

Abstract
Computational analysis of cancer pharmacogenomics data has resulted in biomarkers predictive of drug response, but the majority of response is not captured by current methods. Methods typically select single biomarkers or groups of related biomarkers, but do not account for response that is strictly dependent on many simultaneous genetic alterations. This shortcoming reflects the combinatorics and multiple-testing problem associated with many-body biological interactions. We developed a novel approach, MOCA (Multivariate Organization of Combinatorial Alterations), to partially address these challenges. Extending on previous work that accounts for pairwise interactions, the approach rapidly combines many genomic alterations into biomarkers of drug response, using Boolean set operations coupled with optimization; in this framework the union, intersection, and difference Boolean set operations are proxies of molecular redundancy, synergy, and resistance, respectively. The algorithm is fast, broadly applicable to cancer genomics data, is of immediate utility for prioritizing cancer pharmacogenomics experiments, and recovers known clinical findings without bias. Furthermore, the results presented here connect many important, previously isolated observations.
Keywords: MOCA, Multivariate Organization of Combinatorial Alterations, Cancer Cell Line Encyclopedia, Pharmacogenomics
Introduction
Cancer pharmacogenomics studies are important for discovering the molecular determinants of drug response, and thus personalizing cancer treatment (1). Seminal work on the NCI-60 cancer cell lines (2, 3) and many subsequent efforts (4–7) highlighted specific genetic alterations as drug targets or biomarkers of drug response. Recently, the Cancer Cell Line Encyclopedia (CCLE) catalogued genomics and drug response data for nearly 1,000 cancer cells lines (8), which might provide unprecedented power for discovering novel biomarkers. While the clinical utility of these efforts is progressing, the majority of observed drug response is poorly explained by current pharmacogenomics models (9).
One shortcoming of contemporary cancer pharmacogenomics models might be a reliance on single-gene biomarkers (9). There is growing evidence that response to cancer therapeutics can be modulated by the concerted impact of multiple genetic alterations. For example, resistance to targeted therapies can develop from alteration in off-target genes (10). Conversely, additional potentiating alterations can be markers of sensitivity, or even guide drug repositioning efforts (11). Taken together, these observations suggest that accounting for many simultaneous alterations could optimize drugging protocols.
Computational methods are necessary for interpreting large pharmacogenomics datasets, and typically rely on simplifying assumptions to make calculations tractable. Restricting analysis to drug-gene pairwise interactions, clusters of correlated interactions, or the average property of a collection of genes are all successful approaches for determining drug-response biomarkers (3, 8, 12–14). However, these approaches cannot account for response that is strictly dependent on many simultaneous alterations when the constituent pairwise interactions are not statistically significant. Unfortunately, assessing the impact of many simultaneous alterations can be computationally intractable. For instance, enumerating all unique combinations of up to 10 genomic features, for a small genomics dataset containing only 1,000 total features, requires ~1023 comparisons. Furthermore, multiple testing correction for this many comparisons could be unreasonably conservative or unreasonably slow.
Here, we develop a novel conceptual framework for combining many genomic alterations into biomarkers of drug response, and incorporate the relevant functionality into our Multivariate Organization of Combinatorial Alterations (MOCA) algorithm (15). Genomic alterations are combined using the union, intersection, and difference Boolean set operations and optimized to correlate with drug response. We apply the algorithm to a dataset of more than 105 unique genomic features and 24 anticancer drugs across 416 samples from the CCLE. The algorithm captures compelling, novel interactions and known correlates of drug response not highlighted in recent studies using CCLE data with either simple Bayesian modeling, MANOVA, or regularized multivariate regression approaches (8, 12). Additionally, standard pairwise MOCA recovered many known single-gene markers of drug response; however, multi-gene features have substantially higher correlation with drug response than do single-gene features.
Conceptual Framework
Figure 1 illustrates the utility of Boolean set operations for discovering many-gene features of drug response. In Figure 1A, neither f1 nor f2 is significantly correlated with drug sensitivity; however, the union of features f1 and f2 shows significant correlation with drug sensitivity. Here, the union operation suggests redundancy of biological function. For instance, KRAS and BRAF mutation can drive cancer, but mutations in both genes are rarely selected for in a single patient because their downstream output is redundant. Therefore, the union of patients with either BRAF or KRAS activation might share a similar phenotype (such as response to MEK inhibitors (16)). The union interaction might also be relevant for drug repositioning. For example, erlotinib was developed to inhibit EGFR, but subsequent studies found that erlotinib was also effective in some ERBB2-activated cancers (17), suggesting that relevant targets of erlotinib inhibition are EGFR or ERBB2 activation.
Figure 1. Boolean set operations applied to pharmacogenomics features.

Cartoon illustrating the utility of the union (A), intersection (B), and difference (C) operations for comparing the drug-sensitivity feature d, with gene-alteration features f1–f6, across samples S1–S10. Numerical values on the right-hand side are two-tailed Fisher’s exact P-values computed for the correlation of that feature (fx) and sensitivity to drug d. Samples (Sx) sensitive to drug d are checkered and samples (Sx) altered in genomic feature fx are solid.
The union operation is also useful for determining the subset of specific mutations, for a particular gene, that optimally describes drug response. For instance, a single gene can have multiple, unique mutations across samples, which can differentially contribute to drug response. In that case, the predictive value of the feature will be optimal if it includes all mutations that correlate with response, but excludes all mutations that do not.
Figure 1B illustrates the utility of the intersection operation. Here, neither f3 nor f4 are correlated with drug sensitivity. But, the intersection of genomic alterations in f3 and f4 are significantly correlated with drug sensitivity. The intersection operation might be indicative of biological synergy. For example, simultaneous activation of MYC and BLC2 proto-oncogenes can have a transforming potential distinct from activation of either gene in isolation (18).
We also consider the difference operation (Figure 1C). The difference operation can be a proxy for drug resistance. For instance, EGFR mutation sensitizes some tumors to erlotinib, provided KRAS is not mutated (19). See Materials and Methods and the supplemental text (SI text) for a detailed description of all procedures and implementation.
Materials and Methods
We considered all data types available in the Cancer Cell Line Encyclopedia (8), which included gene expression, CNA, mutation, and drug response. There were 416 cell lines common to all four data types, which were considered in this analysis. In addition to genomic features, tissue was considered a feature. P-values were calculated using Fisher’s exact test (two tailed); features with a Benjamini and Hochberg FDR < 0.05 were considered significant. We also calculated the statistical sensitivity, specificity, or a sum of both, for all significant interactions.
MOCA created drug-response-optimized mutation features by taking the union of random collections of site-specific mutations, for a particular gene, and comparing each random collection to a particular drug. MOCA uses an optimization protocol to enrich the pool of site-specific mutation features for those most correlated to the response of that particular drug. A drug-response-optimized mutation feature was only considered for subsequent analysis if it had an FDR < 0.05 and optimization of permuted data always had an FDR > 0.05 (to filter genes that could be optimized below the significance threshold by chance).
Creation of many-gene markers of drug sensitivity began by selecting an appropriate target feature of response. Next, the target feature was combined with every other feature (individually), using each of three Boolean set operations (union, intersection, and difference). If, for instance, the union of feature x and the target feature had substantially greater correlation with drug response than the target feature alone, then feature x was considered for subsequent optimization; this criteria was applied to all union, intersection, and difference feature-target feature combinations. Lastly, union, intersection, and difference features were combined into many-gene features using an optimization protocol similar to the one used to create drug-response-optimization mutation features. The optimization procedure was important for focusing the search on features most correlated with drug response. Employing an entirely random approach, or exhaustive approach, can result in prohibitively conservative multiple testing correction because the number of tests exceeds the significance of any individual interaction.
To validate the utility of many-gene markers for blind prediction of drug response, we randomly divided CCLE data into training (80%) and testing (20%) datasets. We first used the training data to identify putative single- and many-gene markers of drug response (i.e., drug-gene interactions with FDR-corrected Fisher’s P-values < 0.05). Next, we assessed the predictive value (statistical sensitivity and specificity) of those single- and many-gene markers on the holdout testing data. See SI Text for a detailed description of all procedures and the accompanying Supplemental Data file for significant interactions and corresponding statistics.
Results and Discussion
We sought to determine genetic alterations and combinations of genetic alterations that are markers of drug response in the CCLE. Correlations between drug response and genetic alterations were considered significant if the corresponding FDR was less than 0.05. Herein, we highlight alterations that are either known biomarkers in human cancer, or those that present a compelling case based on human biology.
Single-gene biomarkers of drug response
We began with an exhaustive search for all significant pairwise correlations of drug response with gene copy-number alteration (CNA), expression, mutation, and tissue type. Expression-based biomarkers of drug response included many known correlations. EGFR and ERBB2 expression were markers of erlotinib and lapatinib sensitivity, respectively (17). Sensitivity to the IGF1R inhibitor AEW541 was significantly correlated with IGF1 overexpression (20). NQO1 was the most significantly correlated expression feature for 17-AAG sensitivity; NQO1 is involved in the biosynthesis of the natural HSP90 inhibitor 17-AAGH2 (21). HDAC1 and HDAC2 overexpression were exclusively associated with sensitivity to the HDAC inhibitor panobinostat; additionally, HDAC5 and HDAC6 overexpression were significantly associated with panobinostat sensitivity (22). MDM2 and MDM4 overexpression were highly correlated with sensitivity to nutlin-3 (23).
RTK inhibitors are among the most common targeted cancer therapeutics, but are often ineffective owing to resistance (24). One proposed mechanism of resistance is so-called kinase switching (24). In this paradigm, the targeted RTK is rendered nonessential via the up-regulation of an off-target RTK. We find an intriguing network of potential kinase-switching interactions among the significant expression-drug correlations. Figure 2 shows the relationship of EGFR, ERBB2, c-MET, and PDGFRB expression, to the six RTK inhibitors in the CCLE. As an example, EGFR underexpression is co-occurring with sensitivity to five RTK inhibitors, and has mutually exclusive overexpression with sensitivity to the ALK inhibitor TAE684.
Figure 2. Relationship of kinase inhibitor sensitivity to the expression of selected kinases show signatures of kinase switching.
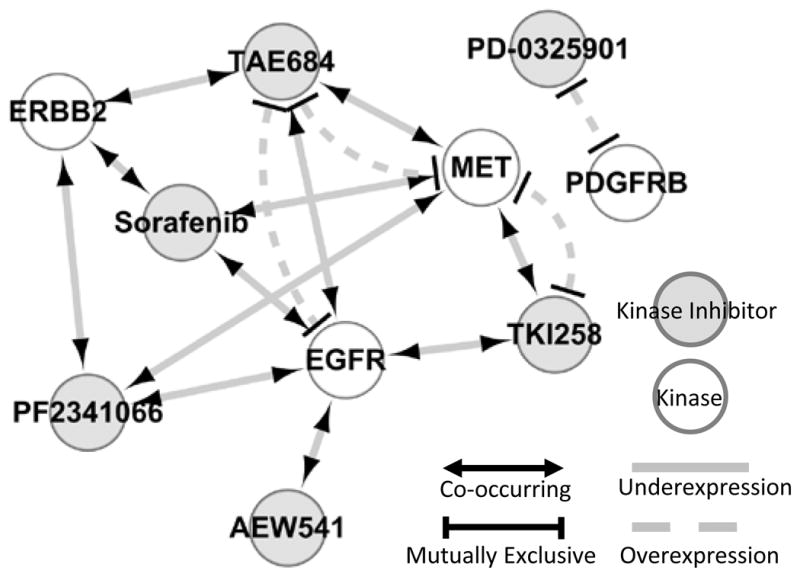
All relationships depicted are with respect to compound sensitivity. For instance, sensitivity to the FGFR inhibitor TKI258 is mutually exclusive with c-MET overexpression and co-occurring with c-MET underexpression.
CNA-based biomarkers included interesting correlates of drug response. KLF5 amplification was exclusively associated sensitivity to the MEK inhibitor PD-0325901; KLF5 activates the MEK/ERK pathway via EGFR stimulation (25). MITF amplification was exclusively associated with sensitivity to PLX4720; MITF amplification and BRAF mutations are co-occurring in human cancers and cancer cell lines (2). JAK3 amplification was mutually exclusive with 17-AAG sensitivity, suggesting JAK3 amplification is a contributor to 17-AAG resistance. Seven SERPINB genes (2, 3, 4, 7, 10, 12, and 13) had copy number deletion mutually exclusive with RAF265 sensitivity, suggesting this family of genes is important for potentiating CCLE cell lines to this RAF drug; this family of protease inhibitors clusters on chromosome 18 and is associated with the malignant phenotype (26).
In some cases, tissue type was significantly correlated with drug sensitivity. Haematopoietic and lymphoid tissue was the broadest marker of drug sensitivity, among tissue features, predicting sensitivity to 15 of the 24 drugs. Similarly, PD-0325901 had the most cross-tissue potency, and was correlated with large intestine, pancreas, and skin tissues. Conversely, ovarian tissue was significantly resistant to the CDK4 inhibitor PD-0332991, and lung tissue was resistant to panobinostat.
We used three types of mutation features: 1) gene-specific mutation features, which include all gene-specific mutations in a single feature. 2) Mutation-specific mutation features, which represent each specific mutation as a unique feature (e.g., TP53 H193R is its own feature). 3) Using an optimization protocol, a subset of mutation-specific mutation features that optimized correlation with drug response, for a specific drug-gene combination, were combined into a single feature; these features are referred to as drug-response-optimized mutation features throughout.
BRAF V600E potentiated cells to the RAF inhibitors PLX4720 and RAF265 and the MEK inhibitors AZD6244 and PD-0329501; these four known interactions (27) were the only significant mutation-specific mutation features identified. Six significant gene-specific mutation features of drug response were identified, which included the four BRAF interactions, as well as AZD624-sensitizing NRAS mutations (27) and mutual exclusivity of nutlin-3 sensitivity and TP53 mutations; nutlin-3 inhibits the TP53 inhibitor MDM2, and is known to selectively target cancers with wild-type TP53 (28).
Drug-response-optimized mutation features
Many drug-response-optimized mutation features are known or compelling correlates of drug response that were not identified in previous studies using CCLE data (8, 12). EGFR optimized mutation features were exclusively associated with sensitivity to all three CCLE EGFR inhibitors. Figure 3A shows consensus EGFR mutations for each of the three EGFR inhibitors; consensus mutations are those seen with the highest frequency during optimization (see Creation of drug-response-optimized mutation features in the SI text). Of the 52 CCLE EGFR mutations, MOCA converges on a few mutations as being primarily responsible for sensitivity to all three CCLE EGFR inhibitors, including two deletions in exon 19 (ELREA746del and ELR746del), Y1069C, and S768I. Notably, deletions at position 746 and 747 create the highest known sensitivity to erlotinib and gefitinib (29). Similarly, The EGFR S768I mutation sensitizes tumors to gefitinib (30). The EGFR Y1069C consensus mutation is interesting because it correlated with sensitivity to all three EGFR inhibitors, a site of phosphorylation, but not yet considered an oncogenic mutation or biomarker of drug sensitivity. Interestingly, L858R and exon 19 deletions are the most prevalent oncogenic EGFR mutations, but EGFR inhibitor response rates are twice as high in tumors with exon 19 deletions (29); remarkably, MOCA recovers this differential sensitivity (see Figure 3A).
Figure 3. Representative ERBB interactions.

Frequency of specific EGFR (A) and ERBB4 (B) mutations from the 100 most correlated drug-response-optimized mutation features shown for all corresponding significant drug interactions (see Creation of drug-response-optimized mutation features in the SI text). Lapatinib-bound EFGR (magenta and beige) and lapatinib-bound ERBB4 (teal and gray) superimposed crystal structures (C) and same orientation for superimposed erlotinib-bound EGFR and ERBB4 (D) showing possible ERBB4 binding pocket for erlotinib. Distribution of selected predictive features of erlotinib sensitivity (E). The Combined feature is feature five from Table 1, Exp superscript is expression (RMA processed), OptMut corresponds to erlotinib-optimized mutation feature, GeneMut is gene-specific mutation feature, and Erlotinib+ means erlotinib sensitivity. Black dashes delineate mutated or drug sensitive samples. Lymph is lymphoid and heamatopoietic tissue.
ERBB4 optimized mutation features correlated with response to the EGFR inhibitors erlotinib and lapatinib, and the HSP90 inhibitor 17-AAG. Of the 89 ERBB4 mutations in the CCLE, the consensus mutations were mostly kinase domain mutations (Figure 3B). Of ERBB family members, ERBB4 shares the greatest sequence homology with EGFR (ERBB1), and is known to interact with some EGFR inhibitors. For instance, lapatinib inhibits EGFR, ERBB2, and ERBB4 (31). Figure 3C shows superimposed crystals structures of lapatinib-bound EFGR and lapatinib-bound ERBB4, highlighting the similar modes of lapatinib interaction for both proteins. Comparing the similar binding orientation adopted by the EGFR-erlontinib complex (Figure 3D), it seems plausible that erlotinib might also interact with ERBB4. Indeed, recent experiments suggest that erlotinib, like lapatinib, is a multi-specificity ERBB inhibitor that interacts with EGFR and ERBB4 (32).
Similar results were obtained for many drug-response-optimized mutation features. NFKB2 was one of the 18 optimized mutation features associated with sensitivity to the XIAP inhibitor LBW242; XIAP is an antiapoptotic gene regulated by NFKB2 (33). EXT2 optimized mutation features were significantly correlated with lapatinib, erlotinib, and AZD0530 sensitivity; EXT2 is involved in heparin sulfate biosynthesis and high serum heparin sulfate concentration is associated with resistance to EGFR inhibitors (34). Of the 37 KRAS and 24 NRAS mutations in the CCLE, consensus mutations of MEK inhibitor sensitivity are known RAS activation sites (16). ERK4 (MAPK4) was exclusively associated with sensitivity to the MEK/ERK inhibitor AZD6244. PTEN optimized mutation features were exclusively associated with sensitivity to the topoisomerase inhibitor irinotecan (35). Remarkably, HDAC1 optimization was exclusively associated with sensitivity to the HDAC inhibitor Panobinostat (22). Of the 18 drug-response-optimized mutation features correlated with the receptor tyrosine kinase inhibitor ZD-6474, seven were kinases (PRKDC, EGFR, PRKAR1B, CLK3, PIK3C2B, PLK3, and EPHA5). MOCA finds that activating KRAS mutations confer resistance to TKI258. This is intriguing because KRAS is downstream of FGFR in the MEK/ERK pathway, and KRAS mutation was recently associated with FGFR inhibitor resistance (36).
MOCA-derived drug-response-optimized mutation features address several limitations. For example, approaches that restrict analysis to mutation- or gene-specific mutation features would not recover these important correlations, including the differential potentiation among EGFR mutations. Furthermore, database-driven approaches, such as restricting analysis to mutations present in COSMIC (37), could miss novel but potentially important findings, such as sensitizing EGFR mutations at the Y1069 phosphorylation site. And, the ERBB4 optimized mutation feature highlights the ability of unbiased approaches to identify relevant targets for drug repositioning. All significant pairwise drug-gene correlations, including drug-response-optimized mutation features, are in the accompanying Supplemental Data file, along with corresponding P-value’s, false discovery rates (FDR’s), and statistical sensitivity and specificity.
Collections of simultaneously altered genes as biomarkers of drug response
As the above results collectively highlight, single-gene markers can recover many known correlations of drug response. However, these interactions fail to properly identify a significant portion of both responders and non-responders. For instance, we found that EGFR overexpression (EGFRExp+) was significantly correlated erlotinib sensitivity (erlotinib+) in the CCLE (see Table 1). But, EGFRExp+ identifies erlotinib+ with a specificity of 76.8% and a statistical sensitivity of 63.4%, meaning the EGFRExp+ marker calls many false negatives and misses many true positives, respectively. A similar result was obtained for the vast number of single-feature markers.
Table 1. Selected biomarkers of erlotinib response.
Fisher’s exact P-Values, statistical sensitivity (Sens), and specificity (Spec) for selected Features, highlighting the increased correlation with drug response obtained from applying Boolean set operations (“∪” is the union, “∩” is the intersection, and “-“ is the difference). Superscript Exp is for expression, OptMut corresponds to erlotinib-optimized mutation feature, GeneMut is gene-specific mutation feature, and CNA copy-number alteration.
| Feature | P-Value | Sens | Spec |
|---|---|---|---|
| EGFRExp+ | 3.0×10−7 | 63.4% | 76.8% |
| EGFRExp+ ∪ ERBB4OptMut | 2.1×10−9 | 70.1% | 76.7% |
| EGFRExp+ ∪ ERBB4OptMut ∪ ERBB2Exp+ | 1.4×10−11 | 85.4% | 69.0% |
| (EGFRExp+ ∪ ERBB4OptMut ∪ ERBB2Exp+) – ERBB3Exp− | 6.2×10−11 | 85.4% | 75.1% |
| ((EGFRExp+ ∪ ERBB4OptMut ∪ ERBB2Exp+) – ERBB3Exp−) - KRASGeneMut | 4.7×10−11 | 85.4% | 82.1% |
| ((((EGFRExp+ ∪ COL14A1OptMut ∪ ERBB4OptMut ∪ TFEBOptMut) (KCNK1Exp+ ∩ KRT6BExp+)) - CRHBPCNA+) - F2RL2CNA+) - KRASGeneMut | 6.3×10−11 | 70.7% | 95.5% |
| ((EGFRExp+ ∪ DSTOptMut ∪ MAD2L2Exp− ∪ TOB2Exp+) ∩ (CASD1Exp− ∩ FAM83HExp+ ∩ MAL2Exp+)) - ANKRD36BCNA− | 4.5×10−11 | 95.1% | 74.1% |
| (EGFRExp+ ∪ NCKIPSDOptMut ∩ (CBLCExp+ ∩ FAM83HExp+ ∩ HPSEExp+ ∩ KCNK1Exp+)) - ANKRD36BCNA−) - KRASGeneMut | 1.1×10−11 | 87.8% | 87.4% |
We sought to determine many-gene features that had increased correlation with drug response, relative to the corresponding target feature alone. Here, we define a target as the gene, or member of the pathway, that the drug was designed to inhibit (see Table S1 and SI text for a list of targets used for each drug). Target features were combined with every other feature, individually, using the union, intersection, and difference set operations, and compared with the relevant drug response vector. Importantly, this approach does not increase the number of interactions (combinatorial space) relative to the number of pairwise interactions. We required these three-body interactions (i.e., drug, target, and another feature) to have a P-value at least two orders of magnitude lower than the constituent pairwise drug-target interaction. This conservative procedure returned a reasonably parsimonious list of features, with many convincing and known correlates of drug response not captured by pairwise MOCA calculations or previous statistical and machine learning approaches applied to the same data (8, 12).
Table 1 highlights the increase in statistical sensitivity and specificity that can be obtained with many-feature biomarkers of drug response. For instance, the union of EGFRExp+ and ERBB4OptMut has a 6.7% increase in statistical sensitivity, relative to EGFRExp+ alone, for identifying erlotinib response. Importantly, the ERBB4OptMut feature was the result of optimizing the union of ERBB4 mutation-specific mutation features, which was required to capture this known interaction. The union of EGFRExp+, ERBB4OptMut, and ERBB2Exp+ increases the statistical sensitivity for identifying erlotinib+ by more than 20% relative to EGFRExp+ alone. This suggests some patients altered in ERBB2, but not necessarily EGFR, might also benefit from erlotinib. Indeed, patients with ERBB2 alteration and not EGFR alteration can benefit from erlotinib treatment (17); notably, the interaction of ERBB2Exp+ and erlotinib+ alone is weakly correlated in the CCLE data, and was only identified when considering many-gene features. Table 1 also shows erlotinib resistance arising from ERBB3 underexpression and/or KRAS mutation; remarkably, ERBB3-deficient cancer cell lines can exhibit erlotinib resistance (38) and KRAS mutation is a strong predictor of erlotinib resistance (19). Importantly, KRAS mutation is weakly correlated with erlotinib resistance in the CCLE, and this known mechanism of resistance would not be recovered without considering many-body interactions.
This five-gene biomarker (fifth row, Table 1) results in a ~20% increase in statistical sensitivity and ~5% increase in specificity. Therefore, this biomarker might be used identify a substantially greater number of patients that could benefit from erlotinib treatment, rather than considering the target (EGFR alteration) alone. And, that fewer non-responders would be subjected to a treatment that might be ineffective. Figure 3E shows the distribution of the individual and combined markers mentioned above.
There is some controversy regarding the role of MDM4 in sensitizing cancers to the MDM2 inhibitor nutlin-3. Some speculation arises from the structural homology of MDM2 and MDM4 proteins, and the similar p53 binding interfaces (Figure 4A). A crystal structure of MDM2 binding a nutlin-3 analog (nutlin-2) revealed a compelling mode of action for nutlin-3, where p53 is displaced from the native p53-MDM2 interface; a similar mode of action has been proposed for nutlin-3 binding MDM4 (see Figure 4A). Laurie et al. used molecular modeling, binding assays, and retinoblastoma killing in rodent models to infer the interaction of MDM4 and nutlin-3 (23). Conversely, using NMR competition experiments and structural comparison, Popowicz et al. concluded that MDM4 and nutlin-3 do not interact (39). And, Hu et al. found that MDM4 expression can cause nutlin-3 resistance in tumor cells (40). In the CCLE, MOCA finds increased correlation of MDM4Exp+ with nutlin-3+, compared with MDM2Exp+ (Table 2). However, taking either the union or intersection of MDM2Exp+ and MDM4Exp+ appears to better resolve the origin of potentiation in these cell lines. Taken together, the results in Table 2 suggest that either MDM2Exp+ or MDM4Exp+ is sufficient to sensitize some cell lines to nutlin-3, but that simultaneous up-regulation of both markers synergize to further sensitize some CCLE cell lines. We highlight this as an open question, and contend that the CCLE contributes to the emerging role of MDM4 expression, with respect to nutlin-3 sensitivity. Figure 4C shows the distribution of the individual and combined MDM2 and MDM4 markers.
Figure 4. Interactions of Nutlin-3 with MDM2 and MDM4.
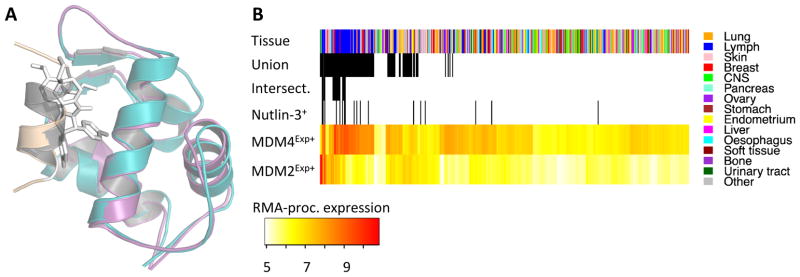
Panel A shows superimposed crystal structures of p53-bound MDM2 (MDM2, teal; p53, beige) and MDM2 binding a nutlin-3 analog (stick representation; MDM2 removed for clarity), and p53-bound MDM4 (MDM4, magenta; p53, gray). Distribution of selected predictive features of nutlin-3 response (B). The superscript Exp+ delineates overexpression (RMA processed) in all panels and Nutlin-3+ delineates samples sensitive nutlin-3. Intersect is the intersection of MDM4Exp+, MDM2Exp+, and Nutlin-3+, and Union is the union of those three features. Lymph is lymphoid and heamatopoietic tissue.
Table 2. Selected biomarkers of nutlin-3 response.
Fisher’s exact P-Values, statistical sensitivity (Sens), and specificity (Spec) for the correlation of nutlin-3 response and MDM2 or MDM4 alteration (“∪” is the union, “∩” is the intersection). Superscript Exp is for expression.
| Feature | P-Value | Sens | Spec |
|---|---|---|---|
| MDM2Exp+ | 6.5×105 | 50.0% | 89.4% |
| MDM4Exp+ | 1.8×107 | 66.7% | 88.4% |
| MDM2Exp+∪ MDM4Exp+ | 4.7×10−6 | 72.2% | 80.2% |
| MDM2Exp+∩ MDM4Exp− | 4.2×10−8 | 44.4% | 97.7% |
There were many other striking multi-genes features, all which significantly improved correlation with drug response, relative to that drugs target alone. For instance, the union of the BRAFV600E feature and the NRAS optimized mutation feature (NRASOptMut) improved the statistical sensitivity for identifying response to the MEK inhibitor AZD6244 by more than 25% relative to NRASOptMut alone. This suggests that the relevant biomarker for response to this MEK inhibitor includes either BRAF or NRAS mutation; indeed, mutation in either gene is known to potentiate cancers to AZD6244 (41). Similar to the erlotinib example (Table 1), correlation with lapatinib response was greatly increased by a feature that includes EGFROptMut, ERBB2Exp+, and ERBB4OptMut; lapatinib is an inhibitor of these three ERBB family members (31). The union of HDAC5Exp+ and HDAC1OptMut increases the statistical sensitivity for identifying response to the HDAC inhibitor panobinostat by ~33% relative to HDAC1OptMut alone; panobinostat is a strong inhibitor of both HDAC1 and HDAC5 (22).
During pairwise calculation, MOCA found significant correlation between IGF1R inhibitor (AEW541) sensitivity and IGF1Exp+, but not IGF1RExp+. However, MOCA finds that the union of IGF1Exp+ and IGF1RExp+ has significantly increased significance with AEW541+, relative to either alone. This result is important for two reasons: 1) it suggests that either IGF1Exp+ or IGF1RExp+ is sufficient to potentiate some cancers for AEW541. 2) It shows that combined features can identify important interactions that are missed comparing only pairwise interactions. Additionally, the intersection of RAD51Exp+ and IGF1Exp+ enhances the specificity of AEW541+ identification; RAD51-mediated DNA repair is stimulated by IGF1, and cells deficient for IGF1R function show significantly less RAD51-mediated DNA repair (42). All significant three-body interactions derived from combining features with set operations are in the accompanying Supplemental Data file, along with corresponding P-value’s, FDR’s, and statistical sensitivity and specificity.
Next, we combined genomic features, determined significant in the previous step, into many-gene biomarkers that optimize correlation with drug response. An important parameter of our new method provides the ability to create many-gene features that optimize a desired predictive value. This type of approach might be useful in a setting where a clinical decision must consider the aggressiveness of the cancer and the toxicity of the relevant drugging protocols. For example, the side effects associated with some chemotherapeutics might warrant a highly specific marker, so that inherent risk is almost certainly coupled with benefit. On the other hand, more aggressive cancers might necessitate a highly sensitive test, so that drugs most broadly applicable can be prioritized. And, in many scenarios, a balance between statistical sensitivity and specificity might be desirable, to balance the post-test risk with the general efficacy of the therapeutic regimen under consideration.
The last three rows of Table 1 show features optimized to correlate with erlotinib response, maximizing either the statistical sensitivity, specificity, or sum of both. The many-gene feature in the sixth row of Table 1 was optimized for maximal specificity (95.5%) while maintaining reasonable statistical sensitivity (70.7%); therefore, this marker identifies the majority of potential responders in the CCLE, and almost never falsely calls non-responders. Similarly, the marker in row seven of Table 1 identifies erlotinib response with a statistical sensitivity of 95.1% and a specificity of 74.1%. And, the final marker in Table 1 achieves a nearly 90% statistical sensitivity and specificity for erlotinib response.
Similar to our method of deriving drug-response-optimized mutation features, when creating many-gene markers of drug response, MOCA reports features observed most frequently in top-predicting combined features (i.e., consensus features). This protocol is useful for highlighting features that most consistently combine to optimize correlation with drug response. For instance, features including the intersection of one or more of nine KRT family genes (KRT5, 6A–6C, and 13–17) were consistently top markers of erlotinib response; KRT genes are known to co-express with EGFR, and other ERBB-family genes, in many human cancers (43). CBLC, KLF2, and KLF5 are all regulators of EGFR, and MOCA found the expression of these genes to be highly correlated consensus features of erlotinib response during optimization; CBLC is also a consensus feature for lapatinib response. ERBB-family members were among the most enriched consensus features for both erlotinib and lapatinib. Interestingly, a TP53 drug-optimized-mutation feature was the most enriched consensus feature for identifying lapatinib sensitivity in union with ERBB2 overexpression. KRAS mutation was the third most enriched consensus feature of erlotinib resistance. MDM4 overexpression was the seventh ranked consensus feature for synergizing with MDM2 overexpression to sensitize cells to nutlin-3. PDGFRB, EGFR, and c-MET overexpression or amplification were all top-ranked consensus features of panobinostat resistance. This might be an important finding because many cancers are up regulated in one or more of these three oncogenes. Haematopoeitic and lymphoid tissue was among the most enriched consensus features of panobinostat sensitivity; this is the only case where a tissue type was a consensus feature during derivation of many-feature markers of drug response. It is noteworthy that many-gene features are more correlated with drug response than tissue type, because molecular markers facilitate a more specific diagnosis than is possible with tissue type alone.
Finally, we assessed the potential utility of many-gene biomarkers for blind prediction. For this validation calculation, CCLE data was divided into 333 samples (~80%) for training and 83 samples (~20%) for testing. First, training data was used to identify putative single- and many-gene predictors of drug response. Next, the predictive value (statistical sensitivity and specificity) of these single- and many-gene predictors was calculated on the holdout testing data. To make the algorithm more amenable to automation, these proof-of-concept calculations were restricted to expression data, and we considered three drugs (erlotinib, lapatinib, and nutlin-3).
The top 25 most predictive, expression-based, single-gene markers of erlotinib response had a mean statistical sensitivity of 0.77 and a standard deviation 0.07. Specificity for this same set of 25 single-gene markers was 0.83 ± 0.06. The top 25 most predictive, expression-based, many-gene markers of erlotinib response had a statistical sensitivity 1.0 ± 0.0 and a specificity of 0.85 ± 0.02. Furthermore, each of the top 25 many-gene markers had a better predictive value (calculated as the sum of statistical sensitivity and specificity) than any of the top 25 single-gene markers.
Similar results were obtained for lapatinib and nutlin-3. For instance, the top 25 single-gene markers of lapatinib response had a statistical sensitivity of 0.67 ± 0.08 and a specificity of 0.83 ± 0.06. Top many-gene predictors of lapatinib response had a sensitivity and specificity of 0.82 ± 0.05 and 0.81 ± 0.05, respectively. Single-gene markers of nutlin-3 response had poor predictive values, with a sensitivity of 0.35 ± 0.09 and a sensitivity of 0.80 ± 0.03. Conversely, many-gene predictors of nutlin-3 response had high predictive value, with a sensitivity 0.80 ± 0.0 and specificity of 0.93 ± 0.01, respectively. And, for both lapatinib and nutlin-3, each of the top 25 many-gene markers had a better predictive value than any of the top 25 single-gene markers.
Proof-of-concept validation calculations yielded many-gene predictors of drug response with high predictive value, similar to those obtained not using a holdout test dataset. Also, predictive value was consistently higher for many-gene predictors, compared to single-gene predictors. Taken together, these results suggest that MOCA, and many-gene predictors, might be useful for improving blind prediction drug response in cancer cell lines.
Conclusion
The ability to identify many known and compelling correlates of drug response, including single and many-gene biomarkers, highlights the utility of MOCA, the CCLE, and the inhibitors considered. MOCA introduces a novel approach to combining many genomic features into biomarkers of phenotype. The abstraction of considering Boolean set operations as proxies of molecular redundancy, synergy, and resistance appears to have some validity. Furthermore, MOCA’s optimization protocols focus the search enough to reduce combinatorics and quickly converge upon meaningful, many-feature markers of response.
It is important to note that the majority of findings highlighted here are known markers or targets of drug response in human cancers. This result implies that cancer cell lines can be useful model systems of human cancers. Additionally, the 24 drugs currently profiled in the CCLE appear to be potent inhibitors of their intended targets. And, as demonstrated here, unbiased computational analysis of such pharmacogenomics datasets might be useful for identifying relevant targets for drug repositioning.
Here, we derived many-gene markers of drug response using a stochastic optimization process. As such, the algorithm is subject to getting trapped in local minima. Furthermore, the astronomic combinatorial space associated the many-body search performed here virtually guarantees the existence of many-gene biomarkers with similar, or better predictive value than those recovered in our study. Indeed, it is computationally intractable to guarantee the recovery of the most optimal solution owing to computational burden and multiple testing problem imparted by the large combinatorial space. The contribution of this work is the ability to identify many-feature predictors of drug response that are an improvement relative to single-feature predictors; but, these represent “good” solutions, not necessarily the “best” solution.
Supplementary Material
Major Findings.
When applied to 416 pharmacogenomically characterized cancer cell lines, MOCA identifies many known and potential markers of drug response. For instance, correlation with ERBB inhibitor response drastically increased when considering EGFR (ERBB1), ERBB2, ERBB3, ERBB4, and KRAS alterations in a single feature. Similarly, a feature combining IGF1, IGF1R, and RAD51 drastically increased correlation with IGF1R inhibitor response, relative to any of these three genetic markers considered in isolation. This approach is also powerful for determining subsets of site-specific mutations, for a particular gene, that increase correlation with drug response. For example, MOCA captures the differential EGFR inhibitor response conferred by common EGFR mutations. Similarly, we find specific HDAC1 mutations cooperate with HDAC5 overexpression to potentiate cells to the HDAC inhibitor panobinostat. Additionally, considering all pairwise gene-drug interactions, MOCA recovers known and compelling correlations, including: RTK inhibitor resistance via c-MET, EGFR, ERBB2, and PDGFRB kinase switching; mutual exclusivity of TP53 mutation and response to the MDM2 inhibitor nutlin-3; greater nutilin-3 potentiation via MDM4, rather than MDM2, overexpression; MEK and RAF inhibitor response in BRAF mutated cell lines; and, MEK inhibitor potentiating NRAS mutations.
Acknowledgments
This work was funded by NIH National Cancer Institute grant CA135877, NSF DBI CAREER award 0845275, and bridge funding from Johns Hopkins University IBBS program to R. Karchin.
Footnotes
The authors declare no conflict of interest
Supporting material for this manuscript includes four supplementary files: a detailed description of all methods and implementation (Supplementary methods); an excel spreadsheet containing all pairwise interactions, grouped by data type (Supplemental pairwise interactions); an excel spreadsheet containing all three-body set interactions, grouped by drug (Supplemental set interactions); and code used to produce all results from this manuscript, including detailed instructions for execution (44).
References
- 1.Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nature Medicine. 2011;17:297–303. doi: 10.1038/nm.2323. [DOI] [PubMed] [Google Scholar]
- 2.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 3.Staunton JE, Slonim DK, Coller HA, Tamayo P, Angelo MJ, Park J, et al. Chemosensitivity prediction by transcriptional profiling. Proceedings of the National Academy of Sciences. 2001;98:10787–92. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proceedings of the National Academy of Sciences. 2007;104:19936–41. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin WM, Baker AC, Beroukhim R, Winckler W, Feng W, Marmion JM, et al. Modeling Genomic Diversity and Tumor Dependency in Malignant Melanoma. Cancer Research. 2008;68:664–73. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, et al. Predicting drug susceptibility of non‚Äìsmall cell lung cancers based on genetic lesions. The Journal of Clinical Investigation. 2009;119:1727–40. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–307. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wistuba II, Gelovani JG, Jacoby JJ, Davis SE, Herbst RS. Methodological and practical challenges for personalized cancer therapies. Nature Reviews Clinical Oncology. 2011;8:135–41. doi: 10.1038/nrclinonc.2011.2. [DOI] [PubMed] [Google Scholar]
- 10.Ellis LM, Hicklin DJ. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clinical Cancer Research. 2009;15:7471–8. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 11.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nature Reviews Drug Discovery. 2004;3:673–83. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 12.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JK, Havaleshko DM, Cho H, Weinstein JN, Kaldjian EP, Karpovich J, et al. A strategy for predicting the chemosensitivity of human cancers and its application to drug discovery. Proceedings of the National Academy of Sciences. 2007;104:13086–91. doi: 10.1073/pnas.0610292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butte AJ, Tamayo P, Slonim D, Golub TR, Kohane IS. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proceedings of the National Academy of Sciences. 2000;97:12182–6. doi: 10.1073/pnas.220392197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masica DL, Karchin R. Correlation of Somatic Mutation and Expression Identifies Genes Important in Human Glioblastoma Progression and Survival. Cancer Research. 2011;71:4550–61. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ball DW, Jin N, Rosen DM, Dackiw A, Sidransky D, Xing M, et al. Selective Growth Inhibition in BRAF Mutant Thyroid Cancer by the Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor AZD6244. Journal of Clinical Endocrinology & Metabolism. 2007;92:4712–8. doi: 10.1210/jc.2007-1184. [DOI] [PubMed] [Google Scholar]
- 17.Cohen EEW, Lingen MW, Martin LE, Harris PL, Brannigan BW, Haserlat SM, et al. Response of Some Head and Neck Cancers to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors May Be Linked to Mutation of ERBB2 rather than EGFR. Clinical Cancer Research. 2005;11:8105–8. doi: 10.1158/1078-0432.CCR-05-0926. [DOI] [PubMed] [Google Scholar]
- 18.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 19.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS Mutations and Primary Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garci3a-Echeverri3a C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, et al. In vivo antitumor activity of NVP-AEW541‚ a novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–9. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 21.Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D. Formation of 17-Allylamino-Demethoxygeldanamycin (17-AAG) Hydroquinone by NAD(P)H:Quinone Oxidoreductase 1: Role of 17-AAG Hydroquinone in Heat Shock Protein 90 Inhibition. Cancer Research. 2005;65:10006–15. doi: 10.1158/0008-5472.CAN-05-2029. [DOI] [PubMed] [Google Scholar]
- 22.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Letters. 2009;280:233–41. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Laurie NA, Donovan SL, Shih C-S, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 24.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of Receptor Tyrosine Kinases Affects the Response of Tumor Cells to Targeted Therapies. Science. 2007;318:287–90. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 25.Dong J-T, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cellular and Molecular Life Sciences. 2009;66:2691–706. doi: 10.1007/s00018-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith SL, Watson SG, Ratschiller D, Gugger M, Betticher DC, Heighway J. Maspin - the most commonly-expressed gene of the 18q21.3 serpin cluster in lung cancer - is strongly expressed in preneoplastic bronchial lesions. Oncogene. 0000;22:8677–87. doi: 10.1038/sj.onc.1207127. [DOI] [PubMed] [Google Scholar]
- 27.Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochemical Pharmacology. 2010;80:624–37. doi: 10.1016/j.bcp.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia. 2008;22:730–9. doi: 10.1038/leu.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudloff U, Samuels Y. A growing family: Adding mutated Erbb4 as a novel cancer target. Cell Cycle. 2010;9:1487–503. doi: 10.4161/cc.9.8.11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YR, Fu YN, Lin CH, Yang ST, Hu SF, Chen YT, et al. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene. 2005;25:1205–15. doi: 10.1038/sj.onc.1209159. [DOI] [PubMed] [Google Scholar]
- 31.Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, Banjade S, et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 2008;16:460–7. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrasco-García E, Saceda M, Grasso S, Rocamora-Reverte L, Conde M, Gómez-Martínez Å, et al. Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Experimental Cell Research. 2011;317:1476–89. doi: 10.1016/j.yexcr.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Bernal-Mizrachi L, Lovly CM, Ratner L. The role of NFKB1 and NFKB2-mediated resistance to apoptosis in lymphomas. Proceedings of the National Academy of Sciences. 2006;103:9220–5. doi: 10.1073/pnas.0507809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishio M, Yamanaka T, Matsumoto K, Kimura H, Sakai K, Sakai A, et al. Serum Heparan Sulfate Concentration is Correlated with the Failure of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment in Patients with Lung Adenocarcinoma. Journal of Thoracic Oncology. 2011;6:1889. doi: 10.1097/JTO.0b013e3182286d41. [DOI] [PubMed] [Google Scholar]
- 35.Saga Y, Mizukami H, Suzuki M, Kohno T, Urabe M, Ozawa K, et al. Overexpression of PTEN Increases Sensitivity to SN-38, an Active Metabolite of the Topoisomerase I Inhibitor Irinotecan, in Ovarian Cancer Cells. Clinical Cancer Research. 2002;8:1248–52. [PubMed] [Google Scholar]
- 36.Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PLoS ONE. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frolov A, Schuller K, Tzeng C-WD, Cannon EE, Ku BC, Howard JH, et al. ErbB3 expression and dimerization with EGFR influence pancreatic cancer cell sensitivity to erlotinib. Cancer Biology & Therapy. 2007;6:548–54. doi: 10.4161/cbt.6.4.3849. [DOI] [PubMed] [Google Scholar]
- 39.Popowicz GM, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, et al. Molecular Basis for the Inhibition of p53 by Mdmx. Cell Cycle. 2007;6:2386–92. doi: 10.4161/cc.6.19.4740. [DOI] [PubMed] [Google Scholar]
- 40.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX Overexpression Prevents p53 Activation by the MDM2 Inhibitor Nutlin. Journal of Biological Chemistry. 2006;281:33030–5. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 41.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Molecular Cancer Therapeutics. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 42.Trojanek J, Ho T, Del Valle L, Nowicki M, Wang JY, Lassak A, et al. Role of the Insulin-Like Growth Factor I/Insulin Receptor Substrate 1 Axis in Rad51 Trafficking and DNA Repair by Homologous Recombination. Molecular and Cellular Biology. 2003;23:7510–24. doi: 10.1128/MCB.23.21.7510-7524.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertucci Fo, Finetti P, Cervera N, Charafe-Jauffret E, Mamessier E, Adelaide J, et al. Gene Expression Profiling Shows Medullary Breast Cancer Is a Subgroup of Basal Breast Cancers. Cancer Research. 2006;66:4636–44. doi: 10.1158/0008-5472.CAN-06-0031. [DOI] [PubMed] [Google Scholar]
- 44.http://www.karchinlab.org/data/MOCA.zip
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.