

Abstract
Reactive oxygen species (ROS) are thought to have effects on T-cell function and proliferation. Low concentrations of ROS in T cells are a prerequisite for cell survival, and increased ROS accumulation can lead to apoptosis/necrosis. The cellular redox state of a T cell can also affect T-cell receptor signaling, skewing the immune response. Various T-cell subsets have different redox statuses, and this differential ROS susceptibility could modulate the outcome of an immune response in various disease states. Recent advances in T-cell redox signaling reveal that ROS modulate signaling cascades such as the mitogen-activated protein kinase, phosphoinositide 3-kinase (PI3K)/AKT, and JAK/STAT pathways. Also, tumor microenvironments, chronic T-cell stimulation leading to replicative senescence, gender, and age affect T-cell susceptibility to ROS, thereby contributing to diverse immune outcomes. Antioxidants such as glutathione, thioredoxin, superoxide dismutase, and catalase balance cellular oxidative stress. T-cell redox states are also regulated by expression of various vitamins and dietary compounds. Changes in T-cell redox regulation may affect the pathogenesis of various human diseases. Many strategies to control oxidative stress have been employed for various diseases, including the use of active antioxidants from dietary products and pharmacologic or genetic engineering of antioxidant genes in T cells. Here, we discuss the existence of a complex web of molecules/factors that exogenously or endogenously affect oxidants, and we relate these molecules to potential therapeutics. Antioxid. Redox Signal. 18, 1497–1534.
I. Introduction
Growing evidence indicates that the cellular reduction/oxidation (redox) status regulates various aspects of cellular function. Oxidative stress can elicit positive responses, such as cellular proliferation or activation, as well as negative responses, such as growth inhibition or cell death, most likely in a concentration-dependent manner (Fig. 1). Multiple cellular components, such as DNA, proteins, and lipids, are affected by oxidative stress, leading to various human diseases, including cancer, neurodegeneration, inflammatory diseases, and aging. The effects of reactive oxygen species (ROS) and reactive nitrogen species (RNS) on immune cells and their roles in promoting or controlling acute and chronic diseases have gained increasing scientific prominence. While T cells are important in the adaptive immune response, ROS play a significant role as important innate effectors, by controlling infection and tumorigenesis as well as by modulating T-cell reactivity and autoimmunity. ROS are also thought to be a third signal, along with proinflammatory cytokines, because they enhance and prolong the antigen-specific proliferative response in T cells (285). Thus, the release of ROS, either exogenously by activated granulocytes and macrophages during inflammation or endogenously by chronically stimulated T cells, is important for balancing T-cell activation versus inactivation and thereby regulating immune outcomes. In addition, the importance of T-cell subsets in tumor immunotherapy has also been recently recognized. However, the persistence of tumor epitope-specific T cells could also be affected by the observed differential susceptibility of T-cell subsets to oxidative stress. In this review, we discuss signaling molecules involved in the regulation of T cells' redox status and the strategies that can be implemented to overcome disease.
FIG. 1.

Fate of peripheral T cell in response to different levels of ROS. Increasing the concentration of ROS leads to a differential T-cell response, including TCR activation and cytokine production. Low concentration of ROS leads to improper signaling and therefore low activation and proliferation. Optimal conditions of ROS are required for proper activation of T cells. Increasing the concentration of ROS can lead to increased apoptosis of T cell as a result of DNA damage and activation of p53 induced-genes and FasL. ROS, reactive oxygen species; TCR, T-cell receptor; Th, T helper.
II. T Cell
T cells are important in regulating the adaptive immune response to specific antigens. Based on the type of T-cell receptor (TCR) expression, T cells are either gamma delta (γδ) or alpha beta (αβ) T cells. γδ T cells comprise <5% of the total T-cell population, found at their highest abundance in the gut mucosa, consistent with their role in mucosal immunity. This review focuses on the αβ TCR-bearing T cells, which have a major role in controlling tumor or infectious disease along with autoimmune disease severity. αβ T cells are further categorized based on the cell surface expression of the co-receptor molecules CD4 and CD8. CD4+ T cells or T helper (Th) cells have low cytotoxic activity and provide help by activating and modulating other immune cells to initiate the body's response to invading microorganisms. CD8+ T cells, on the other hand, are referred to as T cytotoxic (Tc) cells and are known to destroy/kill cells that have been infected with foreign invading microorganisms. Both CD4+ and CD8+ T cells are important in autoimmunity, asthma, and allergic responses as well as in tumor immunity. During TCR activation in a particular cytokine milieu, naïve CD4+ T cells and CD8+ T cells may differentiate into one of several lineages of Th or Tc, including Th1/Tc1, Th2/Tc2, Th9/Tc9, Th17/Tc17, Th22/Tc22, and iTreg (induced regulatory T cells, T regulatory cells induced from CD25− cells), as defined by their pattern of cytokine production and function (Fig. 2) (325). How these variations may relate to differences in response, survival, and persistence of the lymphocyte subsets under oxidative stress is dictated by innate differences among cytokines and signaling requirements for T-cell development, the networks of transcription factors involved in T-cell differentiation, and the epigenetic regulation of key T-cell cytokines.
FIG. 2.

Signature cytokine secreted and transcription factors involved in different T cell subsets.
III. T-Cell Subsets and Susceptibility to Oxidative Stress
ROS are critical across a broad spectrum of cellular processes, including signaling, tumor progression, and innate immunity. However, the role of ROS in mammalian adaptive immune system development and function remains largely unknown. A recent study demonstrated that thymus-specific elevation of mitochondrial O2•− disrupts normal T-cell development and impairs the function of the mammalian adaptive immune system (40). Emerging evidence reveals that the differentiation state of lymphocytes largely determines their fate in terms of function and persistence. Although several studies describe the functional consequences of oxygen radical-related immuneregulation, relatively little is known about the sensitivity of individual lymphocyte subsets to oxidative stress and how this process affects disease progression. The susceptibility of T cell subsets to oxidative stress is discussed below.
In an adaptive immune response, T cells recruit and activate B cells, CD8+ T cells, macrophages, mast cells, neutrophils, eosinophils, and basophils. In the presence of differentiating signals, naïve CD4+ (Th) and CD8+ (Tc) cells could exhibit either of the known major lineages, namely Th1/Tc1, Th2/Tc2, Th9/Tc9, Th17/Tc17, Th22/Tc22, and iTreg (Fig. 2) (324). These T-cell subsets are marked by differential cytokine production and cellular distribution. They also have the ability to switch from one lineage to another under certain inflammatory conditions. Taken together, T cells are therefore described as plastic (209). This T-cell plasticity may be affected by an oxidative microenvironment, thereby shifting the normal immune response. A recent study showed that the oxidative microenvironment exerts an opposing effect on cytokine secretion by Th1 compared to Th2 cells (77). In this study, in vitro derived Th1 and Th2 clones or T cells derived from autoimmune thyroiditis were used to examine the ability of Th1 or Th2 cells to expand and produce cytokines in response to oxidative stress. Results showed that low doses of hydrogen peroxide (H2O2) reduce the interferon-γ (IFN-γ) production of activated Th1 clones and increase IL-4 secretion by activated Th2 clones (Fig. 3). Importantly, another study has shown that mitochondrial ROS can control T-cell activation by regulating IL-2 and IL-4 expression (129). Kaminski et al. (129) report that an oxidative signal originating from the mitochondrial respiratory complex I regulates T-cell activation-induced expression of the cytokines IL-2 and IL-4. Using a complex I inhibitor or small interfering RNA (siRNA)-mediated downregulation of the complex I chaperone NDUFAF1 (NADH dehydrogenase-1 alpha-subcomplex subunit), they demonstrated that TCR-induced ROS production by complex I is crucial for activation-induced IL-2 and IL-4 expression in resting and preactivated human T cells. Moreover, using T cells isolated from patients with atopic dermatitis, these authors suggest that inhibition of complex I-mediated ROS generation blocks disease-associated spontaneous hyperexpression and TCR-induced expression of IL-4. Thus, oxidative stress plays an important role in the pathogenesis of allergic respiratory diseases and can upregulate Th2-driven inflammation, thereby increasing disease severity, bronchial hyper-responsiveness, and airway remodeling. Similarly, a role of superoxide in modulating Th17 and Th1 T cell responses has also been documented (286). This study showed that T-cell polarization was altered when macrophage and T cell superoxide production involving nicotinamide adenine dinucleotide phosphate oxidase (NADPH) oxidase (NOX) was inhibited. A comparison of the cytokine profile after stimulation with immobilized anti-CD3 and anti-CD28 T cells from NOX-deficient mice showed a skewed Th17 phenotype, whereas T cells from NOX-intact mice exhibited the Th1 phenotype. Similar results were observed in vivo where NOX-deficient NOD mice with a Th17 phenotype exhibited concomitant susceptibility to experimental allergic encephalomyelitis (EAE) and significant protection against type 1 diabetes.
FIG. 3.
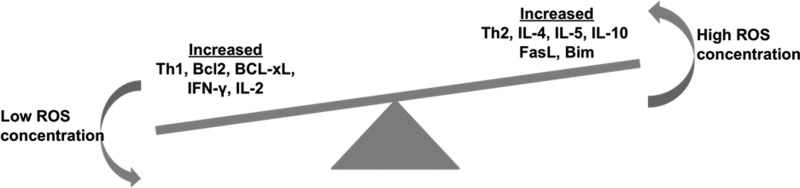
Control of ROS on Th1, Th2 paradigm. Increasing the concentration of ROS suppresses the number of Th1 cells and increases the number of Th2 cells. Moreover, it increases the proapoptotic genes such as FasL and Bim. Lower concentration of ROS/RNS promotes the expression of antiapoptotic genes. RNS, reactive nitrogen species.
Emerging evidence also indicates that the T-cell differentiation state determines T-cell effectiveness in responding to infection and tumor. Determining the differentiation states to characterize effective T cells is important for designing the vaccination protocols of immunotherapy, which primarily target antigen-specific T-cell activation and expansion. The T-cell subsets could be defined by the expression of a set of cell surface molecules, as depicted in Table 1 (248). The difference in phenotype expression is primarily an outcome of the activation status of these T cells, reflecting their ability to secrete cytokines and kill target cells. In the widely accepted classification scheme based on expression of cell surface molecules, central memory T cells (TCM) are antigen-experienced cells that constitutively express CD62L and CCR7, two surface molecules necessary for cellular extravasations in high endothelial venules and migration to the T-cell zones of the peripheral lymph nodes. In contrast, effector memory T cells (TEM) are antigen-experienced T cells that have significantly downregulated these markers, and hence have a propensity to populate peripheral tissues, such as the liver and lung, as well as inflammatory sites. Other important issues with regard to T-cell subsets of varying phenotype and function are their maintenance and the effect of activation-induced cell death (AICD) after re-exposure to an antigen. The extent of cell death of effectors with the memory phenotype may influence the size of the eventual memory T-cell pool. In addition, the ability of T cells to persist in conditions of oxidative stress, in either an infectious disease state or a tumor microenvironment, determines which pool of T cells persists under these conditions.
Table 1.
Phenotype and Function of T-Cell Subsets
| T-cell subset | Phenotype |
|---|---|
| Naïve | CD62L+++ CCR7+++ |
| CD27+++ CD28+++ | |
| CD45RA+++ CD45RO− | |
| KLRG-1−CD57− | |
| TCM | CD62L++CCR7++ |
| CD27++CD28++ | |
| CD45RA−CD45RO+++ | |
| KLRG-1−CD57− | |
| TEM | CD62L−CCR7− |
| CD27++CD28− | |
| CD45RA−CD45RO+++ | |
| KLRG-1++CD57− | |
| Teff | CD62L−CCR7− |
| CD27−CD28− | |
| CD45RA−/+CD45RO++ | |
| KLRG-1+++CD57++ | |
| Treg | CD25+++ |
| FOXP3+++ |
The phenotypic expressions or functional attributes are indicated as low (+), intermediate (++), and high (+++).
RA, rheumatoid arthritis; TCM, central memory T cells; TEM, effector memory T cells; Teff, effector T; Treg, regulatory T cell.
Antigen-specific human CD8+ cytotoxic T lymphocytes (CTLs) with the heterogeneous phenotype (as discussed above) could also be programmed in response to the strength and duration of TCR stimulation. While antigen-mediated repetitive TCR stimulation has been shown to enhance the vaccine efficacy (prime/boost strategies) by increasing the CTL number and function (179), some recent studies have questioned this view (18, 26, 94). In our laboratory, we have characterized the effect of antigen restimulation on T-cell fate in a melanoma epitope MART-127–35-specific self- and influenza epitope MP58–66-specific nonself-reactive CTLs raised from peripheral blood of HLA-A2+ healthy donors as described earlier (183–185). CTLs expanded from T-cell precursors by a single round of stimulation (primary, 1°) or two rounds (secondary, 2°) with cognate peptide-pulsed autologous dendritic cells were compared for T-cell function and death upon restimulation with cognate peptide-loaded surrogate antigen-presenting cells (APCs), T2 cells. As shown in Figure 4, 1° MART-127–35 and MP58–66 CTLs exhibited less translocation of phosphatidylserine (PS) to the outer layer of the CTL membrane than the corresponding 2° CTLs, as measured by annexin V staining. This PS exposure represents an early hallmark in detecting cells undergoing AICD. The data in Figure 4 show that CTLs that encountered antigen once were less sensitive to TCR restimulation-induced cell death as compared to CTLs that encountered antigen twice. This shows that the T cells are intrinsically modulated after each antigen encounter (and similarly by antigen dose, duration of antigen encounter, or cytokines in their microenvironment) that could affect their persistence or function and thus the immune outcome.
FIG. 4.

Cognate antigen stimulation-mediated expansion and cell death in CD8+ T cells. Comparison of AICD in MART-127–35 and MP58–66 epitope-specific primary (1°) and secondary (2°) expanded CTLs. CTL populations were stimulated with control and cognate epitope, and AICD was measured using annexin V staining (histogram). Numbers in the top right corner represent mean fluorescence intensity (MFI) values of annexin V. Percent increase in annexin V+ cells in groups treated with either cognate epitope or control epitope is shown in the middle panel. AICD, activation-induced cell death; CTLs, cytotoxic T lymphocytes.
Furthermore, to ascertain contributing factors that would contribute to increased sensitivity to TCR-mediated cell death in 2° CTLs as opposed to 1° CTLs, we examined the expression of various cell surface markers. A representative example is shown in Figure 5. There was an increase in the percentage of antigen-specific CTLs in the 2° CTLs when compared to the 1° CTLs (upper right quadrant in dot plots). MART-127–35-specific CTLs were tracked by using an MHC class I tetramer reagent, which revealed an increase from 23% to 47%, whereas MP58–66 specific CTLs increased from 44% to 54%. Notably, the phenotype concomitantly drifted toward a TEM-like phenotype, with the loss of CD62L, CD45RA, and CD27 levels and the gain of CD45RO expression. As we have reported earlier (185), the nonself influenza epitope-specific CTLs exhibited a more TEM-like phenotype regardless of the number of antigen stimulations. Thus, while a distinct decrease in CD62L and CD27 expression was observed in MART-127–35-specific CTLs, only a slight decrease in already low levels of expression was noticed in MP58–66 specific 2° CTLs when compared to the 1° CTLs. Nevertheless, antigen restimulation significantly decreased the expression of the naïve cell surface marker CD45RA and increased the expression of the memory marker CD45RO on both MART-127–35- and MP58–66-specific CTLs. Since evidence exists for a role of intracellular thiols in ROS production and of cell surface thiol level in T cells' ability to secrete ROS, we further evaluated whether differences in cell surface phenotypes (thereby reflecting qualitatively distinct T-cell subsets) were correlated with cognate antigen-induced cell death. We characterized the difference in TCR-induced annexin V expression (a marker of early apoptosis) in the CTL population with heterogeneous cell surface expression patterns of markers implicated in defining TCM- versus TEM-like subsets. Antigen-specific CTLs stimulated for 4 h with a cognate or control epitope were stained, in parallel, for a panel of cell surface markers and gated for high or low/negative expression for each of these markers and then evaluated for annexin V expression levels. The CTLs exhibiting a TEM-like phenotype (CD27loCD62LloCCR7loCD45ROhiCD45RAlo) were preferentially sensitive to AICD (stained brighter for annexin V), compared with CTLs with a TCM-like phenotype (CD27hiCD62LhiCCR7hiCD45ROloCD45RAhi) (Fig. 6). Thus, antigen-specific CTLs with a TEM-like phenotype preferentially undergo TCR-mediated AICD compared to CTLs with a TCM-like phenotype. The increased sensitivity of TEM-like T cells to TCR-induced AICD could also be correlated to reduced levels of thiols in CD45RO+ T cells as compared to CD45RA+ T cells, as previously shown in our laboratory (184) (Fig. 6).
FIG. 5.

Antigen restimulation leads to phenotype drift. MART-127–35- and MP58–66 epitope-specific CTLs primed with peptide-pulsed autologous DCs were restimulated and analyzed by flow cytometry after one (1°) or two rounds (2°) of TCR restimulation. Histograms represent the expression of a particular cell surface marker on gated tetramer positive CD8+ T cells. Numbers on the right corner represent MFI values. Data shown in (A) and (B) are from one of three experiments with similar results.
FIG. 6.

Evaluation of the relative sensitivity to AICD in phenotypically different T-cell subsets. After restimulation of the CTLs with cognate epitope, death of specific CTL subpopulations was measured by gating on high and low expression levels of each of the cell surface markers related to T-cell memory. Histograms represent the level of annexin V expression in CTL populations, which are gated for low (Lo) or high (Hi) expression level of the respective cell surface marker. Data shown are from one of two experiments with similar results. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Earlier studies have also shown that human peripheral blood mononuclear cells (PBMCs) stimulated with either HLA-A2-restricted influenza peptide or nonspecific receptor cross-linking, followed by treatment with micromolar (μM) concentrations of H2O2, produce reduced amounts of cytokines (172). This reduction in Th1 cytokines was observed in the memory/effector (CD45RO+) T-cell subset and correlated with a block in nuclear factor kappa B (NFκB) activation. Takahashi et al.(277) have shown that the sensitivity of T-cell subsets of unstimulated human PBMCs to cell death was further investigated by inducing the T cells with low doses of H2O2. This study demonstrated that TEM (CCR7−CD45RA−) cells were particularly sensitive to low doses of H2O2, whereas TCM (CCR7+CD45RA−) are significantly less sensitive (277). Time–kinetic experiments, in which the mitochondrial membrane potential and caspase 3 activities were analyzed, suggested that the mitochondrial pathway is the primary cell death pathway for CD45RO+ T cells when exposed to low levels of H2O2. However, in our laboratory, we reasoned that for an oxidative agent, such as H2O2, to differentially induce cell death, either reduced or oxidized surface molecules must regulate susceptibility to H2O2-mediated apoptosis. Because recent studies have implicated reduced thiols (cysteine–SH) in the function of individual cell surface proteins (81, 245), we evaluated the level of cell surface thiols (cs-SH) on CD45RO+ and CD45RA+ T-cell subsets using a fluorochrome-conjugated maleimide dye, as previously described (245). Our data suggest that CD8+ T cells with a CD45RA+ phenotype express more cs-SH as compared to the T-cell subset with a CD45RO+ phenotype (186). It has also been documented that the overall cs-SH content on cell surface molecules could be correlated to the level of intracellular glutathione (iGSH) (246). Measuring iGSH on CD8+ T cells using the monochlorobimane-staining method (75) revealed higher expression of this key antioxidant molecule in a naïve CD45RAhigh T-cell subset as compared to the antigen-experienced CD45RAlow T-cell subsets (Fig. 7). This finding accords with our data that show that T cells lose thiol expression after proliferation on repeated TCR stimulation. Recent studies have implicated that the levels of cs-SH can be manipulated in vitro by altering the levels of intracellular iGSH or γ-glutamylcysteinylglycine (187). Intracellular GSH is the most prevalent intracellular thiol ubiquitous to all cell types whose function is to maintain the cellular redox state. Other studies have also suggested that a lower capacity to produce ROS is associated with an increased number of reduced thiol groups (–SH) on T-cell membrane surfaces (186). Artificially increasing the number of reduced thiols on T cells either by culturing with reducing agents, such as β–mercaptoethanol (309), or by using N-acetylcysteine (L-NAC), a thiol antioxidant and precursor of GSH, lowered the threshold for T-cell reactivity and enhanced proliferative responses in vitro. Experiments in our laboratory have shown that pretreatment of both tumor epitope-specific primary and TCR-transduced T cells with the thiol donor L-NAC increased cellular thiols and rescued T cells from TCR stimulation-mediated AICD without impairing their functional capacity (Fig. 8). These results suggest that T-cell expansion upon cognate antigen encounter affects the cell surface marker expression, replicative history, and the CTL quality by decreasing its antioxidant capacity.
FIG. 7.

Activated CD8+ T cells were analyzed for expression of iGSH using monochlorobimane (mBCl). Briefly, cells were preloaded for 15 min with 10-μM mBCl, which forms blue fluorescent adducts with iGSH. Immediately before flow cytometry analysis, propidium iodide was added. For mBCl, cells were excited with a violet (UV) 405-nm laser, and emission was acquired with a 440/40 filter. Changes in the iGSH are reflected by the appearance of populations of cells with differences in mBCl fluorescence. Histogram represents the level of fluorescence from stained samples that were gated for high or low cell surface expression of CD45RA. CD45RA is primarily expressed on naïve T cells (CD45RAhi) and is lost after TCR stimulation (CD45RAlo). Data shown are from one of three experiments with similar results. iGSH, intracellular glutathione; RA, rheumatoid arthritis.
FIG. 8.

Susceptibility of melanoma epitope-reactive T cells to AICD and its rescue by increasing thiols. (A) MART-127–35 epitope-reactive CTLs were stimulated with a control or cognate epitope (1 (g/ml) or first preincubated with 5 mM of L-NAC for 30 min and then stimulated with their cognate epitope. Induction of AICD in CTL was evaluated 4 h later by staining for tetramer, anti-CD8 mAb, and annexin V. Histograms depict annexin V expression on CD8+, tetramer+ CTLs under the indicated conditions. gp-100 (control epitope); MART-127–35 (cognate epitope). (B) CD8+ T cells transduced with TIL1383I TCR were either untreated or pretreated with 5 mM of L-NAC for 30 min before stimulated with cognate tyrosinase peptide or control MART-1 peptide-pulsed T2 cells and stained with anti-CD8, CD34 mAbs, and annexin V. Percent increase in annexin V-positive cells upon stimulation with cognate peptide over annexin V-positive cells in culture stimulated with a control peptide is depicted. Data shown in (A) and (B) are from one of three representative experiments. NAC, N-acetylcysteine.
In addition, different T-cell phenotypes have differential susceptibility to oxidative stress-induced apoptosis, and this susceptibility is associated with cell surface thiol expression and iGSH. The data discussed above suggest that increased levels of reduced thiol groups and iGSH in a T-cell subset could be responsible for its increased ability to persist in a tumor-induced oxidative stress microenvironment. Our laboratory is currently exploring whether this differential susceptibility to oxidative stress between T-cell subsets correlates with differences in expression levels of genes involved in oxidative stress/ROS metabolism or can be modulated by factors that play an important role in T-cell development and maintenance (such as cytokines, APC, antigen dose, and Toll-like receptors). Our recent data show that IL-15 increases the level of cell surface thiols and the overall antioxidant capacity of CD8+ T cells (138). We also observed that CD8+ T cells cultured in IL-15 result in an increased mitochondrial mass as compared to T cells cultured in IL-2 (Fig. 9A). The increase in mitochondrial mass signifies a higher mitochondrial copy number in the T cells when cultured in the presence of IL-15. Since the mitochondrion is a key regulator of the metabolic activity of the cell and is an important organelle in both production and degradation of free radicals, an increase in higher mitochondrial mass supports our published data that T cells cultured in the presence of IL-15 showed higher ROS and RNS secretion (138). Since mitochondria are produced from the transcription and translation of genes both in the nuclear genome and in the mitochondrial genome, we further evaluated the genes encoded by the mitochondrial genome that are mostly involved in the electron transport chain mitochondrial cytochrome B (MT-CYB), mitochondrial cyclooxygenase 2 (MT-COX2); NDUFA; (NADH dehydrogenase [ubiquinone]-1 alpha-subcomplex subunit). Our data show that cells cultured in the presence of IL-15 have more expression of these mitochondrion-associated genes (Fig. 9B) (24). If other cytokines that also influence the T-cell phenotype also modulate mitochondrial biogenesis is currently under investigation in our laboratory. Our preliminary data evaluating the effect of IL-12-, IL-21-, and IL-17-secreting T cells also show that the antioxidant capacity of T cells is modulated in the presence of these cytokines (unpublished data). Thus, it is evident that cytokines do have the capacity to modulate the redox status of T cells. Moreover, T cells with a different phenotype have differential susceptibility to oxidative stress based on their redox status. This is further supported by the recent observation that CD8+ memory T cells, but not CD8+ effector T (Teff) cells, possessed substantial mitochondrial spare respiratory capacity (SRC), and SRC is a critical regulator of CD8+ T-cell memory development (285). Mitochondrial SRC is the extra capacity available in cells to produce energy in response to increased stress or work and is associated with cellular survival. Thus, understanding the T-cell phenotype and function should hold importance in modulating a disease state that involves T cells. While increasing the antioxidant capacity by thiol donors prevented AICD, a similar strategy may be used to increase the persistence of T cells or modulate the T-cell phenotype (effector vs. memory) by modulating mitochondrial function. Overall, these results show how cytokines control the T-cell phenotype and bioenergetic stability of T cells, and that regulating T cell mitochondrial metabolism may be key in controlling pathogenic disease states (290).
FIG. 9.

Effect of IL-15 on mitochondrial biogenesis. (A) T cells were cultured in presence of IL-2 (10 ng/ml) and IL-15 (10 ng/ml) for 15 days. Mitochondrial mass was determined by staining the cells with a MitoTracker dye. (B) T cells from two different healthy donors were cultured in presence of IL-2 and IL-15, and expression of mitochondrial genes was determined by real-time PCR.
In addition to Teff cells with different phenotypes, naturally occurring regulatory T cells (Tregs) with a strong immunosuppressive capacity and known to play a critical role in the T-cell homeostasis exhibit reduced sensitivity to oxidative stress-induced cell death while maintaining their suppressive function (279, 293). Human Tregs have several defense mechanisms to counteract oxidative stress. Tregs express high levels of cell surface thiols, which are important reducing agents, as well as increased intracellular antioxidative capacity (195). In addition, Tregs express and secrete high levels of thioredoxin-1 (TRX-1), a major antioxidative molecule (196) that is required for cell surface thiol expression and decreases the susceptibility of Tregs to oxidative stress-induced apoptosis. This enhanced antioxidative capacity of Tregs possibly serves as a feedback inhibition mechanism during inflammation and prevents uncontrolled immune reactions by favoring survival of suppressor rather than effector cells. Moreover, Tregs have been shown to interfere with GSH metabolism in dendritic cells and T cells (314), thereby allowing Tregs to survive oxidative stress signals by diminishing GSH synthesis via decreased expression of γ-glutamylcysteine synthetase, the rate-limiting enzyme for GSH synthesis. Tregs also consume extracellular cysteine and partition it more proficiently to the oxidation product, whereas Teff cells divert more of the cysteine pool toward protein and GSH synthesis (314). Initially, a higher concentration of GSH is seen in the nucleus during cell proliferation; later, GSH is redistributed to the cytoplasm. Tregs block the redistribution of GSH in Teff cells. Together, these data suggest that Tregs can modulate sulfur-based redox metabolism, and as a result of this modulation, T-cell activation and proliferation are suppressed. Recently, the importance of macrophage-produced ROS in inducing Tregs has also been highlighted (149). Based on the results of this study, the induction and immunosuppressive function of Tregs are dependent on ROS, and ROS deficiency may lead to decreased Treg induction and hampered T-cell suppression. This study demonstrates that macrophages significantly express both gp91phox and p47phox proteins, more than DCs, and are better in producing ROS and inducing Tregs. Coculturing of CD4+CD25− (Th cells) cells with macrophages and anti-CD3/CD28 demonstrated induction of Tregs (increase in the number of iTregs- CD4+CD25+FoxP3+), which was attributed to increased ROS secretion, since coculture of the same cells in the presence of apocynin (a NOX inhibitor) lowered the number of iTregs and their proliferation. Thus, these findings together highlight the importance of the redox-regulating molecules in skewing the T-cell subset, which in turn could result in modulating the immune status of an individual. In the following section, we further discuss important redox-regulating molecules that could have implications in T-cell response.
IV. T Cell and Redox Regulators
It is well known that TCR activation triggers a number of signaling cascades. Interestingly, an early event in TCR activation activates lymphocyte protein-tyrosine kinase (Lck) by the TCR/CD3 complex. This is followed by recruitment of ζ-chain-associated protein kinase (Zap-70), which further amplifies the response and leads to the activation of multiple pathways, including ERK, mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK). These pathways ultimately determine the cell fate for differentiation and proliferation (36, 160). In addition, the cellular concentration of ROS can also lead to selective activation of several transcription factors such as NFκB, p53, and AP-1. These transcription factors determine the fate of cells, survival versus death. The following section focuses briefly on the roles of reactive molecules in modulation/regulation of the various signaling molecules (Fig. 10).
FIG. 10.

Redox regulation of signaling pathway leading to T-cell activation/proliferation. ROS are required for trigering different signaling pathways, which are required for proper activation and maturation of T cells. MAPK are known to activate Linker of activated T cells (LAT), which is responsible for TCR engagement. ERK-MAPK forms a negative feedback loop and represses LAT. NrF2 is required for activation of DCs. NFκB is required for maturation of APCs. FoxO1 is responsible for maintenance of naïve T cells by regulating L-selectin (CD62L) and CCR7. p38-MAPK is required for T-cell activation. All the above signaling pathways require ROS for activation. APCs, antigen-presenting cells; ERK, extracellular signal-regulated kinases; FoxO, forkhead transcription factors of class O; MAPK, mitogen-activated protein kinase; NFκB, nuclear factor kappa B; NRF2, nuclear factor erythroid-2-related factor 2.
A. Transcription factors
1. Nuclear factor κB
NFκB is a dimeric transcription factor that is involved in the vital signaling pathway for the activation of proinflammatory cytokines, chemokines, cellular proliferation, and mediators of apoptosis (284). NFκB is also important for the maturation and survival of T lymphocytes, as they are activated upon TCR activation (130). Several studies show that the redox state of a cell can regulate NFκB, which can in turn affect the generation of a robust T-cell immune response (273). NFκB is also shown to be constitutively expressed in acute myeloid leukemia (AML) cells, suggesting that NFκB acts as an antiapoptotic mediator in AML cells. Therefore, when NFκB activation is blocked with pharmacological agents, AML cells undergo targeted apoptosis (76). A study in cardiac dysfunction in type II diabetes demonstrated that enhanced activity of NFκB leads to increased ROS, ONOO−, gp91phox, and NOX1 expression. Therefore, these studies demonstrate that ROS-mediated activation of transcription factor NFκB is a central regulator of T-cell immunity, inflammation, and cell survival.
2. p53. p53, one of the most important tumor suppressor proteins, is vital for protecting cells in oxidative stress by maintaining genomic integrity
DNA damage is the most common outcome in cells after oxidative stress. p53 arrests proliferation of such cells and triggers signals to undergo apoptosis to overcome DNA damage (132, 180, 225). However, studies reveal that p53 can protect against DNA damage from ROS-induced oxidation by upregulating several antioxidant genes such as glutathione peroxidase 1 (GPX1) (243). This study illustrates that p53−/−-deficient mice develop lymphomas by 6 months of age, whereas the p53−/− mice supplemented with the antioxidant L-NAC diet were significantly protected from developing lymphomas. While a direct crosstalk between p53 signaling and ROS production is not clearly understood, it has been shown that T cells require low concentrations of ROS for proper activation and signaling. However, accumulating ROS may lead to activation of p53 and p53-inducible genes, leading to apoptosis (225). Additionally, high levels of ROS may result in the phosphorylation and stabilization of p53 and subsequent activation of apoptotic signals as seen in other biological systems and diseases (283). In vitro and in vivo studies using p53−/− mice suggest that p53 is important in regulating apoptotic signals in T cells (266). Based on recent evidence, p53 is a key regulator of T-cell maturation and of the differentiation of α/β T cells in the thymus (210).
Moreover, p53 plays a significant role in controlling the cell cycle at the mitotic phase of antigen-specific proliferating T cells (17). Interestingly, infiltration of immune cells, specifically the tumor-infiltrating T cells (TILs), Tregs, and tumor-associated macrophages, was associated with p53 mutations in ovarian cancer patients (259). Based on recent DNA microarray studies, Desaint et al. (60) showed that 42 differentially expressed genes were identified in cells in response to H2O2 treatment. Interestingly, 17 of the 42 genes (∼40%) were targets of p53 (60), demonstrating a role for oxidative stress regulation in p53-mediated immune responses. Polyak et al. have shown that p53 can induce the expression of different genes such as proline oxidase, which upregulates ROS production in cancer cells (225, 239). The tumor suppresor p53 has also been shown to regulate mitochondrial ROS production by altering the expression of the mitochondrial antioxidant enzyme, manganese superoxide dismutase (MnSOD) (109, 240). Therefore, an impaired communication between the mitochondria, p53, and the nucleus is implicated in several disease conditions. Taken together, p53 is important in regulating T-cell maturation and proliferation, but the p53-induced expression of mitochondrial ROS in primary T cells warrants further research.
3. Forkhead transcription factors of class O
Forkhead transcription factors are well known for their role in maintaining immune system function (39, 212). Recent data have highlighted the role of the forkhead transcription factors of class O (FoxO) in oxidative stress and in differentially regulating persistence of T-cell subsets (49). This study reports that the Mst1-FoxO-signaling pathway is important in maintaining the survival of naïve T cells by increasing the expression of downstream antioxidant molecules, such as SOD2 and catalase. As a result of the increase in FoxO-mediated expression of SOD2 and catalase, T cells are resistant to oxidative-stress-induced apoptosis.
Riou et al. also demonstrated that survival of ex vivo isolated TCM is increased, because they are more resistant to both spontaneous and Fas-induced apoptosis than cells with the TEM phenotype, resulting in their increased persistence in vitro (238). Moreover, this study reports that ex vivo TCM express increased levels of the transcriptionally inactive form of FoxO3a (phosphorylated FoxO3a) and decreased levels of the downstream proapoptotic molecule, Bim. Recent studies have also demonstrated that TCM are less sensitive to H2O2 in comparison to TEM (277). Therefore, these results indicate a role of FoxO3a in redox regulation of T cells and protection from apoptosis. This type of protective role of FoxO3a is also implicated during human immunodeficiency virus (HIV) infection, where FoxO3a regulates the survival of central memory CD4+ T cells during HIV infection (291). Furthermore, FoxO1 and FoxO3a have been shown to regulate the transcription of p27Kip1, Bim, and L-selectin in response to IL-2 in murine and human T lymphocytes (72, 270). Interestingly, Kerdiles et al. have demonstrated that FoxO1 regulates the development of functional Tregs, and that FoxO1 is required for survival and homing of naïve T cells to secondary lymphoid organs (139).
In a recent study, FoxOs were reported to be critical in regulating cell cycle, in inhibition to apoptosis, and mediators of resistance to physiologic oxidative stress in hematopoietic stem cells (HSCs) (282). This is an important finding, since the results from this study show that there was a marked increase of ROS in FoxO-deficient HSCs compared to wild-type (WT) HSCs, which correlates with changes in the expression of ROS-regulating genes. The importance of the FoxO-signaling pathway in cellular tolerance to increased intracellular ROS and maintenance of peripheral naïve T-cell homeostasis is also substantiated in other studies (49). Thus, FoxO proteins play an essential role in regulating physiologic oxidative stress, and thereby promote enhanced survival of myeloid and lymphoid cells in response to ROS.
4. Nuclear factor erythroid-2-related factor 2
The nuclear factor erythroid-2-related factor 2 (Nrf2) transcription factor regulates the basal and inducible expression of a wide array of antioxidant genes. In a recent study, regulation of the cellular redox level and Th1/Th2 balance was shown to protect against the development of pulmonary fibrosis in a preclinical model of pulmonary fibrosis where C57BL/6 mice and Nrf2-deficient mice were administered bleomycin intratracheally (140). Similarly, the absence of Nrf2 has been shown to exacerbate experimental autoimmune encephalomyelitis, suggesting that activated Nrf2 attenuates autoimmunity and proinflammatory conditions (126). Comparisons of disease severity in WT and Nrf2 knockout mice after immunization with myelin oligodendrocyte glycoprotein (MOG) 35–55 revealed that the clinical progression of the disease is marked by rapid onset and increased severity in the Nrf2 knockout mice. In addition, in the Nrf2-deficient mice, increased immune cell infiltration and glial cell activation in the spine were observed in conjunction with increased levels of expression of inflammatory enzymes (inducible NO synthase [iNOS], phox-47, gp91-phox, and phox-67), cytokines (IFN-γ, IL1-β, tumor necrosis factor [TNF]-α, and IL-12), and chemokines (B lymphocyte chemoattractant [BLC] and monokine induced by IFN-γ [MIG]). These results further substantiate that Nrf2 can modulate an autoimmune neuroinflammatory response by regulating proinflammatory enzymes, the pro-oxidant enzymes.
Nrf2 can also modulate the T-cell response by regulating oxidative stress-induced activation of dendritic cells. A recent study suggested that disruption of the transcription factor Nrf2 in dendritic cells leads to increased oxidative stress, and in response to allergic, airborne pollutants, a Th2-like immune response is elicited in these Nrf2-deficient dendritic cells (305). Nrf2 deficiency has been further implicated in the decline of Th1 immune response in the course of aging (141). Moreover, the observed decrease in Th1 immunity in aged mice could be restored by administering either an Nrf2 agonist, sulforaphane, or a thiol precursor, L-NAC. Mechanistic studies using Nrf2-deficient T cells suggest that Nrf2 deficiency enhances the sensitivity to Fas-mediated apoptosis by regulating the intracellular levels of GSH, a major intracellular antioxidant (194). Taken together, these studies suggest that Nrf2 regulates T-cell immunity by fine-tuning the redox equilibrium.
B. Mitogen-activated protein kinases
1. c-Jun N-terminal kinase
Recent research has suggested that ROS and RNS act as signaling molecules in the activation of JNK, commonly known as stress-activated protein kinase, leading to apoptosis (262). While it is known that CD3 and CD28 co-stimulation is required for JNK activation, IL-2 production (64), and IL-2 gene transcription in Jurkat cells (human CD4+ T cell line) (184), recent studies have demonstrated that JNK signaling is important for Teff cell function, but not for naïve T-cell activation (64). Furthermore, reports by Dong et al. showed that T cells isolated from JNK1 and JNK2 knockout mice can be stimulated to produce IL-2, but they cannot functionally differentiate into Th1 and Th2 lineages (63, 315). Pani et al. also demonstrated that treatment of T cells with the mitogen concanavalin-A (ConA) upregulates ROS and JNK, whereas use of L-NAC (a ROS scavenger) inhibits ConA-stimulated thymocyte proliferation and JNK upregulation (216). Therefore, JNK may play an important role in redox signaling.
Earlier studies from our group have also shown that T cells reactive to melanoma epitope MART-127–35 and influenza matrix protein MP58–66 have increased JNK phosphorylation when reactivated with a cognate epitope (207). Moreover, this upregulation in JNK phosphorylation is inhibited when T cells are pretreated with a superoxide dismutase (SOD) mimetic Mn (III) tetrakis (5,10,15, 20-benzoic acid) porphyrin (MnTBAP). Inhibition of ROS also rescues these antigen-specific T cells from TCR restimulation-induced cell death, suggesting that ROS inhibitors prevent AICD in primary CTLs by blocking JNK activation. Thus, JNK has a crucial role in the signaling cascade leading to T-cell activation and proliferation in response to immunogenic stimuli.
2. Extracellular signal-regulated kinases
It is known that extracellular signal-regulated kinase (ERK) signaling plays an important role in the early phase of naïve T-cell activation and positive selection of T cells (1, 55). Furthermore, MAPK has been identified as a critical regulator of CD95/Fas-mediated apoptosis in T cells (110). CD95/Fas-mediated apoptosis is important in the negative selection of T-cell development and in later stages of T-cell activation when activated T cells are no longer needed and are now triggered to undergo CD95/Fas-mediated AICD. Koike et al. have reported that ERK activation plays a critical role at late stages (after 2 h of activation) of T-cell activation, and that ERK2 has specific functions in the activation, proliferation, and survival of CD8+ T cells (146). As suggested by D'Souza et al., ERK2 is required for proliferation of activated CD8+ T cells without co-stimulation and is responsible for increased T-cell survival (55).
Using a Zap-70-deficient Jurkat T cell line (P116) in their studies, Griffith et al. found that Zap-70 is required for the H2O2-induced activation of ERK in T cells (90). However, they also reported a Zap-70-independent pathway for activation of ERK. TCR activation of T cells has also been shown to activate ERK within 15 min, and moreover, low concentrations of H2O2 may be required for proper ERK activation (61). Thus, low concentrations of ROS may positively regulate ERK activation, and higher concentrations may negatively affect ERK signaling in T cells.
3. p38-MAPK
The p38-MAPK kinase pathway is activated in response to numerous cellular stress stimuli and cytokines. Known for its ability to induce apoptosis, p38-MAPK can regulate the cells' survival signals in response to external stimuli. For example, cellular stress such as DNA damage can elicit a survival signal where p38-MAPK can regulate the G2/M and G1/S cell cycle checkpoints (280). In response to treatment with UV radiation or a DNA-damaging agent (8-MOP), the p38-MAPK pathway was activated in Jurkat T cells, suggesting that p38-MAPK is important for T-cell survival (38). Several studies also show that the p38-MAPK pathway is responsible for differentiation of naïve CD4+ T cells into effector cells. It may also be responsible for Th17 subset differentiation (208). Moreover, it is also required for the production of IFN-γ from CD8+ T cells (237). One study has suggested that T cells infected with Vibrio vulnificus undergo cytotoxic cell death by a p38-mediated production of ROS (143). Jurkat T cells that are exposed to a pathogenic bacterium, V. vulnificus, require NOX, which induces the production of ROS, thereby leading to the death of the infected T cells. This substantiates the role of p38-MAPK in ROS production in infected T cells.
V. Key Molecules in Redox Regulation
To protect against oxidative damage, cells have developed an antioxidant defense mechanism that maintains cellular redox homeostasis. Many antioxidant molecules have been identified to regulate the redox status of a cell and play a major role in regulating immunological functions (Table 2). Under conditions of endogenous or exogenous oxidative stress, the altered expression of these molecules affects cellular function. We briefly introduce these molecules with reference to T cells in the following section and further discuss their role in specific diseases in section VII.
Table 2.
T-Cell and Role of Antioxidant Molecules
| No. | Antioxidants | Protein name | Antioxidant role in T-cell regulation | Reference |
|---|---|---|---|---|
| 1 | GSH | Glutathione | Tregs block GSH redistribution from the nucleus to the cytoplasm in Teff cells | (314) |
| 2 | GST | s-S-Transferase | Activator protein-1 induction by GST gene expression alters intracellular oxidative stress | (224) |
| 3 | GCLC | Glutamate–cysteine ligase catalytic subunit | Mitogen activation leads to: Upregulated GCLC catalytic subunit Depletion of GSH |
(300) |
| 4 | GCLM | Glutamate–cysteine ligase, modifier subunit | Mitogen activation leads to increased GCLM catalytic subunit | (300) |
| 5 | GLUT1 | Glucose transporter 1 | Induced upon TCR activation | (274) |
| 6 | TRX | Thioredoxin | Overexpression blocks NFκB activation by H2O2. Induces IL-2R expression on T cells |
(148) |
NFκB, nuclear factor kappa B; TCR, T-cell receptor; Treg, regulatory T cell.
A. Antioxidant/redox proteins
1. Superoxide dismutase
SOD may protect cells from oxidative damage by catalyzing the dismutation of two molecules of superoxide anion into water and H2O2. SOD has three known isoforms: SOD1, SOD2, and SOD3. While copper/zinc SOD (Cu/ZnSOD or SOD1) is present in the cytosolic fraction, manganese superoxide dismutase (MnSOD or SOD2) is located in the mitochondrial fraction, and SOD3 is extracellular (101). Based on studies using transgenic mice, mutations in the SOD1 gene have been shown to be associated with the familial form of amyotrophic lateral sclerosis (ALS), suggesting that SOD1 is important in preventing oxidative-stress-associated diseases (121). In contrast, using a tetracycline-inducible system, Zou et al. were able to show that the extracellular form of SOD (SOD3) is important in preventing oxidative-stress-induced diseases and injuries (327). In this section, we will focus on the role of SOD2 in protecting cells from oxidative-stress-induced damage.
SOD2 functions primarily to protect mitochondrial components from superoxide anions, which are liberated as a normal byproduct of respiration. Mitochondrial Complex I and Complex III are estimated to produce superoxide anions from about 1%–5% of oxygen consumed as a consequence of normal respiration. MnSOD is the cell's primary defense mechanism against free radical-mediated damage and is important in regulating oxidative and apoptotic signals (217). Another study has suggested that overexpression of the human MnSOD transgene in hematopoietic progenitor cell line 32D cl3 cells and other cell lines results in stabilization of the mitochondria and reduction in radiation-, TNF-α-, or cytokine withdrawal-induced cell death (70). Thus, MnSOD stabilizes the mitochondrial membrane and reduces apoptosis. The experimental data from our laboratory also show that preincubation of CTLs with an antioxidant SOD mimetic, MnTBAP, protected CTLs from AICD (207). Of note, we have found that several other antioxidants (MnTPyP, Tyron, and D1417) also protect the CTLs from AICD.
Furthermore, based on the results of a study using a T-cell-specific manganese SOD2 conditional knockout mouse, SOD2-KO leads to increased O2•− production, apoptosis, and developmental defects in the T-cell population, resulting in immunodeficiency and susceptibility to the influenza A virus H1N1 infection (40). This phenotype was rescued with mitochondrial-targeted superoxide-scavenging drugs. These findings demonstrate that the loss of regulated mitochondrial superoxide production leads to aberrant T-cell development and function, and further suggests that manipulations of mitochondrial superoxide may significantly alter clinical outcomes resulting from viral infections.
2. Glutathione peroxidase
GPX is an important group of antioxidants responsible for reducing peroxides. Their importance arises from their ability to protect cells from ROS-induced damage to lipids, proteins, and nucleic acids (226). This type of protective effect is seen in vitro and in vivo, where GPX1 is responsible for reduction of ROS production in cells after TCR activation and also protects cells from ROS-induced apoptosis (306). A recent study demonstrated that GPX1-deficient CD4+ T cells produced more intracellular ROS and IL-2 than WT cells. Moreover, the study also showed that the GPX1-deficient CD4+ cells have a greater proliferation rate than the WT cells. In support of the pivotal role of GPX in protecting cells from ROS-induced apoptosis, studies using an 8E5 HIV-infected human T-cell line have suggested that these cells are more susceptible to H2O2-induced apoptosis as a result of catalase and GPX deficiency (306). GPX1-deficient cells exhibit Th1 phenotype when exposed to TGF-β and IL-6, anti-IL-4 antibody, and anti-IFN-γ antibody, therefore suppressing the Th2 and Th17 phenotype of these cytokine-treated cells. Thus, these results show that deletion of GPX1 skews the phenotype of T cells toward the Th1 phenotype by suppressing Th2 subset development. Therefore, GPX1 is important not only for reducing intracellular ROS accumulation but also for regulating Th1 cell proliferation and differentiation.
3. NADPH oxidase
NOX, a membrane-bound enzyme complex made up of six subunits, is a major source of ROS that participates in signal transduction of nonphagocytic cells. Recent studies have demonstrated the important role of NOX in shaping Th responses and as a signaling intermediate to modulate Th17 and Th1 cell responses (286). Upon CD3 and CD28 activation, T cells from NOX-deficient mice were primarily of the Th17 phenotype, whereas NOX-intact cells differentiated into the Th1 phenotype. Corroborating evidence is seen in a study by Purushothaman et al., where mice deficient in the NOX catalytic subunit gp91(phox) demonstrated gp91's role as an important regulator of T-cell function (228).
Briefly, IL-2 withdrawal results in a significantly reduced level of apoptotic cell death of gp91(phox)-deficient T (T−/−) cells as compared to the WT T cells (T+/+). That is, gp91 (phox)-deficient T cells are resistant to apoptosis. Furthermore, activated T−/− cells displayed improved survival after activation by superantigens in vivo, where in vitro activated T−/− cells were adoptively transferred into congenic hosts. Thus, NOX is an important regulator of adaptive immunity. Mechanistic studies have shown that NOX-derived ROS direct Treg-mediated suppression of CD4+ Teff cells, a process that is subject to inhibition by thiol-containing antioxidants, NOX inhibitors, or neutrophil cytosolic factor 1 (Ncf1) (p47phox) -deficient Tregs and Teff (68). Thus, NOX play an important role in T-cell susceptibility to ROS and modulate T-cell function.
4. Catalase
Catalase is an important antioxidant enzyme responsible for conversion of H2O2 to H2O and O2. The antioxidant and protective functions of catalase in T-cell lines (CCR-CEM acute lymphoblastic leukemia [ALL] cell lines) have been previously demonstrated (252). Recently, catalase has been identified as a critical factor for maintaining activated T cells at high density during ex vivo expansion of T cells in adoptive T-cell immunotherapy (168). This study highlighted the importance of cell density in T-cell activation, proliferation, survival, and apoptosis in ex vivo cultures. In addition, the study implicated a role for increased catalase secretion in high-cell-density culture supernatants that were able to rescue oxidative stress-mediated, activated T-cell death. The importance of catalase in protecting T cells from ROS was further substantiated in a recent study where T cells transduced with catalase, using a retroviral vector, enhanced the survival of CD4+ T cells under oxidative stress (11). Taken together, these results show that catalase is an important modulator of T-cell-mediated immunity.
5. Glutathione and cell surface thiol
GSH, a nonprotein thiol, acts as an antioxidant and prevents cellular damage from ROS. The synthesis of GSH in cells may be disrupted during aging and under pathologic conditions such as diabetes mellitus (251) and cystic fibrosis (37). Apoptotic stimuli result in the decrease of cellular GSH, generation of ROS, and RNS, and is important in downstream signaling involving the Fas ligand (75). Maintaining optimal GSH levels in the cellular microenvironment is critical, as its depletion may lead to the progression of certain diseases, including HIV (246) and arthritis (81, 116). The GSH level has been shown to decrease as HIV disease progresses, and low GSH in subjects with advanced HIV disease predicts poor survival and impaired T-cell function. These studies suggest that the decreased production of ROS is associated with an increased number of reduced thiol groups on the T-cell surface, thereby modifying T-cell function in an immune response.
Interestingly, our data show that the TEM subset (known to be more sensitive to AICD or apoptosis) expressed less cs-SH, while the T-cell subset with TCM phenotype (relatively resistant to AICD or apoptosis) expressed more cs-SH (unpublished observation). Furthermore, a decrease in the percentage of T cells with reduced thiols is noticeable after treatment with increasing dose of H2O2 along with a concomitant decrease in the number of viable cells, as seen in the scatter plot (Fig. 11). These data suggest that the high cell surface thiol-bearing cells would survive better in an oxidative microenvironment, and thiol levels on T cells could play an important role in T-cell persistence and immune outcomes.
FIG. 11.

Reduction in cell surface thiols in cells undergoing H2O2-induced apoptosis. Human T cells were exposed to different concentrations of hydrogen peroxide (H2O2), and cell surface thiols were measured using ALM-633 dye (Invitrogen) by flow cytometry. Briefly, cells were incubated with 5 μM ALM-633 for 15 min on ice. Cells were washed with cold phosphate buffered saline, labeled with antibodies against cell surface markers, and analyzed by flow cytometry. Left panel shows forward- and side-scatter plot with numerical values representing the percent of gated live cells. Right panel shows the cell surface thiols on gated cells.
6. Thioredoxin
TRXs are a large family of proteins with cysteine-containing redox-active centers that are important mediators of scavenging H2O2 (235). TRX has been shown to protect lymphoid cells from apoptosis by its peroxide-scavenging activity (120). In a recent study, 28 children with T-cell acute lymphoblastic leukemia (T-ALL) were evaluated for total lymphocyte count, and the results suggested that a higher lymphocyte count is associated with higher level of TRX expression in T-ALL patients. Moreover, exogenous treatment with TRX increased proliferation of lymphocytes in vitro, suggesting that TRX is important in T-cell proliferation (261). Our recent work shows that CD8+ cells cultured in presence of IL-15 increase ROS and TRX1, therefore increasing T-cell survival (138). Another study illustrated that Tregs produce higher amounts of TRX, which may result in increased tolerance to oxidative stress (196). Therefore, TRX expression in response to oxidative stress is important for T-cell function and survival.
7. Vitamins
Vitamins are important nonenzymatic antioxidants that play an important role in immunological responses (Table 3). A recent report suggests that vitamins play an important role not only in normal growth but also in strengthening immunological responses (170). For example, β-carotene, a precursor of Vitamin A, is known to enhance production of IL-2 and IFN-γ in T lymphocytes in an in vivo model system (312). Vitamin A and D are required for proper homing of T cells to the gut and skin, respectively (182). DCs in the gut-associated lymphoid organs produce enzymes to convert Vitamin A to retinoic acid, which induces the surface expression of the T-cell gut-homing receptor, CCR9 (119). However, retinoic acid suppressed the expression of the skin-homing receptors, CCR4, E-lig, and P-lig. Similarly, DCs in the skin draining lymph nodes have the ability to convert Vitamin D to 1,25(OH)2D3, where 1,25(OH)2D3 is known to induce the expression of the skin-homing receptor, CCR10 (264). Interestingly, Vitamin D and the Vitamin D receptor (VDR) are important for a TCR response to antigen stimulation. VDR is highly expressed on active T cells, whereas naïve T cells do not express VDR (298). Induction of VDR expression and phospholipase C isoenzyme is required for T-cell signaling and activation. Vitamin D deficiency has been shown to increase Th17 and Th9 cells and reduce the Treg population, thereby promoting autoimmune diseases, such as multiple sclerosis (MS), type I diabetes, systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) (44, 178, 215). Taken together, Vitamin A and D are important for regulating T-cell-mediated immunity in the gut and skin.
Table 3.
Antioxidant Compounds of Natural Dietary Products with Role in T Cell Function
| No. | Key antioxidants | Food products | Antioxidant role | T cell function | Reference |
|---|---|---|---|---|---|
| 1 | Catechin hydrate (CH) | Green Tea | Scavenger of free radicals, represses NO production | Reduces TNF-α production and increases the secretion of IL-2 in hPBMCs | (8) |
| 2 | Aliphatic C(17)-polyacetylenes | Carrot, celeriac, parsnip, and parsley | Potential antioxidant | Improves the helper T-cell function and shifts Th1/Th2 balance toward Th1 | (5, 50, 227) |
| 3 | Curcumin (diferuloylmethan) | Yellow component of turmeric | Antioxidant | Decreases Bax level and increases Bcl-2 in T cells by downregulation of the cytokine receptor γ-chain | (27, 142) |
| 4 | Epigallocatechin 3-gallate (EGCG) | Green Tea | Increases expression and activities of MnSOD, catalase, and GPX | Inhibits proliferation of T cell by blocking IL-2/IL2-R signaling. It also arrests T-cell growth by blocking 20S/26S proteasome complex, and subsequent inhibition of Ikk B |
(6, 158, 308) |
| 5 | Ajoene | Garlic | Antioxidant | At higher concentrations, it has inhibitory effects. At lower concentration, it increases lymphocyte proliferation |
(52, 134) |
| 6 | Chalcones (precursors of flavones) | Responsible for yellow pigmentation of plants | LPS-stimulated increase of iNOS expression and inhibits COX2, scavenger of hydroxyl radical | Inhibits proliferation of lymphocytes and blocks Th1/Th2 cytokine production. Cytoprotective activity for Jurkat cells in the G1 phase and cytotoxic for cells in the G2/M phase |
(21, 114, 242) (311) |
| 7 | OptiBerry (anthocyanin) | Wild blueberry, wild bilberry, and cranberry | MCP-1 and inducible NFκB transcriptions as well as the inflammatory biomarker IL-8 | Reduces T-cell proliferation and also reduces IL-2 and IL2-R | (52, 320) |
| 8 | Proanthocyanidin | Jamapa bean and grape seed | High anti-free-radical activity | Reduces H2O2-induced cell apoptosis and ROS. Reduces Tregs and CD4− Th17 cells |
(12, 125, 218) |
| 9 | Resveratrol | Peanuts | Antioxidant and anti-inflammatory agent | It suppresses the Treg in tumor-bearing mice and enhances the expression of IFN-γ in CD8− T cells | (42, 112, 316) |
| 10 | Lycopene | Tomatoes | Strong antioxidant effect | It increases antioxidant capacity and attenuates T-cell-dependent adaptive immune response | (41, 62, 205) |
| 11 | Beta-Carotene (Precursor of Vitamin A) | Carrots and other orange/yellow vegetables and fruit | Oxidizing hydroxyl radical, super oxide, and reducing free radicals | Treatment with beta-carotene enhances IL-2 and IFN-γ production by T lymphocytes | (62, 193, 312) |
| 12 | Vitamin A | Carrots, cheese, eggs, and meat | Antioxidant | Tropism of T cells to gut | (182) |
| 13 | Vitamin B6 | Whole grain, vegetables, and meat | Antioxidant | Suppresses proinflammatory cytokines | (113) |
| 14 | Vitamin C | Citrus fruit, the cabbage family, tomatoes, peppers, and greens | Attacks free radicals, including those from overexposure to sunlight. Boosts immunity and helps produce anti-inflammatory steroids | Vitamin C shifts immune responses toward Th1, which may be due to Vitamin C uptake by DCs increased IL-12p70 secretion | (62, 124) |
| 15 | Vitamin D | Cod liver oil and egg | Antioxidant | Tropism of T cell to skin. Important for TCR response. Naïve T cells do not express vitamin D receptor. It upregulates expression of phospholipase C-γ1, which is important for TCR activation |
(182, 298) |
| 16 | Vitamin E | Wheat germ oil and sunflower oil | Free-radical scavenger | Vitamin E suppresses CD95L and protects T cells of HIV-1-infected individuals from CD95-mediated AICD | (157) |
AICD, activation induced cell death; GPX, glutathione peroxidase; HIV, human immunodeficiency virus; hPBMCs, human peripheral blood mononuclear cells; IFN-γ, interferon-γ; iNOS, inducible NO synthase; MnSOD, manganese superoxide dismutase; ROS, reactive oxygen species; Th, T helper; TNF, tumor necrosis factor.
Vitamin C, another important nonenzymatic antioxidant, is associated with increased production of IL-12 by DCs when activated with ascorbic acid (AA). Moreover, T cells activated with AA-treated DCs produce higher amounts of Th1-polarizing cytokines (IL-2 and IFN-γ) as compared to the Th2-polarizing cytokines (IL-5 and IL-10) (124). Therefore, Vitamin C can modulate the T-cell responses by increasing Th1 bias. In addition, in vitro and in vivo studies have suggested that Vitamin E can enhance IL-2 production, T cell proliferation, and redistribution of ζ-chain-associated protein kinase 70 (Zap70) protein–tyrosine kinase, and its substrates (linker of activated T cells [LAT] and Vav) to immune synapses in aged mice compared to young mice (177, 191, 307). Zap70, LAT, and VAV redistribution is involved in downstream signaling in T cells, which are required for cytokine gene transcription. Vitamin E has also been shown to be important in neutralizing the ROS-mediated damage of membrane lipids in age-associated, impaired CD4+ T cell function. Taken together, the immune system is enhanced with Vitamin E supplementation. These studies therefore suggest that vitamins with antioxidant capacity may be important molecules involved in redox regulation of T cells.
VI. Web of Redox Regulation Molecules
Redundant pathways that regulate the production of ROS and RNS are known to exist, and a balance between pro-oxidant and antioxidant molecules maintains low cellular oxidative stress. However, due to exogenous factors (pollutants or ionizing radiation) or endogenous factors (inflammation or chronic viral stimulation), genes that regulate the cells' susceptibility to oxidative stress are differentially expressed, leading to pathogenesis.
Recent studies have demonstrated that engagement of the TCR induces rapid production of ROS (Fig. 12). Moreover, ROS production modulates T-cell signal transduction and gene expression (16, 61). Under conditions of normal TCR activation, T cells contain relatively low-to-moderate levels of ROS (61, 105). Since ROS have the ability to diffuse short distances, they are characterized as small signaling molecules. However, a repetitive stimulation of the TCR or stimulation with a high dose of antigen, or exposure to H2O2, could increase ROS concentrations, leading to T-cell hyporesponsiveness or death (106). Furthermore, data from other cell types have suggested that ROS could promote either apoptosis or necrosis, depending upon the amount of cellular ROS (74, 326). Abundant ROS promote lipid peroxidation (LPO), macromolecular damage, and necrotic cell death, and a decreased amount of ROS promotes changes in cell signaling and cell function (131).
FIG. 12.

Web of redox regulation and potential targets for T-cell therapy. Various molecules that interact with each other at some stage to regulate oxidative stress are shown. Dark arrows indicate well-known pathways; dotted arrow shows pathways relatively unstudied. Thick dark arrows show possible targets that may be deployed in T-cell adoptive therapy (SOD, NF(B, CAT, and iNOS). Activation of T cell with antigen when presented with MHC (present on an APC) produces ROS and superoxide by the NADPH oxidase (NOX) system and activates NF(B. NF(B activates the transcription of various genes such as Th1 cytokines and Fas ligand (FasL). The mitochondria and the cytoplasm have superoxide dismutase (SOD2 and SOD1, respectively), which metabolizes superoxide to peroxide (H2O2), and CAT converts it to nonharmful products (H2O and O2). H2O2 is also known to damage DNA; however, it may downregulate cFOS and BCL2 by unknown mechanisms. It also activates various signaling pathways such as MAPK, p38-MAPK, and ERK-MAPK. Superoxide also forms peroxynitrite radicals (ONOOˉ) that may upregulate formation of the EGF receptor (EGFR) by phosphorylation of various protein kinases (MAPK, ERK, p38, and JNK). CAT, catalase; iNOS, inducible NO synthase; JNK, c-Jun N-terminal kinase; MHC, major histocompatibility complex; NADPH, nicotinamide adenine dinucleotide phosphate-oxidase.
Interestingly, T cells activated in vivo and subsequently cultured briefly in vitro exhibit features of necrosis (i.e., rapid membrane permeability) and apoptosis (i.e., caspase-dependent DNA fragmentation and nuclear condensation) (292). When viewed under an electron microscope, these activated T cells clearly exhibit many of the hallmarks of apoptosis (i.e., nuclear condensation, cell shrinkage, and loss of the nuclear membrane) (292). Notably, all of these cell death characteristics (both apoptotic and necrotic) can be prevented by culture with antioxidants, thereby suggesting a role for ROS in different forms of cell death (105, 252, 254). However, the molecular mechanisms that regulate T-cell fate and downstream signaling upon ROS exposure are largely unknown and are currently major topics of interest in T-cell research.
The mitochondrion is a primary source of cellular energy and ROS, and is functionally important in regulating CD8+ T cell activity (318). When transgenic CD8+ T cells were incubated with increasing doses of rotenone (an electron transport complex I inhibitor used to block mitochondrial function), production of H2O2, calcium flux, and ERK1/2 phosphorylation were decreased within minutes of T-cell activation. Failure to undergo signal transduction resulted in decreased T-cell division initiated by peptide-coated cells, CD3/CD28 Abs, and PMA/ionomycin stimulation. Decreased function after rotenone incubation was not phenotype specific. That is, naïve, effector, and memory CD8+ T cells exhibited decreased production of both IFN-γ and TNF-α after peptide stimulation. Furthermore, incubation with rotenone decreased degranulation of effector and memory cells, a critical step in the cytolysis of infected cells. These data suggest that electron transport complex I is required for CD8+ T-cell function.
Moreover, activation of T cells is associated with an increase in ROS and an increase in the level of reversible cysteine sulfenic acid production. Reversible cysteine sulfenic acid is a reactive oxygen intermediate (ROI), which is important in the formation of disulfide bonds and GSH conjugation, suggesting that reversible cysteine sulfenic acid is an important ROI in regulating multiple cell-signaling processes associated with T-cell activation (188). Analysis of specific proteins revealed that the protein tyrosine phosphatases SHP-1 and SHP-2, as well as actin, underwent increased sulfenic acid modification after TCR stimulation. Subsequent experiments revealed that the reversible formation of cysteine sulfenic acid was critical for ERK1/2 phosphorylation, calcium flux, cell growth, and proliferation of naïve CD8+ and CD4+ T cells. Inhibition of reversible cysteine sulfenic acid formation decreased TNF-α production more than IFN-γ. Taken together, these results suggest that reversible cysteine sulfenic acid formation is an important regulatory mechanism by which CD8+ T cells can modulate signaling, proliferation, and function.
Analysis of individual intracellular signaling molecules in the redox state has demonstrated the importance of redox regulation of signaling molecules in vivo. In addition to AP-1 and NFκB, the binding of several transcription factors is modulated by redox status, including c-Myb (199) and p53 (96), JNK (Jun NH2-terminal kinase)/SAPK (stress-activated protein kinase) (104) and MAPK/ERK (244), and src family PTK kinase, such as Lck (100). Our group recently identified a role for ROS in regulating T-cell death upon repetitive TCR stimulation (207). We have demonstrated that an SOD mimetic compound, MnTBAP, protects MART-127–35-specific CTLs from AICD without interfering with the IFN-γ response and without inhibiting JNK phosphorylation (207). Other studies have also shown that mitochondria have a role in ROS-mediated upregulation of FasL (92). While ROS are known to participate in AICD regulation by induction of CD95L expression, the mechanism and signal transduction pathways necessary for ROS-induced AICD are still areas of intense research. Kaminski et al. have shown that the proximal TCR-signaling machinery, including Zap70, LAT, SH2 domain-containing leukocyte protein of 76 kDa (SLP76), phospholipase Cgamma1 (PLCgamma1), and protein kinase C theta (PKC-theta), are crucial for ROS production (128). Kaminski et al. (128) further report that PKC-theta is translocated to the mitochondria and suggest that PKC-theta-dependent ROS generation by mitochondrial complex I is essential for inducing AICD. Furthermore, in cells depleted of mitochondrial DNA, mitochondria were identified as the source of activation-induced ROS (128). Briefly, inhibition of the mitochondrial electron transport complex I assembly by siRNA-mediated knockdown of the chaperone NDUFAF1 blocked ROS production. This signal was found to be essential for CD95L expression, where inhibition of complex I assembly by NDUFAF1-specific siRNA prevents AICD.
Interestingly, studies indicate a role of MnSOD inactivation during Fas (CD95)-mediated apoptosis in Jurkat T cells (217). This study illustrated that after oligomerization of the Fas receptor, MnSOD is selectively degraded during apoptosis. In the presence of cycloheximide, an inhibitor of protein synthesis, the cell death rate and MnSOD degradation were accelerated. MnSOD cleavage was partially inhibited in the presence of the pan-caspase inhibitor, z-VAD-fmk, suggesting that caspases may mediate cellular protection by stabilizing antioxidant enzymes and thereby controlling oxidative stress (217). Therefore, functional MnSOD is important in regualting oxidative-stress-mediated apoptosis of T cells.
T cells also have the capacity to generate NO upon antigen stimulation, which may affect signal transduction, Fas ligand surface expression, and apoptotic cell death of mature T lymphocytes. The involvement of iNOS in the generation of T-lymphoctye-derived NO and its role in inhibiting T lymphocyte proliferation have been documented (108, 135). Vig et al. have shown that iNOS production in activated T cells regulates T-cell death and immune memory (295). Thus, it has become clear that iNOS activity also regulates the development, differentiation, and/or function of T cells and B cells and also affects natural killer (NK) cells (30). The role of NO in mediating T-cell cytokine production and signal transduction in histidine decarboxylase knockout mice (HDC-KO mice) has also been recently documented (147). In the HDC-KO mice, with a defect in endogenous histamine production, an increase in IFN-γ mRNA and protein expression levels of splenocytes was reported to be associated with a significant increase (2.5-fold) in NO production, compared to the WT animals. Furthermore, histamine treatment decreased NO production from both WT and HDC-KO mouse T lymphocytes. These results indicate that, in addition to its direct effects on T-lymphocyte function, histamine regulates cytokine production and T-cell signal transduction through its effects on NO production.
While these observations describe causes and effects of various ROS species, it should also be noted that the quantitative and qualitative effects of ROS are affected by other factors that include age, gender, ethnicity (linked to gene polymorphism of antioxidant genes), external environment, and degree of inflammation. These factors form a complex web that regulates the susceptibility of an individual to oxidative-stress-mediated diseases.
VII. Role of Redox Regulation in Human Diseases
As discussed in previous sections, ROS are essential for efficient and proper execution of a large number of cellular processes, including signaling induced by exogenous factors. However, ROS are highly reactive, where excessive and prolonged ROS production, disturbance in redox regulators, or an imbalance in the expression of antioxidant enzymes can considerably damage cellular constituents. Thus, ROS balance is implicated in the onset of various diseases (Fig. 13). However, this review focuses on diseases related to T-cell pathogenesis arising from direct effects of a pro-oxidant microenvironment.
FIG. 13.

Various human disorders and effected antioxidant systems. Based on recent reports, a deficiency in the antioxidant system can lead to increased susceptibility to certain diseases and disorders. The diagram shows different human disorders such as carcinoma, leukemia, cardiovascular diseases, neurological disorders, and autoimmune disorders that are associated with a deficiency in redox regulators such as SOD, CAT, TRX, and GSH. TRX, thioredoxin.
A. Autoimmune disorders
1. Rheumatoid arthritis
RA is a common autoimmune disease characterized by chronic inflammation of the synovial joints, where T cells have a defined role in the pathogenesis of this disease (73). While the exact mechanism of ROS-associated RA has not been established, recent studies suggest that oxygen free radicals play an important role in such chronic inflammatory diseases (213). Based on studies using animal models for human RA, Olofsson et al. demonstrated that a polymorphism in the Ncf1 (p47phox) gene leads to reduced oxidative burst and increased activation of arthritogenic T cells, thereby increasing the severity of arthritis (211). Furthermore, NOX2-derived ROS can suppress antigen-dependent T-cell reactivity and reduce the severity of the disease in animal models (82, 189). Interestingly, an oxidative burst-inducing pharmaceutical compound known as phytol (3,7,11,15-tetramethyl-2-hexadecene-1-ol) has been used to treat arthritis-prone Ncf1DA rats (115). The investigators in this study propose that the higher oxidative burst by phagocytes may lead to apoptosis of autoreactive arthritogenic T cells in this model of RA. Increased concentrations of ROS activate phagocytes, and activated phagocytes are known to increase the overall production of TNF-α and IL-1β. These cytokines are then responsible for activation of NFκB (197). The resulting constitutive feedback loop leads to the activation of NOX-signaling pathways, increased oxidative stress, and cytokine production. This attracts leukocytes, memory T cells, macrophages, and other inflammatory cells in synovial joints, leading to degradation of cartilage and bone. The high oxidative stress levels in the synovial fluid of RA patients lead to the hyporesponsiveness of the T cells in the synovial fluid in these inflamed joints (91). This finding was later demonstrated to result from oxidative-stress-mediated membrane displacement of a protein called LAT. Proper anchorage of LAT and the activation of T cells were seen upon supplementing the cell with GSH. Furthermore, persistent ROS production in RA synovial fluid T cells is associated with Ras and Ras-proximate-1 (Rap1)-signaling proteins (233). These signaling molecules play an important role in T-cell stimulation. It was demonstrated that activation of Ras is necessary for intracellular ROS production, while activation of Rap1 can block Ras-dependent ROS production. These studies suggest that impaired redox regulation of synovial fluid T cells may be associated with the pathogenesis of RA.
2. Systemic lupus erythematosus
SLE is an autoimmune disease characterized by acute and chronic inflammation of various tissues, prominently the kidneys, skin, joints, or the central nervous system. SLE mainly occurs due to production of autoantibodies due to an imbalance in cytokine secretion and antioxidant molecules in SLE patients (15). Interestingly, ROS-modified IgG has been associated with the induction of circulating SLE autoantibody where neoepitopes elicit an immune response in SLE patients (7). Other studies have suggested that increased endogenous NOS (201), reduced oxidative burst (58), and decreased expression of SOD, catalase (CAT), and GSH (260) could potentially lead to T-cell-mediated immune responses in SLE. Earlier studies have demonstrated that oxidative stress leads to the downregulation of TCR ζ-chain expression in patients who have undergone surgery (117). This type of oxidative stress-induced downregulation is seen in T cells isolated from SLE patients (83, 84). While it is known that the ζ-chain is important for TCR signaling and activation, lower GSH levels, elevated levels of ROS, subsequent T-cell activation, together, lead to prolonged mitochondrial hyperpolarization and ATP depletion in SLE. Furthermore, decreased expression of ζ-chain and low levels of GSH prevent the regeneration of ζ-chain in SLE T-cells. Taken together, these fundamental studies in TCR signaling suggest a critical role of oxidative stress-induced downregulation of TCR signaling via mitochondrial transmembrane potential regulation in SLE patients.
3. Type 1 diabetes
Type 1 diabetes is an autoimmune-induced disease in which T cells recognize pancreatic β-cell antigens and initiate leukocyte infiltration. The ROS generated by the initial insult to the islets stimulate the activation of redox-dependent NFκB and other transcription factors. This stimulation leads to an increase in the production of proinflammatory cytokines, such as TNF-α, IL-1β, and ROS, which eventually damage the pancreatic β-cells. Moreover, innate derived ROS are critical for T-cell activation, as they stimulate production of these cytokines from APCs while enhancing T-cell proliferation and activation (285). Therefore, oxidative stress has a high impact in the development of type 1 diabetes. Moreover, there are several other factors associated with the etiology of type 1 diabetes, including environmental factors, genetic susceptibility, deficiency, and diet.
Islets contain low levels of antioxidant enzymes such as SOD1, GPX1, and CAT, suggesting that pancreatic β-cells are highly susceptible to oxidative stress (156). Adoptive transfer of the diabetogenic T-cell clone (BDC-2.5) has been shown to induce diabetes in young NOD-SCID mice, which can be delayed or prevented altogether by treating the recipient mice with the SOD mimic AEOL-10113 before the adoptive transfer of the BDC-2.5 clone (223). Therefore, this study supports the finding that the pancreatic β-cells are highly susceptible to oxidative-stress-induced apoptosis. Moreover, oxidative stress has been shown to induce diabetes-associated vascular dysfunction leading to diabetic nephropathy, retinopathy, and cardiomyopathy (85, 278). Thus, modulating ROS can serve as an important therapeutic tool to treat type 1 diabetes and its vascular complications.
B. Cardiovascular disorders
1. Atherosclerosis
Atherosclerosis is a cardiovascular disease marked by chronic inflammation of the blood vessels. Oxidative stress and inflammation result in the infiltration of activated inflammatory cells from the coronary circulation into the arterial wall, leading to the pathogenesis of atherosclerosis (25). It is known that accumulation of low-density lipoprotein (LDL) on the internal membrane of arteries (intima) and prolonged oxidative modification of LDL molecules by ROS lead to formation of atheroslerotic lesions. Oxidative modification of LDL can initiate an inflammatory response (99). Ammirati et al. have recently reported a significant correlation between increased circulating TEM and the generation of atherosclerotic lesions and high LDL production (9). Activated T cells are known to contribute to the proinflammatory components of atherosclerosis. Ammirati et al. suggest that TEM are responsible for atherosclerotic lesions (9). These authors further suggest that TEM subsets that express HLA-DR, CXCR3, and CCR5 are recruited into the atherosclerotic plaques. Frostegard et al. demonstrated that exposing monocytes and T cells to oxidized LDL for 72 h leads to an increase in IL-2 receptor expression and DNA synthesis (78). The results indicate that oxidized LDL may induce migration of monocytes, thereby leading to the formation of atherosclerotic plaques. Based on murine in vitro and in vivo studies, elevated homocysteine (Hcy) levels were thought to be an independent risk factor for atherosclerosis (321). The autooxidation of Hcy-generated ROS significantly promoted ConA-induced proliferation and partially inhibited apoptosis of cultured, activated mouse splenic T lymphocytes. Furthermore, the in vivo studies using ApoE-knockout mice, with hyperhomocysteinemia, have significant T-cell proliferation in response to ConA and increased intracellular ROS. These data suggest that oxidative stress is involved in the chronic inflammatory progression of atherosclerosis and suggest that ROS production is a key factor in the progression of cardiovascular diseases.
2. Cerebral ischemia/stroke
A stroke is an interruption of the blood supply to any part of the brain leading to the rapid loss of brain function. Oxidative state plays a key role in the regulation and control of numerous signal transduction pathways in neurons (53, 198). Microglial cells and astroglial cells are direct effector cells of the CNS. Several studies present evidence that activated microglial cells, in response to ischemia, have the potential to release several proinflammatory cytokines such as TNF-α, IL-1β, and IL-6, as well as other potential cytotoxic molecules, including NO and ROS (167). IL-1-induced inflammation initiates the expression of chemokines CXCL1 and CXCL2 (181). Moreover, these chemokines may cause the infiltration of neutrophils. A recent study has identified infiltration of interleukin-17-producing γδ T cells 3 days after the onset of ischemia in a mouse model. The study also demonstrates that blocking T-cell infiltration reduced the infract size (263). Moreover, Liesz et al. have indicated that Tregs may delay brain damage after stroke (159). It is well known that hypertension is a high risk factor for stroke-induced death, and recently, a role for T cells in angiotensin II-induced hypertension has been proposed by Guzik et al. (93). Angiotensin II induces T-cell activity, proinflammatory cytokine production, and infiltration in perivascular fat. Based on these in vivo studies, angiotensin II can stimulate peripheral blood T cells to produce TNF-α and IFN-γ as well as express tissue-homing receptors, all of which lead to the development of hypertension and vascular dysfunction. Hoch et al. have further demonstrated that angiotensin II is important in activating T cells, in inducing the expression of tissue-homing markers, and in producing the proinflammatory cytokine, TNF-α (107). The researchers further suggest that an autocrine loop exists in T cells where T-cell-derived angiontesin II activates T-cell NOX, thereby inducing ROS production, and subsequently TNF-α production. Therefore, angiotensin II-induced hypertension is associated with the accumulation of T cells in perivascular fat, and moreover with T-cell accumulation in atherosclerotic lesions. These studies would then suggest that there is an indirect and direct association of T-cell activity, hypertension, and stroke. Moreover, deletion of SOD3 in the circumventricular organ can increase T-cell activation, leading to increased angiotensin II-induced vascular superoxide production and vascular inflammation because of T cell and leukocytein filtration (164). Therefore, redox regulation of T cell may play an important role in the pathogenesis of stroke.
C. Cancer and metastasis
The role of ROS in cancer cells has long been suspected, and growing evidence suggests that cancer cells are under increased oxidative stress in comparison to normal cells. Szatrowski et al. and other groups have demonstrated enhanced ROS production, along with overproduction of antioxidant enzymes, in cancer cells (267, 275). Moreover, hypoxia-induced ROS production in the tumor milieu is associated with loss of genomic stability and downregulation of DNA repair pathways (31). For example, oncogenes c-myc and an allele of RAS2 (val19) have been shown to induce ROS production in tumor cells (221). While the oncogenic c-myc increases ROS production, induces DNA damage, and regulates p53 function, oncogenic RAS2 constitutively activates the cAMP-PKA pathway resulting in the increase in ROS production. Tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and activated granulocytes produce significant amounts of ROS and NO, which leads to oxidative stress-induced downregulation of immune responses (151, 256, 275). In advanced cancer patients, activation of granulocytes and the granulocyte-derived H2O2 is associated with the systemic T-cell suppression. Increased ROS production is seen in cancer cells as compared to normal cells, including primary leukemia cells isolated from patients with chronic lymphocytic leukemia, CLL (221, 323).
Zhou et al. (323) have further suggested that treating these cells with 2-methoxyestradiol, an SOD inhibitor, has a cytotoxic effect on these cancer cells. This would imply that it would be beneficial if anticancer therapies are directed to regulate ROS production in cancer cells, but not in normal cells. For example, a recent study demonstrates a novel DR5 (TRAIL R-2) monoclonal antibody, AD5–10, generates ROS production in Jurkat leukemia cells. As a result of the ROS production, JNK is activated, and mitochondrial membrane potential is lost, and intracellular GSH and oxidized glutathione (GSSG) levels are decreased, and endonuclease G is translocated from the mitochondria into the cytosol, all of which lead to the selective apoptosis of the leukemia cells in vitro (45). Moreover, Jurkat, a human acute lymphocytic leukemia cell line (Jurkat T-ALL), transfected with a dominant-negative (DN) form of JNK (but not of p38), enhanced NFκB activation, reduced caspase-8 in death-inducing signaling complexes (DISCs), and reduced damage to mitochondria, thereby inhibiting AD5–10-induced cell death in the DN-mutant cells.
The use of pro-oxidants to increase ROS as well as inhibitors of antiapoptotic proteins has been employed to control tumor growth. Several preclinical and clinical studies have used inhibitors of the Bcl-2 family of proteins to induce apoptosis in cancer cells by primarily involving the mitochondrial apoptosis pathways (145). Jurkat and HeLa cells transfected with a tetracycline-regulated Bcl-2 expression system were used in redox signaling studies to determine the role of Bcl-2, ROS, and GSH levels in apoptotic signals. Based on the results presented in this study, ABT-737 (pan-Bcl-2 inhibitor) induces caspase activation and apoptosis by decreasing GSH levels, with a concomitant increase in ROS (111). In summary, these studies validate rational molecular approaches that target antiapoptotic pathways involving ROS when developing novel combination therapies for cancer treatment in synergy with other strategies.
Strategies employed by the tumor cells to evade cell death include immune tolerance to malignant cells, impaired antigen presentation, tumor-induced immune suppression, negative co-stimulatory pathways, and dysfunctional TCR signaling that contributes to the resistance to AICD (230). In addition to these strategies, recent studies have suggested that deregulation of critical signaling pathways, in particular Notch, PI3K/Akt, MAPK, Jak/STAT, and TGF-β, may contribute to T-ALL and T-cell non-Hodgkin's lymphoma. A recent study identified IL-7 as a survival factor required for early T-cell differentiation and normal T-cell proliferation (20). IL-7 induces the activation of the JAK/STAT, MEK/ERK, and PI3K/Akt signaling pathways in T-ALL cells, where PI3K signaling is shown to induce Bcl-2 expression, and is required for the proliferation and survival of T cells. The elevated levels of ROS seen in T-ALL cells is attributed to the PI3K/Akt/mTOR pathway-dependent induction by IL-7, and this overexpression of ROS requires NOX activity and mitochondrial respiration (265). Furthermore, the same group of researchers have shown that T-ALL cells require glucose uptake for ROS production in T-ALL cells treated with IL-7. Moreover, the study also demonstrates that IL-7 increases the expression of the glucose transporter GLUT1 in a PI3K-dependent manner. These observations suggest that substrates of molecular pathways activated by microenvironmental factors regulate the viability and proliferation of T-ALL cells and provide the means for the development of novel treatment strategies.
Interestingly, immunosuppressive cells known as MDSCs can impair CD8+ T-cell immunity by increased production of ROS, which inhibits ζ-chain expression in T cells (200). In addition, MDSC induced the nitrosylation of the TCR tyrosine residues on the surface of antigen-specific CD8+ T cells, thereby inducing T-cell dysfunction. Tacke et al. have demonstrated that chronic Hepatitis C virus infection can lead to MDSC-mediated suppression of T-cell response by generating ROS, suggesting that MDSCs are important in regulating T-cell-mediated immune responses (276). Moreover, based on this report, MDSCs suppress T-cell responsiveness by upregulating the expression of p47phox, a component of the NOX2 complex, which is important in ROS production. A recent study demonstrated that Nrf2 deficiency creates a responsive microenvironment for metastasis to the lung, where MDSCs of Nrf2-deficient mice induced overproduction of ROS compared to the WT mice (255). Therefore, the increased pulmonary metastatic potential of tumor cells in Nrf2-deficient mice can be attributed to the overproduction of ROS. Moreover, greater numbers of inflammatory cells, including MDSCs, were identified in the lung and bone marrow of Nrf2-deficient mice with metastatic cancer. Cancer cells have efficient antioxidant mechanisms to survive high levels of ROS. Some of these defense mechanisms include upregualtion of peroxirodoxins (Prx) and TRX in response to oxidative injury (43, 229). In addition, the predominance of other suppressive immune cells (such as Tregs and MDSCs) that are innately high in antioxidant molecules aid in the development of cancer and evade effector immune response. Redox-modulating strategies thus are important as controls of cancer development and progression.
D. Neurological disorders
1. Amyotrophic lateral sclerosis
ALS is marked by the progressive loss of motor neurons, neurofilament inclusions, astrocytosis and muscle atrophy, inflammation, and T-cell infiltration. ALS is caused by a mutation in the SOD1 gene, which renders SOD1 inactive, resulting in the oxidative damage of motor neurons. To date, there are several transgenic mouse models for human ALS with specific mutations in the Cu2+/Zn2+ SOD1 gene, including SOD1G93A, SOD1G37A, SOD1G85R, SOD1D90A, and SOD1G93A transgenic rat model. Currently, the SOD1G93A transgenic mouse model is used to test therapeutic agents to treat ALS and to understand ALS disease progression at the molecular level (123). While earlier studies have demonstrated reduced counts of Tregs and monocytes in ALS, a recent study suggests that Tregs (CD4+CD25+FoxP3+) are recruited to the CNS to prevent or attenuate the neurodegenerative process by stimulating the secretion of anti-inflammatory cytokines (144). On the contrary, Tregs have been shown to help in protecting ALS from progression, and FoxP3 is lost in later stages of this disease. Furthermore, injecting CD4+ T cells has been shown to increase the survival of mSOD1 mice, a murine model of human ALS (23). Another study has identified that CD154 (co-stimulatory CD40 ligand on T cell) can further activate the immune response when bound to CD40 on APCs, a promising therapeutic target for ALS (22). Taken together, these studies would suggest that understanding the molecular mechanisms of microglial-derived inflammatory signals would be important in treating this neurodengerative disease, and further research in regulating microglial-derived ROS and cytokine expression is warranted.
2. Multiple sclerosis
MS is a CNS demyelinating, inflammatory disease marked by a significant decrease in the number of oligodendrocytes (OLs). MS is associated with axonal degeneration and irreversible neurological disabilities (29). EAE is a well-established animal model for MS with a Th1 cytokine-induced active phase of the disease (69). That is, during the active phase of this disease in EAE, TNF-α, IL-2, and myelin/OL glycolprotein-specific antibodies are actively produced. Moreover, oxidative stress-induced heme oxygenase-1 (HO-1), a heat shock protein, is overexpressed in the CNS of EAE mice (163). Other studies have shown that erythropoietin (EPO) and EPO-induced upregulation of endogenous HO-1 repress immune responses to EAE in MOG-EAE mice to protect the neuronal cells from oxidative stress-triggered cytotoxicity (46). Intraperitoneal administration of EPO was shown to reduce clinical severity by significantly repressing immune responses by the Th1 and Th17 lymphocyte populations at the site of the lesions and increasing systemic Th2 and Treg populations. The initial infiltration of autoreative CD4+ Th1 cells in the brain lesions of MS is followed by the upregulation of proinflammatory cytokines, TNF-α as well as IL-17, a proinflammatory cytokine secreted by Th17 cells (214). Based on studies using uncoupling protein 2 (UCP2)-deficient mice, UCP-2 normally functions to reduce the production of ROS during infection, and in EAE, UCP-2 has a protective response (297). This study further illustrates that UCP-2-deficient mice have modified their immune response where CD4+ T cells produce significantly higher levels of proinflammatory cytokines; for example, TNF-α and IL-2 and the CD4+ and CD8+ T cells upregulate ROS production. Further research to limit demyelination in MS by developing antioxidant therapies directed toward Th1- and Th17-mediated immune responses in MS is warranted.
3. Parkinson's disease
Similar to ALS, Parkinson's disease (PD) is a neurodegenerative disease marked by activated microglia (innate immune cells of the CNS) and infiltrating T cells at sites of neuronal injury. In PD, predominantly, the M1 microglial phenotypes are present in the injured neurons, where M1 microglia are responsible for secreting proinflammatory cytokines (i.e., TNF-α and IL-1β), ROS, and NO, all of which lead to neurotoxicity, (13). Inflammation of phagocytic microglia, release of cytokines, T cells, and complement activation play a role in damaging neurons, in particular, the dopaminergic neurons. In PD patients, the pigmented dopaminergic neurons in the substantia nigra part of the midbrain are triggered to undergo apoptosis; based on recent evidence, two mutations in the α-synuclein gene have been linked to the autosomal dominant form of this disease (165). Interestingly, the α-synuclein gene mutation is associated with accumulation of dopamine in the dopaminergic neurons in PD patients. The instability of dopamine's catechol ring structure can apparently undergo spontaneous oxidation, leading to excessive ROS production and increasing oxidative-stress-induced death of dopaminergic neurons in PD patients. A greater understanding of the T-cell population that mediates these effects, as well as the molecular signals involved in PD progression, should identify new targets for neuroprotective immunomodulation to treat these devastating neurodegenerative diseases.
E. Skin disorders
The skin is constantly exposed to endogenous and environmental pro-oxidant agents, which lead to the generation of harmful ROS. Although healthy skin is equipped with a large number of defense mechanisms, including antioxidant systems that attenuate the harmful effects of ROS, the increased or prolonged presence of free radicals can override ROS defense mechanisms and mediate numerous cellular responses that can contribute to the development of a variety of skin disorders.
1. Psoriasis
Psoriasis is a common inflammatory disease of the skin and joints, triggered by an immune response to endogenous (self) and exogenous factors (bacterial or viral). Formation of psoriatic lesions is thought to be elicited by the complex cross-talk between keratinocytes and components of the innate and acquired immune system. Psoriatic keratinocytes possess an enhanced ability to resist apoptosis, which might be one of the key pathogenic mechanisms in psoriasis. In addition, statistically significant decreases in erythrocyte catalase, GSH, SOD levels, and GPx activities were noted in psoriatic patients (322). The observed increase in erythrocyte malondialdehyde suggests that the cells were in a perioxidative state. Based on these results, oxidative stress plays a significant role in the progression of psoriasis.
Furthermore, subsequent studies have suggested that antioxidant strategies could prove to be beneficial treatments for psoriasis. The cellular signaling pathways, such as MAPK/AP-1, NFκB, and JAK-STAT signal transducers and activators of transcription, are known to be redox sensitive and are involved in the progression of psoriasis (299). Interestingly, psoriasis has been suggested to be an autoimmune response to streptococcal throat infection. Patients with psoriasis have CD4+ and CD8+ T cells that are reactive to antigen determinants on streptococcal M-protein and type-1 keratins. Therefore, molecular mimicry has been proposed as the method of eliciting autoimmunity in psoriatic lesions (288).
2. Scleroderma
Scleroderma is a chronic autoimmune disease characterized by fibrosis, vascular injury, and increased collagen deposition in the skin (313). There are two forms of scleroderma: limited cutaneous systemic sclerosis (SSc) and diffuse cutaneous SSc (dcSSc). While hands, arms, and face are affected in patients with SSc, dcSSc involves fibrosis of a large area of the skin and at least one other internal organ: the kidney, heart, or lung (51). Murine models of SSc and dcSSc were used to understand the role of ROS in the pathogenesis of SSc (258). Briefly, Balb/c and immunodeficient Balb/c SCID mice were injected with pro-oxidative agents, bleomycin, or phosphate buffered saline. Based on histological, biochemical, and immunological studies, ROS played a significant role in SSc, and the specific oxidation of autoanitgen has been shown to induce systemic sclerosis. Moreover, Servettaz et al.(258), demonstrate that the type of ROS determines the form of SSc. Peroxynitrites induced limited SSc, which exhibit skin fibrosis and serum anti-CENP-B IgG, whereas hypochlorite or hydroxyl radicals induced diffuse SSc with cutaneous and lung fibrosis and serum anti-DNA topoisomerase I antibodies. This finding suggests that decreased antioxidant levels and increased accumulation of ROS are key components in the progression of scleroderma.
Recent investigations have suggested that in addition to ROS production, apoptosis is involved in the progression of scleroderma (313). Patients with SSc have high serum levels of soluble Fas, which would protect autoreactive T cells from FasL-induced apoptosis. In these same patients, spontaneous apoptosis of CD8+ T cells was also observed, suggesting that the T-cell imbalance leads to autoimmunity in patients with SSc. However, this is based on the stage of the SSc disease. Based on recent findings, effector peripheral blood CD8+ T cells upregulate the expression of profibrotic cytokine IL-13, thereby leading to the observed cellular toxicity in SSc patients (79). Interestingly, current immunotherapy regimens focus on regulating the immune system in scleroderma with conventional methods (nonselective immunosuppression), T-cell- or B-cell-targeted therapies, HSC transplantation, antifibrotic treatments, or immunomodulatory agents (175).
3. Vitiligo
Vitiligo is a condition caused by the loss of pigmentation of the skin as a result of apoptosis of epidermal melanocytes. Biochemical, neurological, environmental, genetic, and immunological factors have all been associated with the activation of vitiligo (87). Moreover, a melanocyte-specific T-cell-mediated immune response has been implicated in the development of vitiligo. Recently, activated, type-1 Teff cells were histologically identified in the perilesional margins of the depigmented skin, suggesting that skin-homing melonocyte-specific CTLs are responsible for the depigmentation in vitiligo patients (289). Furthermore, using an explant model for vitiligo, Van Den Boorn et al. (289) were able to demonstrate that the perilesional T cells induce apoptosis of melaoncytes only in the pigmented skin explants. Other studies have suggested that ROS and H2O2 production impairs biological processes (87). Tyrosinase is a rate-limiting enzyme required for melanogenesis, and in the presence of ROS and/or H2O2, the activity of this enzyme is impaired, resulting in the loss of melanocytes in vitiligo.
F. Viral diseases
1. Human immunodeficiency virus
Oxidative stress appears to contribute to HIV infection in humans, where ROS may play a critical role in HIV replication. A number of studies have demonstrated that GSH levels and acid-soluble thiol levels are decreased in AIDS patients, while plasma glutamate concentrations are increased (34, 65, 66). Moreover, GSH levels are decreased in plasma, lung epithelial lining fluid, and PBMCs in HIV-infected patients. In addition to confirming earlier findings that HIV-infected individuals have decreased GSH levels, recent studies suggest that LPO, which is associated with oxidative stress, is significantly higher in HIV-infected patients (301). These findings suggest that HIV-infected patients have increased oxidative stress, and the efficacy of treating these patients with antioxidants remains to be determined. Rhesus monkeys infected with simian immunodeficiency virus (SIVmac251), a closely related animal model of HIV, have significant loss of plasma thiol levels (i.e., decreased cysteine levels) and increased glutamate concentrations within 2 weeks of infection (67). Based on these studies, GSH depletion and the concomitant increase in ROS are correlated with the pathogenesis of HIV-infected patients.
This knowledge was exploited to initiate clinical trials employing antioxidant therapy to treat AIDS patients. The first study of this kind used the antioxidant NAC, a cysteine prodrug that was shown to block HIV expression in chronic and acute infection models as well as block HIV replication in normal PBMCs. NAC restores GSH levels and maintains normal levels of thiols during oxidative stress (241). The observed antiviral effect of NAC is due to inhibition of viral stimulation by ROS that are produced in response to chronic stimulation of HIV-specific T cells and inflammatory cytokines (241, 269). An earlier study by Sandstrom et al. demonstrated that T-cell lines infected with HIV are highly susceptible to apoptosis. Moreover, they also demonstrated that GPx deficiency in HIV-infected human leads to apoptosis of CD4+ T cells (253). Other studies have supported the above finding, and moreover, these studies also demonstrated that CD4+ T cells have lower concentration of active GPx and SOD and have a high concentration of H2O2 (86). Therefore, other strategies to replenish antioxidant molecules are being actively pursued by researchers worldwide.
2. Influenza
Influenza virus, an orthomyxovirus family of RNA viruses, infects the epithelial cells lining the throat and lung. Several studies show that influenza virus infection leads to changes in pro-oxidant levels and oxidative stress (204). Based on animal studies, cells lavaged from the lungs of influenza virus-infected mice have been shown to possess an enhanced superoxide generation capacity (222). Moreover, xanthine oxidase is overexpressed in these mice, suggesting that influenza infection leads to oxidative stress. Reports also suggest that influenza virus-infected cells show lower levels of GSH (203).
Influenza virus is also known to cause neutrophil dysfunction and activate neutrophil respiratory-burst responses. Teff cells are responsible for controlling inflammation in the lungs during influenza infections by producing anti-inflammatory cytokines such as IL-10 (271). Changes in redox regulation in Teff cells during influenza infection may affect the onset of disease. When transgenic mice expressing human Trx1 were infected with influenza virus, NFκB activation and production of higher amount of TNF-α and IL-6 were shown to increase inflammation and severity of the disease (88). Another study using NOX2-deficient mice infected with influenza virus showed a decrease in titers of virus in comparison to WT. However, NOX2 deletion does not show any difference in CD8+ Teff cells. Moreover, this study confirmed that treatment with the NOX2 inhibitor, apocynin, has the ability to reduce viral titer, inflammation of the respiratory tract, and cell superoxide production (296). Taken together, these studies suggest that influenza infections change the redox balance of T cells.
G. Aging
Aging and age-related diseases, including cancer, Alzheimer's disease, atherosclerosis, and metabolic disorders, are associated with low-grade inflammation, oxidative stress, and production of NFκB-dependent proinflammatory cytokines, such as IL-1, IL-6, and TNF-α. While the aging process has been associated with the decline of adaptive immunity, referred to as immunosenescence, innate immunity appears to be upregulated, leading to the expression of proinflammatory cytokines. Moreover, NFκB signaling, NAD-dependent deacetylase sirtuin 1 (SIRT1), and p53 have been implicated as key regulators of aging and age-related diseases (249, 317). NFκB has been further shown to act as a link between the innate and adaptive immunity in the aging process, and age-dependent activation of NFκB signaling is tissue specific (2, 250).
The aging process appears to shift the T-cell population from CD45RA+ naïve T cells to CD45RO+ memory cells, but these T cells have impaired antigen responsiveness (161, 250). Furthermore, CD4+ T-cell-mediated activation of B-lymphocytes and the decrease in the number of CD8+ T cells are observed in the aging process, contributing to the downregulation of adaptive immunity in the aged and age-related diseases. Recently, an in vivo model of spontaneous arthritis in aged mice suggested that NAPDH oxidase 2 (NOX2) deficiency induced the production of proinflammatory cytokines, reduced ROS, and increased the number of IL-17-producing Th17 cells while decreasing the Treg population (155). Several other studies show aging-associated shift in production of proinflammatory cytokines (IL-17, IL-1β) associated with coronary heart diseases (54). Moreover, a study by Daynes et al. has shown that this imbalance, increased proinflammatory cytokine production, may be due to an impairment of peroxisome proliferator-activated receptor-α (57). Taken together, aging may induce imbalance in production of ROS, which leads to cytokine imbalance, thereby increasing susceptibility to various disorders. Moreover, the Th17 and Treg balance may be important in regulating the downstream immune response associated with aging. As previously mentioned, aging is associated with impaired T-cell function due to increased protein and DNA damage (220). T cells exhibit diminished TCR signaling in aged individuals, and this T-cell dysfunction has been attributed to ROS-mediated damage to cellular components.
During the aging process, adaptive immune response is decreased, which has also been attributed to the oxidative stress-induced shortening of the telomeres of T- and B-lymphocytes (136). Similarly, chronic oxidative stress has been shown to reduce the telomere length in human endothelial cells, leading to the premature senescence of these cells. Based on these studies, it is possible that age-related T-cell dysfunction may be due to this shortening of the telomere in T cells (268). Furthermore, CD4+ and CD8+ T and B cells have shortened telomeres in association with age progression. That is, with age, telomere shortening is observed in T cells, and more importantly, the rate at which telomeres are lost is different between the two T-cell populations. Based on these recent findings, telomere integrity of T cells as well as oxidative stress induction in these cells are important in regulating the aging process and in regulating the progression of age-related disease.
H. Gender-related differences in the oxidative stress response and T-cell immunity
Gender disparity in redox imbalance and susceptibility to apoptotic signals results in the increased risk of cardiovascular pathogenesis (174), asthma (171), melanoma (127), and lung cancer (287). Whether or not the gender-dependent regulation of oxidative stress and apoptosis is associated with T-cell immune response in these diseases is unknown and warrants further investigation. However, these studies on gender-related pathogenesis further suggest that female hormones play a significant role in regulating autoimmunity. For example, T-cell chemokine response is regulated by estrogen expression, and the chemoreceptors are important for T-cell homing (i.e., to the spleen) as well as T-cell response to antigen stimulation (190). Based on the results of this study, female mouse-derived CD4+ T cells overexpress CCR1-CCR5 and exhibit increased tyrosine phosphorylation in response to estrogen and MIP-1β stimulation, respectively. However, the molecular mechanisms involved in inducing expression of chemokine receptors on T cells in response to estrogen stimulation are currently under investigation.
It is well known that estrogen is an important factor in determining female gender bias in autoimmune diseases. Estrogen has further been implicated in regulating iNOS mRNA, iNOS protein, and nitric oxide expression in activated T cells (133). Moreover, an increased level of nitric oxide is associated with tissue injury in autoimmune diseases that predominantly manifest in females, such as RA, MS, SLE, and glomerulonephritis. While moderately high levels of nitric oxide protect immune cells from apoptosis, chronic infections or pathologies can induce very high levels of nitric oxide expression, resulting in immune cell apoptosis or necrosis. Interestingly, after injury, estrogen stimulation can lead to the dysregulation of cell-mediated immune responses by modulating proinflammatory cytokine expression, such as TNF-α, IL-1β, and IL-6 (28). Similarly, estrogen binds to the estrogen receptor on T cells, and T-cell activation is enhanced in female patients with SLE (236). This upregulation of T-cell activation results in impaired regulation of autoantibody production, thereby enhancing the overall immune response in female SLE patients. These studies, therefore, support gender-dependent responses in susceptibility to oxidative stress, apoptosis, and T-cell immune response.
VIII. Gene Mutation and Polymorphism in Oxidative Stress and Diseases
Recent discoveries of potentially causal single-nucleotide polymorphisms (SNPs) and advancements in the use of high-throughput genotyping techniques for complex diseases hold great promise. This has led to large-scale genetic association studies such as the HapMap (haplotype mapping) Project and the Cancer Genome Project. Such studies should be helpful in the future in the prognosis of human diseases and should guide clinical decisions in treating human diseases. It is well known that SNPs can modulate protein expression, thereby effecting disease susceptibility. SNPs and mutations in antioxidant genes may lead to the modulation of the cellular redox state, effecting T-cell activation and/or T-cell proliferation. Moreover, individuals with diverse SNPs will respond differentially to T-cell therapy. Therefore, investigating SNPs in antioxidant and redox-regulating genes is of critical importance in the diagnosis, prognosis, and treatment of human diseases.
Genetic studies have shown that polymorphic variants of antioxidant genes may also increase susceptibility to lymphoma (Table 4). Ishikawa et al. showed that two mutations (G13886A and 13884insC) in ND6 (the gene responsible for coding NADH subunit 6) resulted in a deficiency in respiratory complex I activity and is associated with overproduction of ROS (118). The mutation-bearing mice had greater tumor progression and metastasis; however, when treated with ROS scavengers, metastasis was suppressed. Mutations in antioxidant genes have been shown to be associated with ROS imbalance and are therefore associated with increased disease susceptibility (89). For example, mutations in the catalase gene are associated with diabetes mellitus, hypertension, and vitiligo. Interestingly, gene polymorphisms in redox regulation genes such as cytochrome b light chain (CYBA) and aldoketo reductase family 1, member A1 (AKR1A1) show increased risk to T-cell and non-Hodgkin's lymphoma (153). Moreovoer, the ser/leu polymorphism in the NOS2A gene modulates risk for B- and T-cell lymphoma by twofold (302). Similarly, mutations in human SOD1 have been shown to cause an inherited form of ALS (257). Moreover, a variant of the promoter region of the catalase gene shows increased risk for vitligo in Chinese people, suggesting that catalase is important in regulating apoptosis of melanocytes (162). Another recent study implicated mitochondrial ROS in the production of proinflammatory cytokines in an autoinflammatory disorder caused by missense mutations in the TNFR1 gene, known as type 1 TNF receptor (TNFR1)-associated periodic syndrome (TRAPS) (35). This study suggested that TNFR1-mutant cells exhibit altered mitochondrial function with enhanced oxidative capacity and mitochondrial ROS generation. Pharmacological blockade of mitochondrial ROS efficiently reduced inflammatory cytokine production after LPS stimulation, suggesting that mitochondrial ROS may be a novel therapeutic target for TRAPS and other inflammatory diseases. The above studies suggest that a mutation or polymorpism in antioxidant genes could play an important role in disease susceptibility.
Table 4.
Gene Polymorphisms in Anti-/Pro-Oxidant Molecules That May Impair or Modulate T-Cell Function in Human Diseases
| No. | Anti/prooxidant gene | Polymorphism | Effect on T-cell redox state | Reference |
|---|---|---|---|---|
| 1 | AKR1A1 | IVS5+282T>C | 1.7-fold risk for NHL and T-cell lymphoma | (153) |
| 2 | CAT | −89A>T | Increases susceptibility to vitligo by 1.9-fold | (162) |
| 3 | CYBA | C242T | Reduced respiratory burst in human neutrophils | (310) |
| 4 | CYBA | C242T | CC genotype exhibits NOX-mediated oxidative stress and endothelial damage. | (192) |
| 5 | CYBA | Ex4+11C>T | 1.6-fold risk for NHL and T-cell lymphoma | (153) |
| 6 | CYP2E1 | CYP2E15 | 2.8-fold higher risk of acute lymphoblastic leukemia | (150) |
| 7 | GCLM | GCLM-588 C>T | Increased risk for carotid atherosclerosis in type 2 diabetic patients | (137) |
| 8 | MnSOD | Ala16Val | Risk for development of type 2 diabetes | (202) |
| 9 | MPO | G463A | Related to reduced generation of ROS, are associated with decreased breast cancer risk | (4) |
| 10 | MPO | −463 G>A | Increased risk for carotid atherosclerosis in type 2 diabetic patients | (137) |
| 11 | NOX p22phox | 242 C>T | Increased risk for carotid atherosclerosis in type 2 diabetic patients | (137) |
| 12 | Ncf1 | DBA.Vβ12 mutant | Increased susceptibility for arthritis and autoimmunity by following mechanisms: Increased autoantibody production Expansion of IL-33R-expressing T cells Impaired T-cell tolerance |
(95) |
| 13 | ND6 | G13886A and 13884insC | Deficient respiratory complex I activity and ROS overproduction | (118) |
| 14 | NFκB | Deletion in NFκB gene | Susceptibility to RA, systemic lupus erythematosus, psoriatic arthritis, giant cell arthritis, type 1 diabetes, multiple sclerosis, celiac disease, and Parkinson's disease, as well as susceptibility of several cancers, such as oral squamous cell carcinoma, colorectal cancer, hepatocellular carcinoma, breast cancer, and myeloma | (272) |
| 15 | NFκB | 94 base pair deletion | 1.45-fold higher risk for colorectal cancer | (10) |
| 16 | NOS2 A | Ser 608 Leu | 2-fold risk increase for NHL in B- and T-cell lymphoma | (302) |
| 17 | NQO1 | alleles 2 and 3 | 1.7-fold risk for acute lymphoblastic leukemia | (150) |
| 18 | Nrf2 | Nfe2l2 (gene for Nrf2) | Locus in Nrf2 gene linkage to susceptibility to hyperoxia and lung diseases | (48) |
| 19 | p22phox | Promoter | Susceptibility to NADPH-mediated oxidative stress in humans and animals with hypertension. | (71) |
| 20 | PON1 | PON1(192), PON1(55), PON1(−162), PON1(−832), PON1(−909), PON1(−1076), and PON1(−1741) | Haplotype (AATTCCT) was associated with better CD4+ cell count in HIV-infected patients | (219) |
| 21 | SOD1 | Cu2+/Zn2+ superoxide dismutase (SOD1) | Risk for amyotrophic lateral sclerosis | (123) |
| 22 | SOD2 | G1677T | Susceptibility to human cancer | (102) |
AKR1A1, Aldoketo reductase family 1, member A1; CAT, catalase; CYBA, cytochrome b-245, alpha-polypeptide; MPO, myeloperoxidase; NADPH, nicotinamide adenine dinucleotide phosphate-oxidase; Ncf1, neutrophil cytosolic factor 1; NHL, non-Hodgkins lymphoma; NOX, NADPH oxidase; NQO1, NADPH quinone oxidoreductase-1; NRF2, nuclear factor erythroid-2-related factor 2; PON1, paraoxonase-1.
Antioxidant gene polymorphisms may also increase susceptibility to several autoimmune diseases and lymphomas (Table 4). Polymorphic variants in antioxidants and redox-regulating genes lead to differential susceptibility to diseases. This may be due to up- or downregulation of antioxidants. For example, cytochrome P450 2E1 (variant -CYP2E1*5) is associated with increased risk to ALL by 2.8-fold, when compared to individuals with the WT alleles (150). Moreover, individuals with gene variants of paraoxonase-1 (PON1) enzyme have shown to have better CD4+ T-cell counts in comparison to individuals with the WT allele of PON1 (219). Table 4 briefly discusses gene polymorphisms in redox regulators that are associated with increased susceptibility to the corresponding disease.
IX. Therapeutic Strategies to Overcome Oxidative Stress-Induced Diseases
Recent studies have also focused on development of therapeutics by targeting the antioxidants that regulate the cellular redox state to restore proper T-cell function. In fact, we and others have used antioxidants to rescue T cells from oxidative stress (207). Our data suggest that pretreatment of melanoma epitope-reactive MART-127–35 and influenza matrix protein MP58–66 with MnTBAP rescues them from repetitive TCR stimulation-induced cell death by quenching ROS species and inhibiting JNK phosphorylation (207). In another study, in vivo administration of MnTBAP protected antigen-specific T cells from contraction after chronic antigen exposure in primary lymphocytic choriomeningitis virus (LCMV) infection (154). Another widely used antioxidant compound is NAC, a cysteine prodrug that replenishes intracellular GSH (14). NAC has been used successfully to treat GSH deficiency in acetaminophen toxicity, bronchitis, Alzheimer's, diabetes, cystic fibrosis (232), and HIV infection, (103, 176). Impaired T-cell function in cancer patients is associated with decreased expression of IL-2R as well as impaired responsiveness to anti-CD3, all of which could be restored with NAC treatment as seen in in vitro experiments. Therefore, restoration of GSH levels may restore T-cell function and antigen responsiveness with the treatment of NAC.
With recent advances in molecular techniques, efforts have been made to increase the cellular expression of antioxidant genes in T cells. Based on a recent study, CD4+ and CD8+ T cells retrovirally transduced with a catalase gene preserved the function of the T cells and evaded H2O2-mediated cell death, with marked resistance to ROS (11). These results have raised promise for gene therapy-based strategies to elevate the antioxidant capacity of Teff cells. Doing so could be important for increasing cell persistence in an oxidative tumor microenvironment, which may subsequently improve immunotherapy for patients with advanced cancer or chronic viral infections. Similar strategies have been used to protect cells from oxidative-stress-induced injury, where adenovirus-delivered SOD and CAT genes are overexpressed in ischemic lung tissue (56), cortical neuronal cells (80), and retinal pigment epithelial cells (234). Targeted delivery of catalase has also been used as a strategy to inhibit tumor metastasis, since H2O2, at sub-lethal concentrations, can act as intracellular second messengers to increase the transcription of genes that regulate the proliferation of tumor cells in metastatic lesions (206). Therefore, catalase can regulate ROS-mediated tissue injury and tumor metastasis. In addition to catalase, inhibitors of molecules involved in oxidative stress signaling have been used in studies where inhibitor treatment results in impaired T-cell function and promotes tumor progression. Inhibitors that target arginase 1 (Arg 1), NOS2, or peroxynitrate scavengers are able to block the immune suppressive capacity of MDSCs, and thereby restore specific T-cell responses in mouse tumor models (32, 33, 59, 152). Interestingly, inhibitors of the iNOS-peroxynitrate pathway have also been shown to enhance memory responses and block AICD in both mouse and human T cells (305). Moreover, the data presented in this study are consistent with other data, suggesting that free radicals are important in the maintenance of immune memory, and that iNOS is important in maintaining T cell homeostasis (305).
Dietary products and supplements are also known for their antioxidant properties and could potentially be useful as therapeutic agents (3, 122). The active antioxidants in dietary products, such as green tea, carrots, turmeric, and other vitamins, can modulate the T-cell immune response (Table 3). The well-studied curcumin (extracted from turmeric) induces antitumor activity in T-cell leukemia by blocking the JAK-STAT pathway (231). Based on in vivo studies, curcumin is known to restore circulating CD4+ T cells and IL-2 production (142) and to induce the STAT5a-mediated expression of the antiapoptotic protein Bcl2 (27), suggesting that reduced rates of apoptosis may be an underlying mechanism by which restoration of circulating T cells is achieved.
Similarly, specific vitamins have antioxidant function (Table 3). Vitamins A and D are required for proper homing of T cells to the gut and skin, respectively, which requires the expression of specific chemokine receptors (182). Another example of a strategy to target the harmful effects of ROS on T-cell function is the administration of high doses of dietary Vitamin E, a known antioxidant (98, 173). Based on these results, dietary Vitamin E supplementation, followed by chemotherapy treatment, increased the number of IL-2- and IFN-γ-producing T cells as well as increased NK-cell activation, thereby suggesting that Vitamin E supplement is helpful in improving immune function in these patients. Moreover, Vitamin E has been shown to suppress the activity of transcription factors NF-κB and AP-1, and suppressed CD95L mRNA expression leading to the protection of T cells in HIV-1-infected individuals (173). Therefore, the use of dietary supplements as antioxidants is a major area of research, and their role as immune modulators, as well as anti-inflammatory and anticancer agents, makes them promising candidates for future therapeutics.
Another useful method for modulating oxidative stress in T cells could be through micro-RNA-regulated expression of ROS-associated genes. miRNA are short noncoding RNA, 18–24 nucleotides in length that post-transcriptionally regulate gene expression by affecting the degradation and translation of target mRNAs (97). Efforts to gain new insights into understanding the role of miRNAs in regulating oxidative stress-related gene expression in different cell systems are being conducted, including auditory cells (303) and cardiac myocytes (47). Similarly, the effects of ROS on miRNA expression, the role of miRNAs in ROS-mediated gene expression and T-cell function, and as potential therapeutic targets are of current interest. Interestingly, a recent study has indicated that miRNA 128a regulates production of ROS by inhibiting the Bim-1 oncogene in cancer stem cells and thereby promotes cellular senescence (294). A role for miRNA-21 in controlling NO-mediated apoptosis has also been shown in endothelial cells that were subjected to shear stress (304). In addition, miR144/miR-451 protects erythrocytes from oxidative stress by a 14-3-3z- (phospho-serine/threonine-binding protein) and FoxO3-mediated mechanism (319). Megenta et al. demonstrated that miR-200c and miR-141 are upregulated in oxidative stress and play key roles in ROS-induced apoptosis of endothelial cells (169). While it is not known if miR144/miR-451- or miR-200/miR-141-mediated pathways are involved in protecting T cells from oxidative stress, recent evidence suggests that miR-155 inhibits IFN-γ signaling in CD4+ T cells (19). Interestingly, miR155 is also important in maintaining Treg homeostasis by targeting the suppressor of cytokine signaling 1 (SOCS1) protein in a Foxp3-mediated pathway, suggesting that miRNAs are important in regulating T-cell function (166). Moreover, ROS-regulated miRNA (miR-27b) overexpression has been shown to suppress NFκB activation in mouse macrophage cells, RAW264.7 (281). Whether or not miR27b is involved in regulating NFκB activation in T cells remains to be determined. These studies highlight that miRNA may be important for maintaining redox homeostasis by regulating the expression of target mRNAs associated with ROS production. Thus, targeting miRNA to regulate oxidative stress and influence expression of targeted genes that are important in disease pathogenesis could be an important future strategy.
X. Conclusion
The studies described in this review reveal that redox balance is critical for modulating T-cell activation, proliferation, and persistence. A balance of various antioxidants is required for the regulation of the cellular redox status, such as GSH, vitamins, SOD, GPX, NOX, and p53. Proper redox status will lead to proper T-cell signaling and activation. However, an imbalance in redox molecules can activate different signaling pathways, ultimately shifting the T-cell phenotype (61). In addition, the differential susceptibility of T-cell populations based on the cell surface marker expression (as identified in sections II and III) is an area that needs further investigation in most disease states. Earlier studies have looked broadly at either CD4+ or CD8+ T cells without considering the vast heterogeneity (effector, memory, and regulatory) that exists in the T-cell lineage. Therefore, it would be important to consider a comprehensive characterization of these T-cell subsets that could potentially be correlated to disease pathogenesis and progression.
The studies cited in this review lead us to conclude that, despite the redundancy in factors that regulate cellular oxidative stress in other model systems, a combination of basic, translational, and clinical research may provide further insight in T-cell immunomodulation and immunotherapeutic strategies. Manipulation of the cellular redox state could be a strategy that results in the modulation of T-cell subsets, thereby improving current immunotherapeutic approaches to treat cancer, autoimmunity, and infectious diseases.
Abbreviations Used
- AA
ascorbic acid
- AICD
activation induced cell death
- AKR1A1
aldoketo reductase family 1, member A1
- ALL
acute lymphoblastic leukemia
- ALS
amyotrophic lateral sclerosis
- APC
antigen-presenting cell
- CAT
catalase
- CMV
cytomegalo virus
- CTLs
cytotoxic T lymphocytes
- CYBA
cytochrome b-245 alpha-polypeptide
- DISC
death-inducing signaling complexes
- ERK
extracellular signal-regulated kinases
- FoxO
forkhead transcription factors of class O
- GCLM
glutamate-cysteine ligase modifier subunit
- GPX
glutathione peroxidase
- GSH
glutathione
- HDC-KO
histidine decarboxylase knockout
- HIV
human immunodeficiency virus
- •HO−
hydroxyl radical
- HO-1
heme oxygenase-1
- H2O2
hydrogen peroxide
- HSC
hematopoietic stem cell
- IFN-γ
interferon-γ
- iGSH
intracellular glutathione
- iNOS
inducible NO synthase
- JNK
c-Jun N-terminal kinase
- LAT
linker of activated T cells
- LDL
low-density lipoprotein
- MAPK
mitogen-activated protein kinase
- MDSC
myeloid-derived suppressor cell
- MHC
major histocompatibility complex
- MPO
myeloperoxidase
- MS
multiple sclerosis
- Mst1
mammalian sterile 20-like 1
- MT-COX2
mitochondrial cyclooxygenase 2
- MT-CYB
mitochondrial cytochrome B
- mTOR
mammalian target of rapamycin
- L-NAC
N-acetylcysteine
- NADPH
nicotinamide adenine dinucleotide phosphate-oxidase
- Ncf1
neutrophil cytosolic factor 1
- NDUFA
NADH dehydrogenase [ubiquinone]-1 alpha-subcomplex subunit
- NFκB
nuclear factor kappa B
- NK
natural killer
- nNOS
neuronal NOS
- NO−
nitric oxide
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- NQO1
NADPH quinone oxidoreductase-1
- NRF2
nuclear factor erythroid-2-related factor 2
- O2•−
superoxide
- ONOO−
peroxynitrite
- PBMC
peripheral blood mononuclear cell
- PD
Parkinson's disease
- PI3K
phosphoinositide 3-kinase
- PKC-theta
protein kinase C theta
- PLCgamma1
phospholipase Cgamma1
- PON1
paraoxonase-1
- Prx
pyroredoxins
- PS
phosphatidylserine
- RA
rheumatoid arthritis
- Rap1
Ras-proximate-1
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SLE
systemic lupus erythematosus
- SLP76
SH2 domain-containing leukocyte protein of 76 kDa
- SNP
single-nucleotide polymorphism
- SOD
superoxide dismutase
- TAMs
tumor-associated macrophages
- Tc
T cytotoxic
- TCM
central memory T cells
- TCR
T-cell receptor
- Teff
effector T
- TEM
effector memory T cells
- Th
T helper
- TNF
tumor necrosis factor
- TNFR1
type 1 TNF receptor
- TRAPS
TNFR1-associated periodic syndrome
- Treg
regulatory T cell
- TRX
thioredoxin
- Zap70
ζ-chain-associated protein kinase 70
Acknowledgments
This work was supported by a grant from the NIH (R01CA138930 to SM) and start-up funds from Department of Surgery at MUSC. We apologize to our colleagues for not citing all primary research articles owing to space restrictions. We are thankful to Dr. Christina V. Johnson in the Department of Microbiology & Immunology at MUSC and Dr. Thomas G. Smith in Center for Academic Excellence/Writing Center, and Dr. Jennifer G. Schnellmann within the Scientific Editing and Publications Office at MUSC for their critical suggestions in preparation of this article.
References
- 1.Adachi K. Davis MM. T-cell receptor ligation induces distinct signaling pathways in naïve vs. antigen-experienced T cells. Proc Natl Acad Sci U S A. 2011;108:1549–1554. doi: 10.1073/pnas.1017340108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adler AS. Sinha S. Kawahara TL. Zhang JY. Segal E. Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal BB. Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71:1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Ahn J. Gammon MD. Santella RM. Gaudet MM. Britton JA. Teitelbaum SL. Terry MB. Neugut AI. Josephy PD. Ambrosone CB. Myeloperoxidase genotype, fruit and vegetable consumption, and breast cancer risk. Cancer Res. 2004;64:7634–7639. doi: 10.1158/0008-5472.CAN-04-1843. [DOI] [PubMed] [Google Scholar]
- 5.Akiyama H. Hoshino K. Tokuzumi M. Teshima R. Mori H. Inakuma T. Ishiguro Y. Goda Y. Sawada J. Toyoda M. The effect of feeding carrots on immunoglobulin E production and anaphylactic response in mice. Biol Pharm Bull. 1999;22:551–555. doi: 10.1248/bpb.22.551. [DOI] [PubMed] [Google Scholar]
- 6.Aktas O. Prozorovski T. Smorodchenko A. Savaskan NE. Lauster R. Kloetzel PM. Infante-Duarte C. Brocke S. Zipp F. Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J Immunol. 2004;173:5794–5800. doi: 10.4049/jimmunol.173.9.5794. [DOI] [PubMed] [Google Scholar]
- 7.Al-Shobaili HA. Al Robaee AA. Alzolibani A. Khan MI. Rasheed Z. Hydroxyl radical modification of immunoglobulin g generated cross-reactive antibodies: its potential role in systemic lupus erythematosus. Clin Med Insights Arthritis Musculoskelet Disord. 2011;4:11–19. doi: 10.4137/CMAMD.S6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alshatwi AA. Catechin hydrate suppresses MCF-7 proliferation through TP53/Caspase-mediated apoptosis. J Exp Clin Cancer Res. 2010;29:167. doi: 10.1186/1756-9966-29-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ammirati E. Cianflone D. Vecchio V. Banfi M. Vermi AC. De Metrio M. Grigore L. Pellegatta F. Pirillo A. Garlaschelli K. Manfredi AA, et al. Effector memory T cells are associated with atherosclerosis in humans and animal models. J Am Heart Assoc. 2012;1:27–41. doi: 10.1161/JAHA.111.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen V. Christensen J. Overvad K. Tjonneland A. Vogel U. Polymorphisms in NFkB, PXR, LXR and risk of colorectal cancer in a prospective study of Danes. BMC Cancer. 2010;10:484. doi: 10.1186/1471-2407-10-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ando T. Mimura K. Johansson CC. Hanson MG. Mougiakakos D. Larsson C. Martins da Palma T. Sakurai D. Norell H. Li M. Nishimura MI, et al. Transduction with the antioxidant enzyme catalase protects human T cells against oxidative stress. J Immunol. 2008;181:8382–8390. doi: 10.4049/jimmunol.181.12.8382. [DOI] [PubMed] [Google Scholar]
- 12.Aparicio-Fernandez X. Reynoso-Camacho R. Castano-Tostado E. Garcia-Gasca T. Gonzalez de Mejia E. Guzman-Maldonado SH. Elizondo G. Yousef GG. Lila MA. Loarca-Pina G. Antiradical capacity and induction of apoptosis on HeLa cells by a Phaseolus vulgaris extract. Plant Foods Hum Nutr. 2008;63:35–40. doi: 10.1007/s11130-007-0066-4. [DOI] [PubMed] [Google Scholar]
- 13.Appel SH. Beers DR. Henkel JS. T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol. 2010;31:7–17. doi: 10.1016/j.it.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atkuri KR. Mantovani JJ. Herzenberg LA. N-Acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avalos I. Chung CP. Oeser A. Milne GL. Morrow JD. Gebretsadik T. Shintani A. Yu C. Stein CM. Oxidative stress in systemic lupus erythematosus: relationship to disease activity and symptoms. Lupus. 2007;16:195–200. doi: 10.1177/0961203306075802. [DOI] [PubMed] [Google Scholar]
- 16.Babior BM. The respiratory burst oxidase. Adv Enzymol Relat Areas Mol Biol. 1992;65:49–95. doi: 10.1002/9780470123119.ch2. [DOI] [PubMed] [Google Scholar]
- 17.Baek KH. Shin HJ. Yoo JK. Cho JH. Choi YH. Sung YC. McKeon F. Lee CW. p53 deficiency and defective mitotic checkpoint in proliferating T lymphocytes increase chromosomal instability through aberrant exit from mitotic arrest. J Leukoc Biol. 2003;73:850–861. doi: 10.1189/jlb.1202607. [DOI] [PubMed] [Google Scholar]
- 18.Bahl K. Kim SK. Calcagno C. Ghersi D. Puzone R. Celada F. Selin LK. Welsh RM. IFN-induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol. 2006;176:4284–4295. doi: 10.4049/jimmunol.176.7.4284. [DOI] [PubMed] [Google Scholar]
- 19.Banerjee A. Schambach F. DeJong CS. Hammond SM. Reiner SL. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur J Immunol. 2010;40:225–231. doi: 10.1002/eji.200939381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barata JT. Cardoso AA. Boussiotis VA. Interleukin-7 in T-cell acute lymphoblastic leukemia: an extrinsic factor supporting leukemogenesis? Leuk Lymphoma. 2005;46:483–495. doi: 10.1080/10428190400027852. [DOI] [PubMed] [Google Scholar]
- 21.Barfod L. Kemp K. Hansen M. Kharazmi A. Chalcones from Chinese liquorice inhibit proliferation of T cells and production of cytokines. Int Immunopharmacol. 2002;2:545–555. doi: 10.1016/s1567-5769(01)00202-8. [DOI] [PubMed] [Google Scholar]
- 22.Beers DR. Henkel JS. Zhao W. Wang J. Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beers DR. Henkel JS. Zhao W. Wang J. Huang A. Wen S. Liao B. Appel SH. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134:1293–1314. doi: 10.1093/brain/awr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beeson CC. Beeson GC. Schnellmann RG. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem. 2010;404:75–81. doi: 10.1016/j.ab.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett MR. Reactive oxygen species and death: oxidative DNA damage in atherosclerosis. Circ Res. 2001;88:648–650. doi: 10.1161/hh0701.089955. [DOI] [PubMed] [Google Scholar]
- 26.Berner V. Liu H. Zhou Q. Alderson KL. Sun K. Weiss JM. Back TC. Longo DL. Blazar BR. Wiltrout RH. Welniak LA, et al. IFN-gamma mediates CD4+ T-cell loss and impairs secondary antitumor responses after successful initial immunotherapy. Nat Med. 2007;13:354–360. doi: 10.1038/nm1554. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharyya S. Mandal D. Saha B. Sen GS. Das T. Sa G. Curcumin prevents tumor-induced T cell apoptosis through Stat-5a-mediated Bcl-2 induction. J Biol Chem. 2007;282:15954–15964. doi: 10.1074/jbc.M608189200. [DOI] [PubMed] [Google Scholar]
- 28.Bird MD. Karavitis J. Kovacs EJ. Sex differences and estrogen modulation of the cellular immune response after injury. Cell Immunol. 2008;252:57–67. doi: 10.1016/j.cellimm.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjartmar C. Wujek JR. Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- 30.Bogdan C. Regulation of lymphocytes by nitric oxide. Methods Mol Biol. 2011;677:375–393. doi: 10.1007/978-1-60761-869-0_24. [DOI] [PubMed] [Google Scholar]
- 31.Bristow RG. Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 32.Bronte V. Serafini P. De Santo C. Marigo I. Tosello V. Mazzoni A. Segal DM. Staib C. Lowel M. Sutter G. Colombo MP, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–278. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 33.Bronte V. Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 34.Buhl R. Jaffe HA. Holroyd KJ. Wells FB. Mastrangeli A. Saltini C. Cantin AM. Crystal RG. Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet. 1989;2:1294–1298. doi: 10.1016/s0140-6736(89)91909-0. [DOI] [PubMed] [Google Scholar]
- 35.Bulua AC. Simon A. Maddipati R. Pelletier M. Park H. Kim KY. Sack MN. Kastner DL. Siegel RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burbach BJ. Medeiros RB. Mueller KL. Shimizu Y. T-cell receptor signaling to integrins. Immunol Rev. 2007;218:65–81. doi: 10.1111/j.1600-065X.2007.00527.x. [DOI] [PubMed] [Google Scholar]
- 37.Cacciatore I. Cornacchia C. Pinnen F. Mollica A. Di Stefano A. Prodrug approach for increasing cellular glutathione levels. Molecules. 2010;15:1242–1264. doi: 10.3390/molecules15031242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cappellini A. Tazzari PL. Mantovani I. Billi AM. Tassi C. Ricci F. Conte R. Martelli AM. Antiapoptotic role of p38 mitogen activated protein kinase in Jurkat T cells and normal human T lymphocytes treated with 8-methoxypsoralen and ultraviolet-A radiation. Apoptosis. 2005;10:141–152. doi: 10.1007/s10495-005-6069-4. [DOI] [PubMed] [Google Scholar]
- 39.Carrette F. Fabre S. Bismuth G. FOXO1, T-cell trafficking and immune responses. Adv Exp Med Biol. 2009;665:3–16. doi: 10.1007/978-1-4419-1599-3_1. [DOI] [PubMed] [Google Scholar]
- 40.Case AJ. McGill JL. Tygrett LT. Shirasawa T. Spitz DR. Waldschmidt TJ. Legge KL. Domann FE. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free Radic Biol Med. 2011;50:448–458. doi: 10.1016/j.freeradbiomed.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cassileth B. Lycopene. Oncology (Williston Park) 2010;24:296. [PubMed] [Google Scholar]
- 42.Cecchinato V. Chiaramonte R. Nizzardo M. Cristofaro B. Basile A. Sherbet GV. Comi P. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem Pharmacol. 2007;74:1568–1574. doi: 10.1016/j.bcp.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Cha MK. Suh KH. Kim IH. Overexpression of peroxiredoxin I and thioredoxin1 in human breast carcinoma. J Exp Clin Cancer Res. 2009;28:93. doi: 10.1186/1756-9966-28-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang SH. Chung Y. Dong C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. J Biol Chem. 2010;285:38751–38755. doi: 10.1074/jbc.C110.185777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen C. Liu Y. Zheng D. An agonistic monoclonal antibody against DR5 induces ROS production, sustained JNK activation and Endo G release in Jurkat leukemia cells. Cell Res. 2009;19:984–995. doi: 10.1038/cr.2009.60. [DOI] [PubMed] [Google Scholar]
- 46.Chen SJ. Wang YL. Lo WT. Wu CC. Hsieh CW. Huang CF. Lan YH. Wang CC. Chang DM. Sytwu HK. Erythropoietin enhances endogenous haem oxygenase-1 and represses immune responses to ameliorate experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:210–223. doi: 10.1111/j.1365-2249.2010.04238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng AM. Byrom MW. Shelton J. Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho HY. Jedlicka AE. Reddy SP. Zhang LY. Kensler TW. Kleeberger SR. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am J Respir Cell Mol Biol. 2002;26:42–51. doi: 10.1165/ajrcmb.26.1.4536. [DOI] [PubMed] [Google Scholar]
- 49.Choi J. Oh S. Lee D. Oh HJ. Park JY. Lee SB. Lim DS. Mst1-FoxO signaling protects Naïve T lymphocytes from cellular oxidative stress in mice. PLoS One. 2009;4:e8011. doi: 10.1371/journal.pone.0008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christensen LP. Aliphatic c(17)-polyacetylenes of the falcarinol type as potential health promoting compounds in food plants of the apiaceae family. Recent Pat Food Nutr Agric. 2011;3:64–77. doi: 10.2174/2212798411103010064. [DOI] [PubMed] [Google Scholar]
- 51.Clements PJ. Roth MD. Elashoff R. Tashkin DP. Goldin J. Silver RM. Sterz M. Seibold JR. Schraufnagel D. Simms RW. Bolster M, et al. Scleroderma lung study (SLS): differences in the presentation and course of patients with limited versus diffuse systemic sclerosis. Ann Rheum Dis. 2007;66:1641–1647. doi: 10.1136/ard.2007.069518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colic M. Vucevic D. Kilibarda V. Radicevic N. Savic M. Modulatory effects of garlic extracts on proliferation of T-lymphocytes in vitro stimulated with concanavalin A. Phytomedicine. 2002;9:117–124. doi: 10.1078/0944-7113-00093. [DOI] [PubMed] [Google Scholar]
- 53.Crack PJ. Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med. 2005;38:1433–1444. doi: 10.1016/j.freeradbiomed.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 54.Csiszar A. Ungvari Z. Koller A. Edwards JG. Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. 2003;17:1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 55.D'Souza WN. Chang CF. Fischer AM. Li M. Hedrick SM. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol. 2008;181:7617–7629. doi: 10.4049/jimmunol.181.11.7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Danel C. Erzurum SC. Prayssac P. Eissa NT. Crystal RG. Herve P. Baudet B. Mazmanian M. Lemarchand P. Gene therapy for oxidant injury-related diseases: adenovirus-mediated transfer of superoxide dismutase and catalase cDNAs protects against hyperoxia but not against ischemia-reperfusion lung injury. Hum Gene Ther. 1998;9:1487–1496. doi: 10.1089/hum.1998.9.10-1487. [DOI] [PubMed] [Google Scholar]
- 57.Daynes RA. Enioutina EY. Jones DC. Role of redox imbalance in the molecular mechanisms responsible for immunosenescence. Antioxid Redox Signal. 2003;5:537–548. doi: 10.1089/152308603770310185. [DOI] [PubMed] [Google Scholar]
- 58.de la Fuente H. Richaud-Patin Y. Jakez-Ocampo J. Gonzalez-Amaro R. Llorente L. Innate immune mechanisms in the pathogenesis of systemic lupus erythematosus (SLE) Immunol Lett. 2001;77:175–180. doi: 10.1016/s0165-2478(01)00220-6. [DOI] [PubMed] [Google Scholar]
- 59.De Santo C. Serafini P. Marigo I. Dolcetti L. Bolla M. Del Soldato P. Melani C. Guiducci C. Colombo MP. Iezzi M. Musiani P, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A. 2005;102:4185–4190. doi: 10.1073/pnas.0409783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Desaint S. Luriau S. Aude JC. Rousselet G. Toledano MB. Mammalian antioxidant defenses are not inducible by H2O2. J Biol Chem. 2004;279:31157–31163. doi: 10.1074/jbc.M401888200. [DOI] [PubMed] [Google Scholar]
- 61.Devadas S. Zaritskaya L. Rhee SG. Oberley L. Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med. 2002;195:59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donaldson MS. Nutrition and cancer: a review of the evidence for an anti-cancer diet. Nutr J. 2004;3:19. doi: 10.1186/1475-2891-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong C. Yang DD. Wysk M. Whitmarsh AJ. Davis RJ. Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 64.Dong C. Yang DD. Tournier C. Whitmarsh AJ. Xu J. Davis RJ. Flavell RA. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- 65.Eck HP. Frey H. Droge W. Elevated plasma glutamate concentrations in HIV-1-infected patients may contribute to loss of macrophage and lymphocyte functions. Int Immunol. 1989;1:367–372. doi: 10.1093/intimm/1.4.367. [DOI] [PubMed] [Google Scholar]
- 66.Eck HP. Gmunder H. Hartmann M. Petzoldt D. Daniel V. Droge W. Low concentrations of acid-soluble thiol (cysteine) in the blood plasma of HIV-1-infected patients. Biol Chem Hoppe Seyler. 1989;370:101–108. doi: 10.1515/bchm3.1989.370.1.101. [DOI] [PubMed] [Google Scholar]
- 67.Eck HP. Stahl-Hennig C. Hunsmann G. Droge W. Metabolic disorder as early consequence of simian immunodeficiency virus infection in rhesus macaques. Lancet. 1991;338:346–347. doi: 10.1016/0140-6736(91)90482-5. [DOI] [PubMed] [Google Scholar]
- 68.Efimova O. Szankasi P. Kelley TW. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+ effector T cells. PLoS One. 2011;6:e16013. doi: 10.1371/journal.pone.0016013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emerson MR. LeVine SM. Heme oxygenase-1 and NADPH cytochrome P450 reductase expression in experimental allergic encephalomyelitis: an expanded view of the stress response. J Neurochem. 2000;75:2555–2562. doi: 10.1046/j.1471-4159.2000.0752555.x. [DOI] [PubMed] [Google Scholar]
- 70.Epperly MW. Bray JA. Esocobar P. Bigbee WL. Watkins S. Greenberger JS. Overexpression of the human manganese superoxide dismutase (MnSOD) transgene in subclones of murine hematopoietic progenitor cell line 32D cl 3 decreases irradiation-induced apoptosis but does not alter G2/M or G1/S phase cell cycle arrest. Radiat Oncol Investig. 1999;7:331–342. doi: 10.1002/(SICI)1520-6823(1999)7:6<331::AID-ROI3>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 71.Escobales N. Crespo MJ. Oxidative-nitrosative stress in hypertension. Curr Vasc Pharmacol. 2005;3:231–246. doi: 10.2174/1570161054368643. [DOI] [PubMed] [Google Scholar]
- 72.Fabre S. Carrette F. Chen J. Lang V. Semichon M. Denoyelle C. Lazar V. Cagnard N. Dubart-Kupperschmitt A. Mangeney M. Fruman DA, et al. FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol. 2008;181:2980–2989. doi: 10.4049/jimmunol.181.5.2980. [DOI] [PubMed] [Google Scholar]
- 73.Feldmann M. Brennan FM. Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 74.Fiers W. Beyaert R. Declercq W. Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 75.Franco R. Panayiotidis MI. Cidlowski JA. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem. 2007;282:30452–30465. doi: 10.1074/jbc.M703091200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frelin C. Imbert V. Griessinger E. Peyron AC. Rochet N. Philip P. Dageville C. Sirvent A. Hummelsberger M. Berard E. Dreano M, et al. Targeting NF-kappaB activation via pharmacologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood. 2005;105:804–811. doi: 10.1182/blood-2004-04-1463. [DOI] [PubMed] [Google Scholar]
- 77.Frossi B. De Carli M. Piemonte M. Pucillo C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol Immunol. 2008;45:58–64. doi: 10.1016/j.molimm.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 78.Frostegard J. Wu R. Giscombe R. Holm G. Lefvert AK. Nilsson J. Induction of T-cell activation by oxidized low density lipoprotein. Arterioscler Thromb. 1992;12:461–467. doi: 10.1161/01.atv.12.4.461. [DOI] [PubMed] [Google Scholar]
- 79.Fuschiotti P. CD8+ T cells in systemic sclerosis. Immunol Res. 2011;50:188–194. doi: 10.1007/s12026-011-8222-1. [DOI] [PubMed] [Google Scholar]
- 80.Gaspar T. Domoki F. Lenti L. Institoris A. Snipes JA. Bari F. Busija DW. Neuroprotective effect of adenoviral catalase gene transfer in cortical neuronal cultures. Brain Res. 2009;1270:1–9. doi: 10.1016/j.brainres.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gelderman KA. Hultqvist M. Holmberg J. Olofsson P. Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A. 2006;103:12831–12836. doi: 10.1073/pnas.0604571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gelderman KA. Hultqvist M. Olsson LM. Bauer K. Pizzolla A. Olofsson P. Holmdahl R. Rheumatoid arthritis: the role of reactive oxygen species in disease development and therapeutic strategies. Antioxid Redox Signal. 2007;9:1541–1567. doi: 10.1089/ars.2007.1569. [DOI] [PubMed] [Google Scholar]
- 83.Gergely P., Jr. Grossman C. Niland B. Puskas F. Neupane H. Allam F. Banki K. Phillips PE. Perl A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:175–190. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gergely P., Jr. Niland B. Gonchoroff N. Pullmann R., Jr. Phillips PE. Perl A. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol. 2002;169:1092–1101. doi: 10.4049/jimmunol.169.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giacco F. Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gil-DelValle L. Gonzalez I. Tarinas A. Al-Verez A. Molina R. Tapanes R. Perez J. Evaluation of oxidative stress in AIDS pediatric patients. Acta Farm Bonaerense. 2004;23:466–471. [Google Scholar]
- 87.Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond) 2011;120:99–120. doi: 10.1042/CS20090603. [DOI] [PubMed] [Google Scholar]
- 88.Go YM. Kang SM. Roede JR. Orr M. Jones DP. Increased inflammatory signaling and lethality of influenza H1N1 by nuclear thioredoxin-1. PLoS One. 2011;6:e18918. doi: 10.1371/journal.pone.0018918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goth L. Rass P. Pay A. Catalase enzyme mutations and their association with diseases. Mol Diagn. 2004;8:141–149. doi: 10.1007/BF03260057. [DOI] [PubMed] [Google Scholar]
- 90.Griffith CE. Zhang W. Wange RL. ZAP-70-dependent and -independent activation of Erk in Jurkat T cells. Differences in signaling induced by H2o2 and Cd3 cross-linking. J Biol Chem. 1998;273:10771–10776. doi: 10.1074/jbc.273.17.10771. [DOI] [PubMed] [Google Scholar]
- 91.Gringhuis SI. Leow A. Papendrecht-Van Der Voort EA. Remans PH. Breedveld FC. Verweij CL. Displacement of linker for activation of T cells from the plasma membrane due to redox balance alterations results in hyporesponsiveness of synovial fluid T lymphocytes in rheumatoid arthritis. J Immunol. 2000;164:2170–2179. doi: 10.4049/jimmunol.164.4.2170. [DOI] [PubMed] [Google Scholar]
- 92.Gulow K. Kaminski M. Darvas K. Suss D. Li-Weber M. Krammer PH. HIV-1 trans-activator of transcription substitutes for oxidative signaling in activation-induced T cell death. J Immunol. 2005;174:5249–5260. doi: 10.4049/jimmunol.174.9.5249. [DOI] [PubMed] [Google Scholar]
- 93.Guzik TJ. Hoch NE. Brown KA. McCann LA. Rahman A. Dikalov S. Goronzy J. Weyand C. Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haanen JB. Schumacher TN. Vaccine leads to memory loss. Nat Med. 2007;13:248–250. doi: 10.1038/nm0307-248. [DOI] [PubMed] [Google Scholar]
- 95.Hagenow K. Gelderman KA. Hultqvist M. Merky P. Backlund J. Frey O. Kamradt T. Holmdahl R. Ncf1-associated reduced oxidative burst promotes IL-33R+ T cell-mediated adjuvant-free arthritis in mice. J Immunol. 2009;183:874–881. doi: 10.4049/jimmunol.0900966. [DOI] [PubMed] [Google Scholar]
- 96.Hainaut P. Milner J. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res. 1993;53:4469–4473. [PubMed] [Google Scholar]
- 97.Hammond SM. RNAi, microRNAs, and human disease. Cancer Chemother Pharmacol. 2006;58(Suppl 1):s63–s68. doi: 10.1007/s00280-006-0318-2. [DOI] [PubMed] [Google Scholar]
- 98.Hanson MG. Ozenci V. Carlsten MC. Glimelius BL. Frodin JE. Masucci G. Malmberg KJ. Kiessling RV. A short-term dietary supplementation with high doses of vitamin E increases NK cell cytolytic activity in advanced colorectal cancer patients. Cancer Immunol Immunother. 2007;56:973–984. doi: 10.1007/s00262-006-0261-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hansson GK. Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 100.Hardwick JS. Sefton BM. Activation of the Lck tyrosine protein kinase by hydrogen peroxide requires the phosphorylation of Tyr-394. Proc Natl Acad Sci U S A. 1995;92:4527–4531. doi: 10.1073/pnas.92.10.4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hernandez-Saavedra D. Zhou H. McCord JM. Anti-inflammatory properties of a chimeric recombinant superoxide dismutase: SOD2/3. Biomed Pharmacother. 2005;59:204–208. doi: 10.1016/j.biopha.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 102.Hernandez-Saavedra D. McCord JM. Association of a new intronic polymorphism of the SOD2 gene (G1677T) with cancer. Cell Biochem Funct. 2009;27:223–227. doi: 10.1002/cbf.1560. [DOI] [PubMed] [Google Scholar]
- 103.Herzenberg LA. De Rosa SC. Dubs JG. Roederer M. Anderson MT. Ela SW. Deresinski SC. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci U S A. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hibi M. Lin A. Smeal T. Minden A. Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 105.Hildeman DA. Mitchell T. Teague TK. Henson P. Day BJ. Kappler J. Marrack PC. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- 106.Hildeman DA. Mitchell T. Kappler J. Marrack P. T cell apoptosis and reactive oxygen species. J Clin Invest. 2003;111:575–581. doi: 10.1172/JCI18007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoch NE. Guzik TJ. Chen W. Deans T. Maalouf SA. Gratze P. Weyand C. Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–R216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hoffman RA. Mahidhara RS. Wolf-Johnston AS. Lu L. Thomson AW. Simmons RL. Differential modulation of CD4 and CD8 T-cell proliferation by induction of nitric oxide synthesis in antigen presenting cells. Transplantation. 2002;74:836–845. doi: 10.1097/00007890-200209270-00018. [DOI] [PubMed] [Google Scholar]
- 109.Holley AK. Dhar SK. St Clair DK. Manganese superoxide dismutase vs. p53: regulation of mitochondrial ROS. Mitochondrion. 2010;10:649–661. doi: 10.1016/j.mito.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 110.Holmstrom TH. Schmitz I. Soderstrom TS. Poukkula M. Johnson VL. Chow SC. Krammer PH. Eriksson JE. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J. 2000;19:5418–5428. doi: 10.1093/emboj/19.20.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Howard AN. Bridges KA. Meyn RE. Chandra J. ABT-737, a BH3 mimetic, induces glutathione depletion and oxidative stress. Cancer Chemother Pharmacol. 2009;65:41–54. doi: 10.1007/s00280-009-1001-1. [DOI] [PubMed] [Google Scholar]
- 112.Huang CP. Au LC. Chiou RY. Chung PC. Chen SY. Tang WC. Chang CL. Fang WH. Lin SB. Arachidin-1, a peanut stilbenoid, induces programmed cell death in human leukemia HL-60 cells. J Agric Food Chem. 2010;58:12123–12129. doi: 10.1021/jf102993j. [DOI] [PubMed] [Google Scholar]
- 113.Huang SC. Wei JC. Wu DJ. Huang YC. Vitamin B(6) supplementation improves pro-inflammatory responses in patients with rheumatoid arthritis. Eur J Clin Nutr. 2010;64:1007–1013. doi: 10.1038/ejcn.2010.107. [DOI] [PubMed] [Google Scholar]
- 114.Huang YC. Guh JH. Cheng ZJ. Chang YL. Hwang TL. Lin CN. Teng CM. Inhibitory effect of DCDC on lipopolysaccharide-induced nitric oxide synthesis in RAW 264.7 cells. Life Sci. 2001;68:2435–2447. doi: 10.1016/s0024-3205(01)01035-9. [DOI] [PubMed] [Google Scholar]
- 115.Hultqvist M. Olofsson P. Gelderman KA. Holmberg J. Holmdahl R. A new arthritis therapy with oxidative burst inducers. PLoS Med. 2006;3:e348. doi: 10.1371/journal.pmed.0030348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hultqvist M. Backlund J. Bauer K. Gelderman KA. Holmdahl R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J Immunol. 2007;179:1431–1437. doi: 10.4049/jimmunol.179.3.1431. [DOI] [PubMed] [Google Scholar]
- 117.Ichihara F. Kono K. Sekikawa T. Matsumoto Y. Surgical stress induces decreased expression of signal-transducing zeta molecules in T cells. Eur Surg Res. 1999;31:138–146. doi: 10.1159/000008632. [DOI] [PubMed] [Google Scholar]
- 118.Ishikawa K. Takenaga K. Akimoto M. Koshikawa N. Yamaguchi A. Imanishi H. Nakada K. Honma Y. Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 119.Iwata M. Hirakiyama A. Eshima Y. Kagechika H. Kato C. Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 120.Iwata S. Hori T. Sato N. Hirota K. Sasada T. Mitsui A. Hirakawa T. Yodoi J. Adult T cell leukemia (ATL)-derived factor/human thioredoxin prevents apoptosis of lymphoid cells induced by L-cystine and glutathione depletion: possible involvement of thiol-mediated redox regulation in apoptosis caused by pro-oxidant state. J Immunol. 1997;158:3108–3117. [PubMed] [Google Scholar]
- 121.Jaarsma D. Haasdijk ED. Grashorn JA. Hawkins R. van Duijn W. Verspaget HW. London J. Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- 122.Jagetia GC. Aggarwal BB. “Spicing up” of the immune system by curcumin. J Clin Immunol. 2007;27:19–35. doi: 10.1007/s10875-006-9066-7. [DOI] [PubMed] [Google Scholar]
- 123.Jain MR. Ge WW. Elkabes S. Li H. Amyotrophic lateral sclerosis: protein chaperone dysfunction revealed by proteomic studies of animal models. Proteomics Clin Appl. 2008;2:670–684. doi: 10.1002/prca.200780023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jeong YJ. Hong SW. Kim JH. Jin DH. Kang JS. Lee WJ. Hwang YI. Vitamin C-treated murine bone marrow-derived dendritic cells preferentially drive naïve T cells into Th1 cells by increased IL-12 secretions. Cell Immunol. 2011;266:192–199. doi: 10.1016/j.cellimm.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 125.Jia Z. Song Z. Zhao Y. Wang X. Liu P. Grape seed proanthocyanidin extract protects human lens epithelial cells from oxidative stress via reducing NF-small ka, CyrillicB and MAPK protein expression. Mol Vis. 2011;17:210–217. [PMC free article] [PubMed] [Google Scholar]
- 126.Johnson DA. Amirahmadi S. Ward C. Fabry Z. Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Joosse A. De Vries E. van Eijck CH. Eggermont AM. Nijsten T. Coebergh JW. Reactive oxygen species and melanoma: an explanation for gender differences in survival? Pigment Cell Melanoma Res. 2010;23:352–364. doi: 10.1111/j.1755-148X.2010.00694.x. [DOI] [PubMed] [Google Scholar]
- 128.Kaminski M. Kiessling M. Suss D. Krammer PH. Gulow K. Novel role for mitochondria: protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol. 2007;27:3625–3639. doi: 10.1128/MCB.02295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kaminski MM. Sauer SW. Klemke CD. Suss D. Okun JG. Krammer PH. Gulow K. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol. 2010;184:4827–4841. doi: 10.4049/jimmunol.0901662. [DOI] [PubMed] [Google Scholar]
- 130.Kane LP. Lin J. Weiss A. It's all Rel-ative: NF-kappaB and CD28 costimulation of T-cell activation. Trends Immunol. 2002;23:413–420. doi: 10.1016/s1471-4906(02)02264-0. [DOI] [PubMed] [Google Scholar]
- 131.Kannan K. Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153–163. doi: 10.1016/s0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- 132.Karawajew L. Rhein P. Czerwony G. Ludwig WD. Stress-induced activation of the p53 tumor suppressor in leukemia cells and normal lymphocytes requires mitochondrial activity and reactive oxygen species. Blood. 2005;105:4767–4775. doi: 10.1182/blood-2004-09-3428. [DOI] [PubMed] [Google Scholar]
- 133.Karpuzoglu E. Ahmed SA. Estrogen regulation of nitric oxide and inducible nitric oxide synthase (iNOS) in immune cells: implications for immunity, autoimmune diseases, and apoptosis. Nitric Oxide. 2006;15:177–186. doi: 10.1016/j.niox.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 134.Kaschula CH. Hunter R. Parker MI. Garlic-derived anticancer agents: structure and biological activity of ajoene. Biofactors. 2010;36:78–85. doi: 10.1002/biof.76. [DOI] [PubMed] [Google Scholar]
- 135.Kasic T. Colombo P. Soldani C. Wang CM. Miranda E. Roncalli M. Bronte V. Viola A. Modulation of human T cell functions by reactive nitrogen species. Eur J Immunol. 2011;41:1843–1849. doi: 10.1002/eji.201040868. [DOI] [PubMed] [Google Scholar]
- 136.Kaszubowska L. Telomere shortening and ageing of the immune system. J Physiol Pharmacol. 2008;59(Suppl 9):169–186. [PubMed] [Google Scholar]
- 137.Katakami N. Sakamoto K. Kaneto H. Matsuhisa M. Shimizu I. Ishibashi F. Osonoi T. Kashiwagi A. Kawamori R. Hori M. Yamasaki Y. Combined effect of oxidative stress-related gene polymorphisms on atherosclerosis. Biochem Biophys Res Commun. 2009;379:861–865. doi: 10.1016/j.bbrc.2008.12.154. [DOI] [PubMed] [Google Scholar]
- 138.Kaur N. Naga OS. Norell H. Al-Khami AA. Scheffel MJ. Chakraborty NG. Voelkel-Johnson C. Mukherji B. Mehrotra S. T cells expanded in presence of IL-15 exhibit increased antioxidant capacity and innate effector molecules. Cytokine. 2011;55:307–317. doi: 10.1016/j.cyto.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kerdiles YM. Beisner DR. Tinoco R. Dejean AS. Castrillon DH. DePinho RA. Hedrick SM. Foxo1 links homing and survival of naïve T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kikuchi N. Ishii Y. Morishima Y. Yageta Y. Haraguchi N. Itoh K. Yamamoto M. Hizawa N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010;11:31. doi: 10.1186/1465-9921-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kim HJ. Barajas B. Wang M. Nel AE. Nrf2 activation by sulforaphane restores the age-related decrease of T(H)1 immunity: role of dendritic cells. J Allergy Clin Immunol. 2008;121:1255–1261, e1257. doi: 10.1016/j.jaci.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim W. Fan YY. Smith R. Patil B. Jayaprakasha GK. McMurray DN. Chapkin RS. Dietary curcumin and limonin suppress CD4+ T-cell proliferation and interleukin-2 production in mice. J Nutr. 2009;139:1042–1048. doi: 10.3945/jn.108.102772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim WH. Goo SY. Shin MH. Chun SJ. Lee H. Lee KH. Park SJ. Vibrio vulnificus-induced death of Jurkat T-cells requires activation of p38 mitogen-activated protein kinase by NADPH oxidase-derived reactive oxygen species. Cell Immunol. 2008;253:81–91. doi: 10.1016/j.cellimm.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 144.Kipnis J. Avidan H. Caspi RR. Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14663–14669. doi: 10.1073/pnas.0404842101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kline MP. Rajkumar SV. Timm MM. Kimlinger TK. Haug JL. Lust JA. Greipp PR. Kumar S. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21:1549–1560. doi: 10.1038/sj.leu.2404719. [DOI] [PubMed] [Google Scholar]
- 146.Koike T. Yamagishi H. Hatanaka Y. Fukushima A. Chang JW. Xia Y. Fields M. Chandler P. Iwashima M. A novel ERK-dependent signaling process that regulates interleukin-2 expression in a late phase of T cell activation. J Biol Chem. 2003;278:15685–15692. doi: 10.1074/jbc.M210829200. [DOI] [PubMed] [Google Scholar]
- 147.Koncz A. Pasztoi M. Mazan M. Fazakas F. Buzas E. Falus A. Nagy G. Nitric oxide mediates T cell cytokine production and signal transduction in histidine decarboxylase knockout mice. J Immunol. 2007;179:6613–6619. doi: 10.4049/jimmunol.179.10.6613. [DOI] [PubMed] [Google Scholar]
- 148.Koura T. Gon Y. Hashimoto S. Azuma A. Kudoh S. Fukuda Y. Sugawara I. Yodoi J. Horie T. Expression of thioredoxin in granulomas of sarcoidosis: possible role in the development of T lymphocyte activation. Thorax. 2000;55:755–761. doi: 10.1136/thorax.55.9.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kraaij MD. Savage ND. van der Kooij SW. Koekkoek K. Wang J. van den Berg JM. Ottenhoff TH. Kuijpers TW. Holmdahl R. van Kooten C. Gelderman KA. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107:17686–17691. doi: 10.1073/pnas.1012016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Krajinovic M. Sinnett H. Richer C. Labuda D. Sinnett D. Role of NQO1, MPO and CYP2E1 genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Int J Cancer. 2002;97:230–236. doi: 10.1002/ijc.1589. [DOI] [PubMed] [Google Scholar]
- 151.Kusmartsev S. Nefedova Y. Yoder D. Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 152.Kusmartsev S. Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–4891. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 153.Lan Q. Zheng T. Shen M. Zhang Y. Wang SS. Zahm SH. Holford TR. Leaderer B. Boyle P. Chanock S. Genetic polymorphisms in the oxidative stress pathway and susceptibility to non-Hodgkin lymphoma. Hum Genet. 2007;121:161–168. doi: 10.1007/s00439-006-0288-9. [DOI] [PubMed] [Google Scholar]
- 154.Laniewski NG. Grayson JM. Antioxidant treatment reduces expansion and contraction of antigen-specific CD8+ T cells during primary but not secondary viral infection. J Virol. 2004;78:11246–11257. doi: 10.1128/JVI.78.20.11246-11257.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee K. Won HY. Bae MA. Hong JH. Hwang ES. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci U S A. 2011;108:9548–9553. doi: 10.1073/pnas.1012645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lei XG. Vatamaniuk MZ. Two tales of antioxidant enzymes on beta cells and diabetes. Antioxid Redox Signal. 2011;14:489–503. doi: 10.1089/ars.2010.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li-Weber M. Weigand MA. Giaisi M. Suss D. Treiber MK. Baumann S. Ritsou E. Breitkreutz R. Krammer PH. Vitamin E inhibits CD95 ligand expression and protects T cells from activation-induced cell death. J Clin Invest. 2002;110:681–690. doi: 10.1172/JCI15073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li W. Nie S. Xie M. Chen Y. Li C. Zhang H. A major green tea component, (−)-epigallocatechin-3-gallate, ameliorates doxorubicin-mediated cardiotoxicity in cardiomyocytes of neonatal rats. J Agric Food Chem. 2010;58:8977–8982. doi: 10.1021/jf101277t. [DOI] [PubMed] [Google Scholar]
- 159.Liesz A. Suri-Payer E. Veltkamp C. Doerr H. Sommer C. Rivest S. Giese T. Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 160.Lin J. Weiss A. T cell receptor signalling. J Cell Sci. 2001;114:243–244. doi: 10.1242/jcs.114.2.243. [DOI] [PubMed] [Google Scholar]
- 161.Linton PJ. Haynes L. Klinman NR. Swain SL. Antigen-independent changes in naïve CD4 T cells with aging. J Exp Med. 1996;184:1891–1900. doi: 10.1084/jem.184.5.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu L. Li C. Gao J. Li K. Zhang R. Wang G. Gao T. Promoter variant in the catalase gene is associated with vitiligo in Chinese people. J Invest Dermatol. 2010;130:2647–2653. doi: 10.1038/jid.2010.192. [DOI] [PubMed] [Google Scholar]
- 163.Liu Y. Zhu B. Luo L. Li P. Paty DW. Cynader MS. Heme oxygenase-1 plays an important protective role in experimental autoimmune encephalomyelitis. Neuroreport. 2001;12:1841–1845. doi: 10.1097/00001756-200107030-00016. [DOI] [PubMed] [Google Scholar]
- 164.Lob HE. Marvar PJ. Guzik TJ. Sharma S. McCann LA. Weyand C. Gordon FJ. Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lotharius J. Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2002;11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- 166.Lu LF. Thai TH. Calado DP. Chaudhry A. Kubo M. Tanaka K. Loeb GB. Lee H. Yoshimura A. Rajewsky K. Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lucas SM. Rothwell NJ. Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ma Q. Wang Y. Lo AS. Gomes EM. Junghans RP. Cell density plays a critical role in ex vivo expansion of T cells for adoptive immunotherapy. J Biomed Biotechnol. 2010;2010:386545. doi: 10.1155/2010/386545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Magenta A. Cencioni C. Fasanaro P. Zaccagnini G. Greco S. Sarra-Ferraris G. Antonini A. Martelli F. Capogrossi MC. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18:1628–1639. doi: 10.1038/cdd.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Maggini S. Wintergerst ES. Beveridge S. Hornig DH. Selected vitamins and trace elements support immune function by strengthening epithelial barriers and cellular and humoral immune responses. Br J Nutr. 2007;98(Suppl 1):S29–S35. doi: 10.1017/S0007114507832971. [DOI] [PubMed] [Google Scholar]
- 171.Malling TH. Sigsgaard T. Andersen HR. Deguchi Y. Brandslund I. Skadhauge L. Thomsen G. Baelum J. Sherson D. Omland O. Differences in associations between markers of antioxidative defense and asthma are sex specific. Gend Med. 2010;7:115–124. doi: 10.1016/j.genm.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 172.Malmberg KJ. Arulampalam V. Ichihara F. Petersson M. Seki K. Andersson T. Lenkei R. Masucci G. Pettersson S. Kiessling R. Inhibition of activated/memory (CD45RO(+)) T cells by oxidative stress associated with block of NF-kappaB activation. J Immunol. 2001;167:2595–2601. doi: 10.4049/jimmunol.167.5.2595. [DOI] [PubMed] [Google Scholar]
- 173.Malmberg KJ. Lenkei R. Petersson M. Ohlum T. Ichihara F. Glimelius B. Frodin JE. Masucci G. Kiessling R. A short-term dietary supplementation of high doses of vitamin E increases T helper 1 cytokine production in patients with advanced colorectal cancer. Clin Cancer Res. 2002;8:1772–1778. [PubMed] [Google Scholar]
- 174.Malorni W. Straface E. Matarrese P. Ascione B. Coinu R. Canu S. Galluzzo P. Marino M. Franconi F. Redox state and gender differences in vascular smooth muscle cells. FEBS Lett. 2008;582:635–642. doi: 10.1016/j.febslet.2008.01.034. [DOI] [PubMed] [Google Scholar]
- 175.Manno R. Boin F. Immunotherapy of systemic sclerosis. Immunotherapy. 2010;2:863–878. doi: 10.2217/imt.10.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mantovani G. Maccio A. Melis G. Mura L. Massa E. Mudu MC. Restoration of functional defects in peripheral blood mononuclear cells isolated from cancer patients by thiol antioxidants alpha-lipoic acid and N-acetyl cysteine. Int J Cancer. 2000;86:842–847. doi: 10.1002/(sici)1097-0215(20000615)86:6<842::aid-ijc13>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 177.Marko MG. Pang HJ. Ren Z. Azzi A. Huber BT. Bunnell SC. Meydani SN. Vitamin E reverses impaired linker for activation of T cells activation in T cells from aged C57BL/6 mice. J Nutr. 2009;139:1192–1197. doi: 10.3945/jn.108.103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Marques CD. Dantas AT. Fragoso TS. Duarte AL. The importance of vitamin D levels in autoimmune diseases. Rev Bras Reumatol. 2010;50:67–80. [PubMed] [Google Scholar]
- 179.Masopust D. Ha SJ. Vezys V. Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 180.Matoba S. Kang JG. Patino WD. Wragg A. Boehm M. Gavrilova O. Hurley PJ. Bunz F. Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 181.McColl BW. Rothwell NJ. Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mebius RE. Vitamins in control of lymphocyte migration. Nat Immunol. 2007;8:229–230. doi: 10.1038/ni0307-229. [DOI] [PubMed] [Google Scholar]
- 183.Mehrotra S. Stevens R. Zengou R. Chakraborty NG. Butterfield LH. Economou JS. Dorsky DI. Mukherji B. Regulation of melanoma epitope-specific cytolytic T lymphocyte response by immature and activated dendritic cells, in vitro. Cancer Res. 2003;63:5607–5614. [PubMed] [Google Scholar]
- 184.Mehrotra S. Chhabra A. Chattopadhyay S. Dorsky DI. Chakraborty NG. Mukherji B. Rescuing melanoma epitope-specific cytolytic T lymphocytes from activation-induced cell death, by SP600125, an inhibitor of JNK: implications in cancer immunotherapy. J Immunol. 2004;173:6017–6024. doi: 10.4049/jimmunol.173.10.6017. [DOI] [PubMed] [Google Scholar]
- 185.Mehrotra S. Chhabra A. Hegde U. Chakraborty NG. Mukherji B. Inhibition of c-Jun N-terminal kinase rescues influenza epitope-specific human cytolytic T lymphocytes from activation-induced cell death. J Leukoc Biol. 2007;81:539–547. doi: 10.1189/jlb.0706479. [DOI] [PubMed] [Google Scholar]
- 186.Mehrotra S. Mougiakakos D. Johansson CC. Voelkel-Johnson C. Kiessling R. Oxidative stress and lymphocyte persistence: implications in immunotherapy. Adv Cancer Res. 2009;102:197–227. doi: 10.1016/S0065-230X(09)02006-5. [DOI] [PubMed] [Google Scholar]
- 187.Meister A. Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 188.Michalek RD. Nelson KJ. Holbrook BC. Yi JS. Stridiron D. Daniel LW. Fetrow JS. King SB. Poole LB. Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 189.Mirshafiey A. Mohsenzadegan M. The role of reactive oxygen species in immunopathogenesis of rheumatoid arthritis. Iran J Allergy Asthma Immunol. 2008;7:195–202. [PubMed] [Google Scholar]
- 190.Mo R. Chen J. Grolleau-Julius A. Murphy HS. Richardson BC. Yung RL. Estrogen regulates CCR gene expression and function in T lymphocytes. J Immunol. 2005;174:6023–6029. doi: 10.4049/jimmunol.174.10.6023. [DOI] [PubMed] [Google Scholar]
- 191.Molano A. Meydani SN. Vitamin E, signalosomes and gene expression in T cells. Mol Aspects Med. 2012;33:55–62. doi: 10.1016/j.mam.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 192.Moreno MU. San Jose G. Fortuno A. Beloqui O. Diez J. Zalba G. The C242T CYBA polymorphism of NADPH oxidase is associated with essential hypertension. J Hypertens. 2006;24:1299–1306. doi: 10.1097/01.hjh.0000234110.54110.56. [DOI] [PubMed] [Google Scholar]
- 193.Mori T. Ohnishi M. Komiyama M. Tsutsui A. Yabushita H. Okada H. Growth inhibitory effect of paradicsompaprika in cancer cell lines. Oncol Rep. 2002;9:807–810. [PubMed] [Google Scholar]
- 194.Morito N. Yoh K. Itoh K. Hirayama A. Koyama A. Yamamoto M. Takahashi S. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene. 2003;22:9275–9281. doi: 10.1038/sj.onc.1207024. [DOI] [PubMed] [Google Scholar]
- 195.Mougiakakos D. Johansson CC. Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood. 2009;113:3542–3545. doi: 10.1182/blood-2008-09-181040. [DOI] [PubMed] [Google Scholar]
- 196.Mougiakakos D. Johansson CC. Jitschin R. Bottcher M. Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood. 2011;117:857–861. doi: 10.1182/blood-2010-09-307041. [DOI] [PubMed] [Google Scholar]
- 197.Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118:4589–4592. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 198.Muhammad S. Bierhaus A. Schwaninger M. Reactive oxygen species in diabetes-induced vascular damage, stroke, and Alzheimer's disease. J Alzheimers Dis. 2009;16:775–785. doi: 10.3233/JAD-2009-0982. [DOI] [PubMed] [Google Scholar]
- 199.Myrset AH. Bostad A. Jamin N. Lirsac PN. Toma F. Gabrielsen OS. DNA and redox state induced conformational changes in the DNA-binding domain of the Myb oncoprotein. EMBO J. 1993;12:4625–4633. doi: 10.1002/j.1460-2075.1993.tb06151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nagaraj S. Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 201.Nagy G. Koncz A. Telarico T. Fernandez D. Ersek B. Buzas E. Perl A. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther. 2010;12:210. doi: 10.1186/ar3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nakanishi S. Yamane K. Ohishi W. Nakashima R. Yoneda M. Nojima H. Watanabe H. Kohno N. Manganese superoxide dismutase Ala16Val polymorphism is associated with the development of type 2 diabetes in Japanese-Americans. Diabetes Res Clin Pract. 2008;81:381–385. doi: 10.1016/j.diabres.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 203.Nencioni L. Iuvara A. Aquilano K. Ciriolo MR. Cozzolino F. Rotilio G. Garaci E. Palamara AT. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003;17:758–760. doi: 10.1096/fj.02-0508fje. [DOI] [PubMed] [Google Scholar]
- 204.Nencioni L. Sgarbanti R. De Chiara G. Garaci E. Palamara AT. Influenza virus and redox mediated cell signaling: a complex network of virus/host interaction. New Microbiol. 2007;30:367–375. [PubMed] [Google Scholar]
- 205.Neyestani TR. Shariatzadeh N. Gharavi A. Kalayi A. Khalaji N. Physiological dose of lycopene suppressed oxidative stress and enhanced serum levels of immunoglobulin M in patients with Type 2 diabetes mellitus: a possible role in the prevention of long-term complications. J Endocrinol Invest. 2007;30:833–838. doi: 10.1007/BF03349224. [DOI] [PubMed] [Google Scholar]
- 206.Nishikawa M. Hashida M. Takakura Y. Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis. Adv Drug Deliv Rev. 2009;61:319–326. doi: 10.1016/j.addr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 207.Norell H. Martins da Palma T. Lesher A. Kaur N. Mehrotra M. Naga OS. Spivey N. Olafimihan S. Chakraborty NG. Voelkel-Johnson C. Nishimura MI, et al. Inhibition of superoxide generation upon T-cell receptor engagement rescues Mart-1(27-35)-reactive T cells from activation-induced cell death. Cancer Res. 2009;69:6282–6289. doi: 10.1158/0008-5472.CAN-09-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Noubade R. Krementsov DN. Del Rio R. Thornton T. Nagaleekar V. Saligrama N. Spitzack A. Spach K. Sabio G. Davis RJ. Rincon M, et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118:3290–3300. doi: 10.1182/blood-2011-02-336552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.O'Shea JJ. Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Okazuka K. Wakabayashi Y. Kashihara M. Inoue J. Sato T. Yokoyama M. Aizawa S. Aizawa Y. Mishima Y. Kominami R. p53 prevents maturation of T cell development to the immature CD4-CD8+ stage in Bcl11b−/− mice. Biochem Biophys Res Commun. 2005;328:545–549. doi: 10.1016/j.bbrc.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 211.Olofsson P. Holmberg J. Tordsson J. Lu S. Akerstrom B. Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet. 2003;33:25–32. doi: 10.1038/ng1058. [DOI] [PubMed] [Google Scholar]
- 212.Ouyang W. Li MO. Foxo: in command of T lymphocyte homeostasis and tolerance. Trends Immunol. 2011;32:26–33. doi: 10.1016/j.it.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ozkan Y. Yardym-Akaydyn S. Sepici A. Keskin E. Sepici V. Simsek B. Oxidative status in rheumatoid arthritis. Clin Rheumatol. 2007;26:64–68. doi: 10.1007/s10067-006-0244-z. [DOI] [PubMed] [Google Scholar]
- 214.Paintlia MK. Paintlia AS. Singh AK. Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. 2011;116:508–521. doi: 10.1111/j.1471-4159.2010.07136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Palmer MT. Lee YK. Maynard CL. Oliver JR. Bikle DD. Jetten AM. Weaver CT. Lineage-specific effects of 1,25-dihydroxyvitamin D3 on the development of effector CD4 T cells. J Biol Chem. 2011;286:997–1004. doi: 10.1074/jbc.M110.163790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pani G. Colavitti R. Borrello S. Galeotti T. Endogenous oxygen radicals modulate protein tyrosine phosphorylation and JNK-1 activation in lectin-stimulated thymocytes. Biochem J. 2000;347(Pt 1):173–181. [PMC free article] [PubMed] [Google Scholar]
- 217.Pardo M. Melendez JA. Tirosh O. Manganese superoxide dismutase inactivation during Fas (CD95)-mediated apoptosis in Jurkat T cells. Free Radic Biol Med. 2006;41:1795–1806. doi: 10.1016/j.freeradbiomed.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 218.Park MK. Park JS. Cho ML. Oh HJ. Heo YJ. Woo YJ. Heo YM. Park MJ. Park HS. Park SH. Kim HY, et al. Grape seed proanthocyanidin extract (GSPE) differentially regulates Foxp3(+) regulatory and IL-17(+) pathogenic T cell in autoimmune arthritis. Immunol Lett. 2011;135:50–58. doi: 10.1016/j.imlet.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 219.Parra S. Marsillach J. Aragones G. Beltran R. Montero M. Coll B. Mackness B. Mackness M. Alonso-Villaverde C. Joven J. Camps J. Paraoxonase-1 gene haplotypes are associated with metabolic disturbances, atherosclerosis, and immunologic outcome in HIV-infected patients. J Infect Dis. 2010;201:627–634. doi: 10.1086/650312. [DOI] [PubMed] [Google Scholar]
- 220.Pawelec G. Solana R. Remarque E. Mariani E. Impact of aging on innate immunity. J Leukoc Biol. 1998;64:703–712. doi: 10.1002/jlb.64.6.703. [DOI] [PubMed] [Google Scholar]
- 221.Pelicano H. Carney D. Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 222.Peterhans E. Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J Nutr. 1997;127:962S–965S. doi: 10.1093/jn/127.5.962S. [DOI] [PubMed] [Google Scholar]
- 223.Piganelli JD. Flores SC. Cruz C. Koepp J. Batinic-Haberle I. Crapo J. Day B. Kachadourian R. Young R. Bradley B. Haskins K. A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes. 2002;51:347–355. doi: 10.2337/diabetes.51.2.347. [DOI] [PubMed] [Google Scholar]
- 224.Pinkus R. Weiner LM. Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-kappaB, and glutathione S-transferase gene expression. J Biol Chem. 1996;271:13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- 225.Polyak K. Xia Y. Zweier JL. Kinzler KW. Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 226.Powers SK. Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Purup S. Larsen E. Christensen LP. Differential effects of falcarinol and related aliphatic C(17)-polyacetylenes on intestinal cell proliferation. J Agric Food Chem. 2009;57:8290–8296. doi: 10.1021/jf901503a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Purushothaman D. Sarin A. Cytokine-dependent regulation of NADPH oxidase activity and the consequences for activated T cell homeostasis. J Exp Med. 2009;206:1515–1523. doi: 10.1084/jem.20082851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Qu Y. Wang J. Ray PS. Guo H. Huang J. Shin-Sim M. Bukoye BA. Liu B. Lee AV. Lin X. Huang P, et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-kappaB signaling. J Clin Invest. 2011;121:212–225. doi: 10.1172/JCI43144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rabinovich GA. Gabrilovich D. Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rajasingh J. Raikwar HP. Muthian G. Johnson C. Bright JJ. Curcumin induces growth-arrest and apoptosis in association with the inhibition of constitutively active JAK-STAT pathway in T cell leukemia. Biochem Biophys Res Commun. 2006;340:359–368. doi: 10.1016/j.bbrc.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 232.Ratjen F. Wonne R. Posselt HG. Stover B. Hofmann D. Bender SW. A double-blind placebo controlled trial with oral ambroxol and N-acetylcysteine for mucolytic treatment in cystic fibrosis. Eur J Pediatr. 1985;144:374–378. doi: 10.1007/BF00441781. [DOI] [PubMed] [Google Scholar]
- 233.Remans PH. Gringhuis SI. van Laar JM. Sanders ME. Papendrecht-van der Voort EA. Zwartkruis FJ. Levarht EW. Rosas M. Coffer PJ. Breedveld FC. Bos JL, et al. Rap1 signaling is required for suppression of Ras-generated reactive oxygen species and protection against oxidative stress in T lymphocytes. J Immunol. 2004;173:920–931. doi: 10.4049/jimmunol.173.2.920. [DOI] [PubMed] [Google Scholar]
- 234.Rex TS. Tsui I. Hahn P. Maguire AM. Duan D. Bennett J. Dunaief JL. Adenovirus-mediated delivery of catalase to retinal pigment epithelial cells protects neighboring photoreceptors from photo-oxidative stress. Hum Gene Ther. 2004;15:960–967. doi: 10.1089/hum.2004.15.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Rhee SG. Yang KS. Kang SW. Woo HA. Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 236.Rider V. Abdou NI. Gender differences in autoimmunity: molecular basis for estrogen effects in systemic lupus erythematosus. Int Immunopharmacol. 2001;1:1009–1024. doi: 10.1016/s1567-5769(01)00046-7. [DOI] [PubMed] [Google Scholar]
- 237.Rincon M. Pedraza-Alva G. JNK and p38 MAP kinases in CD4+ and CD8+ T cells. Immunol Rev. 2003;192:131–142. doi: 10.1034/j.1600-065x.2003.00019.x. [DOI] [PubMed] [Google Scholar]
- 238.Riou C. Yassine-Diab B. Van grevenynghe J. Somogyi R. Greller LD. Gagnon D. Gimmig S. Wilkinson P. Shi Y. Cameron MJ. Campos-Gonzalez R, et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med. 2007;204:79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Rivera A. Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005;280:29346–29354. doi: 10.1074/jbc.M504852200. [DOI] [PubMed] [Google Scholar]
- 240.Robbins D. Zhao Y. Oxidative stress induced by MnSOD-p53 interaction: pro- or anti-tumorigenic? J Signal Transduct. 2012;2012:101465. doi: 10.1155/2012/101465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Roederer M. Ela SW. Staal FJ. Herzenberg LA. N-acetylcysteine: a new approach to anti-HIV therapy. AIDS Res Hum Retroviruses. 1992;8:209–217. doi: 10.1089/aid.1992.8.209. [DOI] [PubMed] [Google Scholar]
- 242.Rozmer Z. Berki T. Perjesi P. Different effects of two cyclic chalcone analogues on cell cycle of Jurkat T cells. Toxicol In Vitro. 2006;20:1354–1362. doi: 10.1016/j.tiv.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 243.Sablina AA. Budanov AV. Ilyinskaya GV. Agapova LS. Kravchenko JE. Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sachsenmaier C. Radler-Pohl A. Muller A. Herrlich P. Rahmsdorf HJ. Damage to DNA by UV light and activation of transcription factors. Biochem Pharmacol. 1994;47:129–136. doi: 10.1016/0006-2952(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 245.Sahaf B. Heydari K. Herzenberg LA. Herzenberg LA. Lymphocyte surface thiol levels. Proc Natl Acad Sci U S A. 2003;100:4001–4005. doi: 10.1073/pnas.2628032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Sahaf B. Heydari K. Herzenberg LA. Herzenberg LA. The extracellular microenvironment plays a key role in regulating the redox status of cell surface proteins in HIV-infected subjects. Arch Biochem Biophys. 2005;434:26–32. doi: 10.1016/j.abb.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 247. This reference has been deleted.
- 248.Sallusto F. Geginat J. Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 249.Salminen A. Huuskonen J. Ojala J. Kauppinen A. Kaarniranta K. Suuronen T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev. 2008;7:83–105. doi: 10.1016/j.arr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 250.Salminen A. Kauppinen A. Suuronen T. Kaarniranta K. SIRT1 longevity factor suppresses NF-kappaB -driven immune responses: regulation of aging via NF-kappaB acetylation? Bioessays. 2008;30:939–942. doi: 10.1002/bies.20799. [DOI] [PubMed] [Google Scholar]
- 251.Samiec PS. Drews-Botsch C. Flagg EW. Kurtz JC. Sternberg P., Jr. Reed RL. Jones DP. Glutathione in human plasma: decline in association with aging, age-related macular degeneration, and diabetes. Free Radic Biol Med. 1998;24:699–704. doi: 10.1016/s0891-5849(97)00286-4. [DOI] [PubMed] [Google Scholar]
- 252.Sandstrom PA. Buttke TM. Autocrine production of extracellular catalase prevents apoptosis of the human CEM T-cell line in serum-free medium. Proc Natl Acad Sci U S A. 1993;90:4708–4712. doi: 10.1073/pnas.90.10.4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sandstrom PA. Tebbey PW. Van Cleave S. Buttke TM. Lipid hydroperoxides induce apoptosis in T cells displaying a HIV-associated glutathione peroxidase deficiency. J Biol Chem. 1994;269:798–801. [PubMed] [Google Scholar]
- 254.Sandstrom PA. Mannie MD. Buttke TM. Inhibition of activation-induced death in T cell hybridomas by thiol antioxidants: oxidative stress as a mediator of apoptosis. J Leukoc Biol. 1994;55:221–226. doi: 10.1002/jlb.55.2.221. [DOI] [PubMed] [Google Scholar]
- 255.Satoh H. Moriguchi T. Taguchi K. Takai J. Maher JM. Suzuki T. Winnard PT., Jr. Raman V. Ebina M. Nukiwa T. Yamamoto M. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis. 2010;31:1833–1843. doi: 10.1093/carcin/bgq105. [DOI] [PubMed] [Google Scholar]
- 256.Schmielau J. Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–4760. [PubMed] [Google Scholar]
- 257.Seetharaman SV. Prudencio M. Karch C. Holloway SP. Borchelt DR. Hart PJ. Immature copper-zinc superoxide dismutase and familial amyotrophic lateral sclerosis. Exp Biol Med (Maywood) 2009;234:1140–1154. doi: 10.3181/0903-MR-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Servettaz A. Goulvestre C. Kavian N. Nicco C. Guilpain P. Chereau C. Vuiblet V. Guillevin L. Mouthon L. Weill B. Batteux F. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol. 2009;182:5855–5864. doi: 10.4049/jimmunol.0803705. [DOI] [PubMed] [Google Scholar]
- 259.Shah CA. Allison KH. Garcia RL. Gray HJ. Goff BA. Swisher EM. Intratumoral T cells, tumor-associated macrophages, and regulatory T cells: association with p53 mutations, circulating tumor DNA and survival in women with ovarian cancer. Gynecol Oncol. 2008;109:215–219. doi: 10.1016/j.ygyno.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 260.Shah D. Kiran R. Wanchu A. Bhatnagar A. Oxidative stress in systemic lupus erythematosus: relationship to Th1 cytokine and disease activity. Immunol Lett. 2010;129:7–12. doi: 10.1016/j.imlet.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 261.Shao L. Diccianni MB. Tanaka T. Gribi R. Yu AL. Pullen JD. Camitta BM. Yu J. Thioredoxin expression in primary T-cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001;61:7333–7338. [PubMed] [Google Scholar]
- 262.Shen HM. Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 263.Shichita T. Sugiyama Y. Ooboshi H. Sugimori H. Nakagawa R. Takada I. Iwaki T. Okada Y. Iida M. Cua DJ. Iwakura Y, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 264.Sigmundsdottir H. Pan J. Debes GF. Alt C. Habtezion A. Soler D. Butcher EC. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8:285–293. doi: 10.1038/ni1433. [DOI] [PubMed] [Google Scholar]
- 265.Silva A. Girio A. Cebola I. Santos CI. Antunes F. Barata JT. Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia. 2011 doi: 10.1038/leu.2011.56. [DOI] [PubMed] [Google Scholar]
- 266.Singh N. Huang L. Qin H. Defective T-cell receptor-induced apoptosis of T cells and rejection of transplanted immunogenic tumors in p53(−/−) mice. Eur J Immunol. 2010;40:559–568. doi: 10.1002/eji.200939736. [DOI] [PubMed] [Google Scholar]
- 267.Sitkovsky M. Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 268.Son NH. Murray S. Yanovski J. Hodes RJ. Weng N. Lineage-specific telomere shortening and unaltered capacity for telomerase expression in human T and B lymphocytes with age. J Immunol. 2000;165:1191–1196. doi: 10.4049/jimmunol.165.3.1191. [DOI] [PubMed] [Google Scholar]
- 269.Staal FJ. Roederer M. Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Stahl M. Dijkers PF. Kops GJ. Lens SM. Coffer PJ. Burgering BM. Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 271.Sun J. Madan R. Karp CL. Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Sun XF. Zhang H. NFKB and NFKBI polymorphisms in relation to susceptibility of tumour and other diseases. Histol Histopathol. 2007;22:1387–1398. doi: 10.14670/HH-22.1387. [DOI] [PubMed] [Google Scholar]
- 273.Suzuki YJ. Forman HJ. Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 274.Swainson L. Kinet S. Manel N. Battini JL. Sitbon M. Taylor N. Glucose transporter 1 expression identifies a population of cycling CD4+CD8+ human thymocytes with high CXCR4-induced chemotaxis. Proc Natl Acad Sci U S A. 2005;102:12867–12872. doi: 10.1073/pnas.0503603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Szatrowski TP. Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 276.Tacke RS. Lee HC. Goh C. Courtney J. Polyak SJ. Rosen HR. Hahn YS. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012;55:343–353. doi: 10.1002/hep.24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Takahashi A. Hanson MG. Norell HR. Havelka AM. Kono K. Malmberg KJ. Kiessling RV. Preferential cell death of CD8+ effector memory (CCR7-CD45RA-) T cells by hydrogen peroxide-induced oxidative stress. J Immunol. 2005;174:6080–6087. doi: 10.4049/jimmunol.174.10.6080. [DOI] [PubMed] [Google Scholar]
- 278.Takayanagi R. Inoguchi T. Ohnaka K. Clinical and experimental evidence for oxidative stress as an exacerbating factor of diabetes mellitus. J Clin Biochem Nutr. 2011;48:72–77. doi: 10.3164/jcbn.11-014FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Tang Q. Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Thornton TM. Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5:44–51. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Thulasingam S. Massilamany C. Gangaplara A. Dai H. Yarbaeva S. Subramaniam S. Riethoven JJ. Eudy J. Lou M. Reddy J. miR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264.7 cells. Mol Cell Biochem. 2011;352:181–188. doi: 10.1007/s11010-011-0752-2. [DOI] [PubMed] [Google Scholar]
- 282.Tothova Z. Kollipara R. Huntly BJ. Lee BH. Castrillon DH. Cullen DE. McDowell EP. Lazo-Kallanian S. Williams IR. Sears C. Armstrong SA, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 283.Trachootham D. Lu W. Ogasawara MA. Nilsa RD. Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Tse HM. Milton MJ. Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med. 2004;36:233–247. doi: 10.1016/j.freeradbiomed.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 285.Tse HM. Milton MJ. Schreiner S. Profozich JL. Trucco M. Piganelli JD. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol. 2007;178:908–917. doi: 10.4049/jimmunol.178.2.908. [DOI] [PubMed] [Google Scholar]
- 286.Tse HM. Thayer TC. Steele C. Cuda CM. Morel L. Piganelli JD. Mathews CE. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol. 2010;185:5247–5258. doi: 10.4049/jimmunol.1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Uppstad H. Osnes GH. Cole KJ. Phillips DH. Haugen A. Mollerup S. Sex differences in susceptibility to PAHs is an intrinsic property of human lung adenocarcinoma cells. Lung Cancer. 2011;71:264–270. doi: 10.1016/j.lungcan.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 288.Valdimarsson H. Thorleifsdottir RH. Sigurdardottir SL. Gudjonsson JE. Johnston A. Psoriasis—as an autoimmune disease caused by molecular mimicry. Trends Immunol. 2009;30:494–501. doi: 10.1016/j.it.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 289.van den Boorn JG. Konijnenberg D. Dellemijn TA. van der Veen JP. Bos JD. Melief CJ. Vyth-Dreese FA. Luiten RM. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. 2009;129:2220–2232. doi: 10.1038/jid.2009.32. [DOI] [PubMed] [Google Scholar]
- 290.van der Windt GJ. Everts B. Chang CH. Curtis JD. Freitas TC. Amiel E. Pearce EJ. Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.van Grevenynghe J. Procopio FA. He Z. Chomont N. Riou C. Zhang Y. Gimmig S. Boucher G. Wilkinson P. Shi Y. Yassine-Diab B, et al. Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med. 2008;14:266–274. doi: 10.1038/nm1728. [DOI] [PubMed] [Google Scholar]
- 292.Vandenabeele P. Galluzzi L. Vanden Berghe T. Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 293.Vence L. Palucka AK. Fay JW. Ito T. Liu YJ. Banchereau J. Ueno H. Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2007;104:20884–20889. doi: 10.1073/pnas.0710557105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Venkataraman S. Alimova I. Fan R. Harris P. Foreman N. Vibhakar R. MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS One. 2010;5:e10748. doi: 10.1371/journal.pone.0010748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Vig M. Srivastava S. Kandpal U. Sade H. Lewis V. Sarin A. George A. Bal V. Durdik JM. Rath S. Inducible nitric oxide synthase in T cells regulates T cell death and immune memory. J Clin Invest. 2004;113:1734–1742. doi: 10.1172/JCI20225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Vlahos R. Stambas J. Bozinovski S. Broughton BR. Drummond GR. Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011;7:e1001271. doi: 10.1371/journal.ppat.1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Vogler S. Pahnke J. Rousset S. Ricquier D. Moch H. Miroux B. Ibrahim SM. Uncoupling protein 2 has protective function during experimental autoimmune encephalomyelitis. Am J Pathol. 2006;168:1570–1575. doi: 10.2353/ajpath.2006.051069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.von Essen MR. Kongsbak M. Schjerling P. Olgaard K. Odum N. Geisler C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol. 2010;11:344–349. doi: 10.1038/ni.1851. [DOI] [PubMed] [Google Scholar]
- 299.Wagner EF. Schonthaler HB. Guinea-Viniegra J. Tschachler E. Psoriasis: what we have learned from mouse models. Nat Rev Rheumatol. 2010;6:704–714. doi: 10.1038/nrrheum.2010.157. [DOI] [PubMed] [Google Scholar]
- 300.Walsh AC. Michaud SG. Malossi JA. Lawrence DA. Glutathione depletion in human T lymphocytes: analysis of activation-associated gene expression and the stress response. Toxicol Appl Pharmacol. 1995;133:249–261. doi: 10.1006/taap.1995.1149. [DOI] [PubMed] [Google Scholar]
- 301.Wanchu A. Rana SV. Pallikkuth S. Sachdeva RK. Short communication: oxidative stress in HIV-infected individuals: a cross-sectional study. AIDS Res Hum Retroviruses. 2009;25:1307–1311. doi: 10.1089/aid.2009.0062. [DOI] [PubMed] [Google Scholar]
- 302.Wang SS. Davis S. Cerhan JR. Hartge P. Severson RK. Cozen W. Lan Q. Welch R. Chanock SJ. Rothman N. Polymorphisms in oxidative stress genes and risk for non-Hodgkin lymphoma. Carcinogenesis. 2006;27:1828–1834. doi: 10.1093/carcin/bgl013. [DOI] [PubMed] [Google Scholar]
- 303.Wang Z. Liu Y. Han N. Chen X. Yu W. Zhang W. Zou F. Profiles of oxidative stress-related microRNA and mRNA expression in auditory cells. Brain Res. 2010;1346:14–25. doi: 10.1016/j.brainres.2010.05.059. [DOI] [PubMed] [Google Scholar]
- 304.Weber M. Baker MB. Moore JP. Searles CD. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun. 2010;393:643–648. doi: 10.1016/j.bbrc.2010.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Williams MA. Rangasamy T. Bauer SM. Killedar S. Karp M. Kensler TW. Yamamoto M. Breysse P. Biswal S. Georas SN. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol. 2008;181:4545–4559. doi: 10.4049/jimmunol.181.7.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Won HY. Sohn JH. Min HJ. Lee K. Woo HA. Ho YS. Park JW. Rhee SG. Hwang ES. Glutathione peroxidase 1 deficiency attenuates allergen-induced airway inflammation by suppressing Th2 and Th17 cell development. Antioxid Redox Signal. 2010;13:575–587. doi: 10.1089/ars.2009.2989. [DOI] [PubMed] [Google Scholar]
- 307.Wu D. Meydani SN. Age-associated changes in immune and inflammatory responses: impact of vitamin E intervention. J Leukoc Biol. 2008;84:900–914. doi: 10.1189/jlb.0108023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wu D. Guo Z. Ren Z. Guo W. Meydani SN. Green tea EGCG suppresses T cell proliferation through impairment of IL-2/IL-2 receptor signaling. Free Radic Biol Med. 2009;47:636–643. doi: 10.1016/j.freeradbiomed.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 309.Wu J. Levy EM. Black PH. 2-Mercaptoethanol and n-acetylcysteine enhance T cell colony formation in AIDS and ARC. Clin Exp Immunol. 1989;77:7–10. [PMC free article] [PubMed] [Google Scholar]
- 310.Wyche KE. Wang SS. Griendling KK. Dikalov SI. Austin H. Rao S. Fink B. Harrison DG. Zafari AM. C242T CYBA polymorphism of the NADPH oxidase is associated with reduced respiratory burst in human neutrophils. Hypertension. 2004;43:1246–1251. doi: 10.1161/01.HYP.0000126579.50711.62. [DOI] [PubMed] [Google Scholar]
- 311.Yadav VR. Prasad S. Sung B. Aggarwal BB. The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer. Int Immunopharmacol. 2011;11:295–309. doi: 10.1016/j.intimp.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Yamaguchi M. Hasegawa I. Yahagi N. Ishigaki Y. Takano F. Ohta T. Carotenoids modulate cytokine production in Peyer's patch cells ex vivo. J Agric Food Chem. 2010;58:8566–8572. doi: 10.1021/jf101295y. [DOI] [PubMed] [Google Scholar]
- 313.Yamamoto T. Scleroderma—pathophysiology. Eur J Dermatol. 2009;19:14–24. doi: 10.1684/ejd.2008.0570. [DOI] [PubMed] [Google Scholar]
- 314.Yan Z. Garg SK. Banerjee R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J Biol Chem. 2010;285:41525–41532. doi: 10.1074/jbc.M110.189944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Yang DD. Conze D. Whitmarsh AJ. Barrett T. Davis RJ. Rincon M. Flavell RA. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 316.Yang Y. Paik JH. Cho D. Cho JA. Kim CW. Resveratrol induces the suppression of tumor-derived CD4+CD25+ regulatory T cells. Int Immunopharmacol. 2008;8:542–547. doi: 10.1016/j.intimp.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 317.Yi J. Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta. 2010;1804:1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Yi JS. Holbrook BC. Michalek RD. Laniewski NG. Grayson JM. Electron transport complex I is required for CD8+ T cell function. J Immunol. 2006;177:852–862. doi: 10.4049/jimmunol.177.2.852. [DOI] [PubMed] [Google Scholar]
- 319.Yu D. dos Santos CO. Zhao G. Jiang J. Amigo JD. Khandros E. Dore LC. Yao Y. D'Souza J. Zhang Z. Ghaffari S, et al. miR-451 protects against erythroid oxidant stress by repressing 14–3-3zeta. Genes Dev. 2010;24:1620–1633. doi: 10.1101/gad.1942110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Zafra-Stone S. Yasmin T. Bagchi M. Chatterjee A. Vinson JA. Bagchi D. Berry anthocyanins as novel antioxidants in human health and disease prevention. Mol Nutr Food Res. 2007;51:675–683. doi: 10.1002/mnfr.200700002. [DOI] [PubMed] [Google Scholar]
- 321.Zhang Q. Zeng X. Guo J. Wang X. Oxidant stress mechanism of homocysteine potentiating Con A-induced proliferation in murine splenic T lymphocytes. Cardiovasc Res. 2002;53:1035–1042. doi: 10.1016/s0008-6363(01)00541-7. [DOI] [PubMed] [Google Scholar]
- 322.Zhou Q. Mrowietz U. Rostami-Yazdi M. Oxidative stress in the pathogenesis of psoriasis. Free Radic Biol Med. 2009;47:891–905. doi: 10.1016/j.freeradbiomed.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 323.Zhou Y. Hileman EO. Plunkett W. Keating MJ. Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–4104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 324.Zhu J. Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Zhu J. Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Zong WX. Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 327.Zou Y. Chen CH. Fike JR. Huang TT. A new mouse model for temporal- and tissue-specific control of extracellular superoxide dismutase. Genesis. 2009;47:142–154. doi: 10.1002/dvg.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]