

Abstract
Four metabolites of okadaic acid were generated by incubation with human recombinant cytochrome P450 3A4. The structures of two of the four metabolites have been determined by MS/MS experiments and 1D and 2D NMR methods using 94 and 133 μg of each metabolite. The structure of a third metabolite was determined by oxidation to a metabolite of known structure. Like okadaic acid, the metabolites are inhibitors of protein phosphatase PP2A. Although one of the metabolites does have an α,β unsaturated carbonyl with the potential to form adducts with an active site cysteine, all of the metabolites are reversible inhibitors of PP2A.
Keywords: Okadaic acid, Dinoflagellate, Algal toxin, Xenobiotic metabolism, Cytochrome P450
1. Introduction
Consumption of the marine algal toxin okadaic acid (OA) results in a syndrome known as diarrheic shellfish poisoning (DSP) which is characterized by severe gastrointestinal symptoms.1 DSP has been associated with the consumption of mussels, scallops or clams tainted with OA and its derivatives. Outbreaks of DSP have been reported in the Americas, Asia and Europe.1–3 Okadaic acid (OA) and the related dinophysistoxins (DTX) are produced by several marine dinoflagellates; a highly diverse group of flagellated, unicellular protists which are responsible for the majority of toxic harmful algal blooms (HABs). Several dinoflagellates of the genera Prorocentrum and Dinophysis including Prorocentrum lima, P. concavum, P. maculosum, D. acuminata, P. rhathymum, and Dinophysis fortii are known to produce OA.4
Of greater concern are the effects of chronic, long-term exposure to these potent hepatotoxins. A growing body of evidence suggests that OA exhibits genotoxic effects.5–8 OA had an aneugenic (chromosome loss) effect on CHO-K1 cells, but only after metabolic activation with a rat liver postmitochondrial S9 fraction.5,9 This effect could be abolished by heat inactivation of the S9 fraction suggesting that OA is metabolically activated to one or more genotoxic products.
Until recently, no information was available on the metabolism of OA by vertebrates. We reported that OA is metabolized by human recombinant cytochrome CYP3A4 to produce four oxidized products.10 Three of these products were tentatively identified on the basis of MS/MS data. We have generated sufficient quantities of two metabolites for characterization by 1D and 2D NMR. The structures of these two metabolites are reported herein as well as their inhibitory activity against protein phosphatase PP2A.
2. Results and discussion
2.1. Structures of metabolites
Proton and carbon assignments for metabolites 1 and 2 (Fig. 1) are listed in Table 1. Previously reported analysis of the MS2 spectrum indicated that metabolite 2 arises from hydroxylation of OA somewhere between C-1 and C-11.10 The molecular formula of metabolite 2 was established as C44H68O14 from the [M+Na]+ ion peak at m/z 843.4495 (calcd for C44H68O14Na, 843.4501) in the HRESIMS. Metabolite 2 could be oxidized with MnO2 to a mixture of metabolites 2 and 4 which showed a new [M+Na]+ at m/z 841.4393 (calcd for C44H66O14Na, 841.4350) in the HRESIMS. This suggested that metabolite 2 is an allylic alcohol and we proposed that the structure of metabolite 2 is 11-hydroxy okadaic acid. However, the C-43 methyl group observed in the 1H NMR of OA and metabolite 1 (δH 1.74 ppm and δC 23.04 ppm) is conspicuously absent in the 1H NMR of metabolite 2, indicating that the oxidation occurred at C-43. A new signal was observed at δH 3.99 ppm which correlated to a new carbon at δC 65.65 ppm in 1H–13C HSQC. A COSY correlation from the new proton signal at δH 3.99 ppm to the olefinic proton on C-9 (δH 5.52 ppm) as well as HMBC correlations to C-9 (δC 122.9 ppm) and C-10 (δC 137.41 ppm) were observed (Fig. 1). Additionally a downfield shift of the C-9 proton (from δH 5.28 ppm to δH 5.52 ppm) and an upfield shift of C-11 (from δC 33.98 ppm to δC 29.41 ppm) were observed. We therefore conclude that metabolite 2 is 43-hydroxyokadaic acid and further that metabolite 4 must be 43-oxo-okadaic acid.
Figure 1.

Molecular structures of okadaic acid and metabolites (top). Diagnostic HMBC correlations for metabolites 1 and 2. Diagnostic COSY correlations (bold lines) for metabolite 1.
Table 1.
NMR spectral data for okadaic acid,11 and metabolites (1) and (2) in methanol-d4
| Unit | Okadaic acid
|
Metabolite 1
|
Metabolite 2
|
|||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | — | 182.8 | — | 181.07 | — | 181.88 |
| 2 | — | 76.3 | — | 76.04 | — | 76.66 |
| 3a/b | 1.65/1.91 | 46.7 | 1.63/2.06 | 46.19 | 1.63/1.97 | 46.22 |
| 4 | 4.08 | 68.9 | 4.09 | 69.20 | 4.11 | 69.03 |
| 5 | 1.3/1.89 | 33.4 | 1.29/1.80 | 33.92 | 1.32/1.80 | 33.15 |
| 6 | 1.66/1.94 | 28.3 | 1.65/1.97 | 27.81 | 1.67/1.97 | 27.91 |
| 7 | 3.35 | 73.4 | 3.37 | 73.23 | 3.41 | 73.09 |
| 8 | — | 107.1 | — | 96.62 | — | 96.83 |
| 9 | 5.26 | 123.8 | 5.28 | 123.49 | 5.52 | 122.90 |
| 10 | — | 139.3 | — | 138.81 | — | 137.41 |
| 11 | 1.82/1.98 | 34 | 1.85/1.97 | 33.98 | 1.97 | 29.41 |
| 12 | 3.87 | 71.8 | 3.80 | 71.81 | 3.81 | 71.79 |
| 13 | 2.35 | 43.2 | 2.37 | 43.29 | 2.36 | 43.16 |
| 14 | 5.95 | 137.2 | 5.91 | 137.68 | 5.93 | 137.52 |
| 15 | 5.5 | 132 | 5.50 | 132.12 | 5.51 | 132.00 |
| 16 | 4.66 | 80.6 | 4.65 | 79.58 | 4.66 | 80.46 |
| 17 | 1.6/.2.21 | 31.5 | 1.59/2.19 | 31.42 | 2.19/1.60 | 31.36 |
| 18 | 1.85/2 | 38.1 | 1.86/2.21 | 37.10 | 2.20 | 37.35 |
| 19 | — | 97.5 | — | 106.14 | — | 106.87 |
| 20 | 1.84/1.87 | 34.2 | 1.85/2.00 | 38.01 | 1.85/2.01 | 37.98 |
| 21 | 1.8/1.9 | 27.7 | 1.78/1.89 | 27.36 | 1.78/1.90 | 27.47 |
| 22 | 3.63 | 71.4 | 3.64 | 71.38 | 3.64 | 71.22 |
| 23 | 3.42 | 78.3 | 3.41 | 78.11 | 3.41 | 77.94 |
| 24 | 4.07 | 72.1 | 4.07 | 72.01 | 4.07 | 71.96 |
| 25 | — | 147.1 | — | 146.28 | — | 146.87 |
| 26 | 3.94 | 86.4 | 3.95 | 85.40 | 3.95 | 86.19 |
| 27 | 4.08 | 66.3 | 4.08 | 66.11 | 4.10 | 65.99 |
| 28 | 0.96/1.37 | 36.8 | 0.93/1.36 | 36.55 | 0.95/1.37 | 36.56 |
| 29 | 1.89 | 32.3 | 1.86 | 32.16 | 1.88 | 32.15 |
| 30 | 3.27 | 76.8 | 3.23 | 76.86 | 3.26 | 76.65 |
| 31 | 1.81 | 28.8 | 1.82 | 28.64 | 1.82 | 28.60 |
| 32 | 1.38/2 | 27.5 | 1.42/2.01 | 27.33 | 1.39/1.99 | 27.32 |
| 33 | 1.361.66 | 31.2 | 1.39 | 30.95 | 1.29 | 30.56 |
| 34 | — | 97 | — | 97.88 | — | 97.29 |
| 35a/b | 1.43/1.61 | 37 | 1.25/1.97 | 46.18 | 1.43/1.62 | 36.85 |
| 36 | 1.53/1.88 | 19.8 | 4.02 | 64.91 | 1.55/1.89 | 19.59 |
| 37a/b | 1.49/1.55 | 26.5 | 1.43/1.84 | 35.99 | 1.52 | 26.33 |
| 38 | 3.51/3.71 | 61.3 | 3.64 | 59.70 | 3.52/3.71 | 61.20 |
| 39 | 0.93 | 11.1 | 0.94 | 10.98 | 0.93 | 10.89 |
| 40 | 1.05 | 16.6 | 0.99 | 16.45 | 1.06 | 16.47 |
| 41 | 5.04/5.38 | 112.6 | 5.06/5.37 | 112.60 | 5.06/5.38 | 112.47 |
| 42 | 1.11 | 17.1 | 1.08 | 16.80 | 1.10 | 16.83 |
| 43 | 1.72 | 23.1 | 1.74 | 23.04 | 3.99 | 65.65 |
| 44 | 1.31 | 27.9 | 1.32 | 27.94 | 1.32 | 27.65 |
The molecular formula of metabolite 1 was established as C44H68O14 from the HRESIMS (m/z calcd for C44H68O14Na [M+Na]+ 843.4501, found: 843.4515). Previously reported analysis of the MS2 spectrum of metabolite 1, indicated that it arises from hydroxylation of OA somewhere between C-27 and C-38.10 The spin system which includes the G-ring from C-35 to C-38 present in OA and in metabolite 2 is not present in the spectra of metabolite 1 (Fig. 1). COSY and TOCSY correlations established a new spin system which included a new multiplet at δH 4.02 ppm and three diastereotopic methylenes. The new multiplet at δH 4.02 ppm correlated to a new carbon at δC 64.91 ppm in the HSQC. This was assigned to C-36. The proton signals from the diastereotopic methylene at C-38 which are distinct in OA and metabolite 2 have converged and overlap with the proton at C-22 (δH 3.64 ppm). An HMBC correlation between the protons on C-38 and C-34 (δC 97.88 ppm) confirmed the location of the spin system.
2.2. Reversibility of protein phosphatase inhibition
Okadaic acid and its analogs are inhibitors of protein phosphatases PP1 and PP2A. The okadaic acid metabolites also inhibit PP2A with IC50 values of 0.94 nM and 1.34 nM for metabolites 1 and 2, respectively10 and 3.98 nM for metabolite 4. Another algal toxin, microcystin-LR from cyanobacteria inhibits protein phosphatases as well. However, unlike OA, microcystin-LR is an irreversible inhibitor. It contains an α,β unsaturated amino acid which acts as a Michael acceptor forming an adduct with a cysteine residue in the enzyme’s active site.12 We reasoned that metabolite 4, the allylic aldehyde might also act as a Michael acceptor towards PP2A. Figure 2 shows recovered PP2A activity after incubating the enzyme with metabolites 2 and 4 for 3 h, relative to recovered activity for OA. Recovered activities for the two metabolites ranged from 113% to 117% when compared to control enzyme with no inhibitor. This indicates that neither metabolite is an irreversible inhibitor of PP2A.
Figure 2.
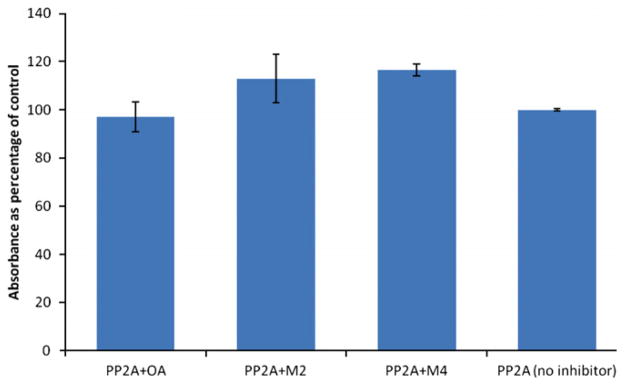
Recovered PP2A activity after incubating with OA (20 μM) and metabolites 2 (100 μM) and 4 (80 μM).
3. Conclusion
The structures of two metabolites (metabolites 1 and 2) of the algal hepatotoxin okadaic acid have been determined by 1D and 2D NMR techniques. The structure of a third metabolite (metabolite 4) is inferred by conversion from metabolite 2. Metabolite 3 is a minor product from the oxidation of OA with CP3A4 and we have been unable to generate sufficient quantities for characterization. Although metabolite 4 is an α,β unsaturated aldehyde with the potential to act as an electrophile, it apparently does not act as a Michael acceptor towards the enzyme PP2A and the OA metabolites identified thus far are reversible inhibitors of PP2A as the PP2A activity can be fully recovered after incubation with these three inhibitors. Identification and characterization of metabolites of OA is the first step toward a better understanding of the full range of effects of OA poisoning as well as the metabolic fate and distribution of this marine algal toxin.
4. Materials and methods
4.1. Preparation of okadaic acid
Okadaic acid was isolated from cultures of Prorocentrum hoffmanianum according to published methods,13 prepared as the ammonium salt and compared by LC–MS to commercial okadaic acid (99.7% purity) which was obtained from Calbiochem (San Diego, CA). HRMS were acquired by the UC
4.2. Preparation of metabolites
Okadaic acid (700 μg) was converted to a mixture of four metabolites by incubation with CYP3A4 as previously described.10 Briefly, reactions were performed at 37 °C for 24 h using OA (50 μM) as the ammonium salt, human recombinant cytochrome P450 3A4 (400 nM), and reaction buffer which contained an NADPH regeneration system provided by the enzyme manufacturer (Codexis, Pasadena, CA). The reactions were quenched with an equal volume of cold methanol, centrifuged to remove precipitated protein and diluted to about 2.5 μM (OA concentration) with methanol. Proteins were precipitated a second time and centrifuged again. The metabolites were separated by HPLC using a 5μm C18, 250 mm × 4.6 mm Discovery column (Supelco, Bellefonte, PA). The chromatographic conditions were as follows: solvent A, CH3CN; solvent B, buffer (1 mM NH4OAc containing 0.1% acetic acid); initial conditions 30% A/70% B (v/v), followed by a linear gradient to 77% A at 20 min; 100% A at 20.5 to 25.5 min; 30% A at 26 to 31 min at a flow rate of 0.5 mL/min. Fractions (0.5 mL) were collected using a Foxy Jr. fraction collector (Teledyne Isco, Lincoln, NE) and analyzed by HPLC–MS as previously described.10 Fractions were pooled to provide metabolite 1 (94 μg), metabolite 2 (133 μg), a mixture of metabolites 2 and 3 (64 μg), and unreacted OA.
4.3. Acquisition of NMR spectra
Structures were determined by analysis of 1H, COSY, TOCSY, ROESY, HSQC and HMBC spectra using a Bruker Avance III 700 spectrometer fitted with a 1.7 mm proton-detect micro-cryoprobe. Approximately 94 μg of M1 and 133 μg of M2 was dissolved in ~30 μL of CD3OD overnight and transferred to 1.7 mm tubes. The TOCSY spectrum was recorded using an MLEV sequence with a 120 ms mixing time. The ROESY spectrum was acquired with a spin-lock pulse of 200 ms. Two HMBC spectra were recorded, optimized for long-range couplings of 8.33 Hz and 5.56 Hz (60 ms and 90 ms evolution times, respectively). All samples were tuned and matched to 50 Ω resistive impedance. Chemical shifts were referenced to the resonance from the residual methyl protons from the solvent, CHD2OD (3.31 ppm).
4.4. Protein phosphatase inhibition assay
Three hundred microliters each of 20 μM OA, 100 μM metabolite 2 and 80 μM metabolite 4 (5% MeOH/H2O) were incubated with PP2A (0.07 Units in 300 μL of enzyme buffer: 50 mM Tris; 2 mM MnCl2; 1.1 mM EDTA; 1.3 mM DTT; 0.125 mg/mL BSA) for 1 h at 37 °C. Three hundred microliters of 5% MeOH/H2O was used as the control. These concentrations of inhibitors were chosen, based on IC50 data, to reduce the PP2A activity by 80%. The incubation reactions were dialyzed separately against 1 L of enzyme buffer for 4 h at 5–7 °C. Recovered enzyme activity was measured in 96-well plates based on a previously described assay.14,15 After incubation with the substrate (p-NPP, 7.8 mM final concentration) for 2 h at 37 °C, absorbance at 405 nm was measured using a Bio-Tek Instruments Synergy 2 plate reader (Winooski, VT).
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) Grant S11 ES11181, the NSF-NIEHS Oceans and Human Health Center Program (National Science Foundation grant 0432368 and NIEHS grant P50 ES12736-01). Assistance with the NMR experiments from J.A. Walter (NRC) is greatly appreciated.
Footnotes
Supplementary data (1H NMR, COSY, HSQC, HMBC and TOCSY spectra for metabolites 1 and 2, a tabulation of COSY and TOCSY correlations for metabolite 1, and HRESIMS data for metabolites 1, 2 and 4) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2012.04.046.
References
- 1.Reguera B, Pizarro G. In: Seafood and freshwater toxins: Pharmacology physiology and detection. 2. Botana L, editor. Taylor & Francis; Londres: 2008. pp. 257–284. Chapter 15. [Google Scholar]
- 2.Swanson KM, Flewelling LJ, Byrd M, Nunez A, Villareal TA. Harmful Algae. 2010;9:190. [Google Scholar]
- 3.Deeds JR, Wiles K, Heideman GB, White KD, Abraham A. Toxicon. 2010;55:1138. doi: 10.1016/j.toxicon.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 4.An T, Winshell J, Scorzetti G, Fell JW, Rein KS. Toxicon. 2010;55:653. doi: 10.1016/j.toxicon.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Hégarat L, Puech L, Fessard V, Poul JM, Dragacci S. Mutagenesis. 2003;18:293. doi: 10.1093/mutage/18.3.293. [DOI] [PubMed] [Google Scholar]
- 6.Fessard V, Grosse Y, Pfohl LA, Puiseux DS. Mutat Res. 1996;361:133. doi: 10.1016/s0165-1161(96)90248-4. [DOI] [PubMed] [Google Scholar]
- 7.Tohda H, Nagao M, Sugimura T, Oikawa A. Mutat Res. 1993;289:275. doi: 10.1016/0027-5107(93)90078-t. [DOI] [PubMed] [Google Scholar]
- 8.Carvalho Pinto-Silva CR, Creppy EE, Matias WG. Arch Toxicol. 2005;79:422. doi: 10.1007/s00204-004-0645-1. [DOI] [PubMed] [Google Scholar]
- 9.Le Hégarat L, Fessard V, Poul JM, Dragacci S, Sanders P. Environ Toxicol. 2004;19:123. doi: 10.1002/tox.20004. [DOI] [PubMed] [Google Scholar]
- 10.Guo F, An T, Rein KS. Toxicon. 2010;55:325. doi: 10.1016/j.toxicon.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miles CO, Wilkins AL, Hawkes AD, Jensen DJ, Cooney JM, Larsen K, Petersen D, Rise F, Beuzenberg V, MacKenzie LA. Toxicon. 2006;48:195. doi: 10.1016/j.toxicon.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 12.MacKintosh RW, Dalby KN, Campbell DG, Cohen PT, Cohen P, MacKintosh C. FEBS Lett. 1995;371:236. doi: 10.1016/0014-5793(95)00888-g. [DOI] [PubMed] [Google Scholar]
- 13.Hu T, Marr J, deFreitas ASW, Quilliam MA, Walter JA, Wright JLC. J Nat Prod. 1992;55:1631. [Google Scholar]
- 14.Simon JF, Vernoux JP. Nat Toxins. 1994;2:293. doi: 10.1002/nt.2620020508. [DOI] [PubMed] [Google Scholar]
- 15.Tubaro A, Florio C, Luxich E, Sosa S, Loggia RD, Yasumoto T. Toxicon. 1996;34:743. doi: 10.1016/0041-0101(96)00027-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.