

Abstract
N-Myristoyltransferase (NMT) is an attractive antiprotozoan drug target. A lead-hopping approach was utilized in the design and synthesis of novel benzo[b]thiophene-containing inhibitors of Plasmodium falciparum (Pf) and Plasmodium vivax (Pv) NMT. These inhibitors are selective against Homo sapiens NMT1 (HsNMT), have excellent ligand efficiency (LE), and display antiparasitic activity in vitro. The binding mode of this series was determined by crystallography and shows a novel binding mode for the benzothiophene ring.
Introduction
Diseases resulting from parasitic infections are a global health crisis, responsible for over 700 000 annual deaths and predominantly affecting developing countries.1 The most serious is malaria, caused by parasites of the genus Plasmodium. The vast majority of malaria infections stem from the species Plasmodium falciparum (Pf) and Plasmodium vivax (Pv). Emerging resistance to current therapies highlights the urgent requirement for new antimalarial medications in the near future.2
N-Myristoyltransferase (NMT) is an enzyme found exclusively in eukaryotes and is responsible for the co- and post-translational attachment of the C14 fatty acid myristic acid to the N-terminal glycine of substrate proteins.3 In malaria, essential proteins such as calcium dependent protein kinase 14 and glideosome associated protein 455 have been shown to require myristoylation to carry out their biological functions. Furthermore, genetic experiments have shown NMT to be essential in Plasmodium berghei in vivo.6 This evidence strongly suggests that NMT is a highly promising antiparasitic drug target.
Results and Discussion
Our previous work has led to the identification of parasite NMT inhibitors via high throughput screening.7,8 As an alternative strategy for hit discovery, NMT has been highlighted as a target for the piggy-back approach.9 We have used this methodology successfully to produce a series of moderate affinity and selective PfNMT inhibitors adapted from antifungal NMT inhibitors developed initially by Roche (1, Table 1).10 Although 1 displays selectivity over the human NMT orthologues (HsNMT) and moderate enzyme affinity, its relatively large size means that the ligand efficiency (LE) is significantly lower than 0.35, the average LE of high-throughput screening hits.11 A poor LE limits the potential of a series in hit to lead development, increasing the chances of later stage attrition. We therefore sought to develop this series with the aim of producing more ligand efficient, selective, and novel hit series for PfNMT and PvNMT.
Table 1. Enzyme Affinity, Parasitic LE, and in Vitro Activity for Compounds 1, 5, 9, 12a,b, 14a,b.
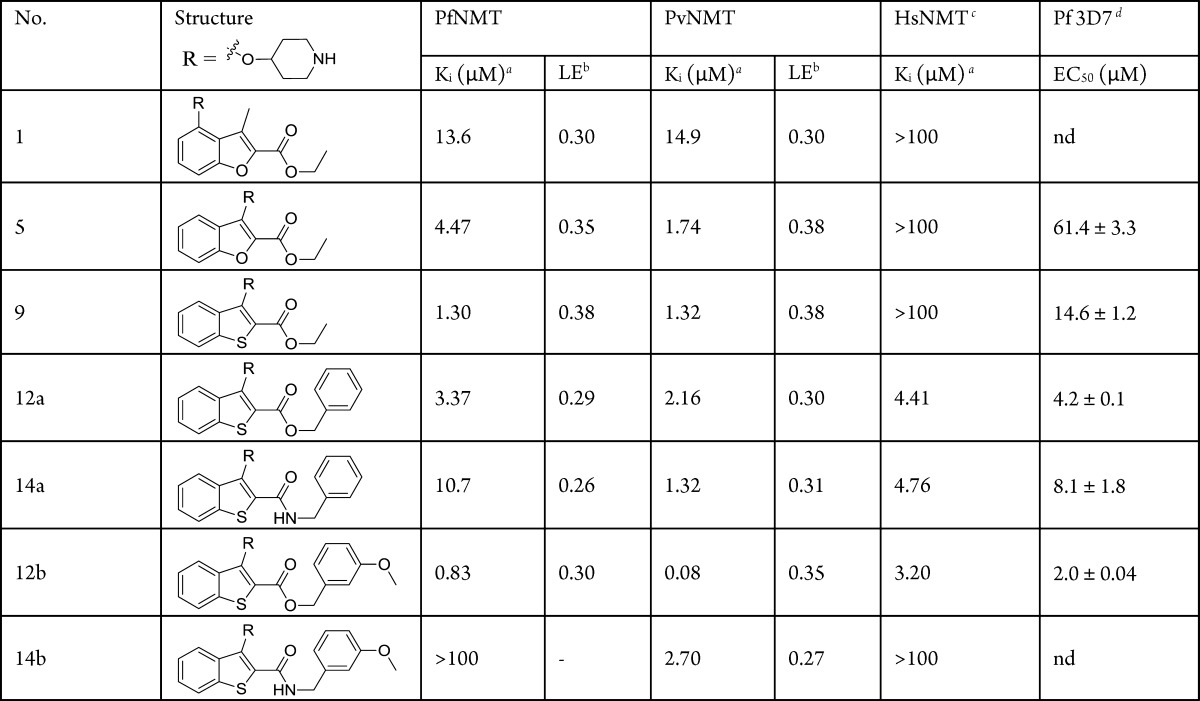
Ki values are quoted in place of IC50 values as a means of expressing the inhibitor affinity while correcting for differing Michaelis constants between enzymes. Enzyme Ki values are calculated from the IC50 values using the Cheng–Prusoff equation (see Supporting Information). IC50 values are the mean of two or more determinations. Standard deviation is within 20% of the IC50.
LE = [−log(Ki)](1.374)/(no. of heavy atoms).
The HsNMT affinities reported in this work refer to HsNMT1; no significant difference in inhibition has been observed between HsNMT1 and HsNMT2 isoforms.
EC50 values for P. falciparum cultured in vitro. 3D7 is a chloroquine-sensitive strain of P. falciparum.
On the basis of the available crystallographic information,12 it was hypothesized that lead hopping by moving the amine substituent from the 4-position on the benzo[b]furan scaffold to the 3-position would be tolerated by the enzyme. This modification would allow the exploration of novel chemical space, facilitating the discovery of novel parasitic NMT inhibitors.
Synthesis of the template was achieved by a Williamson ether synthesis followed by Dieckmann condensation, affording 3 in high yield. A Mitsunobu reaction and deprotection resulted in 5, designed as an analogue of 1 (Scheme 1). Pleasingly, this shift in substitution pattern resulted in a 10-fold affinity improvement against PvNMT, a 3-fold improvement against PfNMT, and no measurable activity against HsNMT up to 100 μM. Coupled with the loss of one heavy atom, this improved the LE to 0.38 for PvNMT and 0.35 for PfNMT (Table 1).
Scheme 1. Synthesis of Benzo[b]furan 1.

Reagents and conditions: (a) ethyl bromoacetate, K2CO3, acetone, reflux, 3 h, 96%; (b) t-BuOK, THF, rt, 15 min, 94%; (c) 1-Boc-4-piperidinol, diisopropyl azodicarboxylate, PPh3, THF, rt, 18 h, 26%; (d) 10% TFA in DCM (v/v), rt, 2 h, 97%.
A range of amines was synthesized to investigate the linker length, basicity, and lipophilicity requirements (see Supporting Information). All changes resulted in complete loss of affinity against all three enzymes, reinforcing previous results from a related series that the piperidine substituent is strongly preferred for affinity.10 For each of these syntheses, the Mitsunobu reaction proceeded with a disappointingly poor yield, typically less than 50%. It was hypothesized that despite the potential for an intramolecular hydrogen bond, the weakly aromatic furan ring13 resulted in significant tautomerism between 3 and the undesired ketone tautomer 6 (Figure 1). Mitsunobu reactions utilizing the disfavored enol tautomer have been previously reported with reactive electrophiles;14 however, it is also known that the presence of carbonyls15 and highly acidic α-carbonyl protons16 can produce side reactions. Furthermore, the unstable heterocycle may lead to problems with stability and toxicity due to the presence of an (albeit hindered) α,β-unsaturated carbonyl in 5 not stabilized by aromaticity.
Figure 1.
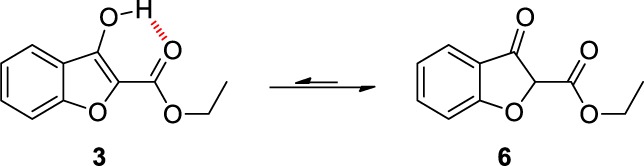
Tautomeric forms of benzo[b]furan 3.
It is known that the aromatic stabilization energy of benzo[b]thiophenes is far greater than that of benzo[b]furans13 and that this results in a higher population of the enol tautomer.17 It was therefore proposed that the bioisosteric replacement of the benzo[b]furan core with benzo[b]thiophene may improve the synthetic efficiency. Furthermore, the benzo[b]furan scaffold is known to form π-interactions with Tyr334 and Ty211 in PvNMT;10 therefore, the increased aromatic character of the benzo[b]thiophene scaffold may result in improved interactions between the scaffold and these residues.
Replacement of oxygen with sulfur enabled modification of the Williamson ether synthesis/Dieckmann condensation into a one-pot cascade procedure and dramatically improved the yield of the Mitsunobu ether synthesis from 26% to 97% (Scheme 2).
Scheme 2. Synthesis of Benzo[b]thiophene 9.

Reagents and conditions: (a) ethyl bromoacetate, t-BuOK, THF, rt, 15 min, 88%; (b) 1-Boc-4-piperidinol, diisopropyl azodicarboxylate, PPh3, THF, rt, 1.5 h, 97%; (c) 10% TFA in DCM (v/v), rt, 2 h, 82%.
9 had 3-fold higher PfNMT affinity than 5, retained HsNMT selectivity, increased structural diversity in the series, and displayed favorable ligand efficiency (Table 1). In addition, 9 displayed significantly improved antiparasitic activity in vitro compared to 5 (Table 1) and exhibited equal binding affinity to PvNMT and PfNMT. 9 was therefore a highly promising plasmodial NMT inhibitor and was selected for further hit to lead development.
Previous work indicated that replacement of the ethyl ester with a m-methoxybenzyl may provide additional affinity (>100-fold in a related series) via hydrophobic interactions and introduction of a methoxy substituent capable of forming a polar interaction with a serine residue in the active site.10 A small selection of benzyl esters and amides was synthesized to probe this possibility within this series of inhibitors (Scheme 3).
Scheme 3. Synthesis of Benzyl Esters and Amides.

Reagents and conditions (a) LiOH·H2O, MeOH, rt, 3 h, 58%; (b) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, hydroxybenzotriazole, N,N-diisopropylethylamine, MeCN, rt, 18 h, 55–66%; (c) benzotriazo-1-yloxytripyrrolidinophosphonium hexafluorophosphate, N,N-diisopropylethylamine, DCM, rt, 18 h, 66–74%; (d) 10% TFA in DCM (v/v), rt, 2 h, 18–81%.
Throughout this series, amides displayed consistently lower affinity than esters, and the benzyl substituent resulted in the loss of a great deal of selectivity compared to ethyl ester 9. Although incorporation of 3-methoxyphenyl (12b, Table 1) produced only a modest improvement in PfNMT enzyme affinity (9 vs 12b, Table 1), the improvement in PvNMT is far more pronounced. In addition, 12b is the most potent antiplasmodial compound of this series with an EC50 of 2.0 μM.
The results in Table 1 indicate that the targeted interactions from the additional aromatic group are not being formed; the benzyl ester 12a is less potent than 9, implying suboptimal interactions for this substituent. Nonetheless, 12b displays excellent PvNMT enzyme affinity and is 10-fold selective over PfNMT. This is in contrast to previous results with a related series, indicating the potential for a novel binding mode of this series compared to the 2,3,4-benzofuran analogues (exemplified by 1).10
The strong enzyme affinity of 12b for PvNMT facilitated determination of the structure of 12b bound to the active site of PvNMT (Figure 2, PDB accession code 4BBH). As implied by the structure–activity relationship in Table 1, 12b adopts an overlapping but distinct binding mode to the 2,3,4-benzofuran series (Figure 2C, PDB accession code 4B14). The interaction between the basic amine moiety and the α-carboxylate of the C-terminal leucine of the protein appears to dominate (Figure 2A), with both inhibitors aligning to form this interaction (Figure 2C). The chemically similar benzofuran and benzothiophene rings occupy distinct locations within the enzyme pocket (Figure 2C). The altered substitution pattern results in the benzothiophene scaffold being buried deeper within a hydrophobic pocket (Figure 2B), providing a rationalization for the high PvNMT enzyme affinity of 12b. As a result of the displacement of the scaffold, the methoxyphenyl group of 12b is unable to reach the pocket occupied by this substituent in the 2,3,4-benzofuran inhibitor (Figure 2C). This results in suboptimal binding of this portion of the molecule, with multiple alternative binding positions visible in the three protein chains present in the asymmetric unit (Figure 2B,C). There is clearly scope for optimizing this substituent, perhaps by extending the linker between the methoxyphenyl group and the scaffold to reach the targeted pocket.
Figure 2.
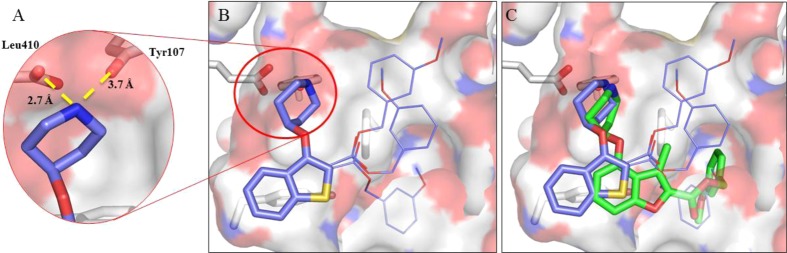
X-ray structure of inhibitors bound to PvNMT.18 (A) A crucial interaction between the enzyme and inhibitor involves the amine of the piperidine moiety of 12b (purple) and a hydrophilic pocket incorporating the enzyme C-terminal carboxylate (Leu410). (B) In this binding mode the benzo[b]thiophene scaffold is deeply buried within the binding pocket. Although this portion of the inhibitor is well-defined, the electron density identifies multiple alternative binding positions for the benzyl ester moiety, as shown by thin bonds (see Supporting Information). This substituent is clearly suboptimal, suggesting an area of future development for this series. (C) The contrasting binding modes are clearly shown by overlay of the PvNMT complex formed by 12b (purple) with that formed by a previously discovered 2,3,4-benzofuran inhibitor (green).10 The novel binding mode explains the difference in SAR between the two series.
The fluctuating selectivity of inhibitors in this series for NMT orthologues is difficult to rationalize based on the crystallographic information; the only residue difference within 5 Å of the active site is the substitution of Tyr334 in PvNMT by Phe334 in PfNMT, with HsNMT and PvNMT displaying a completely homologous active site. The reasons for selectivity are likely to be more subtle, perhaps because of long-range factors modulating stereoelectronic interactions or protein dynamics in the binding site.
The current series contains a potentially biologically labile ester group, and as yet we have no information about the metabolic stability. Further development will focus on compound stability with potency and selectivity to produce a robust lead series for further development.
Conclusion
Analysis of structural information indicated the potential for chemical diversity of a Pf/PvNMT inhibitor via a lead-hopping approach. Altering the positions of scaffold substitution and scaffold-hopping based on aromatic stabilization yielded highly selective and ligand efficient 9 that is built around a benzo[b]thiophene core. Attempts to translate structure–activity relationships developed in a related series yielded 12b. Compound 12b is a structurally novel, high affinity PvNMT inhibitor that displays excellent LE and antiplasmodial activity in vitro. The crystal structure of 12b bound to PvNMT highlighted a novel binding mode for this scaffold.
Experimental Section
Purity of tested compounds was ≥95% unless otherwise specified, as confirmed by LC–MS.
Ethyl 3-Hydroxybenzo[b]thiophene-2-carboxylate 7
To a solution of methyl 2-mercaptobenzoate (1.63 mL, 11.9 mmol) and ethyl bromoacetate (1.32 mL, 11.9 mmol) in dry THF (130 mL) at 0 °C was added potassium tert-butoxide (5.14 g, 71.3 mmol) gradually over 2 min. The mixture was stirred and allowed to warm to rt over 15 min, quenched with 2 M HCl solution to pH 2, and diluted with 75 mL of water. 7 was extracted with 3 × 75 mL portions of EtOAc. The organic layers were combined, washed with 75 mL of brine, dried over MgSO4, and concentrated under reduced pressure to give product 7 as a yellow solid (2.32 g, 88%). 1H NMR (CDCl3, δ, ppm) 10.21 (1H, s), 7.94 (1H, d, J = 8.0), 7.74 (1H, d, J = 8.0), 7.50 (1H, ddd, J = 8.0, 7.5, 1.4), 7.44–7.37 (1H, m), 4.43 (2H, q, J = 7.1), 1.43 (3H, t, J = 7.1).
tert-Butyl 4-((2-(Ethoxycarbonyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate 8
To a solution of 7 (2.70 g, 12.2 mmol) in THF (30 mL) were added tert-butyl 4-hydroxypiperidine-1-carboxylate (4.89 g, 24.3 mmol) and triphenylphosphine (6.38 g, 24.3 mmol). The mixture was stirred under nitrogen for 20 min and cooled to 0 °C. Diisopropyl azodicarboxylate (4.79 mL, 24.3 mmol) in THF (10 mL) was added dropwise over 5 min. The mixture was warmed to rt and stirred for 1.5 h, then concentrated under reduced pressure and purified by flash chromatography, yielding 8 as a pink oil (4.77 g, 97%). 1H NMR (CDCl3, δ, ppm) 7.86 (1H, d, J = 7.9), 7.74 (1H, d, J = 8.0), 7.47 (1H, ddd, J = 8.0, 7.8, 0.8), 7.39 (1H, dd, J = 7.9, 7.8), 4.79–4.69 (1H, m), 4.38 (2H, q, J = 7.2), 4.01–3.86 (2H, m), 3.20–3.07 (2H, m), 2.05–1.95 (2H, m), 1.91–1.79 (2H, m), 1.48 (9H, s), 1.41 (3H, t, J = 7.2).
3-((1-(tert-Butoxycarbonyl)piperidin-4-yl)oxy)benzo[b]thiophene-2-carboxylic Acid 10
To a solution of 8 (2.50 g, 6.17 mmol) in MeOH (30 mL) was added LiOH·H2O (3.35 g, 79.8 mmol). The mixture was stirred at rt for 3 h. The mixture was concentrated under reduced pressure, dissolved in 100 mL water, and acidifed to pH 2 with 2 M HCl solution (30 mL). Precipitate was removed by filtration and washed with 5 × 10 mL of water, then dried in a vacuum desiccator, yielding 10 as an off-white solid (1.36 g, 58%). 1H NMR (CDCl3, δ, ppm) 7.87 (1H, d, J = 8.0), 7.81 (1H, d, J = 8.2), 7.53 (1H, ddd, J = 8.0, 7.0, 0.9), 7.44 (1H, dd, J = 8.2, 7.0), 4.82–4.73 (1H, m), 4.06–3.93 (2H, m), 3.15–3.03 (2H, m), 2.11–2.00 (2H, m), 1.94–1.80 (2H, m), 1.49 (9H, s).
tert-Butyl 4-((2-(((3-Methoxybenzyl)oxy)carbonyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate 11b
To a solution of 10 (50 mg, 0.13 mmol) in MeCN (2 mL) were added hydroxybenzotriazole (27 mg, 0.20 mmol), N,N-diisopropylethylamine (26 μL, 0.16 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (30 mg, 0.16 mmol). The mixture was stirred at rt for 15 min. (3-Methoxyphenyl)methanol (20 μL, 0.16 mmol) was added and the mixture stirred at rt for 18 h. The mixture was concentrated under reduced pressure and dissolved in 10 mL of saturated NH4Cl solution. 11b was extracted with 3 × 10 mL EtOAc. Combined organic layers were washed with 10 mL of brine, dried over MgSO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography, yielding 11b as a colorless oil (37 mg, 60%). 1H NMR (CDCl3, δ, ppm) 7.86 (1H, d, J = 8.0), 7.75 (1H, d, J = 8.2), 7.51–7.46 (1H, m), 7.43–7.37 (1H, m), 7.32 (1H, apparent t, J = 7.9), 7.04 (1H, d, J = 7.8), 7.02–7.00 (1H, m), 6.90 (1H, dd, J = 8.2, 2.3), 5.35 (2H, s), 4.74–4.66 (1H, m), 3.94–3.86 (2H, m), 3.84 (3H, s), 3.07–2.98 (2H, m), 1.98–1.88 (2H, m), 1.85–1.73 (2H, m), 1.48 (9H, s).
3-Methoxybenzyl-3-(piperidin-4-yloxy)benzo[b]thiophene-2-carboxylate 12b
To a solution of 11b (34 mg, 0.07 mmol) in DCM (1.00 mL) was added TFA (100 μL). The solution was stirred at rt for 2 h. The mixture was concentrated under reduced pressure and purified by HPLC, yielding 12b as a colorless oil (5 mg, 18%). tR = 12.3 min; 1H NMR (CDCl3, δ, ppm) 7.82 (1H, d, J = 8.0), 7.76 (1H, d, J = 8.1), 7.56–7.48 (1H, m), 7.47–7.40 (1H, m), 7.33 (1H, apparent t, J = 7.9), 7.04 (1H, d, J = 7.4), 7.01–6.98 (1H, m), 6.91 (1H, dd, J = 8.2, 2.4), 5.34 (2H, s), 4.88–4.81 (1H, m), 3.84 (3H, s), 3.54–3.44 (2H, m), 3.11–3.01 (2H, m), 2.22–2.11 (4H, m). 13C NMR (CDCl3, δ, ppm) 161.48, 159.84, 153.90, 138.23, 136.98, 134.06, 129.82, 128.39, 125.07, 123.14, 122.61, 120.34, 115.87, 113.81, 113.79, 76.10, 66.82, 55.29, 41.01, 28.07. ESI HRMS, found 398.1425 (C22H24NO4S, [M + H]+, requires 398.1426).
Acknowledgments
The authors are grateful to Andrew Bell, Victor Goncalves, Jennie Hickin, and William Heal for valuable discussions. We thank Munira Grainger for providing the parasites used in the in vitro assay. We acknowledge the European Synchrotron Radiation Facility, Grenoble, France, for the provision of data collection facilities and Marek Brzozowski for expert crystal handling. This work was supported by the Engineering and Physical Sciences Research Council (DTA Award), Medical Research Council (Grants 0900278 and U117532067).
Glossary
Abbreviations Used
- nd
not determined
- Pf
Plasmodium falciparum
- Pv
Plasmodium vivax
- NMT
N-myristoyltransferase
- Hs
Homo sapiens
Supporting Information Available
Experimental procedure, characterization of intermediates and target compounds, description of biological assays, determination of Ki values, biological data of supplementary compounds, and crystallographic information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Accession Codes
The coordinates and structure factor files have been deposited in the Protein Data Bank under the accession code 4BBH.
Supplementary Material
References
- TDR Biennial Report 2010/11; TDR: For Research on Diseases of Poverty; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindegardh N.; Socheat D.; White N. J. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.; Bhatnagar R.; Knoll L.; Gordon J. Genetic and Biochemical-Studies of Protein N-Myristoylation. Annu. Rev. Biochem. 1994, 63, 869–914. [DOI] [PubMed] [Google Scholar]
- Möskes C.; Burghaus P. A.; Wernli B.; Sauder U.; Dürrenberger M.; Kappes B. Export of Plasmodium falciparum calcium-dependent protein kinase 1 to the parasitophorous vacuole is dependent on three N-terminal membrane anchor motifs. Mol. Microbiol. 2004, 54, 676–691. [DOI] [PubMed] [Google Scholar]
- Rees-Channer R. R.; Martin S. R.; Green J. L.; Bowyer P. W.; Grainger M.; Molloy J. E.; Holder A. A. Dual acylation of the 45 kDa gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 2006, 149, 113–116. [DOI] [PubMed] [Google Scholar]
- Pino P.; Soldati-Favre D. Personal communication.
- Goncalves V.; Brannigan J. A.; Whalley D.; Ansell K. H.; Saxty B.; Holder A. A.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. Discovery of Plasmodium vivaxN-Myristoyltransferase Inhibitors: Screening, Synthesis, and Structural Characterization of Their Binding Mode. J. Med. Chem. 2012, 55, 3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. S.; Mills J. E.; Williams G. P.; Brannigan J. A.; Wilkinson A. J.; Parkinson T.; Leatherbarrow R. J.; Tate E. W.; Holder A. A.; Smith D. F. Selective Inhibitors of Protozoan Protein N-Myristoyltransferases as Starting Points for Tropical Disease Medicinal Chemistry Programs. PLoS Neglected Trop. Dis. 2012, 6, e1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer P. W.; Tate E. W.; Leatherbarrow R. J.; Holder A. A.; Smith D. F.; Brown K. A. N-Myristoyltransferase: A Prospective Drug Target for Protozoan Parasites. ChemMedChem 2008, 3, 1–8. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Brannigan J. A.; Moss D. K.; Brzozowski A. M.; Wilkinson A. J.; Holder A. A.; Tate E. W.; Leatherbarrow R. J. Design and Synthesis of Inhibitors of Plasmodium falciparumN-Myristoyltransferase, a Promising Target for Anti-Malarial Drug Discovery. J. Med. Chem. 2012, 55, 8879–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseru G. M.; Makara G. M. The Influence of Lead Discovery Strategies on the Properties of Drug Candidates. Nat. Rev. Drug Discovery 2009, 8, 203–212. [DOI] [PubMed] [Google Scholar]
- Kawasaki K.; Masubuchi M.; Morikami K.; Sogabe S.; Aoyama T.; Ebiike H.; Niizuma S.; Hayase M.; Fujii T.; Sakata K.; Shindoh H.; Shiratori Y.; Aoki Y.; Ohtsuka T.; Shimma N. Design and Synthesis of Novel Benzofurans as a New Class of Antifungal Agents Targeting Fungal N-Myristoyltransferase. Part 3. Bioorg. Med. Chem. Lett. 2003, 13, 87–91. [DOI] [PubMed] [Google Scholar]
- Katritzky A.; Karelson M.; Malhotra N. Heterocyclic Aromaticity. Heterocycles 1991, 32, 127. [Google Scholar]
- Redman A. M.; Dumas J.; Scott W. J. Preparation of 5-Substituted 3-Aminofuran-2-carboxylate Esters. Org. Lett. 2000, 2, 2061–2063. [DOI] [PubMed] [Google Scholar]
- Liu Y.-x.; Xu C.-f.; Liu L.-z. The Preparation of Vinylhydrazinedicarboxylates from Ketones via Mitsunobu Reaction. Synthesis 2003, 9, 1335–1338. [Google Scholar]
- Coppola G. M. Cyclization of 4-Hydroxy-3-hydroxyalkylcarbostyrils Under Mitsunobu Conditions. Synth. Commun. 2004, 34, 3381–3387. [Google Scholar]
- Friedrichsen W.; Traulsen T.; Elguero J.; Katritzky A. R. Tautomerism of Heterocycles: Five-Membered Ring with One Heteroatom. Adv. Heterocycl. Chem. 2000, 76, 85–156. [Google Scholar]
- This figure was produced using the program PyMol (www.pymol.org). The protein surfaces are color-coded according to the electrostatic surface potentials: red is negatively charged; blue is positively charged.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.