

Abstract
Renal microvascular (MV) damage and loss contribute to the progression of renal injury in renal artery stenosis (RAS). Hepatocyte growth factor (HGF) is a powerful angiogenic and antifibrotic cytokine that we showed to be decreased in the stenotic kidney. We hypothesized that renal HGF therapy will improve renal function mainly by protecting the renal microcirculation. Unilateral RAS was induced in 15 pigs. Six weeks later, single-kidney RBF and GFR were quantified in vivo using multidetector computed tomography (CT). Then, intrarenal rh-HGF or vehicle was randomly administered into the stenotic kidney (RAS, n = 8; RAS+HGF, n = 7). Pigs were observed for 4 additional weeks before CT studies were repeated. Renal MV density was quantified by 3D micro-CT ex vivo and histology, and expression of angiogenic and inflammatory factors, apoptosis, and fibrosis was determined. HGF therapy improved RBF and GFR compared with vehicle-treated pigs. This was accompanied by improved renal expression of angiogenic cytokines (VEGF, p-Akt) and tissue-healing promoters (SDF-1, CXCR4, MMP-9), reduced MV remodeling, apoptosis, and fibrosis, and attenuated renal inflammation. However, HGF therapy did not improve renal MV density, which was similarly reduced in RAS and RAS+HGF compared with controls. Using a clinically relevant animal model of RAS, we showed novel therapeutic effects of a targeted renal intervention. Our results show distinct actions on the existing renal microcirculation and promising renoprotective effects of HGF therapy in RAS. Furthermore, these effects imply plasticity of the stenotic kidney to recuperate its function and underscore the importance of MV integrity in the progression of renal injury in RAS.
Keywords: kidney, HGF, renal artery stenosis, microcirculation, remodeling, imaging
microvascular (MV) dysfunction, damage, and/or rarefaction are often observed in target organs during the progression of hypertension, diabetes, and cardiovascular disease. In turn, evidence supports a central pathophysiological role for MV dysfunction and damage in promoting cardiovascular disease-induced organ injury (29), underscoring a cause-effect relationship between MV disease and the pathophysiology of organ damage. In the kidney, glomerular and peritubular MV damage and loss have been linked to renal functional impairment and progression of renal damage in chronic renal disease (irrespective of the etiology) (17) and renal graft function (41). Indeed, renal MV disease is an important contributor to the progression of renal injury (23, 40) and an important determinant of whether treatments are effective (7).
Almost 15% of patients with renal artery stenosis (RAS) develop intractable hypertension or progressive loss of kidney function (15). RAS is the main cause of chronic renovascular disease (RVD), a disorder capable of inducing severe, progressive, and irreversible renal dysfunction and damage. We previously showed in a model of experimental RVD that the kidney exhibits progressive MV dysfunction, damage, and gradual loss that correlate with deterioration of renal function and marked renal fibrosis (11, 23, 50). These findings suggest a potential instigating role of MV disease in the initiation and progression of renal damage.
We have recently shown that the renal microvasculature is susceptible to repair and proliferation. For example, a single administration of the proangiogenic vascular endothelial growth factor (VEGF) into the stenotic kidney reversed MV rarefaction, renal dysfunction, and attenuated fibrosis (8, 23). However, the renal MV protection afforded by VEGF is not complete, making the identification of novel therapeutic interventions to modify the progression of renal injury an important clinical goal. Nevertheless, targeted renoprotective interventions in RVD are still limited.
Hepatocyte growth factor (HGF) is a mesenchyme-derived pleiotropic antifibrotic factor and a powerful stimulator of angiogenesis that is constitutively produced in the adult kidney (30). HGF stimulates proliferation and migration of endothelial and vascular smooth muscle cells and promotes vascular proliferation and repair directly and by interactions with other angiogenic cytokines (20). We previously showed that renal expression of HGF is decreased in the stenotic kidney (12, 24), but whether and how administration of HGF may serve as a renoprotective agent in RVD have never been investigated. The current study was designed to determine the potential for using HGF therapy in the stenotic kidney. We hypothesized that a therapeutic intrarenal administration of HGF will improve renal function in the stenotic kidney mainly by protecting the renal microcirculation. Furthermore, this study also aimed to establish the underlying mechanisms of HGF-induced renoprotection.
MATERIALS AND METHODS
In Vivo and Ex Vivo Studies in the Stenotic Kidney
The Institutional Animal Care and Use Committee at the University of Mississippi Medical Center approved all the procedures. Twenty-three prejuvenile domestic pigs were observed for 10 wk. In 15 pigs, unilateral RAS (a surrogate and cause of RVD) was induced at baseline, as previously shown (9, 10, 28). Additional animals were used as normal controls (normal, n = 8). Blood pressure was chronically measured using telemetry (PhysioTel, Data Sciences International) (9, 10, 23, 28).
Six weeks after induction of RAS, the pigs were anesthetized with intramuscular telazol (5 mg/kg) and xylazine (2 mg/kg), intubated, and mechanically ventilated on room air. Anesthesia was maintained with a mixture of ketamine (0.2 mg·kg−1·min−1) and xylazine (0.03 mg·kg−1·min−1) in normal saline and administered via an ear vein cannula (0.05 ml·kg−1·min−1). Renal angiography was performed in all pigs under fluoroscopic guidance, and stenosis was quantified, as previously described (23). After angiography, the catheter was positioned in the superior vena cava, and in vivo helical multidetector computer tomography (MDCT) flow studies were performed for quantification of single-kidney renal blood flow (RBF; ml/min), perfusion (ml·min−1·g tissue−1), and glomerular filtration rate (GFR; ml/min), as previously validated (9, 13, 27).
Immediately after completion of the 6-wk MDCT in vivo studies (and before the values of RBF and GFR were known), all RAS pigs were randomized into two groups: those that received vehicle (RAS, n = 8) and those treated with a single intrarenal infusion of recombinant human (rh)-HGF (2 μg, RAS+HGF, n = 7), as described recently (7, 23). The pigs were then observed for 4 additional wk, in vivo studies were repeated at 10 wk, and renal-derived CT data analysis was completed a few days later. Renal vascular resistance was calculated at 6 and 10 wk (measured during the in vivo studies) as described (8). Blood from the inferior vena cava and renal veins (from the stenotic kidney) were collected at 6 and 10 wk to measure plasma renin activity (PRA) and serum creatinine (SCr), as described (8, 9, 23).
Two days after completion of all the in vivo studies, the pigs were euthanized by an overdose administration of pentobarbital sodium (100 mg/kg iv). The kidneys were then removed and immersed in heparinized saline (10 units/ml). One lobe was used for micro-CT reconstruction, while another lobe was sectioned, snap-frozen in liquid nitrogen, and stored at −80°C to quantify the protein expression of the angiogenic and survival factors hypoxia-induced factor (HIF)-1α, VEGF, angiopoietin (Ang)-1, phosphorylated endothelial nitric oxide synthase (p-eNOS), p-Akt, HGF and its cMet receptor, transforming growth factor (TGF)-β, and the specific smad-4 and -7 receptors, tissue-transglutaminase (tTg), matrix metalloproteinase (MMP)-9, and the specific inhibitor TIMP-1. Furthermore, expression of neutrophil gelatinase-associated lipocalin (Ngal), proinflammatory nuclear-factor-κ (NF-κB), TNF-α, and monocyte chemoattractant protein (MCP)-1 was also measured. Renal tissue was also used to quantify the activity of superoxide dismutase, a potent scavenger of reactive oxygen species (ROS), by ELISA (Superoxide Dismutase Activity Colorimetric Assay Kit, Abcam, Cambridge, MA). Finally, another portion of the kidney was preserved in 10% formalin and used to study renal morphology, MV remodeling, tubular damage, and apoptosis in midhilar renal cross sections stained with trichrome and hematoxylin and eosin (H&E), respectively (23, 25).
MDCT analysis.
Manually traced regions of interest were selected in MDCT images in the aorta, renal cortex, medulla, and papilla, and their densities were sampled. Time-density curves were generated and fitted with extended gamma-variate curve fits. The area under each segment of the curve and its first moment were calculated using curve-fitting parameters and used to calculate single-kidney RBF (ml/min), GFR (ml/min), and regional perfusion (ml·min−1·g tissue min−1) using previously validated methods (13, 27).
Micro-CT.
The stenotic kidney was perfused (Syringe Infusion Pump 22, Harvard Apparatus, Holliston, MA) with an intravascular contrast agent (Microfil MV122, Flow Tech, Carver, MA). The kidney samples were scanned at 0.3-in. increments using a micro-CT scanner and reconstructed at 9-μm resolution for subsequent analysis using the Analyze software package (Biomedical Imaging Resource, Mayo Clinic, Rochester, MN), as previously described (23). The cortex was tomographically divided, and the spatial density and distribution of microvessels (diameters under 200 μm) and vascular volume fraction (the ratio of the sum of cross-sectional areas of all vessels and the total area of the region of interest) were calculated, as previously described (23).
Western blotting of renal cortical tissue homogenates was performed, as previously described (10). Specific polyclonal antibodies against HIF-1α (1:1,000 for both, Cell Signaling, Boston, MA), VEGF, p-Akt, p-eNOS (1:200 for all, Santa Cruz Biotechnology, Santa Cruz, CA), SDF-1 and the specific CXCR4 receptor, and Ang-1 (1:1,000, Abcam, Cambridge, MA,) were used. Furthermore, renal expression of TGF-β (1:1,000, Abcam) and the specific smad-4 and -7 receptors (1:200 for both, Santa Cruz Biotechnology), tTg, MMP-9, and TIMP-1 (1:1,000, Cell Signaling) were also quantified. In addition, renal expression of Ngal (1:200, Santa Cruz Biotechnology) and proinflammatory NF-κB, TNF-α (1:300 for both, Santa Cruz Biotechnology), and MCP-1 (1:200, Abcam) were also measured. β-Actin (1:500, Sigma, St. Louis, MO) served as a loading control. Protein expression (1 band per animal) was quantified by densitometry and averaged per group.
Histology.
Midhilar 5-μm paraffin-embedded cross sections of each kidney (1 per animal) stained with trichrome were examined, staining was semiautomatically quantified (NIS Element 3.0, Nikon Instruments, Melville, NY), and expressed as the percentage of staining of the total surface area, as described (9, 23). The percentage of sclerotic glomeruli was determined by recording the number of sclerotic glomeruli of 100 counted glomeruli, and the MV media-to-lumen ratio was quantified, as previously described (9, 23). The immunoreactivity against HGF and its specific receptor cMet (1:50 for both, Santa Cruz Biotechnology) was determined in renal cross sections (1 per animal) following standard procedures (8). Finally, for quantification of renal tubular casts, 10–15 randomly selected fields per slide (1 per animal) were chosen in a blind manner and quantified separately by two independent observers (N. Stewart and A. R. Chade). Using ×20 magnification, tubular casts were graded as follows: no damage (0), mild (+1, unicellular patchy isolated damage), moderate (+2, damage <25%), severe (3+, damage between 25 and 50%), and very severe (+4, >50% damage) as previously shown (45).
Apoptosis.
The fraction of apoptotic cells was calculated in renal cross sections stained with H&E. Around 15–20 tubule-interstitial and glomerular randomly selected regions in the stenotic (or normal) kidney of each pig were used. The fraction of apoptotic cells was calculated and expressed as a percentage of apoptotic cells (of the total number of cells) per field, as shown (25).
Statistical Analysis
Results are expressed as means ± SE. Comparisons within groups were performed using a paired Student's t-test and among groups using one-way ANOVA, with the Bonferroni correction for multiple comparisons. Statistical significance was accepted for P ≤ 0.05.
RESULTS
In Vivo Studies
General characteristics.
Body weight was similar in all pigs after 6 wk, and they all showed a similar weight gain at 10 wk (Tables 1 and 2). The angiographic degree of stenosis was similar in RAS and RAS+HGF-treated animals after 6 and 10 wk (Tables 1 and 2, respectively, P < 0.01 vs. normal). However, administration of rh-HGF at 6 wk was followed by a lower blood pressure at 10 wk compared with placebo-treated RAS pigs (Table 2). PRA was similar among the groups (Tables 1 and 2), consistent with our previous studies using this model of chronic renovascular hypertension (9, 23). On the other hand, the increased RVR after 6 and 10 wk of RAS (Tables 1 and 2) was decreased in RAS after administration of HGF (Table 2).
Table 1.
Body weight, mean arterial pressure, degree of stenosis, plasma renin activity, serum creatinine, and basal single-kidney hemodynamics and function (means ± SE) after 6 wk of observation in normal, renal artery stenosis disease (RAS), and RAS pigs before treatment with intrarenal HGF
| Parameter | Normal | RAS | RAS pre-HGF |
|---|---|---|---|
| Body weight, kg | 47.6 ± 2.0 | 46.2 ± 1.0 | 45.2 ± 2.0 |
| Mean arterial pressure, mmHg | 89.5 ± 2.1 | 128.7 ± 2.0* | 125.2 ± 3.7* |
| Degree of stenosis, % | 0.0 ± 0.0 | 74.2 ± 4.1* | 73.8 ± 6.2* |
| Plasma renin activity, ng•ml−1•h−1 | 0.29 ± 0.01 | 0.34 ± 0.05 | 0.29 ± 0.03 |
| Serum creatinine, μmol/l | 82.2 ± 3.5 | 96.2 ± 7.0* | 91.8 ± 4.0* |
| Renal vascular resistance, mmHg•ml•min−1 | 0.19 ± 0.03 | 0.9 ± 0.4* | 0.48 ± 0.06* |
| Renal volume, ml | |||
| Cortex | 119.8 ± 8.5 | 62.6 ± 6.2* | 69.5 ± 5.8* |
| Medulla | 36.8 ± 2.3 | 18.4 ± 6.8* | 28.3 ± 3.9* |
| Renal blood flow, ml/min | 504.4 ± 55.6 | 260.8 ± 58.9* | 276.3 ± 35.9* |
| Perfusion, ml•min−1•g tissue−1 | 3.4 ± 0.3 | ||
| Cortex | 3.4 ± 0.4 | 3.2 ± 0.6 | 3.4 ± 0.3 |
| Medulla | 2.6 ± 0.5 | 2.4 ± 0.6 | 1.6 ± 0.8 |
| Glomerular filtration rate, ml/min | 53.9 ± 5.6 | 34.8 ± 8.1* | 38.7 ± 7.4* |
P < 0.05 vs. normal. †P < 0.05 vs. RAS.
Table 2.
Body weight, mean arterial pressure, degree of stenosis, plasma renin activity, serum creatinine, basal single-kidney hemodynamics and function, and superoxide dismutase activity (means ± SE) after 10 wk in normal, RAS and RAS pigs treated with intrarenal HGF at 6 wk (RAS+HGF).
| Parameter | Normal | RAS | RAS+HGF |
|---|---|---|---|
| Body weight, kg | 52.7 ± 2.0 | 53.4 ± 4.0 | 53.7 ± 1.0 |
| Mean arterial pressure, mmHg | 91.3 ± 4.9 | 141.5 ± 6.1* | 129.3 ± 4.7*† |
| Degree of stenosis, % | 0.0 ± 0.0 | 76.3 ± 7.1* | 75.6 ± 6.8* |
| Plasma renin activity, ng•ml−1•h−1 | 0.32 ± 0.06 | 0.38 ± 0.1 | 0.24 ± 0.1 |
| Serum creatinine, μmol/l | 85.1 ± 4.5 | 116.7 ± 10.5*# | 97.2 ± 5.5^ |
| Renal vascular resistance, mmHg•ml•min−1 | 0.2 ± 0.02 | 2.1 ± 0.3* | 0.33 ± 0.03*†# |
| Renal volume, ml | |||
| Cortex | 112.0 ± 3.1 | 58.3 ± 9.3* | 85.2 ± 6.3*†# |
| Medulla | 35.2 ± 1.1 | 19.1 ± 2.0* | 21.6 ± 2.3* |
| Renal blood flow, ml/min | 528.5 ± 23.9 | 243.9 ± 59.6* | 395.8 ± 41.6†# |
| Perfusion, ml•min−1•g tissue−1 | |||
| Cortex | 4.2 ± 0.3 | 3.5 ± 0.8 | 4.0 ± 0.1# |
| Medulla | 2.3± 0.3 | 2.0 ± 0.6 | 1.9 ± 0.4 |
| Glomerular filtration rate, ml/min | 72.3 ± 4.9 | 37.6 ± 8.3* | 60.8 ± 5.3*†# |
| Superoxide dismutase activity, % | 91.7 ± 1.4 | 79.7 ± 2.5* | 85.1 ± 2.0*†$ |
P < 0.05 vs. normal.
P < 0.05 vs. RAS.
P < 0.05 vs. 6 wk.
P = 0.07 vs. normal.
P = 0.06 vs. normal.
MDCT-derived single-kidney hemodynamics and function.
Consistent with our previous work, basal RBF and GFR were reduced to a similar degree in the stenotic kidney of all RAS animals at 6 wk (23) and was accompanied by elevated SCr (Table 1), which remained unchanged in placebo-treated RAS after 10 wk. However, RBF, GFR, and SCr were significantly improved compared with the 6-wk pre-HGF hemodynamics (Table 2). Furthermore, the improvements were also significant compared with placebo-treated RAS animals (albeit not normalized) at 10 wk (Table 2). These beneficial effects on renal hemodynamics were accompanied by an increased activity of superoxide dismutase in the stenotic kidney, suggesting augmented renal scavenging of reactive oxygen species after HGF therapy (Table 2).
Ex Vivo Studies in the Stenotic Kidney
Renal expression of angiogenic factors.
rh-HGF in the stenotic kidney increased the renal expression of HGF and cMet in endothelial cells and renal tubules in the stenotic kidney (Fig. 1). Furthermore, administration of rh-HGF restored the renal expression of HIF-1α, VEGF, p-eNOS, p-Akt, and Ang-1, augmented the expression of SDF-1 and CXCR4, and decreased the expression of endostatin (Fig. 2).
Fig. 1.
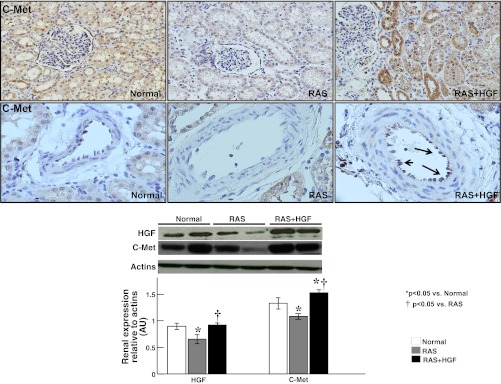
Representative renal cross sections showing immunostaining (×40, top) against HGF and its specific cMet receptor and quantification of renal protein expression (by Western blotting, bottom) in normal, renal artery stenosis (RAS), and RAS+HGF kidneys. The administration of recombinant human (rh)-HGF restored the expression of renal HGF and augmented the tubular and vascular expression of the cMet receptor in the stenotic kidney. *P < 0.05 vs. normal. †P < 0.05 vs. RAS.
Fig. 2.
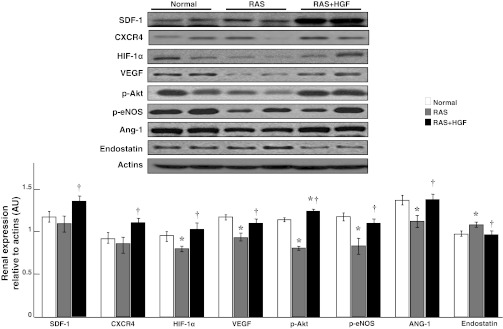
Representative renal protein expression (n = 6/group, 2 representative bands shown) and quantification of SDF-1, CXCR4, hypoxia-induced factor (HIF)-1α, VEGF, phosphorylated (p)-Akt, p-endothelial nitric oxide synthase (eNOS), angiopoietin (Ang)-1, and endostatin. Intrarenal administration of HGF augmented the expression of angiogenic and vasculo-protective factors and reduced the expression of antiangiogenic endostatin. *P < 0.05 vs. normal. †P < 0.05 vs. RAS.
MV 3D architecture.
The density of microvessels with diameters <200 μM were similarly and significantly decreased throughout the renal cortex and medulla in the stenotic kidney of RAS and RAS+HGF animals after 10 wk (P < 0.05 vs. normal, Fig. 3, left). Administration of rh-HGF did not modify MV density, indicating a null effect on renal MV proliferation despite the augmented expression of proangiogenic factors in the HGF-treated kidney.
Fig. 3.
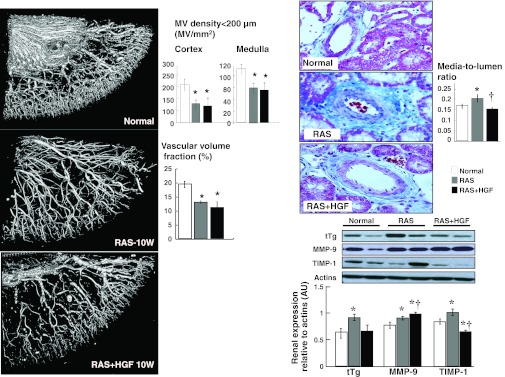
Left: representative 3D micro-computed tomography (CT) reconstruction and quantification of cortical and medullary microvascular (MV) density (diameters under 200 μM) and vascular volume fraction. Right: representative pictures and quantification of MV media-to-lumen ratio and renal protein expression of tTg and MMP-9 in normal, RAS, and RAS+HGF. Intrarenal administration of HGF did not increase MV density in the stenotic kidney. However, it significantly reduced MV remodeling, suggesting a protective effect on the preexisting renal microvasculature. *P < 0.05 vs. normal. †P < 0.05 vs. RAS.
MV remodeling.
Administration of rh-HGF in the stenotic kidney decreased the expression of tTg and improved the expression of MMP-9 and TIMP-1. These were accompanied by a normalized media-to-lumen ratio, suggesting a protective effect on the preexisting vasculature (Fig. 3, right).
Renal morphology.
Fibrosis and patchy inflammatory infiltrates in the stenotic kidney were evident in the cortex, while no significant changes were observed in the renal medulla. Treatment with rh-HGF in the stenotic kidney resulted in a decrease in renal damage, accompanied by decreased expression of TGF-β and smad-4, increased smad-7, and a decreased expression of proinflammatory NF-κB (P < 0.05 vs. RAS, Fig. 4, A and B), although TNF-α and MCP-1 remained similarly elevated in RAS and RAS+HGF kidneys. Finally, the presence of tubular casts was significantly reduced as renal expression of Ngal augmented after HGF therapy, suggesting a protective effect on the renal tubules (Fig. 4C).
Fig. 4.
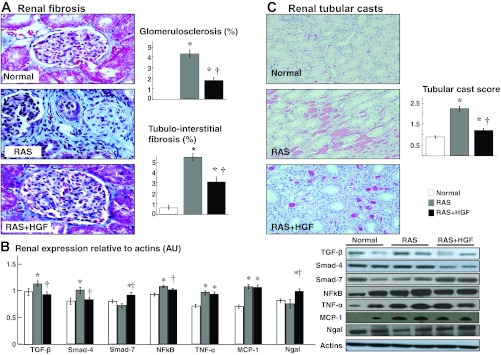
Representative trichrome pictures of glomerular (A, ×40) region (shown as examples to illustrate renal damage), quantification of tubule-interstitial fibrosis and glomerulosclerosis, renal protein expression of transforming growth factor (TGF)-β and the smad-4 and -7 receptors, NF-κB, TNF-α, MCP-1, and Ngal (B), and representative hematoxylin and eosin (H&E) renal cross sections (C, ×40), and quantification of protein tubular casts (score) in normal, RAS, and RAS+HGF kidneys. Administration of HGF into the stenotic kidney reduced proinflammatory and profibrotic activity and the overall renal damage. *P < 0.05 vs. normal. †P < 0.05 vs. RAS.
Renal apoptosis.
Apoptosis was attenuated after administration of rh-HGF therapy in both glomerular and tubule-interstitial compartments in the stenotic kidney, albeit not normalized (normal: 5.5 ± 0.3%, RAS: 13.53 ± 2.3%, and RAS+HGF: 7.4 ± 0.2%; P < 0.05 vs. normal and RAS).
DISCUSSION
The current study shows that a single intrarenal administration of rh-HGF significantly reduced the damage of the renal parenchyma in the stenotic kidney. The HGF-treated kidney showed an improved RBF and GFR compared with not only vehicle-treated pigs but also pre-HGF renal function. These effects were accompanied by a marked decrease in renal MV remodeling and damage despite a similar reduction in cortical or medullary MV density in both placebo- and HGF-treated kidneys. Thus these results suggest that HGF therapy has a protective effect on the preexisting renal microcirculation and can reverse renal injury in the stenotic kidney. Furthermore, they support feasibility and potential of rh-HGF administration as a therapeutic tool in RVD.
HGF is a mesenchyme-derived pleiotropic growth factor. It is a powerful stimulator of angiogenesis and renal epithelial tubule healing that stimulates renal regeneration by promoting cell survival, vascular proliferation, and decreased apoptosis and fibrosis (5, 14, 35). HGF, which is produced constitutively in the adult kidney (30), stimulates proliferation and migration of both endothelial and vascular smooth muscle cells via binding its specific and ubiquitous c-Met receptor. HGF induces angiogenesis and tissue regeneration and seems to be a central player in the angiogenic cascade both by activation of and synergistic interactions with other angiogenic cytokines, such as VEGF (20). HGF has also been shown to increase eNOS, an essential factor in mediating angiogenesis and endothelial function via production of nitric oxide (NO) (31, 44) and is an important regulator of VEGF-mediated angiogenesis. Another factor stimulated by HGF is Akt (32), a key intercellular signaling factor that interacts, regulates, and improves the maturation and permeability of angiogenic vessels, cell survival, and decreases apoptosis. It also closely interacts with eNOS and regulates NO production as well (1). Furthermore, HGF stimulates angiopoietins, a widely expressed family of growth factors that act as promoters of HGF (26) and directly promote proliferation, mobilization, recruitment, and survival of cell progenitors (39, 42, 47, 49). Interaction between angiopoietins and HGF regulates the communication between endothelial and mural cells, critical steps in vascular stabilization. Ang-1 in turn enhances endothelial cell survival and limits MV permeability (2, 6, 38).
We have consistently shown that the stenotic kidney has a marked reduction in the expression of angiogenic factors (23), in addition to the decreased HGF/cMet reported in the current study. We have recently showed that the administration of exogenous VEGF into the stenotic kidney induced a marked renoprotection by promoting MV proliferation and repair and reducing renal damage (8, 23). VEGF plays an important role as a primary defensive mechanism for the adaptive response to ischemia in the kidney (33) by initiating angiogenesis that leads to MV sprouting. In the current study, we observed that an intrarenal administration of HGF restored (and even normalized in some cases) the renal expression of proangiogenic VEGF, p-eNOS, Akt, and Ang-1, which was likely achieved via stimulation of the HGF/cMet pathway since their expression was augmented in the stenotic kidney after rh-HGF therapy. While both VEGF and HGF are powerful prosurvival cytokines (16, 48), their effects on angiogenesis differ. VEGF promotes neovascularization mainly via distinct stimulation of endothelial cells, whereas HGF stimulates both endothelial and vascular smooth muscle cells, thus facilitating MV maturation, diminishing MV permeability, and possibly improving the functionality of the neovessels (18). In addition to augmenting the renal expression of angiogenic factors, administration of HGF reduced the expression of antiangiogenic endostatin (3). Taken together, this evidence supports the notion of a potential proangiogenic effect after HGF administration.
Despite the suggestive renal proangiogenic effect of rh-HGF administration, renal MV density was similarly reduced in RAS and RAS+HGF kidneys, suggesting an apparent null effect on MV proliferation in the stenotic kidney. However, it is possible that the improved expression of p-eNOS and Ang-1, and reduced endostatin after administration of rh-HGF may have stimulated the repair of damaged or dysfunctional preexisting microvessels (3, 46). Furthermore, the accompanying reduction in MV remodeling (as suggested by the normalized media-to-lumen ratio and improved renal expression of tTg and MMP-9) indicates that intrarenal rh-HGF therapy may have had a protective effect on the preexisting renal MV network. The possibility of enhanced MV and tissue repair in the HGF-treated kidney is further supported by the augmented expression of SDF-1/CXCR4, a pivotal signaling axis that stimulates migration and homing of progenitor cells to ischemic tissues to initiate tissue repair (34, 37). In turn, the stimulation of the SDF-1/CXCR4 axis suggests that the prolonged effects of a single dose of HGF therapy may have been initiated and sustained by an enhanced recruitment of cell progenitors that persists beyond the relatively short half-life of HGF (a few minutes). All these effects on the stenotic kidney were functionally consequential and followed by improvements in RBF, GFR, and perfusion compared with placebo-treated kidneys and to pre-HGF renal function as well. HGF therapy also increased the renal activity of SOD, suggesting an augmented scavenging of reactive oxygen species in the RAS+HGF kidney. The improved redox status and increased p-eNOS may have resulted in augmented bioavailability of NO and likely contributed to the improvement in renal hemodynamics of the HGF-treated stenotic kidney at 10 wk. Furthermore, administration of HGF in the stenotic kidney reduced the presence of tubular casts and augmented the expression of Ngal, which has been shown to enhance reparative tubule formation by interactions with HGF, possibly preventing dysregulated branching during tubule repair (22). Overall, these data first imply a cause-effect relationship and a protective role for rh-HGF administration on the renal parenchyma of the stenotic kidney. This is also supported by the improvement in SCr in RAS+HGF and by the halted progression of hypertension after administration of rh-HGF. It is possible that the remaining renal injury in the stenotic (and contralateral) kidney may partly explain the absence of a more profound effect on blood pressure after HGF administration. However, the reasons are not entirely clear and warrant further investigation.
HGF has also been shown to be a powerful antifibrotic cytokine and a critical promoter of extracellular matrix removal in the kidney by counteracting the profibrotic effects of TGF-β and promoting the activity of MMPs (14, 36). MMPs are also pivotal enzymes for the normal expansion and development of MV networks and balance of angiogenic factors that maintain normal renal structure (4, 10, 43). Furthermore, TGF-β has been shown to induce apoptosis in MV and glomerular endothelial cells and to downregulate the signaling of angiogenic survival factors, enhancing the susceptibility of endothelial cells to undergo apoptosis (4). We observed that administration of HGF significantly reduced the renal expression of TGF-β and smad-4. This was accompanied by an increased expression of the inhibitor smad-7, increased expression of cleaved MMP-9, and reduced expression of TIMP-1 in the stenotic kidney, suggesting an overall decreased in profibrotic activity. Furthermore, the augmented renal expression of SDF-1/CXCR4 may suggest that HGF therapy stimulated tissue repair (37). Consequently, we observed a significant attenuation of tubule-interstitial and perivascular fibrosis, glomerulosclerosis, and renal apoptosis, supporting the notion of a significant reduction in renal injury after administration of rh-HGF. On the other hand, administration of HGF therapy in the stenotic kidney reduced the expression of NF-κB, suggesting a decrease in proinflammatory activity. Previous studies showed that anti-inflammatory actions of HGF are partly mediated by disruption of NF-κB signaling (19, 21). However, HGF therapy did not reduce the expression of MCP-1 and TNF-α in the stenotic kidney, which may explain the persistence of some inflammatory infiltrates in the HGF-treated kidneys.
Limitations and Perspectives
A potential limitation of the current study could be that the single intrarenal infusion of rh-HGF was applied at a relatively early stage of RVD. Hence, future studies using HGF therapy at more advanced stages of RVD and renal damage will contribute to determining the full potential of this intervention. However, we believe that the marked improvements in renal function and damage, with no evident adverse effects, are promising and may open new avenues for future research into the clinical utility of HGF as a therapeutic option for renal disease. Finally, while we are aware that additional research is needed to further elucidate the underlying mechanisms, this study represents an important proof of concept that administration of HGF may have clinical value.
In summary, using an established clinically relevant large-animal model that mimics chronic RVD, the current study shows promising therapeutic effects of a targeted renal intervention. The apparent absence of collateral or adverse effects in this study supports the potential of rh-HGF as a feasible therapeutic alternative to protect the stenotic kidney. The distinct effects of rh-HGF on the renal parenchyma implies plasticity of the stenotic kidney to recuperate its function, underscores the importance of MV integrity on the progression of renal injury, and suggests a protective effect of HGF therapy on the existing renal microcirculation. Interventions that slow, stop, or reverse the progression of renal damage (e.g., renal dysfunction, MV remodeling, renal fibrosis) would be valuable to improve the chances of recovery in patients with RVD.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-095638 and HL-051971.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.R.C. provided concept and design of research; A.R.C. and N.S. performed experiments; A.R.C. and N.S. analyzed data; A.R.C. interpreted results of experiments; A.R.C. prepared figures; A.R.C. and N.S. drafted manuscript; A.R.C. and N.S. edited and revised manuscript; A.R.C. and N.S. approved final version of manuscript.
REFERENCES
- 1. Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol 290: H547–H559, 2006 [DOI] [PubMed] [Google Scholar]
- 2. Baffert F, Le T, Thurston G, McDonald DM. Angiopoietin-1 decreases plasma leakage by reducing number and size of endothelial gaps in venules. Am J Physiol Heart Circ Physiol 290: H107–H118, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Bloch W, Huggel K, Sasaki T, Grose R, Bugnon P, Addicks K, Timpl R, Werner S. The angiogenesis inhibitor endostatin impairs blood vessel maturation during wound healing. FASEB J 14: 2373–2376, 2000 [DOI] [PubMed] [Google Scholar]
- 4. Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol 13: 2600–2610, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Breier G, Blum S, Peli J, Groot M, Wild C, Risau W, Reichmann E. Transforming growth factor-beta and Ras regulate the VEGF/VEGF-receptor system during tumor angiogenesis. Int J Cancer 97: 142–148, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res 98: 1014–1023, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chade AR, Kelsen S. Renal microvascular disease determines the responses to revascularization in experimental renovascular disease. Circ Cardiovasc Interv 3: 376–383, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chade AR, Kelsen S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: a novel potential therapeutic approach. Am J Physiol Renal Physiol 302: F1342–F1350, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chade AR, Rodriguez-Porcel M, Grande JP, Krier JD, Lerman A, Romero JC, Napoli C, Lerman LO. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation 106: 1165–1171, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Chade AR, Rodriguez-Porcel M, Grande JP, Zhu X, Sica V, Napoli C, Sawamura T, Textor SC, Lerman A, Lerman LO. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler Thromb Vasc Biol 23: 1295–1301, 2003 [DOI] [PubMed] [Google Scholar]
- 11. Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB J 20: 1706–1708, 2006 [DOI] [PubMed] [Google Scholar]
- 12. Chade AR, Zhu XY, Grande JP, Krier JD, Lerman A, Lerman LO. Simvastatin abates development of renal fibrosis in experimental renovascular disease. J Hypertens 26: 1651–1660, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Daghini E, Primak AN, Chade AR, Krier JD, Zhu XY, Ritman EL, McCollough CH, Lerman LO. Assessment of renal hemodynamics and function in pigs with 64-section multidetector CT: comparison with electron-beam CT. Radiology 243: 405–412, 2007 [DOI] [PubMed] [Google Scholar]
- 14. Dworkin LD, Gong R, Tolbert E, Centracchio J, Yano N, Zanabli AR, Esparza A, Rifai A. Hepatocyte growth factor ameliorates progression of interstitial fibrosis in rats with established renal injury. Kidney Int 65: 409–419, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Dworkin LD, Murphy T. Is there any reason to stent atherosclerotic renal artery stenosis? Am J Kidney Dis 56: 259–263, 2010 [DOI] [PubMed] [Google Scholar]
- 16. Foster RR, Saleem MA, Mathieson PW, Bates DO, Harper SJ. Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am J Physiol Renal Physiol 288: F48–F57, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Futrakul N, Butthep P, Patumraj S, Siriviriyakul P, Futrakul P. Microvascular disease and endothelial dysfunction in chronic kidney diseases: therapeutic implication. Clin Hemorheol Microcirc 34: 265–271, 2006 [PubMed] [Google Scholar]
- 18. Gerritsen ME. HGF and VEGF: a dynamic duo. Circ Res 96: 272–273, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Giannopoulou M, Dai C, Tan X, Wen X, Michalopoulos GK, Liu Y. Hepatocyte growth factor exerts its anti-inflammatory action by disrupting nuclear factor-kappaB signaling. Am J Pathol 173: 30–41, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Golocheikine A, Tiriveedhi V, Angaswamy N, Benshoff N, Sabarinathan R, Mohanakumar T. Cooperative signaling for angiogenesis and neovascularization by VEGF and HGF following islet transplantation. Transplantation 90: 725–731, 2010 [DOI] [PubMed] [Google Scholar]
- 21. Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J Biol Chem 283: 7401–7410, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gwira JA, Wei F, Ishibe S, Ueland JM, Barasch J, Cantley LG. Expression of neutrophil gelatinase-associated lipocalin regulates epithelial morphogenesis in vitro. J Biol Chem 280: 7875–7882, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Iliescu R, Fernandez SR, Kelsen S, Maric C, Chade AR. Role of renal microcirculation in experimental renovascular disease. Nephrol Dial Transplant 25: 1079–1087, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kelsen S, Hall JE, Chade AR. Endothelin-A receptor blockade slows the progression of renal injury in experimental renovascular disease. Am J Physiol Renal Physiol 301: F218–F225, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelsen S, He X, Chade AR. Early superoxide scavenging accelerates renal microvascular rarefaction and damage in the stenotic kidney. Am J Physiol Renal Physiol 303: F576–F583, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi H, DeBusk LM, Babichev YO, Dumont DJ, Lin PC. Hepatocyte growth factor mediates angiopoietin-induced smooth muscle cell recruitment. Blood 108: 1260–1266, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krier JD, Ritman EL, Bajzer Z, Romero JC, Lerman A, Lerman LO. Noninvasive measurement of concurrent single-kidney perfusion, glomerular filtration, and tubular function. Am J Physiol Renal Physiol 281: F630–F638, 2001 [DOI] [PubMed] [Google Scholar]
- 28. Lerman LO, Schwartz RS, Grande JP, Sheedy PF, Romero JC. Noninvasive evaluation of a novel swine model of renal artery stenosis. J Am Soc Nephrol 10: 1455–1465, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Levy BI, Schiffrin EL, Mourad JJ, Agostini D, Vicaut E, Safar ME, Struijker-Boudier HA. Impaired tissue perfusion: a pathology common to hypertension, obesity, and diabetes mellitus. Circulation 118: 968–976, 2008 [DOI] [PubMed] [Google Scholar]
- 30. Liu Y, Rajur K, Tolbert E, Dworkin LD. Endogenous hepatocyte growth factor ameliorates chronic renal injury by activating matrix degradation pathways. Kidney Int 58: 2028–2043, 2000 [DOI] [PubMed] [Google Scholar]
- 31. Lu Y, Xiong Y, Huo Y, Han J, Yang X, Zhang R, Zhu DS, Klein-Hessling S, Li J, Zhang X, Han X, Li Y, Shen B, He Y, Shibuya M, Feng GS, Luo J. Grb-2-associated binder 1 (Gab1) regulates postnatal ischemic and VEGF-induced angiogenesis through the protein kinase A-endothelial NOS pathway. Proc Natl Acad Sci USA 108: 2957–2962, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Makondo K, Kimura K, Kitamura N, Kitamura T, Yamaji D, Jung BD, Saito M. Hepatocyte growth factor activates endothelial nitric oxide synthase by Ca2+- and phosphoinositide 3-kinase/Akt-dependent phosphorylation in aortic endothelial cells. Biochem J 374: 63–69, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manotham K, Tanaka T, Ohse T, Kojima I, Miyata T, Inagi R, Tanaka H, Sassa R, Fujita T, Nangaku M. A biologic role of HIF-1 in the renal medulla. Kidney Int 67: 1428–1439, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Mazzinghi B, Ronconi E, Lazzeri E, Sagrinati C, Ballerini L, Angelotti ML, Parente E, Mancina R, Netti GS, Becherucci F, Gacci M, Carini M, Gesualdo L, Rotondi M, Maggi E, Lasagni L, Serio M, Romagnani S, Romagnani P. Essential but differential role for CXCR4 and CXCR7 in the therapeutic homing of human renal progenitor cells. J Exp Med 205: 479–490, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mizuno S, Matsumoto K, Nakamura T. HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Biosci 13: 7072–7086, 2008 [DOI] [PubMed] [Google Scholar]
- 36. Morishita R, Aoki M, Hashiya N, Yamasaki K, Kurinami H, Shimizu S, Makino H, Takesya Y, Azuma J, Ogihara T. Therapeutic angiogenesis using hepatocyte growth factor (HGF). Curr Gene Ther 4: 199–206, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20: 322–332, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satchell SC, Anderson KL, Mathieson PW. Angiopoietin 1 and vascular endothelial growth factor modulate human glomerular endothelial cell barrier properties. J Am Soc Nephrol 15: 566–574, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Schroder K, Schutz S, Schloffel I, Batz S, Takac I, Weissmann N, Michaelis UR, Koyanagi M, Brandes RP. Hepatocyte growth factor induces a proangiogenic phenotype and mobilizes endothelial progenitor cells by activating nox2. Antioxid Redox Signal 15: 915–923, 2010 [DOI] [PubMed] [Google Scholar]
- 40. Sila-asna M, Bunyaratvej A, Futrakul P, Futrakul N. Renal microvascular abnormality in chronic kidney disease. Ren Fail 28: 609–610, 2006 [DOI] [PubMed] [Google Scholar]
- 41. Steegh FM, Gelens MA, Nieman FH, van Hooff JP, Cleutjens JP, van Suylen RJ, Daemen MJ, van Heurn EL, Christiaans MH, Peutz-Kootstra CJ. Early loss of peritubular capillaries after kidney transplantation. J Am Soc Nephrol 22: 1024–1029, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tajima F, Tsuchiya H, Nishikawa K, Kataoka M, Hisatome I, Shiota G. Hepatocyte growth factor mobilizes and recruits hematopoietic progenitor cells into liver through a stem cell factor-mediated mechanism. Hepatol Res 40: 711–719, 2011 [DOI] [PubMed] [Google Scholar]
- 43. Tran ED, Yang M, Chen A, Delano FA, Murfee WL, Peutz-Kootstra CJ. Matrix metalloproteinase activity causes VEGFR-2 cleavage and microvascular rarefaction in rat mesentery. Microcirculation 18: 228–237, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uruno A, Sugawara A, Kanatsuka H, Arima S, Taniyama Y, Kudo M, Takeuchi K, Ito S. Hepatocyte growth factor stimulates nitric oxide production through endothelial nitric oxide synthase activation by the phosphoinositide 3-kinase/Akt pathway and possibly by mitogen-activated protein kinase kinase in vascular endothelial cells. Hypertens Res 27: 887–895, 2004 [DOI] [PubMed] [Google Scholar]
- 45. Yamasowa H, Shimizu S, Inoue T, Takaoka M, Matsumura Y. Endothelial nitric oxide contributes to the renal protective effects of ischemic preconditioning. J Pharmacol Exp Ther 312: 153–159, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Yang Z, Xia WH, Zhang YY, Xu SY, Liu X, Zhang XY, Yu BB, Qiu YX, Tao J. Shear stress-induced activation of Tie2-dependent signaling pathway enhances reendothelialization capacity of early endothelial progenitor cells. J Mol Cell Cardiol 52: 1155–1163, 2012 [DOI] [PubMed] [Google Scholar]
- 47. Yang ZJ, Xu SL, Chen B, Zhang SL, Zhang YL, Wei W, Ma DC, Wang LS, Zhu TB, Li CJ, Wang H, Cao KJ, Gao W, Huang J, Ma WZ, Wu ZZ. Hepatocyte growth factor plays a critical role in the regulation of cytokine production and induction of endothelial progenitor cell mobilization: a pilot gene therapy study in patients with coronary heart disease. Clin Exp Pharmacol Physiol 36: 790–796, 2009 [DOI] [PubMed] [Google Scholar]
- 48. Yo Y, Morishita R, Nakamura S, Tomita N, Yamamoto K, Moriguchi A, Matsumoto K, Nakamura T, Higaki J, Ogihara T. Potential role of hepatocyte growth factor in the maintenance of renal structure: anti-apoptotic action of HGF on epithelial cells. Kidney Int 54: 1128–1138, 1998 [DOI] [PubMed] [Google Scholar]
- 49. Yu X, Song M, Chen J, Zhu G, Zhao G, Wang H, Hunag L. Hepatocyte growth factor protects endothelial progenitor cell from damage of low-density lipoprotein cholesterol via the PI3K/Akt signaling pathway. Mol Biol Rep 37: 2423–2429, 2011 [DOI] [PubMed] [Google Scholar]
- 50. Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, Lerman LO. Cortical microvascular remodeling in the stenotic kidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol 24: 1854–1859, 2004 [DOI] [PubMed] [Google Scholar]