

Abstract
The potentially oncogenic Epstein-Barr virus (EBV) is carried by almost all humans in a well equilibrated coexistence. The phenotype of the cells that carry EBV genomes is determined by virally-encoded and cellular proteins. B lymphocyte is the main target of the virus and latent infection of this cell induces proliferation. Nine virus-encoded genes participate in the “growth program” that is expressed in a narrow differentiation window of the B cell. Such cells have the potential to develop malignant proliferations. However, several control mechanism eliminate this danger and the general chronic virus carrier state is most often asymptomatic. One mechanism exploits the normal regulation in the immune system, the T cell mediated modulation of the B cell differentiation state. Another is based on cognate recognition and elimination of the infected cells. The expression of EBV encoded genes in B lymphocytes can be also “restricted,” they do not express all components of the viral growth program. Here, we discuss a rare viral expression in B cells that has not been connected with malignant transformation yet.
Keywords: EBV, Type II, Type IIa, Type IIb, EBNA-2, LMP-1
Introduction
EBV shows a high degree of B cell tropism. It binds to a B lymphocyte specific surface molecule, CD21 (receptor for the C3d fragment of complement).1 Latent virus infection of B lymphocytes induces proliferation. In vitro, these immunoblasts can grow into lymphoblastoid cell lines (LCLs).2,3 They maintain the viral genome as multiple episomes that express nine proteins. Six of them are localized in the nucleus, EBNA 1–6, whereas the remaining three are associated with the cell membrane, LMP-1, -2A and -2B. This expression pattern is referred to as latency Type III or growth program.4,5
EBV infected B cells that express the growth program, are potentially malignant. This danger is counteracted by immunological surveillance in healthy individuals. It involves recognition and elimination of the cells by cell mediated immunity. The details of this immune response have been well studied.6-8 EBV encoded antigenic proteins expressed on the viral genome carrying B cell have been identified and the effector cell subsets were characterized. Recognition of the immunoblasts in a MHC Class I restricted manner by cytotoxic CD8 T cells was believed to play the main role in the control of Type III cells.9,10 But there is also evidence for an additional immunological mechanism that curbs the growth potential of the infected B cells.11-15 It is based on the requirement of B cell specific differentiation factors for the expression of the viral encoded genes that constitute viral growth program.16 These proteins are present only in a defined differentiation window of the B cell. When the Type III lymphocytes proceed in the differentiation pathway, some of the genes in the growth program are downregulated. This mechanism is probably important during the primary infection, prior to the mobilization of the adaptive EBV specific response.11,17 Expression of the viral genes is also determined by the differentiation state of the B cell at the occasion of infection. When the infection occurs in a B cell that is outside the critical differentiation window that allows the expression of the full growth program, the acquisition of the viral genome does not induce proliferation.18-21
We discuss one type of viral latent expression pattern in B lymphocytes that does not induce proliferation. Although such cells were encountered in several experimental systems, they have received relatively little attention.
EBV Gene Expression in Latent Infection of B Lymphocytes
Human B lymphocyte populations can be infected with EBV in vitro. Latent infection leads to transformation and proliferation of the cells that is induced by the complex interaction of EBV encoded proteins and virally induced cellular proteins. The functions of the EBV-encoded proteins in the transformation event and in the maintenance of proliferation have been characterized in the in vitro generated LCLs.5 The set of EBV encoded proteins expressed in these cells comprises six proteins, localized in the nucleus, EBNA 1–6, and three cell membrane associated proteins, LMP-1, -2A and -2B (Fig. 1). This expression pattern is usually referred to as Type III latency or growth program. The expression of EBNA-2 and LMP-1 are pivotal for proliferation and therefore they can be exploited as markers for the proliferating cells.22 Importantly, as an innate immune response, naïve CD4 T cells are activated by encounter of EBV transformed B blasts before the adaptive response develops.17
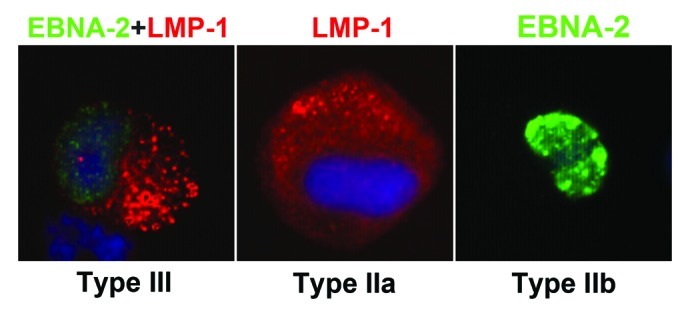
Figure 1. EBNA-2 and LMP-1 expression 7 days after in vitro EBV infection of cord blood mononuclear cells. DAPI staining (Vectashield) for nuclear DNA; EBNA-2 staining with mouse anti-EBNA-2 mAb clone PE2, followed by goat anti-mouse IgG1-Alexa Fluor 488; LMP-1 staining with anti-LMP-1 mAb clone S-12, followed by anti-mouse IgG2a-Alexa Fluor 594.22
Studies of malignancies revealed that the expression of EBV encoded proteins can occur in various combinations, and they impose different phenotypic and functional properties on the viral genome carrying B cell.4 In EBV positive Burkitt’s lymphoma (BL), only one nuclear protein, EBNA-1, is expressed, referred to as Type I latency. The main function of EBNA-1 is the maintenance of the episomal form of the viral genome. A low number of B lymphocytes, with Type I latency is responsible for the EBV carrier state.23 The phenotype of the Type I cell enables its existence in the face of the highly efficient EBV specific immune response in healthy individuals because it lacks activation associated changes which are required for the interaction with T cells. Furthermore, the resident viral genome does not alter the response of B cells to differentiation inducing stimuli.
The expression of the limited assortment of EBV encoded proteins, EBNA-1 and LMP-1 but not EBNA-2, was first detected in nasopharyngeal carcinoma, thus in epithelial cells.24 It was designated as latency Type II. We modified the Type II designation to Type IIa in order to accommodate another restricted expression pattern, see later. The Type IIa assortment of EBV encoded genes is typical for EBV positive Reed/Sternberg cells (R/S) of Hodgkin’s lymphoma (HL) that is of B cell origin.25,26 Type IIa expressor HL cells have no inherent proliferation capacity. Type II latency does not induce proliferation of B cells in vitro.22 The in vivo proliferation of R/S cells is induced by the growth-promoting factors produced in the microenvironment. In spite of considerable effort, B-cell lines with Type IIa EBV latency have not been established from HLs.27 The few established and available HL-derived cell lines are all EBV-negative, except for the L591 line that expresses Type III latency.28 However Type III cells do not occur in the HLs. Thus there is no evidence that the Type IIa EBV expression induces cell proliferation unless signals from the microenvironment contribute. The HL tumor mass is composed of granulomatous tissue in which only a small proportion, 1–10%, of the cells are the characteristic R/S cells.29 The granuloma is created by complex interaction between the R/S cells and components of the immune system. Humoral factors regulate the phenotypes in autocrine and paracrine manners. Importantly, typical HL can arise also without the contribution of EBV. The proportion of EBV positive HL cases varies with age and with geography. Critical contribution of EBV to the pathogenesis of HL is indicated by the occurrence of Type IIa cells in the lymphoid tissues of the patients of infectious mononucleosis (IM) and by the elevated risk for EBV positive HL when primary infection in children elicits the IM syndrome.30 It was proposed that the R/S cells are originally faulty differentiated B cells that are normally destined for elimination by apoptosis but EBV infection would save them.31 The Type IIa assortment of the EBV encoded genes is explained by the inadequate differentiation of B cell due to lack of specific transcription factors required for expression of EBNA-2.32,33
Recently, we have detected cells with latency Type IIa EBV expression pattern early after in vitro infection of cord blood lymphocyte populations and in LCL cells exposed to activated CD4 T lymphocytes or their products IL-21 and CD40 ligand. In the culture of EBV infected cord blood mononuclear cells, the proliferating Type III cells dominate and finally establish LCLs.22 In the latter experiment the Type III cells are induced for plamacytoid differentiation that is accompanied by downregulation of EBNA-2 and cessation of proliferation.13,14,22
For easy reference, we modified thus the original designation of Type II latency to Type IIa and refer to the reciprocal pattern that lacks LMP-1 but expresses EBNA-2 (and the other nuclear proteins) as Type IIb latency.16 This expression type is the subject of this review. The phenotype and the fate of the cells with Type IIa vs. Type IIb exhibiting expression patterns differ considerably; importantly the former but not the latter can contribute to the development of malignancy.
Type IIb Latency Program
In Type IIb latency, the EBNAs are expressed, but LMP-1 is not. Type IIb cells were first seen in B-Chronic Lymphocytic Leukemia (B-CLL) cells infected with EBV in vitro.34-37 B-CLL arises by the clonal expansion of long-lived resting B lymphocytes.38 EBV is not involved in its pathogenesis.39 Only rare EBV-positive cells were detected in the lymph nodes and in the bone marrow of the CLL patients.40,41 Blood derived CLL cells do not survive in culture and their response to B cell activating signals is weak.
Chronic lymphocytic leukemia (CLL)
CLL cells regularly express CD21, the EBV receptor, and can be infected in vitro. The events following infection of normal blood derived B cells and CLL cells differ.39,42 In contrast to the EBV infected normal B lymphocyte population, blast transformation was not induced, the nuclear chromatin remained dense and the activation of the immediate-early genes, c-myc, ATF-2, and c-Jun did not occur in the CLL cells. Critical steps in B cell transformation such as phosphorylation of Rb and decline of p27 expression did not occur either.43 However, activation of the CLL cells by CD40L led to blast transformation and in these cells the EBNA-2 staining resembled that of infected normal B lymphocytes. The initial steps of growth transformation thus occurred, but they were not followed by continuous proliferation. In some CLL clones the infected cells were activated and underwent one or two division cycles.18 Occasional CLL clones yielded LCLs with Type III latency. They emerged after prolonged culture period and the cells lost the CD5 surface marker, a hallmark of B-CLL cells. We assume that CLL cells changed in the culture condition and became competent for transformation.44 We conclude that the expression of Type IIb EBV latency is typical for the CLL cells and is determined by the differentiation window of B lymphocytes. However, the cells do not express the growth program, and the infection does not induce proliferation.
We found that EBV carrying Type IIb cells do not stimulate autologous T cells in spite of expression of the immunogenic proteins (EBNA-2, -3, -4, -6). Moreover they were not recognized by CTLs generated against autologous LCLs.45 We ascribed this to the cell phenotype that remained similar to resting B cells, lacking the surface molecules for efficient interaction with T lymphocytes such as critical levels of MHC and expression of co- stimulatory molecules. This assumption was validated by recognition of the EBV infected CLL cells by the autologous CTLs when they were additionally activated with CD40L and IL-2. Their recognition was EBV specific, because non-infected but activated CLL cells were not targeted by the CTLs. Thus, due to the maintenance of the phenotype of a resting B lymphocyte, the CLL cells were not recognized by the immune response even when they harbored the virus against which a highly efficient immune response regularly develops.
We have encountered an unusual CLL patient who had an EBV-positive subclone in the blood.46 The EBV carrying CLL cells proliferated promptly when explanted and the protein expression pattern was the Type III. Such cells were not detected in the ex vivo sample and the patient did not have a proliferating B cell disease. We assume therefore that the EBV genome carrying subclone did not express the growth program in vivo but underwent phenotypic change in vitro which involved acquisition of factors that allowed its expression. Lines could be established from the virus genome subclone in the explants on repeated occasions even from samples collected from the recurrent disease after drug induced remissions. The derived LCLs and the ex vivo B-CLL cells had identical Ig rearrangement and cytogenetic markers. The EBV-positive subclone persisted for several years. This patient showed thus that the virus genome carrying CLL cells (1) followed the dynamics of the disease and were not disturbed by the EBV specific immune response, (2) had no selective advantage in vivo and (3) the EBV genome carrying cells changed to Type III and proliferated in vitro. We assume that if this change would have occurred in vivo, the cells would have been eliminated by the immune response.
Establishment of two LCLs from the CLL cells with one year interval was reported in a case that showed change in the disease phenotype.47 At the time of the establishment of the two LCLs, the CLL showed prolymphocytoid transformation, (PLL). The outgrowth of proliferating lines in the explants is probably related to the change in the phenotype of the disease. In an earlier comparative study, PLL but not CLL cells proliferated when infected with EBV in vitro.35 If infection of PLL cells would have occurred in vivo latency Type III should have expressed. Since the patient survived at least one year after the establishment of first cell line, we speculate cells with latency Type III were not present in vivo. Moreover ex vivo samples on two occasions before the establishment of cell lines were found to be EBNA-2 negative. The authors concluded that the malignant cells were most likely infected in the culture by the virus released from normal B cells. We found that both of the lines were infected with the same virus strain but the event of infection differed as proven by the terminal repeat analysis (to be published).
Burkitt’s lymphoma
EBV carrying BL express EBNA-1, the protein required for the maintenance of the viral episome, latency Type I. The pivotal proteins of the growth program, EBNA-2 and LMP-1, are not expressed. Both EBV positive and negative BLs carry translocation of the myc oncogene to an Ig locus. Almost all “endemic” BLs that occur in Africa and New Guinea are EBV positive but only fractions of the “sporadic” BLs that occur elsewhere carry EBV. Myc driven proliferation was initially thought to be the key factor in the genesis of BLs and therefore the role of EBV remained unclear.48 Kennedy et al. emphasized that the viral genomes can be lost in vitro but not in vivo from the EBV positive tumors.49 They regarded therefore EBV, particularly the EBNA-1 protein to be essential for the virus carrying tumors probably due to anti-apoptotic function. Indeed, as recognized later, in addition to driving proliferation, activation of myc could render cells prone to apoptosis and evidence was presented that EBV counteracted this effect.50
One unusual endemic BL case, regarding EBV protein expression, with heterogeneous cell population was described by Kelly et al.51 Four types of clones were derived from the early in vitro passages. A few clones were EBV negative. The EBV positive clones were of three latency types. (1) Typical, only EBNA-1 expressor Type I. (2) EBNA-2 negative LMP-1 positive expressor, carrying a deletion of the EBNA-2 sequence. These cells were EBNA -3, -4, -6 positive and were referred to as Wp-restricted latency. They differed thus from latency Type IIa in which all the nuclear proteins are absent due to cell differentiation determined regulation of the EBV encoded protein expression. (3) EBNA-2 positive LMP-1 negative, Type IIb. All clones carried the myc translocation. The Type IIb clones carried single integrated EBV genome. Type IIb cells (5–10%) were detected in the biopsy of original tumor tissue and in the first in vitro passages by immunofluorescence with high variations in the intensities of EBNA-2 staining among the clones.
Activation of c-myc was probably responsible for the proliferation of these clones. The clones differed in apoptotic propensity when elicited by two different triggers and this could be correlated with variations in the expression of EBV encoded genes. The authors conclude that EBV counterbalanced the apoptotic function of myc in these cells.
Infectious mononucleosis and Post-transplant lymphoproliferative disorders (PTLD)
Simultaneous visualization of EBNA-2 and LMP-1 proteins in B cells of IM, PTLD and AIDS associated lymphoma tissues characterized cells with Type IIa, IIb, and III latencies.52-55 Considerable proportion of the EBV positive cells with small to medium size expressed the Type IIb latency. The Type IIa cells were larger and resembled R/S cells in EBV positive HL tissues. Morphologically intermediate sized cells expressed Type III.52-54 Evaluation of the quantitative expression of these EBV encoded proteins revealed the high degree of variations in these tissues; including cells with extremely weak reactivity with the LMP-1 specific reagent.
Kurth et al. substantiated these results in IM tonsil samples and provided details about the differentiation stage, localization and the clonality of EBV infected B cells in relation to the germinal centers.53 Type I, classical Type II and Type III cells were detected. Their relative frequencies were as follows: fewer than 10%, 20–30%, 10–20% respectively. In addition, 50–60% of small to medium-sized cells expressed EBNA-2 but not LMP-1, thus Type IIb latency. Sequence analysis of V region genes showed that cells with both unmutated and mutated genes carried EBV and their clones could expand without somatic hypermutation.
These careful studies did not provide biological classification of the Type IIb cells. The nature of the Type IIb cells was “undefined” and they were regarded either as transitional stages between the various latency types or proceeding to the virus productive stage.52,53
Humanized mouse
Type IIb cells were detected in two studies that employed EBV infected “humanized mice.” In such mice, the functional human immune system develops from administered human stem/progenitor cells. The recipient Rag2−/− γc−/− mice or the NOD/LtSz-scid/IL2Rγnull mice are highly immunodeficient.56,57 EBV infected humanized mice can be regarded as small animal models in which the details of the virus-target cell interaction and the contribution of immune regulation can be studied as the mouse cells are not susceptible for EBV infection. It can be exploited for experimental manipulation of the immunological functions e.g., elimination of T cell subsets, administration of immunomodulators, establishing the importance of the viral dose and the effect of viral variants. Yajima M et al. emphasized that the consequence of EBV infection, histology and phenotype of the infected cells in such mice is similar to that seen in immunocompromised humans.58 In one study, immunoblastic lymphomas with Type IIa latency were induced but from explanted samples Type III cells grew.59 Considering the great variation of the EBV related parameters in humans, it is not surprising that in the complex experimental system using humanized mice the details vary. Variation is evident even in single experiment as results may vary in mice of one litter infected with the same virus dose.56 In addition, the emphasis on the studied parameters varies with the interest of investigator. However, it can be concluded that the dominant latencies were Type I and Type II.
Type IIb cells were detected in EBV infected Rag2−/− γc−/− humanized mice.56 The morphology of the lymphoid tissue, topographical distribution, latency programs, immunophenotype, Ig mutation status of the infected cells and the composition of the T cell subsets were studied. Generally, these parameters were reminiscent of those seen in patients with infectious mononucleosis. In situ hybridization demonstrated efficient B cell infection as indicated by high proportions of EBER positive B lymphocytes and immunohistochemistry detected Type III, Type IIa and Type IIb programs. The relative frequencies were similar to that seen in IM tissues.
The lymphoid tissue of the uninfected mice showed follicular hyperplasia with primary follicle and only few germinal centers. EBV infected mice that had two different morphological patterns were analyzed. Cells with Type IIb latency were detected in both. In the lymphoid tissues of the infected mice with follicular hyperplasia and intensive germinal center formation, the majority of EBV carrying B cells, based on EBER expression, were localized outside and at the periphery of the follicles. These were mainly small-medium-sized Type I cells. Type IIb cells were detected in the mantle zone and within the germinal center. In both latency types, the B cells were naïve; they did not carry somatic mutations. In contrast, the few cells with latency Type IIa program that were localized in the germinal center carried few mutations.
The EBV positive cell populations in the lymphoid tissue of mice with nodular and diffuse lymphoid proliferation without germinal center formation were polymorphic, containing both mutated and unmutated Type III cells. A considerable fraction of the B cells exhibited the Type IIb program. In these mice, the CD8 T cells were enriched, as it occurs in infectious mononucleosis.
Type IIb cells were demonstrated in EBV infected humanized mice in the study, in which the contribution of the virus spread in the development of lymphomas was the main subject. Therefore mice were also infected with lytic replication defective virus. In addition to the CD34 positive stem cells, isolated from human fetal liver, the immunodeficient mice received thymus and liver tissue from the same source. Six of the 11 control virus infected mice developed Type I and Type IIb “lymphomas” detected in serial sections of the spleen. LMP-1 positive cells were rare. All lymphomas were infiltrated with CD4 and CD8 T cells; however the Type IIb “tumors” had more robust CD4 cell infiltration. The authors emphasize that this is the first model in which EBV induced lymphomas with Type I and IIb latency were induced.57
Conclusions
EBV Latency Type IIb B lymphocytes were detected in several experimental systems. It was first seen and occurred regularly in cultures of in vitro EBV infected blood derived CLL cells. This finding has two important aspects. First, it showed convincingly that the latency type i.e., the assortment of expressed EBV encoded proteins can be determined by the differentiation window of the B cell at the event of infection. The other aspect concerns the role of EBNA-2 in the expression of LMP-1. The EBNA-2 protein is part of a molecular complex that binds to LMP-1 promoter. The mechanism responsible for the absence of LMP-1 in the CLL cells when EBNA-2 is present has not been clarified yet.
CLL cells infected with EBV in vitro express Type IIb latency and they do not proliferate. Lines were obtained in rare experiments. However, they were Type III and they emerged from the infected cultures with longer latency period compared with the emergence of LCLs in infected cultures of normal B cell populations. We assumed that some cells in the EBV infected CLL population changed in the culture condition and this involved also expression of cellular factors critical for the establishment of Type III latency.
The Type IIb proliferating clones selected from one unique BL sample carried Ig/myc translocation, as it is the case with EBV negative BLs. The EBV encoded proteins probably modified but they were not responsible for the proliferating potential of the cells.
CLL develops as an expansion of CD5 positive B cell with mature phenotype. Its exact origin and pathogenesis are unknown. EBV positive Type IIb expresser cells were detected in the lymphoid tissue during the acute phase of IM. We propose that the characteristic expression of EBV encoded genes, EBNA-2 positive but LMP-1 negative, may serve as marker for identification of the original B cell representing a particular differentiation window. The fate of the Type IIb cells in the IM tissue is not known. While they have no indigenous proliferative potential they may undergo a few divisions, perhaps under the influence of factors in the microenvironment. The experiments with the humanized mice suggest that CD4 T cells could provide the stimulus. In IM, the Type IIb cells disappear upon recovery probably by apoptosis. The CLL disease that is not related to EBV may arise from corresponding B lymphocytes by unknown mechanism and the leukemia cells may maintain certain features of their origin.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Department of Obstetrics and Gynecology, Karolinska University Hospital, for the provision of cord blood samples. This work was supported by the Swedish Cancer Society and by the Cancer Research Institute (New York, NY)/Concern Foundation (Los Angeles, CA).
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/23035
References
- 1.Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A. 1984;81:4510–4. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Küppers R. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol. 2003;3:801–12. doi: 10.1038/nri1201. [DOI] [PubMed] [Google Scholar]
- 3.Pope JH, Horne MK, Scott W. Transformation of foetal human keukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int J Cancer. 1968;3:857–66. doi: 10.1002/ijc.2910030619. [DOI] [PubMed] [Google Scholar]
- 4.Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat Rev Microbiol. 2008;6:913–24. doi: 10.1038/nrmicro2015. [DOI] [PubMed] [Google Scholar]
- 5.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–68. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 6.Klein G. Epstein-Barr virus strategy in normal and neoplastic B cells. Cell. 1994;77:791–3. doi: 10.1016/0092-8674(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 7.Rickinson AB, Moss DJ. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu Rev Immunol. 1997;15:405–31. doi: 10.1146/annurev.immunol.15.1.405. [DOI] [PubMed] [Google Scholar]
- 8.Yao QY, Rickinson AB, Epstein MA. A re-examination of the Epstein-Barr virus carrier state in healthy seropositive individuals. Int J Cancer. 1985;35:35–42. doi: 10.1002/ijc.2910350107. [DOI] [PubMed] [Google Scholar]
- 9.Khanna R, Moss DJ, Burrows SR. Vaccine strategies against Epstein-Barr virus-associated diseases: lessons from studies on cytotoxic T-cell-mediated immune regulation. Immunol Rev. 1999;170:49–64. doi: 10.1111/j.1600-065X.1999.tb01328.x. [DOI] [PubMed] [Google Scholar]
- 10.Pearson G, Dewey F, Klein G, Henle G, Henle W. Relation between neutralization of Epstein-Barr virus and antibodies to cell-membrane antigens-induced by the virus. J Natl Cancer Inst. 1970;45:989–95. [PubMed] [Google Scholar]
- 11.Tosato G, Magrath IT, Blaese RM. T cell-mediated immunoregulation of Epstein Barr virus- (EBV) induced B lymphocyte activation in EBV-seropositive and EBV-seronegative individuals. J Immunol. 1982;128:575–9. [PubMed] [Google Scholar]
- 12.Rochford R, Hobbs MV, Garnier JL, Cooper NR, Cannon MJ. Plasmacytoid differentiation of Epstein-Barr virus-transformed B cells in vivo is associated with reduced expression of viral latent genes. Proc Natl Acad Sci U S A. 1993;90:352–6. doi: 10.1073/pnas.90.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kis LL, Salamon D, Persson EK, Nagy N, Scheeren FA, Spits H, et al. IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc Natl Acad Sci U S A. 2010;107:872–7. doi: 10.1073/pnas.0912920107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagy N, Adori M, Rasul A, Heuts F, Salamon D, Ujvári D, et al. Soluble factors produced by activated CD4+ T cells modulate EBV latency. Proc Natl Acad Sci U S A. 2012;109:1512–7. doi: 10.1073/pnas.1120587109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wendel-Hansen V, Rosén A, Klein G. EBV-transformed lymphoblastoid cell lines down-regulate EBNA in parallel with secretory differentiation. Int J Cancer. 1987;39:404–8. doi: 10.1002/ijc.2910390322. [DOI] [PubMed] [Google Scholar]
- 16.Klein E, Kis LL, Klein G. Epstein-Barr virus infection in humans: from harmless to life endangering virus-lymphocyte interactions. Oncogene. 2007;26:1297–305. doi: 10.1038/sj.onc.1210240. [DOI] [PubMed] [Google Scholar]
- 17.Klein E, Liu A, Claesson HE. Activation of innate immunity by the leukotriene B 4 inhibits EBV induced B-cell transformation in cord-blood derived mononuclear cultures. Immunol Lett. 2008;116:174–7. doi: 10.1016/j.imlet.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Teramoto N, Gogolák P, Nagy N, Maeda A, Kvarnung K, Björkholm T, et al. Epstein-Barr virus-infected B-chronic lymphocyte leukemia cells express the virally encoded nuclear proteins but they do not enter the cell cycle. J Hum Virol. 2000;3:125–36. [PubMed] [Google Scholar]
- 19.Kurth J, Hansmann ML, Rajewsky K, Küppers R. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc Natl Acad Sci U S A. 2003;100:4730–5. doi: 10.1073/pnas.2627966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kis LL, Nishikawa J, Takahara M, Nagy N, Matskova L, Takada K, et al. In vitro EBV-infected subline of KMH2, derived from Hodgkin lymphoma, expresses only EBNA-1, while CD40 ligand and IL-4 induce LMP-1 but not EBNA-2. Int J Cancer. 2005;113:937–45. doi: 10.1002/ijc.20654. [DOI] [PubMed] [Google Scholar]
- 21.Anastasiadou E, Vaeth S, Cuomo L, Boccellato F, Vincenti S, Cirone M, et al. Epstein-Barr virus infection leads to partial phenotypic reversion of terminally differentiated malignant B cells. Cancer Lett. 2009;284:165–74. doi: 10.1016/j.canlet.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 22.Rasul AE, Nagy N, Sohlberg E, Ádori M, Claesson HE, Klein G, et al. Simultaneous detection of the two main proliferation driving EBV encoded proteins, EBNA-2 and LMP-1 in single B cells. J Immunol Methods. 2012;385:60–70. doi: 10.1016/j.jim.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci U S A. 2004;101:239–44. doi: 10.1073/pnas.2237267100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fåhraeus R, Fu HL, Ernberg I, Finke J, Rowe M, Klein G, et al. Expression of Epstein-Barr virus-encoded proteins in nasopharyngeal carcinoma. Int J Cancer. 1988;42:329–38. doi: 10.1002/ijc.2910420305. [DOI] [PubMed] [Google Scholar]
- 25.Deacon EM, Pallesen G, Niedobitek G, Crocker J, Brooks L, Rickinson AB, et al. Epstein-Barr virus and Hodgkin’s disease: transcriptional analysis of virus latency in the malignant cells. J Exp Med. 1993;177:339–49. doi: 10.1084/jem.177.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pallesen G, Hamilton-Dutoit SJ, Rowe M, Young LS. Expression of Epstein-Barr virus latent gene products in tumour cells of Hodgkin’s disease. Lancet. 1991;337:320–2. doi: 10.1016/0140-6736(91)90943-J. [DOI] [PubMed] [Google Scholar]
- 27.Staratschek-Jox A, Wolf J, Diehl V. Hodgkin's disease. In: Masters JRW, Palsson B, ed. Human cell culture Vol 3 Lymphoma and leukemia cell lines: Dordrecht: Kluwer Academic Press, 2000:339-53. [Google Scholar]
- 28.Vockerodt M, Belge G, Kube D, Irsch J, Siebert R, Tesch H, et al. An unbalanced translocation involving chromosome 14 is the probable cause for loss of potentially functional rearranged immunoglobulin heavy chain genes in the Epstein-Barr virus-positive Hodgkin’s lymphoma-derived cell line L591. Br J Haematol. 2002;119:640–6. doi: 10.1046/j.1365-2141.2002.03894.x. [DOI] [PubMed] [Google Scholar]
- 29.Küppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9:15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- 30.Hjalgrim H, Smedby KE, Rostgaard K, Molin D, Hamilton-Dutoit S, Chang ET, et al. Infectious mononucleosis, childhood social environment, and risk of Hodgkin lymphoma. Cancer Res. 2007;67:2382–8. doi: 10.1158/0008-5472.CAN-06-3566. [DOI] [PubMed] [Google Scholar]
- 31.Bräuninger A, Schmitz R, Bechtel D, Renné C, Hansmann ML, Küppers R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. Int J Cancer. 2006;118:1853–61. doi: 10.1002/ijc.21716. [DOI] [PubMed] [Google Scholar]
- 32.Tierney R, Kirby H, Nagra J, Rickinson A, Bell A. The Epstein-Barr virus promoter initiating B-cell transformation is activated by RFX proteins and the B-cell-specific activator protein BSAP/Pax5. J Virol. 2000;74:10458–67. doi: 10.1128/JVI.74.22.10458-10467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Contreras-Brodin B, Karlsson A, Nilsson T, Rymo L, Klein G. B cell-specific activation of the Epstein-Barr virus-encoded C promoter compared with the wide-range activation of the W promoter. J Gen Virol. 1996;77:1159–62. doi: 10.1099/0022-1317-77-6-1159. [DOI] [PubMed] [Google Scholar]
- 34.Doyle MG, Catovsky D, Crawford DH. Infection of leukaemic B lymphocytes by Epstein Barr virus. Leukemia. 1993;7:1858–64. [PubMed] [Google Scholar]
- 35.Walls EV, Doyle MG, Patel KK, Allday MJ, Catovsky D, Crawford DH. Activation and immortalization of leukaemic B cells by Epstein-Barr virus. Int J Cancer. 1989;44:846–53. doi: 10.1002/ijc.2910440517. [DOI] [PubMed] [Google Scholar]
- 36.Rickinson AB, Finerty S, Epstein MA. Interaction of Epstein-Barr virus with leukaemic B cells in vitro. I. Abortive infection and rare cell line establishment from chronic lymphocytic leukaemic cells. Clin Exp Immunol. 1982;50:347–54. [PMC free article] [PubMed] [Google Scholar]
- 37.Takada K, Yamamoto K, Osato T. Analysis of the transformation of human lymphocytes by Epstein-Barr virus. II. Abortive response of leukemic cells to the transforming virus. Intervirology. 1980;13:223–31. doi: 10.1159/000149129. [DOI] [PubMed] [Google Scholar]
- 38.Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol. 1999;17:399–408. doi: 10.1200/JCO.1999.17.1.399. [DOI] [PubMed] [Google Scholar]
- 39.Klein E, Nagy N. Restricted expression of EBV encoded proteins in in vitro infected CLL cells. Semin Cancer Biol. 2010;20:410–5. doi: 10.1016/j.semcancer.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 40.Kanzler H, Küppers R, Helmes S, Wacker HH, Chott A, Hansmann ML, et al. Hodgkin and Reed-Sternberg-like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell-derived clones: potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin’s disease. Blood. 2000;95:1023–31. [PubMed] [Google Scholar]
- 41.Tsimberidou AM, O’Brien S, Kantarjian HM, Koller C, Hagemeister FB, Fayad L, et al. Hodgkin transformation of chronic lymphocytic leukemia: the M. D. Anderson Cancer Center experience. Cancer. 2006;107:1294–302. doi: 10.1002/cncr.22121. [DOI] [PubMed] [Google Scholar]
- 42.Avila-Cariño J, Andersson J, Mellstedt H, Klein E. B-CLL cells experimentally infected with EBV enter DNA synthesis, produce cytokines and stimulate T-lymphocytes. Immunol Lett. 1996;54:45–52. doi: 10.1016/S0165-2478(96)02643-0. [DOI] [PubMed] [Google Scholar]
- 43.Maeda A, Bandobashi K, Nagy N, Teramoto N, Gogolák P, Pokrovskaja K, et al. Epstein-barr virus can infect B-chronic lymphocytic leukemia cells but it does not orchestrate the cell cycle regulatory proteins. J Hum Virol. 2001;4:227–37. [PubMed] [Google Scholar]
- 44.Rosén A, Bergh AC, Gogok P, Evaldsson C, Myhrinder AL, Hellqvist E, et al. Lymphoblastoid cell line with B1 cell characteristics established from a chronic lymphocytic leukemia clone by in vitro EBV infection. Oncoimmunology. 2012;1:18–27. doi: 10.4161/onci.1.1.18400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomita Y, Avila-Cariño J, Yamamoto K, Mellstedt H, Klein E. Recognition of B-CLL cells experimentally infected with EBV by autologous T lymphocytes. Immunol Lett. 1998;60:73–9. doi: 10.1016/S0165-2478(97)00142-9. [DOI] [PubMed] [Google Scholar]
- 46.Lewin N, Minarovits J, Weber G, Ehlin-Henriksson B, Wen T, Mellstedt H, et al. Clonality and methylation status of the Epstein-Barr virus (EBV) genomes in in vivo-infected EBV-carrying chronic lymphocytic leukemia (CLL) cell lines. Int J Cancer. 1991;48:62–6. doi: 10.1002/ijc.2910480112. [DOI] [PubMed] [Google Scholar]
- 47.Stacchini A, Aragno M, Vallario A, Alfarano A, Circosta P, Gottardi D, et al. MEC1 and MEC2: two new cell lines derived from B-chronic lymphocytic leukaemia in prolymphocytoid transformation. Leuk Res. 1999;23:127–36. doi: 10.1016/S0145-2126(98)00154-4. [DOI] [PubMed] [Google Scholar]
- 48.Nagy N, Klein G, Klein E. To the genesis of Burkitt lymphoma: regulation of apoptosis by EBNA-1 and SAP may determine the fate of Ig-myc translocation carrying B lymphocytes. Semin Cancer Biol. 2009;19:407–10. doi: 10.1016/j.semcancer.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 49.Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas. Proc Natl Acad Sci U S A. 2003;100:14269–74. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allday MJ. How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt’s lymphoma? Semin Cancer Biol. 2009;19:366–76. doi: 10.1016/j.semcancer.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelly GL, Milner AE, Baldwin GS, Bell AI, Rickinson AB. Three restricted forms of Epstein-Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 2006;103:14935–40. doi: 10.1073/pnas.0509988103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brink AA, Dukers DF, van den Brule AJ, Oudejans JJ, Middeldorp JM, Meijer CJ, et al. Presence of Epstein-Barr virus latency type III at the single cell level in post-transplantation lymphoproliferative disorders and AIDS related lymphomas. J Clin Pathol. 1997;50:911–8. doi: 10.1136/jcp.50.11.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, et al. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity. 2000;13:485–95. doi: 10.1016/S1074-7613(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 54.Niedobitek G, Agathanggelou A, Herbst H, Whitehead L, Wright DH, Young LS. Epstein-Barr virus (EBV) infection in infectious mononucleosis: virus latency, replication and phenotype of EBV-infected cells. J Pathol. 1997;182:151–9. doi: 10.1002/(SICI)1096-9896(199706)182:2<151::AID-PATH824>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 55.Oudejans JJ, Jiwa M, van den Brule AJ, Grässer FA, Horstman A, Vos W, et al. Detection of heterogeneous Epstein-Barr virus gene expression patterns within individual post-transplantation lymphoproliferative disorders. Am J Pathol. 1995;147:923–33. [PMC free article] [PubMed] [Google Scholar]
- 56.Cocco M, Bellan C, Tussiwand R, Corti D, Traggiai E, Lazzi S, et al. CD34+ cord blood cell-transplanted Rag2-/- gamma(c)-/- mice as a model for Epstein-Barr virus infection. Am J Pathol. 2008;173:1369–78. doi: 10.2353/ajpath.2008.071186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol. 2011;85:165–77. doi: 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yajima M, Imadome K, Nakagawa A, Watanabe S, Terashima K, Nakamura H, et al. A new humanized mouse model of Epstein-Barr virus infection that reproduces persistent infection, lymphoproliferative disorder, and cell-mediated and humoral immune responses. J Infect Dis. 2008;198:673–82. doi: 10.1086/590502. [DOI] [PubMed] [Google Scholar]
- 59.Islas-Ohlmayer M, Padgett-Thomas A, Domiati-Saad R, Melkus MW, Cravens PD, Martin MdelP, et al. Experimental infection of NOD/SCID mice reconstituted with human CD34+ cells with Epstein-Barr virus. J Virol. 2004;78:13891–900. doi: 10.1128/JVI.78.24.13891-13900.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]