

In politics and economics, “black swan” events are highly improbable, unforeseeable, once-in-a-lifetime developments that are so powerful as to cause unexpected but long-lasting changes in the trajectory of an election or of a country’s fiscal status. In biology, black swan events change our view of the possible, pointing us toward novel interpretations that take us in new and previously unconsidered directions.
The most recent black swan event in Alzheimer’s research occurred in mid-2012 with the discovery in Iceland of a mutation in the Alzheimer’s amyloid precursor protein (APP) that dramatically reduces the catalytic efficiency of cleavage of APP by beta-site APP-cleaving enzyme, the rate-limiting step in amyloid-beta (Aβ) generation [for review, see (1)]. Persons blessed with this Icelandic APP mutation at the time of their conception are apparently destined to live and develop normally, but, more importantly, they are protected from the development of Alzheimer’s disease (AD) in late life, even if they harbor two copies of the high-risk apolipoprotein E (APOE) ε4 allele, a situation that usually associates with enhanced cerebral amyloidosis and a 10-fold increase in AD risk.
In this issue, Thambisetty et al. (2) report the next black swan event in AD research. To date, approximately two dozen pathogenic APP mutations and approximately 200 pathogenic presenilin 1 (PS1) mutations (not to mention APOE ε4) all enhance cerebral amyloidosis and either cause or increase the risk for AD. When any of these AD-linked genes is overexpressed in the brains of laboratory mice, all increase Aβ accumulation. Now Thambisetty et al. (2) found what appears to be an exception to the high-risk-equals-enhanced-amyloid-accumulation rule. Using 11C Pittsburgh compound B (11C-PiB) amyloid imaging, Thambisetty et al. (2) shows that subjects with both an APOE ε4 allele and the high-risk CR1 alleles have a reduced burden of fibrillar brain amyloid as compared with APOE ε4 carriers that lack the CR1 polymorphism.
One possible explanation is that the CR1-associated amyloid plaques, however attenuated in density, may be hyperinflamed and thus hypertoxic to the neurons nearby. Perhaps these hyperinflamed plaques could also be hyperlytic to amyloid fibrils, thereby explaining the reduced 11C-PiB signal. Taken in context with the recent linkage of TREM2 to AD risk (3), one must consider the possibility that, in some forms of sporadic AD, immune-inflammatory processes may be the most important drivers of pathogenesis. Perhaps a more comprehensive formulation of AD would place neurotoxicity, neuroinflammation, and amyloidogenesis into a feed-forward, self-amplifying cycle (Figure 1) (4). Such a reformulation is especially timely, given the evidence of the genetic linkage to TREM2 (3) and CR1 (2), which dovetails with new biologic data regarding interleukin-12/23 (5) and NLRP3 (6).
Figure 1.
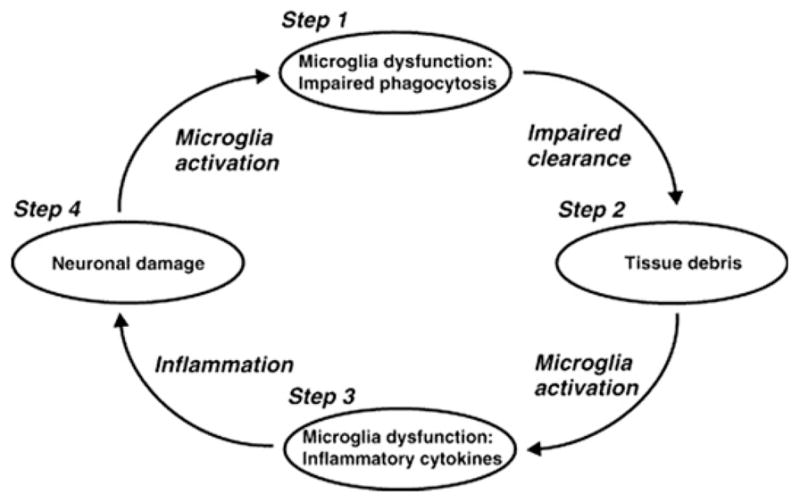
A model of Alzheimer’s disease as a feed-forward cycle of neuropathology and immunoinflammatory pathology. (Reproduced with permission from Neumann and Takahashi [4]).
Other scenarios that we might not anticipate are also possible: CR1 linkage might act through unknown pathways to cause the supersensitivity of neurons to Aβ-induced toxicity and/or to cause hyperaccumulation of tauopathy. Indeed, unpublished observations from the Haroutunian laboratory indicate that CR1 mRNA levels correlate better with neurofibrillary tangle density and phosphorylated tau abundance than with neuritic plaque density. Especially given this lead linking CR1 expression to tauopathy, the next step should be careful and comprehensive studies of brains from genetically and clinically characterized patients to determine whether the molecular pathology in brains from patients with the CR1 linkage differs in important ways from that observed in the typical or CR1-negative sporadic AD.
Are there other ways to reconcile the result from Thambisetty et al. (2) with existing dogma? 11C-PiB is unable to detect Aβ oligomers, and Schöll et al. (7) recently found that AD patients harboring the oligomerogenic Arctic APP familial AD mutation have low 11C-PiB retention (Figure 2). The low 11C-PiB retention in Arctic FAD was not entirely unexpected because Aβ-Arctic forms oligomers well but forms fibrils poorly. Perhaps the effect of the CR1 linkage is to shift the equilibrium away from APOE ε4-modulated formation of 11C-PiB–retaining Aβ fibrils, favoring instead the formation of 11C-PiB–negative Aβ oligomers. Abnormalities in another cell surface inflammatory/immunologic receptor, CD45, has been associated with promotion of Aβ oligomerization, providing a precedent for the formulation that CR1 might act by favoring Ab oligomerization over fibrillogenesis (8).
Figure 2.
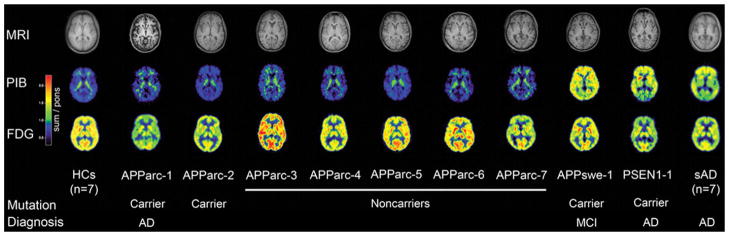
Arctic APP mutations are associated with reduced 11C-PiB retention. Transaxial sections of individual magnetic resonance imaging (upper row), Pittsburgh compound B (PiB) (middle row), and 18F-fluorodeoxyglucose (FDG) positron emission tomography (lower row) scans of all participants. Images of patients with sporadic Alzheimer’s disease (sAD) and healthy controls (HCs) are mean images. The Arctic APP (APParc) mutation carriers APParc-1 and APParc-2 showed very low cortical PiB retention, comparable with the five noncarriers APParc 3–7 and HCs. APParc-1 revealed globally decreased glucose metabolism and brain atrophy, and APParc-2 regionally decreased glucose metabolism. MCI, mild cognitive impairment. (Reproduced with permission from Schöll et al. [7]).
As noted by Thambisetty et al. (2), we also do not yet know whether the association of the CR1 risk polymorphism is applicable to all of the many diverse cohorts of elderly persons with AD. Thambisetty et al. (2) note that their study cohort was characterized by upper-middle socioeconomic status and above-average education. The study cohort was also relatively young (mean age of ~78 years). Several recent studies suggest that age may be a significant variable when assessing AD neuropathology (9) and the contribution of immune/inflammatory processes to dementia. In one study, the relationship between inflammatory markers and dementia changed in opposite directions, depending on whether the cohort was comprised of persons under the age of 86 years or above that age (i.e., the young-old versus the oldest-old [10]). The association of the inflammatory marker CRP with cognitive impairment in young-old elderly individuals is reversed in the oldest-old, and this apparent cognition protective property of high CRP in the oldest-old appears to be heritable because the protection can be demonstrated in their young first-degree relatives (11). Similarly, the frequency of the CC genotype of the lipid transfer gene MTP decreases significantly with increasing age at death up to the age of 85 years, but then this same CC genotype is enriched in centenarians and their offspring (12).
The recent advent of whole-genome sequencing and network analysis of genome-wide association studies (GWAS) and other data sets promises to begin to account for the multitude of downstream molecular events associated with any perturbation (Figure 3) (13–15). One beauty of genetics and whole-exome sequencing is that such data-driven, hypothesis-free approaches enable discovery of molecules and pathways that one would never even think to look for in more traditional hypothesis-based studies (e.g., no one would have ever looked for or found an AD endophenotype that associated high risk with reduced amyloid plaque burden). Analysis of the effects of CR1 polymorphisms on the network profiles of APOE ε4 carriers might well be revealing in terms of providing clues toward the effects of the CR1 polymorphism on downstream pathways and molecules.
Figure 3.

Inflammatome gene regulatory (Bayesian) network enriched for AD genes identified and highly replicated in genetic studies. (A) A probabilistic causal network constructed from human omental adipose tissue collected in a cohort of morbidly obese patients (13). The nodes in the network are gene expression traits monitored in the omental adipose tissue from this cohort. The directed links between the nodes are derived via a Bayesian network reconstruction algorithm that leverages DNA variation as a systematic perturbation source to resolve causality (14). The pink nodes highlighted in this network are the inflammatome signature genes we have previously identified as strongly causally associated with a number of diseases (13–15). (B) A zoomed-in view of the subnetwork highlighted in panel (A) by the pink nodes. This inflammatome-based network is enriched for inflammatory and immune response gene ontology categories (color-coded pathways are indicated; all enrichments are significant at a 1% false discovery rate). In addition, genes previously identified in genetic studies and extensively replicated as associated with AD are represented in this network, including the genes highlighted: TREM2, CR1, CD33, MS4A4A, MS4A6A, and HCK
Conceivably, clinical trials that combine Aβ-reducing therapies with antiinflammatory drugs or biologics may represent a future wave of clinical trials. Coincidentally, intravenous immunoglobulin (IVIg), a biologic that contains naturally occurring anti-Aβ oligomer antibodies as well as novel antiinflammatory activities, is currently being evaluated in two clinical trials. If there is a positive signal from those trials, the naturally occurring anti-Aβ oligomer antibodies, the novel antiinflammatory activities, and many of the multiple components of IVIg will be the subject of intense scrutiny to identify the specific molecular identities of any anti-Alzheimer’s properties. Regardless of the outcome of the IVIg trials, the immune/inflammatory pathogenesis of AD is rapidly achieving prominence equal to that currently enjoyed by that centered around structural neuropathology (Aβ oligomers, amyloid plaques, neurofibrillary tangles, etc). However, if recent discoveries are harbingers of future trends, an immunoinflammatory amyloid hypothesis of AD (Figure 1) may turn out to be a more accurate and complete formulation of the true nature of the disease.
Acknowledgments
The authors acknowledge the support of National Institutes of Health Grant AG05138 (to SG, VH, MS), Grant AG02219 (to VH), Grant NS075685 (to SG), Grant AG042965 (to SG and Scott Noggle), grant AG05133 (to S.T.D.), grant AG14449 (to S.T.D.), and grant AG025204 (to S.T.D.), Veterans Affairs Award MIRECC (to VH), Veterans Affairs MERIT Review Grant 5I01BX000348 (to SG), and the Cure Alzheimer’s Fund (to SG). The authors also thank Frank Heppner (Charité, Berlin, Germany) for his helpful discussions and suggestions.
SG acknowledges research support from Amicus Therapeutics, Baxter Pharmaceuticals and S.A.B. Service for Diagenic, Inc., and Balance Therapeutics. MS serves on a scientific advisory board for Medivation, Inc; as a consultant for Bayer Schering Pharma, Bristol-Meyers Squibb, Elan Corporation, Genentech, Inc., Medivation, Inc., Medpace Inc, Pfizer Inc., Janssen, Takeda Pharmaceutical Company Limited, and United Biosource Corporation; and receives research support from the National Institutes of Health (National Institute on Aging/National Center for Research Resources). STD acknowledges consultation for or research support from the National Institute on Aging, Elan, Novartis, Janssen, Pfizer, Baxter, Myriad, Neurochem, and GlaxoSmithKline in the past 5 years. EES holds stock with, and serves as a consultant to, Pacific Biosciences, Inc.
Footnotes
VH reports no biomedical financial interests or potential conflicts of interest.
References
- 1.Gandy S, DeKosky ST. Toward the treatment and prevention of Alzheimer’s disease: rational strategies and recent progress. Ann Rev Med. doi: 10.1146/annurev-med-092611-084441. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thambisetty M, An Y, Nalls M, Sojkova J, Swaminathan S, Zhou Y, et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann H, Takahashi K. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol. 2007;184:92–99. doi: 10.1016/j.jneuroim.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 5.Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med. 2012;18:1812–1819. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- 6.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. doi: 10.1038/nature11729. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229–236. doi: 10.1212/WNL.0b013e31825fdf18. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Hou H, Rezai-Zadeh K, Giunta B, Ruscin A, Gemma C, et al. CD45 deficiency drives amyloid-β peptide oligomers and neuronal loss in Alzheimer’s disease mice. J Neurosci. 2011;31:1355–1365. doi: 10.1523/JNEUROSCI.3268-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haroutunian V, Schnaider-Beeri M, Schmeidler J, Wysocki M, Purohit DP, Perl DP, et al. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch Neurol. 2008;65:1211–1217. doi: 10.1001/archneur.65.9.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katsel P, Tan W, Haroutunian V. Gain in brain immunity in the oldest-old differentiates cognitively normal from demented individuals. PLoS ONE. 2009;4:e7642-1–15. doi: 10.1371/journal.pone.0007642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silverman JM, Schmeidler J, Beeri MS, Rosendorff C, Sano M, Grossman HT, et al. C-reactive protein and familial risk for dementia: a phenotype for successful cognitive aging. Neurology. 2012;79:1116–1123. doi: 10.1212/WNL.0b013e3182698c89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huffman DM, Deelen J, Ye K, Bergman A, Slagboom EP, Barzilai N, Atzmon G. Distinguishing between longevity and buffered-deleterious genotypes for exceptional human longevity: the case of the MTP gene. J Gerontol A Biol Sci Med Sci. 2012;67:1153–1160. doi: 10.1093/gerona/gls103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenawalt DM, Dobrin R, Chudin E, Hatoum IJ, Suver C, Beaulaurier J, et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res. 2011;21:1008–1016. doi: 10.1101/gr.112821.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Sova P, Xu Q, Dombek KM, Xu EY, Vu H, et al. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 2012;10:e1001301. doi: 10.1371/journal.pbio.1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, Deignan JL, Qi H, Zhu J, Qian S, Zhong J, et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41:415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]