mRNA fragments would correspond to the nucleotide that is complementary to the 10th nucleotide of the miRNA. Therefore, the cleaved RNA targets should have distinct peaks in the degradome sequence reads at the predicted cleavage site relative to other regions of the transcript [50,51]. CleaveLand pipeline [60] was used to identify cleaved targets for both known and new miRNAs in B. napus. The abundance of the sequenced tags was plotted for each transcript, and the results are shown in Figures 3, Additional file 6: Figure S3 and Additional file 7: Figure S4. The cleaved target transcripts were categorized into five classes (categories 0, 1, 2, 3 and 4). There were 31 non-redundant ESTs identified as known miRNAs targets, covering 17 miRNA families (Table 4). Nine target ESTs were classified as category 0 (Figure 3a). Category 0 targets are transcripts where the degradome reads corresponding to the expected miRNA-mediated cleavage site were the most abundant reads matching the transcript and there is only one peak on the transcript with more than one raw read at the position. Transcripts of one target (EV184491, for Bna-miR156a) fall into category 1 (Figure 3b), where the total abundance of degradome sequences at the cleavage position is equal to the maximum on the transcript, and there is more than one raw read at the position and more than one maximum position on the transcript. 3 target ESTs were classified as Category 2 (Figure 3c), where abundance at the cleavage position is less than the maximum but higher than the median for the transcript and more than one raw read at the position. 2 target ESTs were classed as Category 3 (Figure 3d), where abundance at the cleavage position is equal to or less than the median for the transcript and more than one raw read at the position. Among the identified targets the most abundant category was category 4 (18 target ESTs), where there is only one raw read at the cleavage position (Figure 3e). Using these classifications we identified targets for 17 conserved miRNA families out of 25. Many highly conserved miRNAs were identified in B. napus (Table 2) did not have detectable sliced targets in the degradome sequencing data (e.g. miR161, miR166, miR168 and miR397). It is possible that the levels of conserved miRNAs (e.g. miR161) or sliced targets are below the detection level in this growth stage, and may be present in other specific stages or tissues that have not yet been analyzed. Alternatively, these miRNAs inhibit target gene expression through translational arrest rather than mRNA cleavage.
Figure 3.
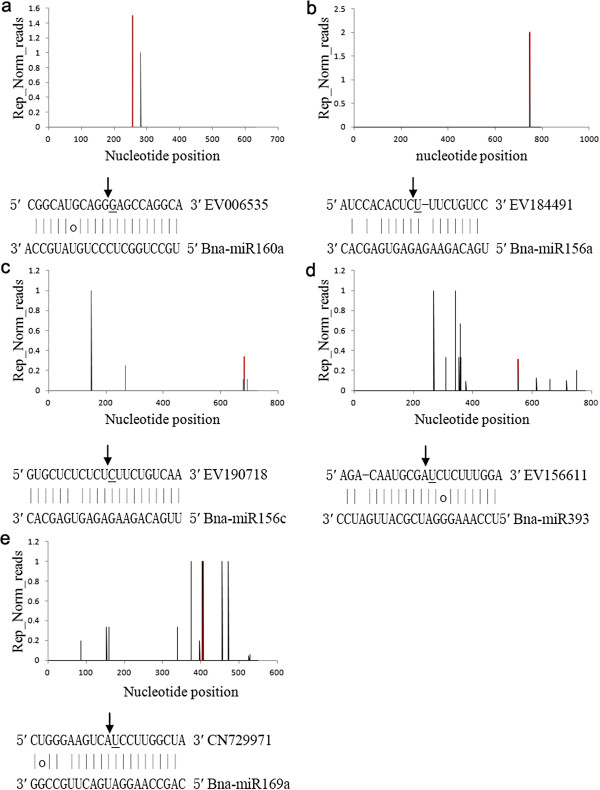
Confirmed microRNA (miRNA) targets using degradome sequencing are presented in the form of target plots (t-plots). We used normalized numbers in plotting the cleavages on target mRNAs, which were referred to as ‘target plots’ (t-plots) by German et al. [51]. Signature abundance throughout the length of the indicated transcripts is shown. Representative t-plots for class 0 (a), class I (b), class II (c), class III (d), and class VI (e) categories are shown. Arrows indicate signatures consistent with miRNA-directed cleavage. miRNA:mRNA alignments along with the detected cleavage frequencies (normalized numbers) are shown. The frequencies of degradome tags with 5′ends at the indicated positions are shown in black, with the frequency at position 10 of the inset miRNA target alignment highlighted in red. The underlined nucleotide on the target transcript indicates the cleavage site detected in the degradome.
Table 4.
Targets of conserved B. napus miRNAs
| miRNA | Target EST | Category | Cleavge site | Reads mapping to the expected cleavage site | Percentage of expected reads to total reads mapped to the full length of EST (%) | Target site location | Target annotation |
|---|---|---|---|---|---|---|---|
| Bna-miR156a |
EL625881 |
4 |
455 |
5 |
19 |
3′UTR |
A. thaliana SPL3 transcription factor |
| |
EV190718 |
2 |
681 |
3 |
38 |
3′UTR |
A. thaliana SPL10 transcription factor |
| |
EV184491 |
1 |
747 |
2 |
100 |
3′UTR |
A. thaliana O-fucosyltransferase family protein |
| Bna-miR156c |
EV190718 |
2 |
682 |
3 |
38 |
3′UTR |
A. thaliana SPL10 transcription factor |
| Bna-miR159 |
EV087133 |
4 |
439 |
2 |
20 |
ORF |
A. thaliana MYB65 |
| |
EV223870 |
4 |
279 |
5 |
25 |
ORF |
metallo-beta-lactamase family protein |
| |
EV136053 |
4 |
685 |
3 |
21 |
ORF |
pyruvate, phosphate dikinase (PPDK) |
| Bna-miR160a |
EV006535 |
0 |
256 |
2 |
50 |
ORF |
auxin response factor |
| Bna-miR164b |
ES906443 |
4 |
68 |
3 |
20 |
ORF |
NAC domain-containing protein |
| Bna-miR167b |
ES962471 |
4 |
264 |
3 |
25 |
ORF |
Auxin response factor 8 |
| Bna-miR167c |
EV208388 |
4 |
657 |
3 |
27 |
ORF |
B. rapa IAA-amino acid hydrolase 3 |
| Bna-miR169a |
EE543166 |
0 |
247 |
3 |
50 |
3′UTR |
A. thaliana NF-YA3 |
| |
CN729971 |
4 |
406 |
3 |
17 |
3′UTR |
A. thaliana NF-YA5 |
| Bna-miR169e |
ES980547 |
2 |
459 |
5 |
36 |
3′UTR |
A. thaliana NF-YA3 |
| |
EE543166 |
0 |
247 |
3 |
50 |
3′UTR |
A. thaliana NF-YA3 |
| |
CN729971 |
4 |
406 |
4 |
22 |
3′UTR |
A. thaliana NF-YA5 |
| Bna-miR169l |
ES980547 |
2 |
459 |
5 |
36 |
3′UTR |
A. thaliana NF-YA3 |
| |
ES959135 |
4 |
352 |
3 |
27 |
3′UTR |
B. napus clone bncbf-b2 |
| |
EE543166 |
0 |
247 |
3 |
50 |
3′UTR |
A. thaliana NF-YA3 |
| |
CN729971 |
4 |
406 |
4 |
22 |
3′UTR |
A. thaliana NF-YA5 |
| Bna-miR171b |
ES907976 |
3 |
609 |
3 |
33 |
ORF |
B. napus SCL1 |
| |
ES902868 |
3 |
673 |
4 |
34 |
ORF |
A. thaliana SCL6-IV |
| Bna-miR171f |
ES902868 |
4 |
676 |
3 |
21 |
ORF |
A. thaliana SCL6-IV |
| |
ES907976 |
4 |
612 |
4 |
25 |
ORF |
B. napus SCL1 |
| Bna-miR171g |
ES902868 |
4 |
676 |
3 |
23 |
ORF |
A. thaliana SCL6-IV |
| |
ES907976 |
4 |
612 |
2 |
18 |
ORF |
B. napus SCL1 |
| Bna-miR172f |
FG568924 |
4 |
488 |
2 |
20 |
ORF |
B. napus APETALA2 |
| |
EV197066 |
0 |
677 |
6 |
50 |
ORF |
A. thaliana AP2-like protein (At2g28550) |
| |
DY025256 |
4 |
609 |
5 |
24 |
ORF |
A. thaliana AP2-like protein (SMZ) |
| Bna-miR390a |
EV220086 |
0 |
433 |
4 |
50 |
ORF |
cytochrome P450 family 78, subfamily A |
| Bna-miR390d |
EV220086 |
0 |
433 |
4 |
50 |
ORF |
cytochrome P450 family 78, subfamily A |
| Bna-miR393 |
EL628991 |
2 |
246 |
10 |
17 |
ORF |
A. thaliana auxin signaling F-box 3 |
| |
EV176346 |
0 |
564 |
6 |
50 |
ORF |
A. thaliana auxin signaling F-box 3 |
| Bna-miR396a |
ES923674 |
4 |
571 |
1 |
20 |
ORF |
A. thaliana bHLH74 transcription factor |
| Bna-miR397b |
ES906654 |
4 |
736 |
3 |
20 |
ORF |
A. thaliana laccase-4 (IRX12) |
| |
EE460611 |
4 |
445 |
5 |
25 |
ORF |
A. thaliana 60S ribosomal protein L15 |
| Bna-miR399 |
EV157460 |
4 |
268 |
3 |
23 |
5′UTR |
A. lyrata PHO2/UBC24 |
| |
CX281881 |
4 |
581 |
5 |
23 |
ORF |
B. napus genes for ITS1, ITS2 |
| Bna-miR408a |
EE417826 |
4 |
457 |
2 |
20 |
ORF |
A. thaliana peptide chain release factor 1 |
| Bna-miR408b |
EV075738 |
4 |
63 |
2 |
22 |
5′UTR |
A. thaliana plantacyanin |
| Bna-miR824 |
EV112524 |
0 |
319 |
4 |
50 |
ORF |
A. thaliana MADS-box protein AGL16 |
| miR1140b |
EV217683 |
0 |
473 |
3 |
50 |
ORF |
T. aestivum mRNA for glycosyltransferase |
| |
ES912747 |
0 |
119 |
1 |
50 |
ORF |
A. thaliana two-component response regulator ARR8 (RR3) |
| Bna-miR2111b | EV221566 | 0 | 337 | 3 | 50 | ORF | A. thaliana F-box/kelch-repeat protein |
Most of the identified targets of the conserved B. napus miRNAs belong to diverse gene families of transcription factors, such as SPLs, ARFs, MYBs, NF-Y subunits, NAC-domain proteins, AP2-like factors, SCLs and MADS-box factors (Table 4). Many of these transcription factors are known to regulate diverse aspects of plant growth and development. For example, SPLs and AP2-like factors targeted by miR156 and miR172, respectively, have been shown to play an important role in phase changes (from juvenile to adult and from vegetative to the reproductive phase) in Arabidopsis[21]. Another important family of transcription factors is the MADS-box gene family,which is known to play a critical role in determining organ specificity during flower development in Arabidopsis[61]. One MADS-box gene (AtAGL16-like protein) was validated as a target for miR824 in B. napus (Table 4). MADS-box factors in B. napus have also been identified to play important roles in petal identity [62]. Similarly, three SCL6s targeted by miR171 play an important role in the regulation of shoot branch production in Arabidopsis[63]. Besides their possible involvement in plant development, miRNA targets identified in this study could also play fundamental roles in biotic and abiotic stress resistance in B. napus. NF-YA transcription factor genes were validated as targets of for miR169 family numbers. The AtNF-YA5 transcription factor, whose transcript is a target of miR169, has been implicated in drought stress responses in Arabidopsis[64]. Over-expression of a miR169-resistant AtNF-YA5 transgene significantly improves drought resistance by promoting stomatal closure under drought stress [64]. Furthermore, NF-YA factors in Petunia hybrida and Antirrhinum majus were validated to play important roles in floral organ identity [65]. NF- YA mRNAs were identified as targets of miR169 in B. napus (Table 4). In addition, laccases (enzymes involved in cell wall metabolism), plantacyanin-like proteins (involved in reproduction and seed setting) and F-box proteins involved in auxin-stimulated protein degradation (TIR1-like) were among the confirmed targets in B. napus (Table 4). Bna-miR1140 is a brassica-specific miRNA identified in our previous work.
Brassica-specific miRNA targets
Out of the 62 candidate new miRNAs, we only identified targets for only 17 miRNAs from the degradome sequencing data, plus 19 non-redundant target ESTs for candidate new brassica-specific miRNAs (Table 5). The abundance of the sequence tags for candidate brassica-specific miRNA target transcripts was plotted as a function of its position in the target genes (Additional file 7: Figure S4). We found there was no clear correlation between the expression level of the new miRNAs and their ability to target an mRNA for cleavage. We found candidate new miRNAs, such as Bna-miRC8, Bna-miRC13, Bna-miRC16, target mRNAs despite their low abundance and that target mRNAs. Consistent with our observation, no clear inverse correlations between the miRNA abundance and the cleavage frequency of target transcripts in Arabidopsis, rice and grapevine have been reported [53,66,67]. The new B. napus miRNAs target different genes with a wide variety of predicted functions. For instance, Bna-miRC16 targets chlorophyll a/b-binding protein gene, Bna-miRC20-1 targets photosystem II reaction center W-like protein gene and Bna-miRC21 targets photosystem I subunit XI gene, which are all involved in photosynthesis. Bna-miRC17a-1 targets cinnamyl alcohol dehydrogenase (CAD), which is likely to be involved in pathogen resistance and plant development [68]. Several specific targets, such as PPR-containing protein (required for normal plant development), ferrochelatase (involved in the heme biosynthetic pathway), GF14 omega proteins (potential roles in signaling), FtsH-like protease (an ATP-dependent zinc metalloprotease, related to photo-oxidative damage), glycosyl hydrolase family proteins (involved in plant cell wall architecture), Histone H2A and Histone H2B (involved in compacting DNA strands and chromatin regulation) were found as targets of rape-specific miRNAs in B. napus.
Table 5.
Targets of candidate novel B. napus miRNAs
| miRNA | Target EST | Category | Cleavage site | Reads mapping to the expected cleavage site | Percentage of expected reads to total reads mapped to the full length of EST (%) | Target site location | Target annotation |
|---|---|---|---|---|---|---|---|
| Bna-miRC2 |
EV142354 |
1 |
347 |
6 |
30 |
ORF |
A. lyrata PPR-containing protein |
| Bna-miRC5-2 |
EV077017 |
0 |
326 |
4 |
50 |
ORF |
A. lyrata exostosin family protein |
| Bna-miRC5-5Bna-miRC8 |
EV154449 FG574835 |
2 4 |
296 119 |
3 2 |
27 20 |
ORF 5′UTR |
A. thaliana alpha-tubulin 6 A. thaliana uncharacterized protein |
| Bna-miRC9 |
EV006535 |
0 |
256 |
2 |
50 |
ORF |
A. thaliana auxin response factor 17 |
| Bna-miRC13 |
EV022057 |
1 |
105 |
3 |
30 |
ORF |
A. thaliana protein PIR |
| Bna-miRC15-1 |
EV191962 |
0 |
132 |
5 |
50 |
5′UTR |
A. thaliana ferrochelatase 1 |
| Bna-miRC15-2 |
EV054423 |
2 |
615 |
3 |
33 |
ORF |
A. lyrata ferrochelatase 1 |
| Bna-miRC16 |
GR445128 |
3 |
416 |
4 |
27 |
ORF |
B.juncea chlorophyll a/b-binding protein |
| Bna-miRC17a-1 |
CD818234 |
1 |
647 |
5 |
36 |
ORF |
A. thaliana cinnamyl alcohol dehydrogenase |
| Bna-miRC18 |
GT074945 |
2 |
341 |
12 |
30 |
ORF |
B.napus GF14 omega |
| Bna-miRC20-1 |
GR442870 |
0 |
578 |
3 |
50 |
ORF |
A. thaliana histone H2B-like protein |
| |
ES987065 |
0 |
39 |
3 |
50 |
ORF |
B.rapa photosystem II center W-like protein |
| Bna-miRC21 |
GT079646 |
2 |
65 |
3 |
33 |
5′UTR |
A. thaliana photosystem I subunit XI |
| Bna-miRC22a-1 |
EV044066 |
3 |
505 |
4 |
27 |
ORF |
A. thaliana OST3/OST6 family protein |
| Bna-miRC26 |
EV077764 |
0 |
593 |
5 |
50 |
ORF |
A.thaliana uncharacterized protein (AT3G51610) |
| Bna-miRC30 |
EV025081 |
2 |
329 |
4 |
36 |
ORF |
Glycosyl hydrolase family protein |
| |
CX189212 |
3 |
269 |
3 |
27 |
ORF |
Glycosyl hydrolase family protein |
| Bna-miRC47 | ES992448 | 0 | 517 | 6 | 50 | ORF | A. thaliana prenylcysteine oxidase (FCLY) |
Verification of miRNA-guided cleavage of target mRNAs in B. napus
To verify the miRNA-guided target cleavage, RLM-RACE experiment was performed to detect cleavage product of 5 predicted Bna-miRNAs (primers were listed in Additional file 8: Table S4). As shown in Figure 4, all five of the Bna-miRNAs guided the target cleavage, often at the tenth nucleotide, or eleventh nucleotide (Figure 4). Thus, all the five predicted targets were found to have specific cleavage sites corresponding to the complementary sequences of miRNA.
Figure 4.
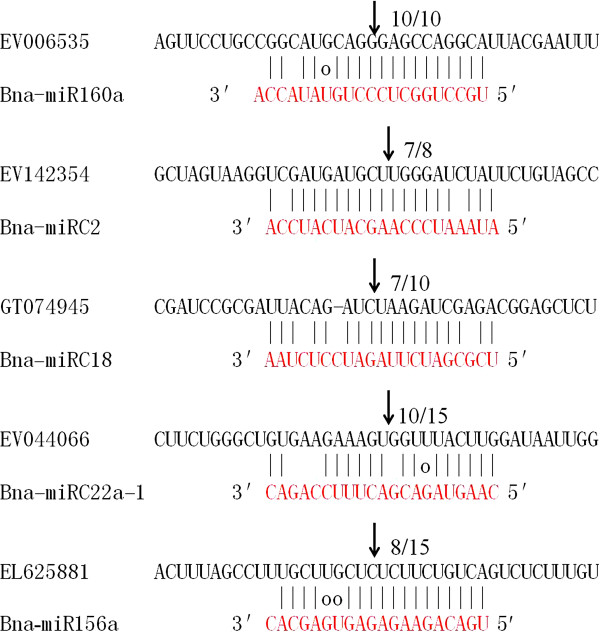
Mapping of the mRNA cleavage sites by RNA ligase-mediated 5′RACE. Each top strand (black) depicts a miRNA complementary site, and each bottom strand depicts the miRNA (red). Watson-Crick pairing (vertical dashes) and G:U wobble pairing (circles) are indicated. The arrows indicate the 5′ termini of mRNA fragments isolated from B. napus, as identified by cloned 5′ RACE products, with the frequency of clones with the predicted cleavage site shown.
Conclusion
Here, 41 conserved data and 62 brassica-specific candidate miRNAs, including 20 miRNA* sequences were firstly identified. The sequencing results were further confirmed using stem-loop quantitative RT-PCR. The data will be updated to incorporate future miRBase updates. Our approach leads to the identification of several conserved and specific brassica miRNA targets in the available EST and genomic databases. 33 non-redundant mRNA targets for the conserved brassica miRNAs and 19 non-redundant mRNA targets of new brassica-specific miRNAs were identified. Validated miRNA targets in B. napus are potentially involved in diverse biological processes, including phase transitions, flowering, hormone signaling, photosynthesis, metabolism and biotic and abiotic stress resistance. Our data will be a useful resource for further investigation of the evolution of small RNA-based regulation in Brassica napus and related species. More importantly, this study will serve as a foundation for future research on the functional roles of miRNAs and their target genes in this important oil crop.
Methods
Plant materials
The dihaploid B. napus line, Westar, was grown in a glasshouse at 22–25°C with a 16 h light/8 h dark photoperiod and light intensity of >8000 lx. Leaves, petiole, stalk, roots and shoot apices from one month-old seedlings were collected and used for RNA extraction. A balanced RNA mix was used for small RNA expression and degradome analysis.
RNA extraction and preparation of sRNA and degradome cDNA libraries for Solexa sequencing
B. napus total RNA from different tissues was extracted using Trizol (Invitrogen). The total RNA balanced mix sample was size-fractionated by 15% denaturing polyacrylamide gel electrophoresis, after which the small RNA fragments of 18–28 nt were isolated from the gel and purified. The small RNA molecules were then ligated to a 5' adaptor and a 3' adaptor sequentially and then converted to cDNA by RT-PCR following the Illumina protocol. The concentration of the sample was adjusted to ~10 nM and a total of 10 μL was used in a sequencing reaction. The purified cDNA library was sequenced on an Illumina GAIIx.
The degradome library was constructed as previously described [51]. Similarly to the short RNA libraries, the degradome cDNA library was sequenced on an Illumina GAIIx.
Bioinformatic analyses
After masking adaptor sequences and removal of contaminated reads the clean reads were filtered for miRNA prediction with the ACGT101-miR-v3.5 package (LC Sciences, Houston, USA). First, reads that matched rRNA, tRNA, snRNA, snoRNA, repeat sequences, and other ncRNAs deposited in Rfam (http://www.sanger.ac.uk/software/Rfam) [48] and the GenBank noncoding RNA database (http://www.ncbi.nlm.nih.gov/) were discarded. The retained 15–26 nt reads were mapped onto the the genome and ESTs of Brassica napus, Brassica rapa and Brassica oleracea using MapMi software under default parameters. Sequences with up to two mismatches were retained for miRNA prediction. After rigorous screening, all retained sequences of 15–26 nt with three or more copies in frequency were considered as potential miRNAs. We then attempted to align the predicted miRNAs to all rape known mature miRNA sequences in miRBase Version 17.0 [48] to identify novelty. Finally, Secondary structure prediction of individual miRNA was performed by MFOLD software (Version 2.38, http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) using the default folding conditions. [69].
The degradome analysis and the classification of target categories were performed using CleaveLand 2.0 [60]. Small RNA targets prediction was run against the transcriptome of interest. The alignment scores (using the [70] rubric) for each hit up to a user-defined cutoff were calculated, full RNA-RNA alignments were printed, and the 'cleavage site' associated with each prediction was also calculated. The cleavage site is simply the 10th nt of complementarity to the aligned small RNA. For randomized queries, no alignments were retained. However, concise records of each predicted target for the random queries were retained, including the predicted cleavage sites.
End-point and SYBR Green I real-time PCR assays of B. napus miRNAs
End-point and Real-time looped RT-PCR [57] were used to validate and measure the levels of B. napus miRNA. Stem–loop RT primers, universal reverse primer and miRNA-specific forward primers for Bna-miR159, Bna-miR159b, Bna-miR160a, Bna-miR162a, Bna-miR165a, Bna-miR166e, Bna-miR167f, Bna-miR169a, Bna-miR171a*, Bna-miR390d, Bna-miR400, Bna-miR1140b, Bna-miRC2, Bna-miRC5-1, Bna-miRC5-6, Bna-miRC9, Bna-miRC17a-1, Bna-miRC18, Bna-miRC21, Bna-miRC22a-1, Bna-miRC30and Bna-miRC45 were designed according to Varkonyi-Gasic et al.[57]. (Additional file 4: Table S1). 1 μg of total RNA was reverse-transcribed to cDNA using ReverTra Ace (TOYOBO, Osaka, Japan). Stem-loop pulsed reverse transcription and end-point PCR was performed according to Varkonyi-Gasic et al. [57]. Advantage 2 PCR Polymerase Mix (Clontech, Mountain View, CA, USA) was used to perform end-point PCR. qRT-PCR was performed using SYBR Premix Ex TaqTM of TaKaRa (TaKaRa Code: DRR041A) on an Applied Biosystems 7500 thermocycler (Applied Biosystems, Foster City, CA, USA). All reactions were run in triplicate. After the reaction, the threshold cycle (Ct) was determined using default threshold settings. The Ct is defined as the fractional cycle number at which the fluorescence passes the fixed threshold.
Modified 5′ RNA ligase-mediated RACE for the mapping of mRNA cleavage sites
Total RNA from different tissues was extracted using Trizol (Invitrogen). Poly(A) + mRNA was purified from all kinds of pooled tissue RNA using the PolyA kit (Promega, Madison, WI), according to manufacturer’s instructions. A small RNA adapter (5′GUUCAGAGUUCUACAGUCCGACGAUC- 3) was ligated to Poly(A) + mRNA. A modified procedure for 5′ RNA ligase-mediated RACE (RLM-5 RACE) was followed with the 5-Full RACE Kit (TaKaRa, Dalian), according to manufacturer’s instructions. Nested PCR amplifications were performed using the 5 small RNA nested primer (5 AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA 3) and gene-specific nested primers (Additional file 8: Table S4). The amplification products were gel purified, cloned, and sequenced, and at least 10 independent clones were sequenced.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MYX and LW designed the study. MYX and YD performed experiments, analysed data and drafted the manuscript. QXZ and JS assisted with bioinformatic analysis and interpreting analysis results. YZL and LZ performed RLM-RACE experiment. YLF supervised the project and edited the manuscript. All authors discussed the results and implications and commented on the manuscript at all stages. All authors read and approved the final manuscript.
Supplementary Material
Secondary structures of 41 conserved B.napus miRNAs and miRNAs*. Pink and red section represents miRNA; yellow section represents miRNA*.
Known miRNAs in B. napus.
Four conserved miRNAs in B. napus.
miRNA and primer sequences.
Secondary structures of 62 putative novel B.napus miRNAs and miRNAs*.
T-plots for targets of known miRNAs. Only one T-plot for target of one miRNA with same category was made.
T-plots for targets of brassica-specific miRNAs. Only one T-plot for target of one miRNA with same category was made.
5RACE primer sequences.
Contributor Information
Miao Y Xu, Email: xumiaoyun76@163.com.
Yun Dong, Email: dongyungs@163.com.
Qiu X Zhang, Email: qiuxue198598@126.com.
Lan Zhang, Email: zhanglan20000@163.com.
Yan Z Luo, Email: yanzhongluo@163.com.
Jie Sun, Email: sunjiezh@vip.sina.com.
Yun L Fan, Email: fan_yunliu@163.com.
Lei Wang, Email: caaswlwl@163.com.
Acknowledgements
This work was supported by the National Key Basic Research Program (Grant No. 2010CB125903) and National Natural Science Foundation of China (Grant No. 31270318). We gratefully acknowledge the English language editing from Dr. Mingbo Wang.
References
- Schwach F, Moxon S, Moulton V, Dalmay T. Deciphering the diversity of small RNAs in plants: the long and short of it. Brief Funct Genomic Proteomic. 2009;8(6):472–481. doi: 10.1093/bfgp/elp024. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Chuck G, Candela H, Hake S. Big impacts by small RNAs in plant development. Curr Opin Plant Biol. 2009;12(1):81–86. doi: 10.1016/j.pbi.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Dugas DV, Bartel B. MicroRNA regulation of gene expression in plants. Curr Opin Plant Biol. 2004;7(5):512–520. doi: 10.1016/j.pbi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Kidner CA, Martienssen RA. The developmental role of microRNA in plants. Curr Opin Plant Biol. 2005;8(1):38–44. doi: 10.1016/j.pbi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Watanabe Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci U S A. 2004;101(34):12753–12758. doi: 10.1073/pnas.0403115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory AC, Elmayan T, Vaucheret H. MicroRNA maturation and action–the expanding roles of ARGONAUTEs. Curr Opin Plant Biol. 2008;11(5):560–566. doi: 10.1016/j.pbi.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320(5880):1185–1190. doi: 10.1126/science.1159151. [DOI] [PubMed] [Google Scholar]
- Llave C, Kasschau KD, Rector MA, Carrington JC. Endogenous and silencing-associated small RNAs in plants. Plant Cell. 2002;14(7):1605–1619. doi: 10.1105/tpc.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110(4):513–520. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17(1):49–63. doi: 10.1101/gad.1048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294(5543):862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Arazi T, Talmor-Neiman M, Stav R, Riese M, Huijser P, Baulcombe DC. Cloning and characterization of micro-RNAs from moss. Plant J. 2005;43(6):837–848. doi: 10.1111/j.1365-313X.2005.02499.x. [DOI] [PubMed] [Google Scholar]
- Axtell MJ, Bartel DP. Antiquity of microRNAs and their targets in land plants. Plant Cell. 2005;17(6):1658–1673. doi: 10.1105/tpc.105.032185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuck G, Meeley R, Hake S. Floral meristem initiation and meristem cell fate are regulated by the maize AP2 genes ids1 and sid1. Development. 2008;135(18):3013–3019. doi: 10.1242/dev.024273. [DOI] [PubMed] [Google Scholar]
- Chuck G, Meeley R, Irish E, Sakai H, Hake S. The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat Genet. 2007;39(12):1517–1521. doi: 10.1038/ng.2007.20. [DOI] [PubMed] [Google Scholar]
- Chuck G, Whipple C, Jackson D, Hake S. The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development. 2010;137(8):1243–1250. doi: 10.1242/dev.048348. [DOI] [PubMed] [Google Scholar]
- Wang JW, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138(4):738–749. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009;138(4):750–759. doi: 10.1016/j.cell.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Woo HR, Kim J, Lim PO, Lee IC, Choi SH, Hwang D, Nam HG. Trifurcate feed-forward regulation of age-dependent cell death involving miR164 in Arabidopsis. Science. 2009;323(5917):1053–1057. doi: 10.1126/science.1166386. [DOI] [PubMed] [Google Scholar]
- Lim PO, Lee IC, Kim J, Kim HJ, Ryu JS, Woo HR, Nam HG. Auxin response factor 2 (ARF2) plays a major role in regulating auxin-mediated leaf longevity. J Exp Bot. 2010;61(5):1419–1430. doi: 10.1093/jxb/erq010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schommer C, Palatnik JF, Aggarwal P, Chetelat A, Cubas P, Farmer EE, Nath U, Weigel D. Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol. 2008;6(9):e230. doi: 10.1371/journal.pbio.0060230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RS, Li J, Alonso-Peral MM, White RG, Gubler F, Millar AA. MicroR159 regulation of most conserved targets in Arabidopsis has negligible phenotypic effects. Silence. 2010;1(1):18. doi: 10.1186/1758-907X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Peral MM, Li J, Li Y, Allen RS, Schnippenkoetter W, Ohms S, White RG, Millar AA. The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 2010;154(2):757–771. doi: 10.1104/pp.110.160630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood DH, Nogueira FT, Howell MD, Montgomery TA, Carrington JC, Timmermans MC. Pattern formation via small RNA mobility. Genes Dev. 2009;23(5):549–554. doi: 10.1101/gad.1770009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner TJ, Sherr I, Scarpella E. Regulation of preprocambial cell state acquisition by auxin signaling in Arabidopsis leaves. Development. 2009;136(19):3235–3246. doi: 10.1242/dev.037028. [DOI] [PubMed] [Google Scholar]
- Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D, Palatnik JF. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development. 2010;137(1):103–112. doi: 10.1242/dev.043067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpella E, Barkoulas M, Tsiantis M. Control of leaf and vein development by auxin. Cold Spring Harb Perspect Biol. 2010;2(1):a001511. doi: 10.1101/cshperspect.a001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todesco M, Rubio-Somoza I, Paz-Ares J, Weigel D. A collection of target mimics for comprehensive analysis of microRNA function in Arabidopsis thaliana. PLoS Genet. 2010;6(7):e1001031. doi: 10.1371/journal.pgen.1001031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmann H, Mica E, Todesco M, Long JA, Weigel D. On reconciling the interactions between APETALA2, miR172 and AGAMOUS with the ABC model of flower development. Development. 2010;137(21):3633–3642. doi: 10.1242/dev.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Cai X, Luo J, Sato S, Jiang Q, Yang J, Cao X, Hu X, Tabata S, Gresshoff PM. et al. The REDUCED LEAFLET genes encode key components of the trans-acting small interfering RNA pathway and regulate compound leaf and flower development in Lotus japonicus. Plant Physiol. 2010;152(2):797–807. doi: 10.1104/pp.109.140947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant L, Mathieu J, Dinh TT, Ott F, Lanz C, Wollmann H, Chen X, Schmid M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell. 2010;22(7):2156–2170. doi: 10.1105/tpc.110.075606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Kim Y, Dinh TT, Chen X. miR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007;51(5):840–849. doi: 10.1111/j.1365-313X.2007.03181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez L, Bussell JD, Pacurar DI, Schwambach J, Pacurar M, Bellini C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell. 2009;21(10):3119–3132. doi: 10.1105/tpc.108.064758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krouk G, Lacombe B, Bielach A, Perrine-Walker F, Malinska K, Mounier E, Hoyerova K, Tillard P, Leon S, Ljung K. et al. Nitrate-regulated auxin transport by NRT1.1 defines a mechanism for nutrient sensing in plants. Dev Cell. 2010;18(6):927–937. doi: 10.1016/j.devcel.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Marin E, Jouannet V, Herz A, Lokerse AS, Weijers D, Vaucheret H, Nussaume L, Crespi MD, Maizel A. miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell. 2010;22(4):1104–1117. doi: 10.1105/tpc.109.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Risueno MA, Van Norman JM, Moreno A, Zhang J, Ahnert SE, Benfey PN. Oscillating gene expression determines competence for periodic Arabidopsis root branching. Science. 2010;329(5997):1306–1311. doi: 10.1126/science.1191937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Somoza I, Cuperus JT, Weigel D, Carrington JC. Regulation and functional specialization of small RNA-target nodes during plant development. Curr Opin Plant Biol. 2009;12(5):622–627. doi: 10.1016/j.pbi.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Vidal EA, Araus V, Lu C, Parry G, Green PJ, Coruzzi GM, Gutierrez RA. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2010;107(9):4477–4482. doi: 10.1073/pnas.0909571107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon EK, Yang JH, Lim J, Kim SH, Kim SK, Lee WS. Auxin regulation of the microRNA390-dependent transacting small interfering RNA pathway in Arabidopsis lateral root development. Nucleic Acids Res. 2010;38(4):1382–1391. doi: 10.1093/nar/gkp1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd SK, Bowman JL. Gene regulation: ancient microRNA target sequences in plants. Nature. 2004;428(6982):485–486. doi: 10.1038/428485a. [DOI] [PubMed] [Google Scholar]
- Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL. et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2(2):e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxon S, Jing R, Szittya G, Schwach F. Rusholme Pilcher RL, Moulton V, Dalmay T: Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008;18(10):1602–1609. doi: 10.1101/gr.080127.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20(24):3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szittya G, Moxon S, Santos DM, Jing R, Fevereiro MP, Moulton V, Dalmay T. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics. 2008;9:593. doi: 10.1186/1471-2164-9-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:154–158. doi: 10.1093/nar/gkn221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14(6):787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008;18(10):758–762. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R. et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26(8):941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- Gregory BD, O'Malley RC, Lister R, Urich MA, Tonti-Filippini J, Chen H, Millar AH, Ecker JR. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev Cell. 2008;14(6):854–866. doi: 10.1016/j.devcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62(6):960–976. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]
- Zhou M. GL, Li PC, Song XW, Wei LY, Chen ZY, Cao XF: Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica) Front Biol. 2010;5(1):67–90. doi: 10.1007/s11515-010-0007-8. [DOI] [Google Scholar]
- Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu JK, Sun Q. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.) Genome Biol. 2007;8(6):96. doi: 10.1186/gb-2007-8-6-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods. 2007;3:12. doi: 10.1186/1746-4811-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang MB, Tu JX, Helliwell CA, Waterhouse PM, Dennis ES, Fu TD, Fan YL. Cloning and characterization of microRNAs from Brassica napus. FEBS Lett. 2007;581(20):3848–3856. doi: 10.1016/j.febslet.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Xie FL, Huang SQ, Guo K, Xiang AL, Zhu YY, Nie L, Yang ZM. Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 2007;581(7):1464–1474. doi: 10.1016/j.febslet.2007.02.074. [DOI] [PubMed] [Google Scholar]
- Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25(1):130–131. doi: 10.1093/bioinformatics/btn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, Muino JM, Jauregui R, Airoldi CA, Smaczniak C, Krajewski P, Angenent GC. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 2009;7(4):e1000090. doi: 10.1371/journal.pbio.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Zhang W, Yu F, Tian J, Li D, Guo A. Functional analysis of the two Brassica AP3 genes involved in apetalous and stamen carpelloid phenotypes. PLoS One. 2011;6(6):e20930. doi: 10.1371/journal.pone.0020930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mai YX, Zhang YC, Luo Q, Yang HQ. MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol Plant. 2010;3(5):794–806. doi: 10.1093/mp/ssq042. [DOI] [PubMed] [Google Scholar]
- Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell. 2008;20(8):2238–2251. doi: 10.1105/tpc.108.059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartolano M, Castillo R, Efremova N, Kuckenberg M, Zethof J, Gerats T, Schwarz-Sommer Z, Vandenbussche M. A conserved microRNA module exerts homeotic control over Petunia hybrida and Antirrhinum majus floral organ identity. Nat Genet. 2007;39(7):901–905. doi: 10.1038/ng2056. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Riechmann JL, Meyerowitz EM. Transcriptome-wide analysis of uncapped mRNAs in Arabidopsis reveals regulation of mRNA degradation. Plant Cell. 2008;20(10):2571–2585. doi: 10.1105/tpc.108.062786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang W, Sunkar R. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010;62(5):742–759. doi: 10.1111/j.1365-313X.2010.04187.x. [DOI] [PubMed] [Google Scholar]
- Sattler SE, Saathoff AJ, Haas EJ, Palmer NA, Funnell-Harris DL, Sarath G, Pedersen JF. A nonsense mutation in a cinnamyl alcohol dehydrogenase gene is responsible for the Sorghum brown midrib6 phenotype. Plant Physiol. 2009;150(2):584–595. doi: 10.1104/pp.109.136408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra-Assuncao JA, Enright AJ. MapMi: automated mapping of microRNA loci. BMC Bioinformatics. 2010;11:133. doi: 10.1186/1471-2105-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121(2):207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Secondary structures of 41 conserved B.napus miRNAs and miRNAs*. Pink and red section represents miRNA; yellow section represents miRNA*.
Known miRNAs in B. napus.
Four conserved miRNAs in B. napus.
miRNA and primer sequences.
Secondary structures of 62 putative novel B.napus miRNAs and miRNAs*.
T-plots for targets of known miRNAs. Only one T-plot for target of one miRNA with same category was made.
T-plots for targets of brassica-specific miRNAs. Only one T-plot for target of one miRNA with same category was made.
5RACE primer sequences.