

Abstract
The role of protonated nucleotides in modulating the pH-dependent properties of nucleic acids is one of the emerging frontiers in the field of nucleic acid biology. The recent development of a constant pH molecular dynamics simulation (CPHMDMSλD) framework for simulating nucleic acids has provided a tool for realistic simulations of pH-dependent dynamics. We enhanced the CPHMDMSλD framework with pH-based replica exchange (pH-REX), which significantly improves the sampling of both titration and spatial coordinates. The results from our pKa calculations for the GAAA tetraloop, which was predicted with lower accuracy previously due to sampling challenges, demonstrates that pH-REX reduces the average unsigned error (AUE) to 0.7 pKa units, and the error of the most poorly predicted residue A17 was drastically reduced from 2.9 to 1.2 pKa unit. Lastly, we show that pH-REX CPHMDMSλD simulations can be used to identify the dominant conformation of nucleic acid structures in alternate pH environments. This work suggests that pH-REX CPHMDMSλD simulations provide a practical tool for predicting nucleic acid protonation equilibrium from first-principles, and offering structural and mechanistic insight into the study of pH-dependent properties of nucleic acids.
Keywords: CPHMD, pKa, nucleic acids, electrostatics, dynamics, λ-dynamics, pH-dependent, molecular dynamics
The role of protonated nucleotides in modulating the pH-dependent properties of nucleic acids is at the frontier of questions in the field of nucleic acid biology.1,2 Protonated nucleotides serve as key catalytic residues in many ribozymes,3–10 and protonated base pairs are known to stabilize RNA loop structures.11,12 They also control the pH-dependent dynamics of numerous RNA systems, such as the retrovirus pseudoknot,13 peptidyl-transferance center of the ribosome,14–19 helix 69 of the 50S ribosomal subunit,20,21 and the U6 intramolecular stem-loop of the spliceosome complex.22 In DNA systems, the presence of protonated A+•C base pairs are known to cause mutagenic and carcinogenic effects.23,24 Measuring the pH-dependence of such properties allows one to infer a macroscopic pKa of the overall nucleic acid system, but limited information can be obtained about the specific residues that control its activity. Given the ubiquity of protonated nucleotides in regulating pH-dependent activity, the ability to measure the microscopic pKa value associated with the protonation event at a specific residue is invaluable in identifying key nucleotides of interest, which is often necessary to obtain a deeper mechanistic insight into these pH-dependent processes.
Recently, Al-Hashimi and co-workers discovered the existence of low population transient state conformations that are functionally important in both RNA and DNA systems, some of which are known to exhibit pH-dependent behavior.25,26 Conventional experimental techniques such as NMR spectroscopy,27–29 pH-dependent fluorescent nucleobase analogs30–32 and Raman spectroscopy33,34 have not been able to directly characterize such pH-dependent transient states, although progress has been made through the development of novel NMR techniques.35 Computational methods such as molecular dynamics (MD) simulations that have been traditionally used to provide mechanistic insight to the role of protonated residues,36–38 do not model realistic pH-dependent dynamics because the protonation states are fixed throughout the simulation. Therefore, there is a need for pH-coupled MD simulations, where the protonation state of titrating residues changes dynamically in response to their microenvironment. In addition, since no a priori information on the pKa value of key titrating residues is required, pH-coupled MD simulations are uniquely suited to investigate pH-dependent transient states and other systems where there is limited experimental data.
One form of pH-coupled MD simulations, known as constant pH MD simulations has been developed for proteins,39–43 and successfully applied to investigate numerous pH-dependent properties.44–50 However, it was only recently that a newer framework of constant pH MD simulations based on multi-site λ-dynamics (CPHMDMSλD) was established to address questions related to the pH-dependent properties of nucleic acids.51 Blind pKa prediction on the lead-dependent ribozyme has shown that the direction of pKa shifts were accurately predicted, with an average unsigned error of 1.3 pKa units relative to experimental pKa values.52 However, for residues in a GAAA tetraloop, which presents significant sampling challenges because of conformation-dependent pKa behavior and coupled titrating interactions, the calculated pKa values were predicted with lower accuracy,52 hampering the usefulness of CPHMDMSλD simulations for predictive studies. In this article, we describe the application of pH-based replica exchange (pH-REX) to augment the sampling capabilities of CPHMDMSλD simulations. Using pH-REX significantly improves sampling of titration and spatial coordinates of the residues in the GAAA tetraloop, reducing the error of A17, the most poorly predicted residue, from −2.9 to −1.2 pKa units. Our work provides evidence that pH-REX CPHMDMSλD simulations allow one to achieve accurate pKa predictions, to around 1 pKa unit, even for residues that are problematic in conventional CPHMDMSλD simulations.
We first present our results on the performance of pH-REX CPHMDMSλD simulations on the lead-dependent ribozyme. To judge the quality of a computational model, pH-dependent experimental observables, such as microscopic pKa values, may be used as an indicator of how accurately the CPHMDMSλD simulation reproduces pH-dependent properties. Unlike proteins, where the microscopic pKa value of multiple ionizable residues for numerous proteins have been measured,53 the literature of nucleic acid pKa research is much sparser, with only a single pKa value measured for a handful of RNA systems. The lead-dependent ribozyme is, to the best of our knowledge, the most thoroughly-studied RNA system from the standpoint of experimentally-measured microscopic pKa values.54 Therefore, we use it as a model system for benchmarking the performance of pH-REX CPHMDMSλD simulations. The microscopic pKa values calculated from pH-REX simulations, as summarized in Table 1, are consistent with previous work that utilized CPHMDMSλD with conventional MD simulations.52 As illustrated in Fig. 1, up to an 8-fold improvement in the transition rates in λ-space is observed in our pH-REX simulations. The sampling improvement of titration coordinates results in faster convergence, which is demonstrated by fact that pH-REX sampling achieves the same level of accuracy using a total simulation time that is 5-fold shorter than conventional CPHMDMSλD simulations. In addition, we also observe that the improvement in λ-space sampling for the residues of the lead-dependent ribozyme is higher than that of the 3-fold improvement in single nucleotide compounds (see Fig. S1).
Table 1.
Calculated pKa values from conventional and pH-REX CPHMDMSλD simulations of the lead-dependent ribozyme demonstrate a similar level of accuracy.
| Residue | Exp pKa | Conventional CPHMDMSλD (3×5ns) | pH-REX CPHMDMSλD (3ns) | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| n | pKa | Error | n | pKa | Error | ||
|
| |||||||
| A4 | <3.1 | 0.4 ± 0.1 | 0.6 ± 0.1 | 1.3 ± 0.5 | 0.9 ± 0.4 | ||
| A8 | 4.3 ± 0.3 | 0.7 ± 0.3 | 3.7 ± 0.3 | −0.6 | 0.9 ± 0.4 | 3.8 ± 0.6 | −0.5 |
| A12 | <3.1 | 1.1 ± 0.3 | 0.7 ± 0.3 | 1.0 ± 0.3 | 0.6 ± 0.2 | ||
| A16 | 3.8 ± 0.4 | 0.7 ± 0.1 | 2.6 ± 0.1 | −1.2 | 0.7 ± 0.1 | 2.6 ± 0.0 | −1.2 |
| A17 | 3.8 ± 0.4 | 0.4 ± 0.0 | 0.9 ± 0.5 | −2.9 | 1.0 ± 0.6 | 1.1 ± 0.5 | −2.7 |
| A18 | 3.5 ± 0.6 | 0.6 ± 0.0 | 3.8 ± 0.1 | 0.3 | 0.8 ± 0.1 | 3.6 ± 0.0 | 0.1 |
| A25 | 6.5 ± 0.1 | 0.4 ± 0.1 | 4.8 ± 0.5 | −1.7 | 0.5 ± 0.1 | 4.5 ± 0.2 | −2.0 |
|
| |||||||
| AUE | 1.3 | 1.3 | |||||
Figure 1.

pH-REX CPHMDMSλD simulations accelerates sampling of titration coordinates by up to 8-fold in the lead-dependent ribozyme.
In complex RNA structures, where multiple residues are titrated simultaneously, the coupled interactions between these titrating groups lead to slower convergence, because the sampling of titration coordinates is hindered by the protonation states of adjacent interacting titrating groups.52 The variable biases applied in conventional CPHMDMSλD simulations only serve to flatten the potential energy surface of each λ variable, but the orthogonal barriers that arise from these coupled titrating interactions are not addressed. Unlike the recent methodological advances in enhanced sampling strategies reported by Yang and co-workers,55,56 pH-REX sampling does not directly address these orthogonal barriers per se. However, it does periodically shuffle conformations to a higher or lower pH where all residues adopt a uniform protonation state. We suggest that this process effectively decouples the protonation states of interacting residues, allowing one to ameliorate the sampling issues related to these orthogonal barriers.
Having demonstrated that pH-REX accelerates sampling of titration coordinates, we now explore the apparent underprediction of the pKa value of residue A17, which is situated in the GAAA tetraloop of the lead-dependent ribozyme. We performed an initial 15 ns simulation of the excised GAAA tetraloop for pH values between 1 to 4, and compared the results to conventional CPHMDMSλD simulations. As summarized in Table 2 and Table S1, the calculated pKa of residue A17 from the conventional simulations is 1.4, compared to the pKa of 2.3 obtained using pH-REX sampling. Extending our simulations for an additional 15 ns confirmed that the pKa value has converged (Fig. S2). On the whole, pH-REX sampling yields systematic improvement of the predicted pKa values of the GAAA tetraloop, where the average unsigned error (AUE) was reduced to 0.7 pKa units, which is 50% lower than our previous work.52
Table 2.
pH-REX CPHMDMSλD simulations of the GAAA tetraloop of lead-dependent ribozyme improved the accuracy of calculated pKa values compared to straightforward MD simulations.
| Residue | GAAA | AAA | |||
|---|---|---|---|---|---|
|
| |||||
| Exp pKa | Conventional CPHMDMSλD (0–15ns) | pH-REX CPHMDMSλD (0–15ns) | pH-REX CPHMDMSλD (16–30ns) | pH-REX CPHMDMSλD (0–15ns) | |
|
| |||||
| A16 | 3.8±0.4 | 3.2 ± 0.2 | 3.1 ± 0.1 | 3.3 ± 0.1 | 3.5 ± 0.1 |
| A17 | 3.8±0.4 | 1.4 ± 0.3 | 2.3 ± 0.6 | 2.6 ± 0.4 | 3.5 ± 0.1 |
| A18 | 3.5±0.6 | 3.9 ± 0.1 | 3.9 ± 0.1 | 4.0 ± 0.1 | 3.9 ± 0.1 |
|
| |||||
| AUE | 1.1 | 0.9 | 0.7 | ||
The apparent underprediction of the pKa value of residue A17 originates from the anti-cooperative interactions between residues A17 and A18, which artificially suppresses the ability of A17 to adopt the protonated state at low pH conditions.52 This arises from the triply stacked conformation of the GAAA tetraloop, which is characterized by short interatomic distances between the N1 atoms of the two residues. We analyzed this interatomic distance in our simulations of the GAAA tetraloop at pH 1, in the context of another reference simulation of the AAA trinucleotide sequence, which has no structural elements imposing conformational restrictions on it. As shown in Fig. 2, the N1-N1 distances sampled in our pH-REX simulations of the GAAA tetraloop ranged from 2 to 10 Å, which are intermediate between the conventional GAAA tetraloop simulations ( 2 to 6 Å ) and the AAA trinucleotide simulations ( 6 to 18 Å ). The conformational space sampled using pH-REX is reasonable as it does not exhibit more dynamical behavior than the free AAA trinucleotide, but it also samples more conformations than conventional CPHMDMSλD simulations of the GAAA tetraloop. This trend of exploring progressively larger N1-N1 distances results in more weakly coupled interactions that is reflected in the pKa value of the central adenine residue, which increases from 1.4 to 2.3 to 3.5 (Table 2). In addition, we also observed a slight difference in the distribution of the N1-N1 distances between the first 15 ns and the subsequent 15 ns trajectory of the excised GAAA tetraloop. Specifically, the “close contact” region of 3 to 6 Å that describes the initial stacked conformation was partially populated in the first 15 ns, which suggests that the system was still equilibrating. This suggests that sufficient equilibration on the order of a few ns may be required, and metrics such as RMSD relative to the initial structure may be used to determine when equilibration is complete, particularly if one is expecting a significant conformational change in an alternate pH environment.
Figure 2.

Distribution of the interatomic distance of the N1 atoms of residue A17 and A18 of the GAAA tetraloop at pH 1 for a conventional 15ns MD simulation (red trace), the first 15 ns pH-REX simulation (blue trace) and the next 15 ns pH-REX simulation (green trace), compared to a 15 ns pH-REX simulation of the AAA trinucleotide (orange trace).
The experimentally measured pKa values are a superposition of the microscopic pKa values of the various conformations visited by the GAAA tetraloop on the timescale accessible to NMR measurements,52 and the various pH values at which such measurements were recorded. We clustered the conformations sampled by pH-REX simulations at pH 1 (low pH) and 4 (high pH), and the representative structures are illustrated in Fig. 3 The initial triply-stacked conformation (Fig. 3b), which is representative of the NMR structure solved at physiological pH is known to lower the pKa value of residue A17. While it may be the dominant conformation sampled at high pH, this conformation is populated only 20% of the time at low pH. We observe that the dominant conformation sampled at low pH is partially unstacked, where the N1-N1 distance is increased to 9.3 Å (Fig. 3a), and a fully unstacked conformation is observed 10% of the time (Fig. 3c). Interestingly, these unstacked conformations are populated 21% of the time at higher pH. The significant improvement in our pKa predictions for residues in the GAAA tetraloop correspond to the sampling of these alternative conformations, suggesting that sampling using pH-REX provides a more accurate model of the tetraloop’s pH-dependent dynamics. Lastly, our results also indicate that pH-REX CPHMDMSλD simulations can be used to identify the dominant conformation of nucleic acid systems in different pH environments or low population conformations at physiological pH. With the discovery of pH-dependent transient states in both RNA and DNA systems that have been suggested to be functionally important,25,26 we anticipate that pH-REX CPHMDMSλD simulations will provide further structural and mechanistic insight into the findings gleaned from experimental studies, especially in situations where direct experimental characterization of such transient states are challenging.
Figure 3.
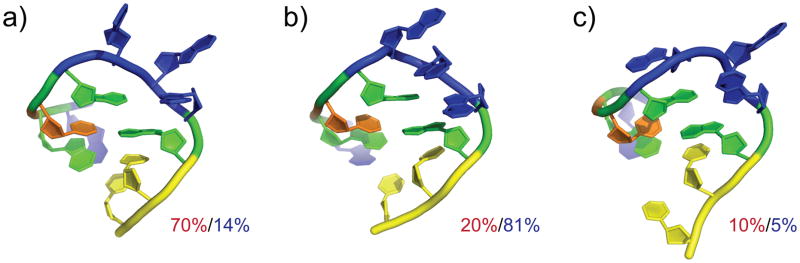
Representative conformations from a cluster analysis of the pH-REX trajectory of the GAAA tetraloop and their relative populations sampled at pH 1 (in red) and pH 4 (in blue).
Thus far, we have shown that pH-REX CPHMDMSλD simulations are effective in modeling accurate pH-dependent dynamics of small nucleic acid motifs like the GAAA tetraloop. In this final section, we demonstrate the scalability of pH-REX CPHMDMSλD simulations to larger systems. Our initial pKa calculations on the full-length ribozyme (Table 1) yielded similar results to conventional CPHMDMSλD simulations, which suggest that the sampling efficiency is not as high as in our simulations of the excised GAAA tetraloop. Since conformational diffusion across pH space is responsible for enhancing sampling, increasing the total number of accepted Monte Carlo (MC) moves should improve the accuracy of calculated pKa values. In the full-length ribozyme, the majority (i.e., 10 out of 15) of the residues are base paired and have pKa values of less than 3.52 Thus, unlike high pH conditions where most of the titrating residues adopt a uniform protonation state, at low pH conditions the majority of residues would be titrating. This implies that there would be more pronounced potential energy difference between adjacent replicas, and consequently a lower MC exchange rate is expected at low pH. Under low pH conditions, the MC exchange rate of the excised tetraloop was at least 30%, which is 3 times higher than that of the full-length ribozyme (SI Fig S3). This lower exchange rate in the full-length ribozyme is correlated with a reduction in the sampling of titration coordinates, particularly for residue A17 (Fig. 4). As shown in Fig. 4a, increasing the frequency of MC attempts from every 1.0 ps to 0.1 ps significantly increased λ-transitions to ~350–650 ns−1. Prior work by Baptista and co-workers has shown that re-equilibration of the solvent induced by the introduction of a charged protonation state requires 1 to 3 ps,39 and we have observed a similar solvent reorganization triggered by a protonation state change in explicit solvent CPHMDMSλD simulation.51 Using the mean solvent relaxation time of 2 ps, one arrives at a transition rate of ~500 ns−1, which balances the need for titration coordinates sampling and solvent relaxation. In some instances, such as residue A8, the transition rate was above this value. We acknowledge that the pH-REX metropolis criterion maintains detailed balance and the results should in principle be unaffected by the MC exchange frequency. However, we observed significant variation in transition rates between the 3 independent runs, which could cause issues in reproducibility and convergence. Therefore, in practice it may be useful to allow for sufficient solvent relaxation occur.
Figure 4.
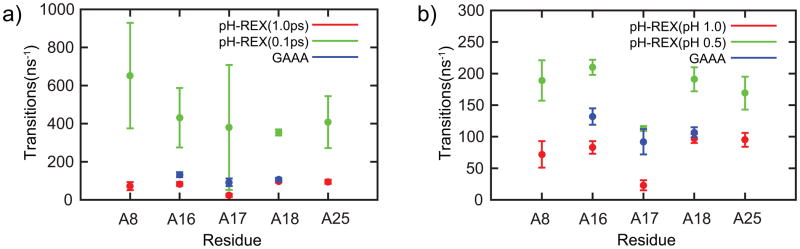
Effects on titration coordinates sampling by (a) increasing the MC attempt frequency and (b) reducing pH window spacing from 1.0 to 0.5.
Our observations on increasing the MC exchange frequency differs with the findings reported by Roitberg and co-workers, where no performance degradation was observed at higher MC exchange frequencies.57 This difference is likely due to the fact that our model uses an explicit solvent representation where solvent reorganization needs to be accounted for, whereas the work of Roitberg and co-workers was performed in implicit solvent, which adiabatically adjusts to the protein conformation. Instead of attempting to increase the MC exchange frequency, one may also increase the probability of exchange by reducing the potential energy difference between adjacent windows (i.e. reduce the pH-spacing). In simulations using a smaller spacing of 0.5 pH units the exchange rate for the full-length ribozyme was increased to 40% as shown in SI Fig. S4. As illustrated in Fig. 4b, reducing the pH-spacing more than doubled the transition rate in λ-space. We observed the most significant improvement in the transition rate of residue A17, which increased from 23ns−1 to 113 ns−1. This is on par with the transition rate of 92 ns−1 observed in the GAAA tetraloop. The transition rate was also uniformly consistent across the 3 independent simulation runs, which ensures the robustness of the calculations. Qualitatively, the titration curves obtained across 3 independent runs also demonstrated better convergence for pH-REX simulations with smaller pH spacing (Fig. S5). Finally, using this smaller pH spacing of 0.5, we reran pH-REX CPHMDMSλD simulations on the full-length ribozyme. After an initial ~10 ns of equilibration, the calculated pKa values started to converge and results comparable to the GAAA tetraloop were achieved within 13 ns (Table 3, Table S2 and Fig. S6), demonstrating that pH-REX CPHMDMSλD simulation scales well to simulate pH-dependent properties of full-sized nucleic acid systems.
Table 3.
pH-REX CPHMDMSλD simulations of the full-length lead-dependent ribozyme at 0.5 pH window spacing demonstrate comparable results to the GAAA tetraloop within 13 ns.
| Residue | Exp pKa | pH-REX CPHMDMSλD
|
||||
|---|---|---|---|---|---|---|
| (0–3ns) | (3–8ns) | (8–13ns) | (13–18ns) | (18–23ns) | ||
|
| ||||||
| A16 | 3.8±0.4 | 2.6 ± 0.1 | 2.7 ± 0.2 | 2.9 ± 0.2 | 2.9 ± 0.1 | 2.8 ± 0.1 |
| A17 | 3.8±0.4 | 1.4 ± 0.3 | 1.5 ± 0.6 | 1.8 ± 0.6 | 2.4 ± 0.1 | 2.4 ± 0.1 |
| A18 | 3.5±0.6 | 3.8 ± 0.1 | 3.8 ± 0.1 | 3.9 ± 0.1 | 3.8 ± 0.1 | 3.7 ± 0.0 |
In conclusion, we have enhanced the framework of constant pH molecular dynamics (CPHMDMSλD) for nucleic acids with pH-based replica exchange (pH-REX) sampling. Using pH-REX significantly improves sampling of titration and spatial coordinates, and the shuffling of conformations across pH space has the effect of decoupling interactions between titrating residues. This allows us to ameliorate some of the sampling issues related to orthogonal barriers that originate from coupled protonation equilibrium and conformational-dependent pKa behavior, as illustrated in our example of the GAAA tetraloop motif. Our pKa calculations on the GAAA tetraloop indicate that pH-REX reduced the average unsigned error (AUE) to 0.7 pKa units, which is 50% lower than previous work,52 and the error of the most poorly predicted residue was drastically reduced from −2.9 to −1.2 pKa unit. The scalability of pH-REX sampling was also demonstrated by showing that similarly accurate pKa values could be achieved when simulating full-sized nucleic acid systems, such as the lead-dependent ribozyme. Our results affirm that pH-REX CPHMDMSλD simulation is maturing into a practical tool for accurate first-principles prediction of nucleic acid protonation equilibrium, where accurate modeling of pH-dependent properties is achieved, even for residues that have hampered convergence due to sampling issues (e.g., slow dynamics and/or coupled titration behavior). Finally, we highlighted that pH-REX CPHMDMSλD simulations can be used to identify the dominant conformation of nucleic acid structures in alternate pH environments or to provide structural characterization of pH-dependent transient states, making it a useful tool to provide structural and mechanistic insight in the study of pH-dependent properties of nucleic acids.
METHODS
Structure Preparation
Input structures of the nucleotide compounds were generated from the CHARMM topology files using the IC facility in CHARMM. The input structure for lead-dependent ribozyme was generated from the PDB file (Accession code: 1LDZ).58 Hydrogen atoms were added using the HBUILD facility in CHARMM.59 The systems were solvated in a cubic box of explicit TIP3P water60 using the convpdb.pl tool from the MMTSB toolset,61 and the ionic strength was simulated by adding the appropriate number of Na+ and Cl− counterions to match experimental salt concentration (100–150mM NaCl). The two isomers of the test nucleotides were constructed using the patch keywords 5PHO and 3PHO. All other nucleic acid structures were hydroxylated using the 5TER and 3TER patches in CHARMM. The excised GAAA tetraloop was constructed by extracting residues 12 to 21 from the lead-dependent ribozyme, and harmonic distance restraints were applied to enforce base pairing between residues A12 and U21. Patches for the protonated forms of adenine and cytosine were constructed as previously reported (see SI Methods).51
Simulation Details
MD simulations were performed within the CHARMM macromolecular modeling program (version c36a6) using the CHARMM36 all-atom force field for RNA62 and TIP3P water.60 The SHAKE algorithm63 was used to constrain the hydrogen-heavy atom bond lengths. The Leapfrog Verlet integrator was used with an integration time step of 2 fs. A non-bonded cutoff of 12 Å was used with an electrostatic force shifting function and a van der Waals switching function between 10 Å and 12 Å. The distance cutoff in generating the list of pairwise interactions was 15 Å. CPHMD simulations utilize an extended Hamiltonian approach, where the protonation state of the residue is described by a continuous variable, λ, which is propagated simultaneously with the spatial coordinates at a specified external pH using multi-site λ-dynamics (see SI Methods).64,65 After an initial minimization, the system was heated for 100 ps and equilibrated for 100ps (for nucleotide compounds) or 400 ps (for RNA structures), followed by a production run of 3 ns, unless otherwise stated. A pH-REX sampling protocol for CPHMD simulations in hybrid and implicit solvent have been previously reported by Wallace and Shen,66 and we have implemented their formalism with our explicit solvent CPHMDMSλD simulations. Replicas were that were simulated at various pH conditions are exchanged based on the following Metropolis criterion:
| (1) |
Exchange attempts were made at every 1 ps unless otherwise stated. CPHMDMSλD simulations were performed from pH 1 to 4 for the GAAA tetraloop and pH 1 to 8 for the full length lead-dependent ribozyme, with integer value pH spacing (unless otherwise specified). All CPHMDMSλD simulations were performed in triplicate.
Calculating pKa values
The populations of unprotonated (Nunprot) and protonated (Nprot) states are defined as the total number of times in the trajectory where conditions λα,1 > 0.8 and λα,2 > 0.8 are satisfied respectively, and are used to derive the unprotonated fraction (Sunprot):
| (2) |
These Sunprot ratios that were computed across the entire pH range were then fitted to a generalized version of the Henderson-Hasselbalch formula to obtain a single pKa value:
| (3) |
The reported pKa value and its error correspond to the mean and standard deviation calculated from 3 sets of independent runs. In the calculation of transition rates, a transition is defined as a move in λ space between physical protonation states using the same definitions for calculating Nunprot and Nprot (i.e., moving between λα,1 > 0.8 and λα,1 < 0.2 constitutes a valid transition). The transition rate statistics reported for each residue was calculated from the simulation where the external pH was closest to the pKa value of the residue.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM037554 and GM057513). We thank Dr. Harish Vashisth for critically reviewing the manuscript and providing helpful comments.
Footnotes
Additional titration curves, table of hill coefficients and analysis of MC exchange rate. This material is available free of charge via the Internet http://pubs.acs.org
References
- 1.Wilcox JL, Ahluwalia AK, Bevilacqua PC. Charged Nucleobases and Their Potential for RNA Catalysis. Acc Chem Res. 2011;44:1270–1279. doi: 10.1021/ar2000452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krishnamurthy R. Role of pK(a) of Nucleobases in the Origins of Chemical Evolution. Acc Chem Res. 2012;45:2035–2044. doi: 10.1021/ar200262x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shih IH, Been MD. Involvement of a Cytosine Side Chain in Proton Transfer in the Rate-determining Step of Ribozyme Self-cleavage. Proc Natl Acad Sci U S A. 2001;98:1489–1494. doi: 10.1073/pnas.98.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wadkins TS, Shih I, Perrotta AT, Been MD. A pH-sensitive RNA Tertiary Interaction Affects Self-cleavage Activity of the HDV Ribozymes in the Absence of Added Divalent Metal Ion. J Mol Biol. 2001;305:1045–1055. doi: 10.1006/jmbi.2000.4368. [DOI] [PubMed] [Google Scholar]
- 5.Ke A, Zhou K, Ding F, Cate JH, Doudna JA. A Conformational Switch Controls Hepatitis Delta Virus Ribozyme Catalysis. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 6.Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC. Mechanistic Characterization of the HDV Genomic Ribozyme: Solvent Isotope Effects and Proton Inventories in the Absence of Divalent Metal Ions Support C75 as the General Acid. J Am Chem Soc. 2008;130:14504–14520. doi: 10.1021/ja801816k. [DOI] [PubMed] [Google Scholar]
- 7.Ravindranathan S, Butcher SE, Feigon J. Adenine Protonation in Domain B of the Hairpin Ribozyme. Biochemistry. 2000;39:16026–16032. doi: 10.1021/bi001976r. [DOI] [PubMed] [Google Scholar]
- 8.Ryder SP, Oyelere AK, Padilla JL, Klostermeier D, Millar DP, Strobel SA. Investigation of Adenosine Base Ionization in the Hairpin Ribozyme by Nucleotide Analog Interference Mapping. RNA. 2001;7:1454–1463. [PMC free article] [PubMed] [Google Scholar]
- 9.Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. Role of an Active Site Adenine in Hairpin Ribozyme Catalysis. J Mol Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Nam K, Gao J, York DM. Quantum Mechanical/Molecular Mechanical Simulation Study of the Mechanism of Hairpin Ribozyme Catalysis. J Am Chem Soc. 2008;130:4680–4691. doi: 10.1021/ja0759141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durant PC, Davis DR. Stabilization of the Anticodon Stem-loop of tRNALys,3 by an A+-C Base-pair and by Pseudouridine. J Mol Biol. 1999;285:115–131. doi: 10.1006/jmbi.1998.2297. [DOI] [PubMed] [Google Scholar]
- 12.Chen G, Kennedy SD, Turner DH. A CA(+) Pair Adjacent to a Sheared GA or AA Pair Stabilizes Size-Symmetric RNA Internal Loops. Biochemistry. 2009;48:5738–5752. doi: 10.1021/bi8019405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nixon PL, Giedroc DP. Energetics of a Strongly pH dependent RNA Tertiary Structure in a Frameshifting Pseudoknot. J Mol Biol. 2000;296:659–671. doi: 10.1006/jmbi.1999.3464. [DOI] [PubMed] [Google Scholar]
- 14.Bayfield MA, Dahlberg AE, Schulmeister U, Dorner S, Barta A. A Conformational Change in the Ribosomal Peptidyl Transferase Center Upon Active/inactive Transition. Proc Natl Acad Sci U S A. 2001;98:10096–10101. doi: 10.1073/pnas.171319598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muth GW, Chen L, Kosek AB, Strobel SA. pH-dependent Conformational Flexibility Within the Ribosomal Peptidyl Transferase Center. RNA. 2001;7:1403–1415. [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong L, Polacek N, Sander P, Bottger EC, Mankin A. pKa of Adenine 2451 in the Ribosomal Peptidyl Transferase Center Remains Elusive. RNA. 2001;7:1365–1369. [PMC free article] [PubMed] [Google Scholar]
- 17.Hesslein AE, Katunin VI, Beringer M, Kosek AB, Rodnina MV, Strobel SA. Exploration of the Conserved A+C Wobble Pair Within the Ribosomal Peptidyl Transferase Center using Affinity Purified Mutant Ribosomes. Nucleic Acids Res. 2004;32:3760–3770. doi: 10.1093/nar/gkh672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beringer M, Bruell C, Xiong L, Pfister P, Bieling P, Katunin VI, Mankin AS, Bottger EC, Rodnina MV. Essential Mechanisms in the Catalysis of Peptide Bond Formation on the Ribosome. J Biol Chem. 2005;280:36065–36072. doi: 10.1074/jbc.M507961200. [DOI] [PubMed] [Google Scholar]
- 19.Beringer M, Rodnina MV. The Ribosomal Peptidyl Transferase. Mol Cell. 2007;26:311–321. doi: 10.1016/j.molcel.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 20.Abeysirigunawardena SC, Chow CS. pH-dependent Structural Changes of Helix 69 from Escherichia Coli 23S Ribosomal RNA. RNA. 2008;14:782–792. doi: 10.1261/rna.779908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakakibara Y, Chow CS. Probing Conformational States of Modified Helix 69 in 50S Ribosomes. J Am Chem Soc. 2011;133:8396–8399. doi: 10.1021/ja2005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiter NJ, Blad H, Abildgaard F, Butcher SE. Dynamics in the U6 RNA Intramolecular Stem-loop: A Base Flipping Conformational Change. Biochemistry. 2004;43:13739–13747. doi: 10.1021/bi048815y. [DOI] [PubMed] [Google Scholar]
- 23.Kim M, Huang T, Miller JH. Competition Between MutY and Mismatch Repair at A × C Mispairs In vivo. J Bacterio. 2003;185:4626–4629. doi: 10.1128/JB.185.15.4626-4629.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giri I, Stone MP. Wobble dC.dA Pairing 5′ to the Cationic Guanine N7 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 adduct: Implications for Nontargeted AFB1 Mutagenesis. Biochemistry. 2003;42:7023–7034. doi: 10.1021/bi020688n. [DOI] [PubMed] [Google Scholar]
- 25.Dethoff EA, Chugh J, Mustoe AM, Al-Hashimi HM. Functional Complexity and Regulation Through RNA Dynamics. Nature. 2012;482:322–330. doi: 10.1038/nature10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolova EN, Kim E, Wise AA, O’Brien PJ, Andricioaei I, Al-Hashimi HM. Transient Hoogsteen Base Pairs in Canonical Duplex DNA. Nature. 2011;470:498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Legault P, Pardi A. In situ Probing of Adenine Protonation in RNA by 13C NMR. J Am Chem Soc. 1994;116:8390–8391. [Google Scholar]
- 28.Wang C, Gao H, Gaffney BL, Jones RA. Nitrogen-15-Labeled Oligodeoxynucleotides .3. Protonation of the Adenine N1 in the A.C and A.G Mispairs of the Duplexes (D[Cg(N-15(1))Agaattcccg])2 and (D[Cgggaattc(N-15(1)Acg])2. J Am Chem Soc. 1991;113:5486–5488. [Google Scholar]
- 29.Moody EM, Brown TS, Bevilacqua PC. Simple Method for Determining Nucleobase pK(a) Values by Indirect Labeling and Demonstration of a pK(a) of Neutrality in dsDNA. J Am Chem Soc. 2004;126:10200–10201. doi: 10.1021/ja047362h. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Cottrell JW, Scott LG, Fedor MJ. Direct Measurement of the Ionization State of an Essential Guanine in the Hairpin Ribozyme. Nat Chem Biol. 2009;5:351–357. doi: 10.1038/nchembio.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cottrell JW, Scott LG, Fedor MJ. The pH Dependence of Hairpin Ribozyme Catalysis Reflects Ionization of an Active Site Adenine. J Biol Chem. 2011;286:17658–17664. doi: 10.1074/jbc.M111.234906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viladoms J, Scott LG, Fedor MJ. An Active-site Guanine Participates in glmS Ribozyme Catalysis in its Protonated State. J Am Chem Soc. 2011;133:18388–18396. doi: 10.1021/ja207426j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. Direct Measurement of a pKa Near Neutrality for the Catalytic Cytosine in the Genomic HDV Ribozyme using Raman Crystallography. J Am Chem Soc. 2007;129:13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 34.Guo M, Spitale RC, Volpini R, Krucinska J, Cristalli G, Carey PR, Wedekind JE. Direct Raman Measurement of an Elevated Base pKa in the Active Site of a Small Ribozyme in a Precatalytic Conformation. J Am Chem Soc. 2009;131:12908–12909. doi: 10.1021/ja9060883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nikolova EN, Gottardo FL, Al-Hashimi HM. Probing Transient Hoogsteen Hydrogen Bonds in Canonical Duplex DNA Using NMR Relaxation Dispersion and Single-Atom Substitution. J Am Chem Soc. 2012;134:3667–3670. doi: 10.1021/ja2117816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ditzler MA, Sponer J, Walter NG. Molecular Dynamics Suggest Multifunctionality of an Adenine Imino Group in Acid-base Catalysis of the Hairpin Ribozyme. RNA. 2009;15:560–575. doi: 10.1261/rna.1416709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banas P, Walter NG, Sponer J, Otyepka M. Protonation States of the Key Active Site Residues and Structural Dynamics of the glmS Riboswitch As Revealed by Molecular Dynamics. J Phys Chem B. 2010;114:8701–8712. doi: 10.1021/jp9109699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mlýnský V, Banás P, Hollas D, Réblová K, Walter NG, Sponer J, Otyepka M. Extensive Molecular Dynamics Simulations Showing that Canonical G8 and Protonated A38H+ Forms are Most Consistent with Crystal Structures of Hairpin Ribozyme. J Phys Chem B. 2010;114:6642–6652. doi: 10.1021/jp1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baptista AM, Teixeira VH, Soares CM. Constant-pH Molecular Dynamics Using Stochastic Titration. J Chem Phys. 2002;117:4184–4200. [Google Scholar]
- 40.Mongan J, Case DA, McCammon JA. Constant pH Molecular Dynamics in Generalized Born Implicit Solvent. J Comput Chem. 2004;25:2038–2048. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- 41.Messer BM, Roca M, Chu ZT, Vicatos S, Kilshtain AV, Warshel A. Multiscale Simulations of Protein Landscapes: Using Coarse-grained Models as Reference Potentials to Full Explicit Models. Proteins: Struct, Funct Bioinf. 2010;78:1212–1227. doi: 10.1002/prot.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee MS, Salsbury FR, Brooks CL., III Constant-pH Molecular Dynamics Using Continuous Titration Coordinates. Proteins: Struct, Funct, Bioinf. 2004;56:738–752. doi: 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- 43.Khandogin J, Brooks CL., III Constant pH Molecular Dynamics with Proton Tautomerism. Biophys J. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khandogin J, Chen J, Brooks CL., III Exploring Atomistic Details of pH-dependent Peptide Folding. Proc Natl Acad Sci U S A. 2006;103:18546–18550. doi: 10.1073/pnas.0605216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khandogin J, Brooks CL., III Linking Folding with Aggregation in Alzheimer’s Beta-amyloid Peptides. Proc Natl Acad Sci U S A. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khandogin J, Raleigh DP, Brooks CL., III Folding Intermediate in the Villin Headpiece Domain Arises From Disruption of a N-terminal Hydrogen-bonded Network. J Am Chem Soc. 2007;129:3056–3057. doi: 10.1021/ja0688880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Machuqueiro M, Baptista AM. The pH-dependent Conformational States of Kyotorphin: A Constant-pH Molecular Dynamics Study. Biophys J. 2007;92:1836–1845. doi: 10.1529/biophysj.106.092445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang BW, Brunetti L, Brooks CL., III Probing pH-dependent Dissociation of HdeA Dimers. J Am Chem Soc. 2011;133:19393–19398. doi: 10.1021/ja2060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace JA, Shen JK. Unraveling A Trap-and-Trigger Mechanism in the pH-Sensitive Self-Assembly of Spider Silk Proteins. J Phys Chem Lett. 2012;3:658–662. doi: 10.1021/jz2016846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Russo NVD, Estrin DA, Martí MA, Roitberg AE. pH-Dependent Conformational Changes in Proteins and Their Effect on Experimental pKas: The Case of Nitrophorin 4. PLoS Comput Biol. 2012;8:1002761–1002761. doi: 10.1371/journal.pcbi.1002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goh GB, Knight JL, Brooks CL., III Constant pH Molecular Dynamics Simulations of Nucleic Acids in Explicit Solvent. J Chem Theory Comput. 2012;8:36–46. doi: 10.1021/ct2006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goh GB, Knight JL, Brooks CL., III pH-dependent Dynamics of Complex RNA Macromolecules. J Chem Theory Comput. 2013 doi: 10.1021/ct300942z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nielsen JE, Gunner MR, Garcia-Moreno EB. The pK(a) Cooperative: A Collaborative Effort to Advance Structure-based Calculations of pK(a) Values and Electrostatic Effects in Proteins. Proteins: Struct, Funct Bioinf. 2011;79:3249–3259. doi: 10.1002/prot.23194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Legault P, Pardi A. Unusual Dynamics and pKa Shift at the Active Site of a Lead-dependent Ribozyme. J Am Chem Soc. 1997;119:6621–6628. [Google Scholar]
- 55.Zheng L, Chen M, Yang W. Random Walk in Orthogonal Space to Achieve Efficient Free-energy Simulation of Complex Systems. Proc Natl Acad Sci U S A. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng LQ, Yang W. Practically Efficient and Robust Free Energy Calculations: Double-Integration Orthogonal Space Tempering. J Chem Theory Comput. 2012;8:810–823. doi: 10.1021/ct200726v. [DOI] [PubMed] [Google Scholar]
- 57.Swails JM, Roitberg AE. Enhancing Conformation and Protonation State Sampling of Hen Egg White Lysozyme Using pH Replica Exchange Molecular Dynamics. J Chem Theory Comput. 2012;8:4393–4404. doi: 10.1021/ct300512h. [DOI] [PubMed] [Google Scholar]
- 58.Hoogstraten CG, Legault P, Pardi A. NMR Solution Structure of the Lead-dependent Ribozyme: Evidence for Dynamics in RNA Catalysis. J Mol Biol. 1998;284:337–350. doi: 10.1006/jmbi.1998.2182. [DOI] [PubMed] [Google Scholar]
- 59.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 61.Feig M, Karanicolas J, Brooks CL., III MMTSB Tool Set: Enhanced Sampling Multiscale Modeling Methods for Applications in Structural Biology. J Mol Graphics Modell. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Denning EJ, Priyakumar UD, Nilsson L, Mackerell AD., Jr Impact of 2′-hydroxyl Sampling on the Conformational Properties of RNA: Update of the CHARMM All-atom Additive Force Field for RNA. J Comput Chem. 2011;32:1929–1943. doi: 10.1002/jcc.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 64.Knight JL, Brooks CL., III Applying Efficient Implicit Non-geometric Constraints in Alchemical Free Energy Simulations. J Comput Chem. 2011;32:3423–3432. doi: 10.1002/jcc.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knight JL, Brooks CL., III Multisite λ Dynamics for Simulated Structure–Activity Relationship Studies. J Chem Theory Comput. 2011;7:2728–2739. doi: 10.1021/ct200444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wallace JA, Shen JK. Continuous Constant pH Molecular Dynamics in Explicit Solvent with pH-Based Replica Exchange. J Chem Theory Comput. 2011;7:2617–2629. doi: 10.1021/ct200146j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.