

Abstract
The non-proteinogenic amino acids capreomycidine and epicapreomycidine are constituents of antibiotically active natural products, but the synthesis of these unusual cyclic guanidine derivatives is challenging. The biosynthesis of capreomycidine has therefore been employed as a guideline to develop a concise biomimetic synthesis of both epimeric amino acids. The resulting domino-guanidinylation-aza-Michael-addition reaction provides the most convenient access to these amino acids in racemic form. Attempts to dissect the domino reaction into two separate transformations for a stereocontrolled version of this synthetic approach have also been made. The synthesized didehydro-arginine derivatives with urethane-protected guanidine moieties did not undergo the aza-Michael-addition anymore. These results may have wider implications for the 1,4-addition of guanidines to α,β-unsaturated carbonyl compounds, particularly to didehydro amino acids.
Electronic supplementary material
The online version of this article (doi:10.1007/s00726-012-1309-8) contains supplementary material, which is available to authorized users.
Keywords: Natural products, Antibiotics, Amino acids, Biomimetic synthesis, Domino reactions, Guanidines
Introduction
Bacterial infections continue to be a major threat to human health, particularly with respect to emerging strains with resistances to established antibiotics (Taubes 2008; Cooper and Shlaes 2011). It is therefore highly desirable to develop novel antibacterial agents which should ideally display new or yet unexploited modes of action (Walsh 2003). Natural products have been used as lead structures for many antibacterial drugs (Mahady et al. 2008). However, their often complex structures represent important challenges to organic synthesis as a prerequisite for detailed structure–activity relationship (SAR) studies. Among the many bacterial secondary metabolites with antibiotic activity, a significant number of compounds are of peptidic nature and contain non-proteinogenic amino acid structures.
The cyclic arginine-derived non-proteinogenic amino acids (2S,3R)-capreomycidine 1 (‘capreomycidine’) and (2S,3S)-capreomycidine 2 (‘epicapreomycidine’) are constituents of several natural products with remarkable antibiotic potencies (Fig. 1). Capreomycidine 1 can be found in tuberactinomycin peptide antibiotics (Nagata et al. 1968; Wakamiya et al. 1970; Yoshioka et al. 1971; Ando et al. 1971; Izumi et al. 1972), e.g., the capreomycins (Herr et al. 1960; Bycroft et al. 1971a; Shiba et al. 1976; Nomoto et al. 1977; Nomoto et al. 1978) and viomycin 3 (Finlay et al. 1951; Bartz et al. 1951; Dyer et al. 1965; Bycroft et al. 1969; Noda et al. 1972), which have both found clinical use as anti-tuberculosis drugs. The 3-epimer epicapreomycidine 2 is a component of the naturally occurring protease inhibitors chymostatin (Umezawa et al. 1970; Tatsuta et al. 1973) and elastatinal (Umezawa et al. 1973; Okura et al. 1975) as well as of the Streptomyces-produced muraymycins, a subclass of nucleoside lipopeptide antibiotics (e.g. muraymycin A1 4, McDonald et al. 2002). Furthermore, β-hydroxy-enduracididine, an amino acid unit with structural similarities to 1 and 2, is a building block of the mannopeptimycins, a novel subclass of glycopeptide antibiotics (He et al. 2002; Singh et al. 2003; Koehn 2008; Schwörer and Oberthür 2009).
Fig. 1.
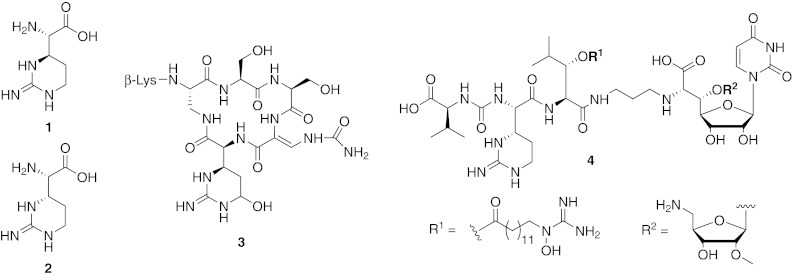
Structures of non-proteinogenic amino acids capreomycidine 1 and epicapreomycidine 2 as well as structures of natural products viomycin 3 and muraymycin A1 4 containing 1 and 2, respectively
Due to their relevance in natural product chemistry, several approaches for the synthesis of amino acids 1 and 2 have been developed. A first non-stereoselective synthesis of 1 and 2 involved the preparation of an aromatic 2-aminopyrimidine precursor, which was then hydrogenated to provide a diastereomeric mixture of 1 and 2, each in racemic form. The separation of the diastereomers was carried out by crystallization of the respective picrates (Bycroft et al. 1971b). A slightly improved version of this synthesis has also been described (Yamashita et al. 2004). The first syntheses of stereochemically pure 1 and 2 employed an aldol reaction for the synthesis of a protected β-hydroxy-ornithine precursor, which then underwent acylase-mediated enzymatic resolution. Further conversion via an aziridine intermediate finally provided either 1 or 2, but the synthesis was lengthy, and the overall yields were rather low (Shiba et al. 1977; Wakamiya et al. 1978; Teshima et al. 1980). The first stereoselective synthesis of capreomycidine 1 involved a Mannich-type reaction with a chiral glycine derivative as the key step. The subsequent guanidinylation reaction only proceeded with one of the two diastereomers obtained from the Mannich transformation, thus finally giving pure 1 after further conversions (DeMong and Williams 2001, 2003). However, this also implies that an according synthesis of 2 using the same strategy is not feasible. An alternative ex-chiral pool synthesis of 13C-labelled 1 starting from Garner’s aldehyde has also been established (Jackson et al. 2002), but investigations in our own laboratory revealed that an application of this approach to the synthesis of 2 is not possible (unpublished data). The first practical synthesis of stereochemically pure epicapreomycidine has been established in recent years. The initial version of this synthetic route involved an ornithine-derived sulfamate which was then employed in a rhodium-catalyzed C–H insertion reaction to construct the stereocenter in the C-3 position. However, this elegant approach suffered from low yields and diastereoselectivities of the C–H activation key step (Tanino et al. 2008, 2010). A superior version of this C–H activation strategy starting from d-tyrosine and displaying perfect stereocontrol has recently been described though (Tanino et al. 2011).
One of the highly useful strategies for the development of efficient synthetic routes is to imitate the biosynthetic pathways found in nature. Such biomimetic syntheses have found widespread applications (Razzak and De Brabander 2011). Our goal was to investigate the feasibility of a biomimetic synthesis of both capreomycidines 1 and 2. The biosynthetic assembly of the cyclic guanidine structure of capreomycidine 1 as part of viomycin biosynthesis in Streptomyces vinaceus has been elucidated (Fig. 2, Yin and Zabriskie 2004; Yin et al. 2004; Ju et al. 2004). Stereoselective 3-hydroxylation of l-arginine 5, catalyzed by the non-heme 2-oxoglutarate (2-OG) dependent Fe(II)-oxygenase VioC, provides (3S)-3-hydroxy-l-arginine 6. The subsequent ring closure reaction is mediated by the pyridoxalphosphate-(PLP)-dependent enzyme VioD and most likely proceeds via the α,β-unsaturated didehydro-arginine intermediate 7, as demonstrated by labelling studies. Michael-type conjugate 1,4-addition of the guanidine moiety then stereoselectively provides capreomycidine 1 with overall formal inversion of the stereocenter at C-3. The biosynthesis of 2 has not been elucidated yet, but the recent analysis of the biosynthetic gene cluster of muraymycin nucleoside antibiotics suggests that it probably occurs in a similar manner, just with a different stereochemical course (Cheng et al. 2011; also see Lemke et al. 2010).
Fig. 2.

Biosynthesis of capreomycidine 1 in Streoptomyces vinaceus
Hence, the goal of this study was to synthesize didehydro-arginine derivatives 8a–h as potential precursors for biomimetic Aza-Michael-additions in order to obtain amino acids 1 and 2 or protected derivatives thereof (Table 1). There is very limited precendent for 1,4-additions of protected guanidines, with a striking example provided by Baran, Seiple and coworkers as part of their synthetic studies on palau’amine and related compounds (Seiple et al. 2011). It was thus envisaged to investigate different protecting group patterns both at the guanidine group (to influence its nucleophilicity) and the didehydro amino acid moiety (to tune its electrophilicity).
Table 1.
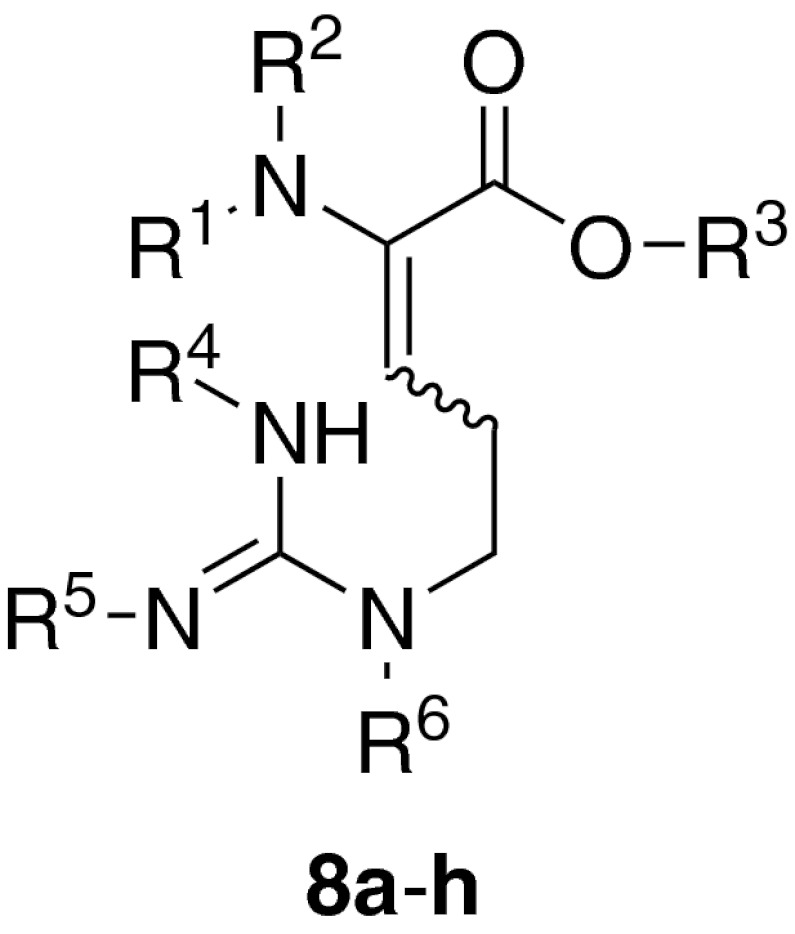
| Compd. | R1 | R2 | R3 | R4 | R5 | R6 |
|---|---|---|---|---|---|---|
| 8a | Boc | H | t-Bu | H | H | H |
| 8b | Boc | Boc | t-Bu | H | H | H |
| 8c | Ac | H | t-Bu | H | H | H |
| 8d | Ac | H | Me | H | H | H |
| 8e | Boc | H | t-Bu | Cbz | Cbz | H |
| 8f | Boc | H | t-Bu | Cbz | Cbz | Bn |
| 8g | Boc | Boc | t-Bu | Cbz | Cbz | H |
| 8h | Boc | Boc | t-Bu | Cbz | Cbz | Bn |
Materials and methods
Chemicals were purchased from Sigma-Aldrich, Alfa Aesar, ABCR and VWR. Amino acid phosphonates 9a–c and 3-azido-propionaldehyde 10 were synthesized as previously reported (Ducho et al. 2009; Schmidt et al. 1984; Davies et al. 1967). Reactions involving oxygen and/or moisture-sensitive reagents were carried out under an atmosphere of argon using anhydrous solvents. Anhydrous solvents were obtained in the following manner: THF was dried over sodium/benzophenone and distilled, MeCN and CH2Cl2 were dried over P2O5 and distilled and DMF was dried over molecular sieves 4 Å. All other solvents were of technical quality and distilled prior to their use, and distilled water was used throughout. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM, VWR) except where indicated under flash conditions. TLC was performed on aluminium plates precoated with silica gel 60 F254 (VWR). Visualisation of the spots was carried out using UV light (254 nm) where appropriate and/or KMnO4 staining under heating (staining solution: 1 g KMnO4, 6 g K2CO3 and 1.5 mL 5 % NaOH(aq) (w/v), all dissolved in 100 mL H2O). 300 MHz-1H as well as 75 MHz-, 76 MHz- and 126 MHz-13C NMR spectra were recorded on Varian UNITY 300, MERCURY 300, INOVA 500 and INOVA 600 spectrometers. All 13C NMR spectra are 1H-decoupled. All spectra were recorded at room temperature except of samples in DMSO-d6 and D2O (standard 35 °C) and where indicated otherwise and were referenced internally to solvent reference frequencies. Chemical shifts (δ) are quoted in ppm. Coupling constants (J) are reported in Hz to the nearest 0.1 Hz. Assignment of signals was carried out using 1H,1H-COSY and HSQC spectra obtained on the spectrometers mentioned above. Low-resolution ESI mass spectrometry was performed on a Varian MAT 311 A spectrometer operating in positive ionization mode. High-resolution (HR) ESI mass spectrometry was carried out on a Bruker microTOF spectrometer or a Bruker 7 T FTICR APEX IV spectrometer. Melting points (mp) were measured on a Büchi instrument and are not corrected. Infrared spectroscopy (IR) was performed on a JASCO FT/IR-4100 spectrometer equipped with an ATR unit. Wavenumbers (ν) are quoted in cm−1. UV spectroscopy was carried out on a JASCO V-630 spectrometer. Wavelengths of maximum absorption (λmax) are reported in nm with the corresponding logarithmic molar extinction coefficient (log ε) given in parenthesis.
General procedure A (synthesis of protected didehydro amino acids by Wittig-Horner reactions)
To a solution of KOt-Bu in THF, a solution of the amino acid phosphonate in THF was added at −78 °C. After 15 min, a solution of 3-azido-propionaldehyde 10 in THF was added dropwise at −78 °C. The reaction mixture was stirred overnight and slowly warmed to rt during this period. The reaction was quenched by addition of MeOH. After the addition of EtOAc, the organic layer was washed with water, dried over Na2SO4 and evaporated under reduced pressure. The resultant crude product was purified by column chromatography.
General procedure B (sequence of Staudinger reduction and guanidinylation reactions with reagent 13)
To a solution of the protected δ-azido-didehydro α-amino acid in THF and water, PPh3 was added at rt. The reaction mixture was stirred overnight at rt. After the addition of EtOAc, the organic layer was washed with aqueous AcOH (10 %, 3 ×). The combined aqueous layers were adjusted to pH 12 by the addition of diluted NaOH solution and extracted with CH2Cl2 (3 ×) and CH2Cl2/i-PrOH (3:1, 1 ×). The combined organics were dried over Na2SO4 and evaporated under reduced pressure. The resultant crude products were sufficiently pure (>95 % as judged by 1H NMR) without further purification. With respect to the limited stability of the didehydro-ornithines, they were prepared freshly and directly used for the subsequent guanidinylation reaction. Thus, the crude didehydro-ornithines were dissolved in DMF, and NEt3 and guanidinylation reagent 13 were added at rt. The reaction mixture was stirred at rt or elevated temperature (to drive the reaction to completion) for the indicated time. The solvent was evaporated under reduced pressure and the resultant crude product was purified by column chromatography.
General procedure C (guanidinylation reactions with reagent 31)
To a solution of the crude ornithine derivative in DMF, guanidinylation reagent 31, NEt3 and AgOTf were added. The reaction mixture was stirred at rt for 3 h, then filtered through Celite and the Celite were washed with EtOAc (3 ×). The filtrate was washed with brine (3 ×), dried over Na2SO4 and evaporated under reduced pressure. The resultant crude product was purified by column chromatography.
Capreomycidine dihydrochloride ((rac)-1 · 2HCl) and epicapreomycidine dihydrochloride ((rac)-2 · 2HCl)
From (rac)-14 and (rac)-15
A solution of a mixture of protected capreomycidine (rac)-14 and epicapreomycidine (rac)-15 (580 mg, 1.77 mmol) in aqueous HCl (6 M, 10 mL, 60 mmol) was stirred at rt for 4 h. The solvent was evaporated under reduced pressure to give a mixture of (rac)-1 · 2HCl and (rac)-2 · 2HCl as a slightly brownish oil (430 mg, 99 %). 1H NMR (300 MHz, D2O): δ = 1.90–2.23 (m, 2 × 2 H, H-4), 3.36–3.55 (m, 2 × 2 H, H-5), 4.18–4.30 (m, 2 × 2 H, H-2, H-3). 13C NMR (76 MHz, D2O): δ = 20.7 (1 × C-4), 21.6 (1 × C-4), 36.0 (1 × C-5), 36.4 (1 × C-5), 48.4 (1 × C-3), 48.7 (1 × C-3), 55.3 (1 × C-2), 55.4 (1 × C-2), 154.5 (2 × guanidine-C), 169.2 (1 × C=O), 169.5 (1 × C=O). MS (ESI+): m/z = 173.1 [M + H]+. HRMS (ESI+): calcd. for C6H12N4O2 ([M + H]+) 173.1033, found 173.1034.
From (rac)-17 and (rac)-18
A solution of protected capreomycidine (rac)-17 and epicapreomycidine (rac)-18 (75 mg, 0.18 mmol) in aqueous HCl (6 M, 1.8 mL, 11 mmol) was stirred at rt for 3 h. The solvent was evaporated under reduced pressure to give a mixture of (rac)-1 · 2HCl and (rac)-2 · 2HCl as a slightly brownish oil (37 mg, 87 %). Analytical data were identical to those given above.
N2-Acetyl-2,3-didehydro-arginine tert-butyl ester (8c)
General procedure B with (Z)-tert-butyl N2-acetyl-2-amino-5-azido-pent-2-enoate (Z)-11b (334 mg, 1.32 mmol), PPh3 (689 mg, 2.63 mmol), water (1.3 mL) and THF (13 mL) for the Staudinger reduction as well as guanidinylation reagent 13 (386 mg, 2.63 mmol), NEt3 (266 mg, 364 μL, 2.63 mmol) and DMF (13 mL) for the guanidinylation reaction. The reaction mixture was stirred at rt for 8 days and at 100 °C for 22 h. Evaporation of the solvent under reduced pressure gave the crude product of 8c as yellow oil (417 mg). 1H NMR (300 MHz, DMSO-d6): δ = 1.39 (s, 9 H, t-Bu-CH3), 1.92 (s, 3 H, Ac-CH3), 2.21–2.34 (m, 2 H, H-4), 2.98–3.08 (m, 2 H, H-5), 6.18 (t, J = 7.5 Hz, 1 H, H-3), 7.09 (sbr, 1 H, 2-NH), 9.39 (s, 1 H, guanidine-NH). MS (ESI+): m/z = 271.2 [M + H]+. HRMS (ESI+): calcd. for C12H22N4O3 ([M + H]+) 271.1765, found 271.1769.
N2-Acetyl-2,3-didehydro-arginine methyl ester (8d)
General procedure B with (Z)-methyl N2-acetyl-2-amino-5-azido-pent-2-enoate (Z)-11c (204 mg, 0.962 mmol), PPh3 (504 mg, 1.92 mmol), water (0.4 mL) and THF (10 mL) for the Staudinger reduction as well as guanidinylation reagent 13 (254 mg, 1.73 mmol), NEt3 (175 mg, 240 μL, 1.73 mmol) and DMF (9 mL) for the guanidinylation reaction. The reaction mixture was stirred at rt for 2 days. Column chromatography (CH2Cl2–MeOH 10:1 → EtOAc–MeOH 1:1 → MeOH) gave impure 8d as a yellow oil (71 mg). 1H NMR (300 MHz, DMSO-d6): δ = 1.93 (s, 3 H, Ac-CH3), 2.26 (td, J = 7.4 Hz, 7.1 Hz, 2 H, H-4), 3.20 (dt, J = 7.1 Hz, 7.0 Hz, 2 H, H-5), 6.18 (t, J = 7.4 Hz, 1 H, H-3), 7.58 (s, 2 H, 2-NH), 8.14 (sbr, 3 H, guanidine-NH), 9.47 (s, 1 H, guanidine-NH). MS (ESI+): m/z = 229.2 [M + H]+. HRMS (ESI+): calcd. for C9H16N4O3 ([M + H]+) 229.1295, found 229.1299.
N2-Boc-N8,N9-bis-Cbz-2,3-didehydro-arginine tert-butyl ester (8e)
General procedure C with crude N2-Boc-2,3-didehydro-ornithine tert-butyl ester 12 (509 mg, obtained from (Z)-11a as described for the synthesis of (rac)-14,15), guanidinylation reagent 31 (828 mg, 2.31 mmol), NEt3 (539 mg, 739 μL, 5.34 mmol), AgOTf (638 mg, 2.49 mmol) and DMF (9 mL). Purification by column chromatography (petroleum ether–EtOAc 6:1 → 3:1) gave 8e as a yellow foam (761 mg, 71 % over 2 steps from (Z)-11a). 1H NMR (300 MHz, DMSO-d6): δ = 1.39 (s, 9 H, t-Bu-CH3), 1.40 (s, 9 H, t-Bu-CH3), 2.36 (dt, J = 7.1 Hz, 6.8 Hz, 2 H, H-4), 3.42 (td, J = 6.8 Hz, 5.9 Hz, 2 H, H-5), 5.04 (s, 2 H, Cbz-CH2), 5.21 (s, 2 H, Cbz-CH2), 6.12 (t, J = 7.1 Hz, 1 H, H-3), 7.27–7.44 (m, 10 H, Cbz-aryl-H), 8.26 (sbr, 1 H, 2-NH), 8.47 (t, J = 5.9 Hz, 1 H, guanidine-NH), 11.59 (s, 1 H, guanidine-NH). 13C NMR (126 MHz, DMSO-d6): δ = 26.8 (C-4), 27.5 (t-Bu-CH3), 27.9 (t-Bu-CH3), 39.0 (C-5), 66.3 (Cbz-CH2), 67.4 (Cbz-CH2), 78.5 (t-Bu-C), 79.9 (t-Bu-C), 127.5 (Cbz-aryl-C-2, Cbz-aryl-C-6), 127.7 (Cbz-aryl-C-2, Cbz-aryl-C-6), 128.1 (Cbz-aryl-C-4), 128.2 (Cbz-aryl-C-3, Cbz-aryl-C-5), 128.3 (Cbz-aryl-C-3, Cbz-aryl-C-5), 130.3 (C-3), 134.9 (C-2), 136.6 (Cbz-aryl-C-1), 152.3 (guanidine-C), 153.2 (Boc-C=O), 154.9 (Cbz-C=O), 163.4 (C-1). MS (ESI+): m/z = 619.3 [M + Na]+. HRMS (ESI+): calcd. for C31H40N4O8 ([M + Na]+) 619.2738, found 619.2742. IR (ATR): ν = 3324, 2977, 1718, 1638, 1620, 1257, 1204, 1137, 1049, 695. UV (MeCN) : λmax (log ε) = 206 (4.54), 237 (4.41). Mp: 57 °C. TLC: Rf = 0.73 (petroleum ether–EtOAc 1:1).
N2-Boc-N5-benzyl-N8,N9-bis-Cbz-2,3-didehydro-arginine tert-butyl ester (8f)
To a solution of crude N2-Boc-2,3-didehydro-ornithine tert-butyl ester 12 (694 mg, obtained from (Z)-11a as described for the synthesis of (rac)-14,15) in CH2Cl2 (20 mL), molecular sieves (4 Å) were added at rt and the mixture was stirred at rt for 30 min. Subsequently, benzaldehyde (259 mg, 247 μL, 2.45 mmol) was added, and the reaction mixture was stirred at rt for 17 h. After filtration, the filtrate was evaporated under reduced pressure. The resultant crude product was dissolved in MeOH (20 mL) and cooled to 0 °C. NaBH4 (229 mg, 6.07 mmol) was added, and the reaction mixture was stirred at 0 °C for 3 h. The reaction was quenched by addition of saturated NH4Cl solution (5 mL). After addition of water (10 mL), the aqueous layer was extracted with EtOAc (3 ×). The combined organics were washed with H2O (25 mL), dried over Na2SO4 and evaporated under reduced pressure to give 29 as a yellow oil (750 mg). Some of this crude product (702 mg) was used without further purification for the guanidinylation reaction according to general procedure C with guanidinylation reagent 31 (869 mg, 2.43 mmol), NEt3 (566 mg, 775 μL, 5.60 mmol), AgOTf (669 mg, 2.61 mmol) and DMF (9.5 mL). Purification by column chromatography (petroleum ether–EtOAc 4:1 → 1:1) gave 8f as a white foam (820 mg, 55 % over 3 steps from (Z)-11a). 1H NMR (300 MHz, DMSO-d6): δ = 1.39 (s, 9 H, t-Bu-CH3), 1.42 (s, 9 H, t-Bu-CH3), 2.39 (dt, J = 7.4 Hz, 7.0 Hz, 2 H, H-4), 3.42 (t, J = 7.0 Hz, 2 H, H-5), 4.62 (s, 2 H, Bn-CH2), 4.95 (s, 2 H, Cbz-CH2), 5.08 (s, 2 H, Cbz-CH2), 6.04 (t, J = 7.4 Hz, 1 H, H-3), 7.19–7.42 (m, 15 H, aryl-H), 8.28 (sbr, 1 H, 2-NH), 10.07 (s, 1 H, guanidine-NH). 13C NMR (126 MHz, DMSO-d6): δ = 25.4 (C-4), 27.5 (t-Bu-CH3), 28.0 (t-Bu-CH3), 46.5 (C-5), 50.5 (Bn-CH2), 66.2 (Cbz-CH2), 78.6 (t-Bu-C), 80.0 (t-Bu-C), 126.9 (aryl-C-2, aryl-C-6), 127.3 (aryl-C-2, aryl-C-6), 127.6 (aryl-C-4), 127.8 (aryl-C-4), 128.0 (aryl-C-3, aryl-C-5), 128.2 (aryl-C-3, aryl-C-5), 128.4 (C-2), 130.3 (C-3), 135.8 (aryl-C-1), 136.8 (aryl-C-1), 151.0 (Boc-C=O), 152.3 (guanidine-C), 153.3 (Cbz-C=O), 163.3 (C-1). MS (ESI+): m/z = 709.3 [M + Na]+. HRMS (ESI+): calcd. for C38H46N4O8 ([M + Na]+) 709.3208, found 709.3206. TLC: Rf = 0.70 (petroleum ether–EtOAc 1:1).
N2,N2-bis-Boc-N8,N9-bis-Cbz-2,3-didehydro-arginine tert-butyl ester (8 g)
General procedure C with crude N2,N2-bis-Boc-2,3-didehydro-ornithine tert-butyl ester 16 (488 mg, obtained from (Z)-11d as described for the synthesis of (rac)-17,18), guanidinylation reagent 31 (588 mg, 1.64 mmol), NEt3 (383 mg, 525 μL, 3.79 mmol), AgOTf (173 mg, 0.675 mmol) and DMF (6.5 mL). Purification by column chromatography (petroleum ether–EtOAc 10:1 → 1:2) gave 8 g as a white foam (642 mg, 50 % over 2 steps from (Z)-11d). 1H NMR (300 MHz, DMSO-d6): δ = 1.31 (s, 18 H, t-Bu-CH3), 1.38 (s, 9 H, t-Bu-CH3), 2.30 (dt, J = 7.0 Hz, 6.6 Hz, 2 H, H-4), 3.46 (td, J = 6.6 Hz, 6.0 Hz, 2 H, H-5), 5.00 (s, 2 H, Cbz-CH2), 5.18 (s, 2 H, Cbz-CH2), 6.70 (t, J = 7.0 Hz, 1 H, H-3), 7.24–7.41 (m, 10 H, Cbz-aryl-H), 8.53 (t, J = 6.0 Hz, 1 H, guanidine-NH), 11.58 (s, 1 H, guanidine-NH). 13C NMR (76 MHz, DMSO-d6): δ = 27.2 (C-4, t-Bu-CH3), 27.5 (t-Bu-CH3), 39.5 (C-5), 66.3 (Cbz-CH2), 67.5 (Cbz-CH2), 80.6 (t-Bu-C), 81.7 (t-Bu-C), 127.7 (Cbz-aryl-C-2, Cbz-aryl-C-6), 127.8 (Cbz-aryl-C-2, Cbz-aryl-C-6), 128.0 (Cbz-aryl-C-4), 128.2 (Cbz-aryl-C-4), 128.4 (Cbz-aryl-C-3, Cbz-aryl-C-5), 131.0 (C-2), 135.1 (Cbz-aryl-C-1), 136.8 (Cbz-aryl-C-1), 137.2 (C-3), 149.6 (Boc-C=O), 152.3 (guanidine-C), 155.1 (Cbz-C=O), 162.0 (Cbz-C=O), 162.9 (C-1). MS (ESI+): m/z = 719.4 [M + Na]+. HRMS (ESI+): calcd. for C36H48N4O10 ([M + Na]+) 719.3263, found 719.3274. IR (ATR): ν = 2978, 1719, 1638, 1366, 1249, 1151, 1094, 1047, 734, 696. UV (MeCN): λmax (log ε) = 206 (4.57), 240 (4.45), 273 (4.48). TLC: Rf = 0.53 (petroleum ether–EtOAc 1:1).
N2,N2-Bis-Boc-N5-benzyl-N8,N9-bis-Cbz-2,3-didehydro-arginine tert-butyl ester (8 h)
To a solution of crude N2,N2-bis-Boc-2,3-didehydro-ornithine tert-butyl ester 16 (1.10 g, obtained from (Z)-11d as described for the synthesis of (rac)-17,18) in CH2Cl2 (25 mL), molecular sieves (4 Å) were added at rt and the mixture was stirred at rt for 30 min. Subsequently, benzaldehyde (504 mg, 478 μL, 4.75 mmol) was added, and the reaction mixture was stirred at rt for 17 h. After filtration, the filtrate was evaporated under reduced pressure. The resultant crude product was dissolved in MeOH (25 mL). NaBH4 (1.79 g, 47.6 mmol) was added in two portions (the second after 4 h), and the reaction mixture was stirred at rt for 22 h. The reaction was quenched by addition of saturated NH4Cl solution (5 mL). After addition of water (10 mL), the aqueous layer was extracted with EtOAc (3 ×). The combined organics were washed with H2O (25 mL), dried over Na2SO4 and evaporated under reduced pressure. The resultant crude product was separated by column chromatography (petroleum ether–EtOAc 1:1) to give impure 30 as a yellow oil (748 mg). This material was used without further purification for the guanidinylation reaction according to general procedure C with guanidinylation reagent 31 (758 mg, 2.12 mmol), NEt3 (493 mg, 676 μL, 4.88 mmol), AgOTf (584 mg, 2.28 mmol) and DMF (8 mL). Purification by column chromatography (petroleum ether–EtOAc 6:1 → 3:1) gave 8 h as a yellow foam (991 mg, 42 % over 3 steps from (Z)-11d). 1H NMR (300 MHz, DMSO-d6): δ = 1.32 (s, 18 H, t-Bu-CH3), 1.39 (s, 9 H, t-Bu-CH3), 2.27 (dt, J = 7.6 Hz, 7.1 Hz, 2 H, H-4), 3.42 (t, J = 7.1 Hz, 2 H, H-5), 4.62 (s, 2 H, Bn-CH2), 4.90 (s, 2 H, Cbz-CH2), 5.03 (s, 2 H, Cbz-CH2), 6.59 (t, J = 7.6 Hz, 1 H, H-3), 7.17–7.33 (m, 15 H, aryl-H), 10.05 (s, 1 H, NH). 13C NMR (126 MHz, DMSO-d6): δ = 25.9 (C-4), 27.3 (t-Bu-CH3), 27.5 (t-Bu-CH3), 45.8 (C-5), 50.4 (Bn-CH2), 66.2 (Cbz-CH2), 80.7 (t-Bu-C), 81.8 (t-Bu-C), 126.2 (aryl-C-2, aryl-C-6), 126.9 (aryl-C-2, aryl-C-6), 127.6 (aryl-C-4), 127.8 (aryl-C-4), 128.0 (aryl-C-3, aryl-C-5), 128.3 (aryl-C-3, aryl-C-5), 130.9 (C-2), 136.0 (aryl-C-1), 136.6 (C-3) 136.8 (aryl-C-1), 149.5 (Boc-C=O), 152.3 (guanidine-C), 153.3 (Cbz-C=O), 161.8 (C-1). MS (ESI+): m/z = 809.4 [M + Na]+. HRMS (ESI+): calcd. for C43H54N4O10 ([M + Na]+) 809.3732, found 809.3736. IR (ATR): ν = 2977, 1718, 1602, 1454, 1366, 1250, 1150, 1090, 733, 696. UV (MeCN): λmax (log ε) = 207 (4.61), 251 (4.26). TLC: Rf = 0.71 (petroleum ether–EtOAc 1:1).
tert-Butyl N2-Boc-2-amino-5-azido-pent-2-enoate (11a)
General procedure A with phosphonate 9a (2.00 g, 5.45 mmol), crude 3-azido-propionaldehyde 10 (1.18 g), KOt-Bu (641 mg, 5.72 mmol) and THF (11 mL (KOt-Bu), 16 mL (9a), 16 mL (10)). Purification by column chromatography (petroleum ether–EtOAc 6:1) gave (Z)-11a as a colorless solid (1.31 g, 77 %) and (E)-11a as a colorless oil (62 mg, 4 %). (Z)-11a: 1H NMR (300 MHz, CDCl3): δ = 1.44 (s, 9 H, t-Bu-CH3), 1.47 (s, 9 H, t-Bu-CH3), 2.45 (dt, J = 7.4 Hz, 6.9 Hz, 2 H, H-4), 3.41 (t, J = 6.9 Hz, 2 H, H-5), 6.19 (sbr, 1 H, NH), 6.38 (t, J = 7.4 Hz, 1 H, H-3). 13C NMR (76 MHz, CDCl3): δ = 28.0 (t-Bu-CH3), 28.2 (t-Bu-CH3), 28.5 (C-4), 50.0 (C-5), 80.5 (t-Bu-C), 82.0 (t-Bu-C), 128.5 (C-3), 128.7 (C-2), 153.1 (Boc-C=O), 163.6 (C-1). MS (ESI+): m/z = 335 [M + Na]+. HRMS (ESI+): calcd. for C14H24N4O4 ([M + Na]+) 335.1690, found 335.1690. IR: ν = 3308, 2980, 2097, 1688, 1493, 1367, 1246, 1146, 847, 778. UV (MeCN): λmax (log ε) = 231 (3.85). Mp: 57 °C. TLC: Rf = 0.42 (petroleum ether–EtOAc 3:1). (E)-11a: 1H NMR (300 MHz, CDCl3): δ = 1.44 (s, 9 H, t-Bu-CH3), 1.52 (s, 9 H, t-Bu-CH3), 2.79 (dt, J = 7.5 Hz, 7.3 Hz, 2 H, H-4), 3.35 (t, J = 7.4 Hz, 2 H, H-5), 6.66 (t, J = 7.5 Hz, 1 H, H-3), 6.74 (sbr, 1 H, NH). 13C NMR (76 MHz, CDCl3): δ = 28.0 (C-4), 28.1 (t-Bu-CH3), 28.3 (t-Bu-CH3), 51.0 (C-5), 80.3 (t-Bu-C), 83.3 (t-Bu-C), 121.3 (C-3), 128.0 (C-2), 153.0 (Boc-C=O), 163.1 (C-1). MS (ESI+): m/z = 335 [M + Na]+. HRMS (ESI+): calcd. for C14H24N4O4 ([M + Na]+) 335.1690, found 335.1690. TLC: Rf = 0.42 (petroleum ether-EtOAc 3:1).
tert-Butyl N2-acetyl-2-amino-5-azido-pent-2-enoate (11b)
General procedure A with phosphonate 9b (1.64 g, 5.83 mmol), crude 3-azido-propionaldehyde 10 (1.15 g), KOt-Bu (686 mg, 6.12 mmol) and THF (11 mL (KOt-Bu), 16 mL (9b), 10 mL (10)). Purification by column chromatography (petroleum ether–EtOAc 3:2) gave (Z)-11b as a yellow oil (910 mg, 61 %) while (E)-11b could not be isolated. 1H NMR (300 MHz, CDCl3): δ = 1.48 (s, 9 H, t-Bu-CH3), 2.10 (s, 3 H, Ac-CH3), 2.40 (dt, J = 7.1 Hz, J = 6.7 Hz, 2 H, H-4), 3.43 (t, J = 6.7 Hz, 2 H, H-5), 6.49 (t, J = 7.1 Hz, 1 H, H-3), 7.13 (sbr, 1 H, NH). 13C NMR (75 MHz, CDCl3): δ = 23.5 (Ac-CH3), 27.9 (t-Bu-CH3), 29.0 (C-4), 49.9 (C-5), 82.3 (t-Bu-C), 127.6 (C-2), 130.8 (C-3), 163.4 (C-1), 168.2 (Ac-C=O). MS: (ESI+): m/z = 531.3 [2 M + Na]+. HRMS (ESI+): calcd. for C11H18N4O3 ([M–H]-) 253.1306, found 253.1312. IR (ATR): ν = 3265, 2979, 2095, 1656, 1509, 1366, 1254, 1135, 848, 735. UV (MeCN): λmax (log ε) = 230 (3.81). TLC: Rf = 0.34 (petroleum ether-EtOAc 3:2).
Methyl N2-acetyl-2-amino-5-azido-pent-2-enoate (11c)
General procedure A with phosphonate 9c (1.62 g, 6.80 mmol), crude 3-azido-propionaldehyde 10 (1.50 g), KOt-Bu (799 mg, 7.14 mmol) and THF (14 mL (KOt-Bu), 15 mL (9c), 10 mL (10)). Purification by column chromatography (petroleum ether–EtOAc 1:1 → 1:3) gave (Z)-11c as a yellow solid (751 mg, 52 %) while (E)-11c could not be isolated. 1H-NMR (300 MHz, CDCl3): δ = 2.13 (s, 3 H, Ac-CH3), 2.43 (dt, J = 7.1 Hz, 6.7 Hz, 2 H, H-4), 3.46 (t, J = 6.7 Hz 2 H, H-5), 3.78 (s, 3 H, OCH3), 6.64 (t, J = 7.1 Hz, 1 H, H-3), 7.09 (sbr, 1 H, NH). 13C NMR (76 MHz, CDCl3): δ = 23.5 (Ac-CH3), 28.9 (C-4), 49.8 (C-5), 52.6 (OCH3), 126.5 (C-2), 132.7 (C-3), 164.8 (C-1), 168.3 (Ac-C=O). MS (ESI+): m/z = 447.2 [2 M + Na]+. HRMS (ESI+): calcd. for C8H12N4O3 ([M + Na]+) 235.0802, found 235.0803. IR (ATR): ν = 3261, 2962, 2086, 1742, 1676, 1372, 1210, 1021, 795, 714. UV (MeCN): λmax (log ε) = 231 (3.80). Mp: 51 °C. TLC: Rf = 0.16 (petroleum ether–EtOAc 1:1).
tert-Butyl N2,N2-bis-Boc-2-amino-5-azido-pent-2-enoate (11d)
To a solution of (Z)-tert-butyl N2-Boc-2-amino-5-azido-pent-2-enoate (Z)-11a (1.29 g, 4.14 mmol) in MeCN 12mL, DMAP (50 mg, 0.41 mmol) and Boc2O (1.98 g, 9.10 mmol) were added at rt. The reaction mixture was stirred at rt for 20 h. The solvent was evaporated under reduced pressure. After the addition of EtOAc, the solution was washed with aqueous KHSO4 solution (1 M, 2 × 10 mL), saturated NaHCO3 solution (1 × 10 mL) and brine (1 × 10 mL), dried over Na2SO4 and evaporated under reduced pressure. The resultant crude product was purified by column chromatography (petroleum ether–EtOAc 10:1) to give (Z)-11d as a colorless oil (1.57 g, 92 %). 1H NMR (300 MHz, DMSO-d6): δ = 1.37 (s, 18 H, t-Bu-CH3), 1.41 (s, 9 H, t-Bu-CH3), 2.29 (dt, J = 7.3 Hz, 6.6 Hz, 2 H, H-4), 3.44 (t, J = 6.6 Hz, 2 H, H-5), 6.65 (t, J = 7.3 Hz, 1 H, H-3). 13C NMR (76 MHz, DMSO-d6): δ = 27.0 (C-4), 27.3 (t-Bu-CH3), 27.5 (t-Bu-CH3), 48.6 (C-5), 80.8 (t-Bu-C), 81.9 (t-Bu-C), 131.5 (C-2), 136.3 (C-3), 149.7 (Boc-C=O), 161.9 (C-1). MS (ESI+): m/z = 847.6 [2 M + Na]+. HRMS (ESI+): calcd. for C19H32N4O6 ([M + Na]+) 435.2214, found 435.2218. IR (ATR): ν = 2979, 2097, 1795, 1717, 1366, 1273, 1250, 1150, 1092, 849. UV (MeCN): λmax (log ε) = 216 (4.01). TLC: Rf = 0.23 (petroleum ether–EtOAc 10:1).
N2-Boc-capreomycidine tert-butyl ester ((rac)-14) and N2-Boc-epicapreomycidine tert-butyl ester ((rac)-15)
General procedure B with (Z)-tert-butyl N2-Boc-2-amino-5-azido-pent-2-enoate (Z)-11a (1.13 g, 3.61 mmol), PPh3 (2.28 g, 8.71 mmol), water (1.7 mL) and THF (44 mL) for the Staudinger reduction as well as guanidinylation reagent 13 (964 mg, 6.58 mmol), NEt3 (1.66 g, 2.28 mL, 16.4 mmol) and DMF (17 mL) for the domino reaction. The reaction mixture was stirred at rt for 20 h and at 70 °C for 7 h. Purification by column chromatography (CH2Cl2–MeOH gradient (3–10 %)) gave a mixture of (rac)-14 and (rac)-15 (ratio 1.0 : 1.0) as a yellow foam (580 mg, 54 % over 2 steps from (Z)-11a). 1H NMR (300 MHz, CDCl3): δ = 1.40 (s, 1 × 9 H, t-Bu-CH3), 1.41 (s, 1 × 9 H, t-Bu-CH3), 1.46 (s, 1 × 9 H, t-Bu-CH3), 1.47 (s, 1 × 9 H, t-Bu-CH3), 1.79–1.93 (m, 1 × 2 H, H-4), 1.93–2.03 (m, 1 × 2 H, H-4), 3.21–3.33 (m, 1 × 2 H, H-5), 3.33–3.47 (m, 1 × 2 H, H-5), 3.76–3.83 (m, 1 × 1 H, H-3), 3.85–3.93 (m, 1 × 1 H, H-3), 4.25–4.35 (m, 2 × 1 H, H-2), 5.71 (dbr, J = 8.2 Hz, 2 × 1 H, NH-2), 7.04 (sbr, 1 × 2 H, guanidine-NH), 7.15 (sbr, 1 × 2 H, guanidine-NH), 7.82 (sbr, 2 × 1 H, guanidine-NH), 8.10 (sbr, 1 × 1 H, guanidine-NH), 8.18 (sbr, 1 × 1 H, guanidine-NH). 13C NMR (76 MHz, CDCl3): δ = 21.7 (1 × C-4), 23.1 (1 × C-4), 28.0 (2 × t-Bu-CH3), 28.2 (1 × t-Bu-CH3), 28.3 (1 × t-Bu-CH3), 36.6 (1 × C-5), 36.7 (1 × C-5), 50.1 (1 × C-3), 51.6 (1 × C-3), 56.4 (1 × C-2), 56.9 (1 × C-2), 80.4 (1 × t-Bu-C), 80.7 (1 × t-Bu-C), 83.6 (1 × t-Bu-C), 83.7 (1 × t-Bu-C), 154.9 (1 × Boc-C=O), 155.1 (1 × Boc-C=O), 155.8 (1 × guanidine-C), 156.0 (1 × guanidine-C), 168.0 (1 × C-1), 168.7 (1 × C-1). MS (ESI+): m/z = 329.3 [M + H]+. HRMS (ESI+) calcd. for C15H28N4O4 ([M + H]+) 329.2183, found 329.2183. TLC: Rf = 0.33 (CH2Cl2–MeOH 9:1).
N2,N2-Bis-Boc-capreomycidine tert-butyl ester ((rac)-17) and N2,N2-Bis-Boc-epicapreomycidine tert-butyl ester ((rac)-18)
General procedure B with (Z)-tert-butyl N2,N2-bis-Boc-2-amino-5-azido-pent-2-enoate (Z)-11d (671 mg, 1.63 mmol), PPh3 (1.32 g, 5.05 mmol), water (1.0 mL) and THF (25 mL) for the Staudinger reduction as well as guanidinylation reagent 13 (388 mg, 2.65 mmol), NEt3 (268 mg, 367 μL, 2.65 mmol) and DMF (13 mL) for the domino reaction. The reaction mixture was stirred at rt for 4 d. Purification by column chromatography (CH2Cl2–MeOH gradient (5–10 %)) gave a mixture of (rac)-17 and (rac)-18 (ratio 1.8:1.0) as a yellow foam (340 mg, 49 % over 2 steps from (Z)-11d). 1H NMR (300 MHz, DMSO-d6): δ = 1.39 (s, 2 × 18 H, t-Bu-CH3), 1.40 (s, 1 × 9 H, t-Bu-CH3), 1.45 (s, 1 × 9 H, t-Bu-CH3), 1.96–2.02 (m, 2 × 2 H, H-4), 3.21–3.28 (m, 2 × 2 H, H-5), 3.98–4.07 (m, 2 × 1 H, H-3), 4.61 (d, J = 8.9 Hz, 1 × 1 H, H-2), 4.70 (d, J = 8.3 Hz, 1 × 1 H, H-2), 7.01 (s, 1 × 2 H, guanidine-NH), 7.09 (s, 1 × 2 H, guanidine-NH), 7.51 (sbr, 1 × 1 H, guanidine-NH), 7.56 (sbr, 1 × 1 H, guanidine-NH), 8.13 (sbr, 1 × 1 H, guanidine-NH), 8.19 (sbr, 1 × 1 H, guanidine-NH). 13C NMR (76 MHz, DMSO-d6): δ = 22.8 (2 × C-4), 27.4 (2 × t-Bu-CH3), 27.5 (2 × t-Bu-CH3), 27.5 (2 × t-Bu-CH3), 34.2 (1 × C-5), 35.0 (1 × C-5), 46.8 (1 × C-3), 47.3 (1 × C-3), 60.0 (1 × C-2), 60.3 (1 × C-2), 81.6 (1 × t-Bu-C), 82.0 (1 × t-Bu-C), 83.0 (1 × t-Bu-C), 83.4 (1 × t-Bu-C), 151.8 (1 × Boc-C=O), 151.8 (1 × Boc-C=O), 153.6 (2 × guanidine-C), 154.0 (2 × Boc-C=O), 166.9 (2 × C-1). MS (ESI+): m/z = 429.3 [M + H]+. HRMS (ESI+): calcd. for C20H36N4O6 ([M + H]+) 429.2708, found 429.2707. TLC: Rf = 0.45 (CH2Cl2–MeOH 9:1).
tert-Butyl N2-Cbz-2-amino-5-azido-pent-2-enoate (21)
General procedure A with phosphonate 20 (3.46 g, 9.27 mmol), crude 3-azido-propionaldehyde 10 (1.84 g), KOt-Bu (1.09 g, 9.73 mmol) and THF (19 mL (KOt-Bu), 28 mL (20), 28 mL (10)). Purification by column chromatography (petroleum ether–EtOAc 8:1) gave (Z)-21 (containing traces of aldehyde 10) as a colorless oil (2.70 g, 81 % as calculated from the 1H NMR spectrum) and (E)-21 as a colorless oil (118 mg, 3 %). (Z)-21: 1H NMR (300 MHz, CDCl3): δ = 1.49 (s, 9 H, t-Bu-CH3), 2.48 (dt, J = 7.2 Hz, 6.8 Hz, 2 H, H-4), 3.44 (t, J = 6.8 Hz 2 H, H-5), 5.14 (s, 2 H, Cbz-CH2), 6.45 (tbr, J = 7.2 Hz, 2 H, H-3, NH), 7.30–7.39 (m, 5 H, Cbz-aryl-H). 13C NMR (76 MHz, CDCl3): δ = 28.0 (t-Bu-CH3), 28.5 (C-4), 49.9 (C-5), 67.4 (Cbz-CH2), 82.3 (t-Bu-C), 128.2 (Cbz-aryl-C-2, Cbz-aryl-C-6), 128.3 (Cbz-aryl-C-4), 128.5 (C-2, Cbz-aryl-C-3, Cbz-aryl-C-5), 129.7 (C-3), 135.9 (Cbz-aryl-C-1), 153.9 (Cbz-C=O), 163.2 (C-1). MS (ESI+): m/z = 715.3 [2 M + Na]+. HRMS (ESI+): calcd. for C17H22N4O4 ([M + Na]+) 369.1533, found 369.1530. IR (ATR): ν = 2971, 2095, 1703, 1498, 1290, 1218, 1147, 1022, 750, 696. UV (MeCN): λma× (log ε) = 204 (4.13), 228 (3.83). TLC: Rf = 0.73 (petroleum ether–EtOAc 1:1). (E)-21: 1H NMR (300 MHz, CDCl3): δ = 1.52 (s, 9 H, t-Bu-CH3), 2.82 (dt, J = 7.5 Hz, 7.2 Hz, 2 H, H-4), 3.36 (t, J = 7.2 Hz 2 H, H-5), 5.11 (s, 2 H, Cbz-CH2), 6.76 (t, J = 7.5 Hz, 1 H, H-3), 6.98 (sbr, 1 H, NH), 7.27–7.39 (m, 5 H, Cbz-aryl-H). 13C NMR (76 MHz, CDCl3): δ = 28.0 (C-4), 28.1 (t-Bu-CH3), 50.9 (C-5), 66.8 (Cbz-CH2), 83.6 (t-Bu-C), 122.2 (C-3), 127.6 (C-2), 128.2 (Cbz-aryl-C-2, Cbz-aryl-C-6), 128.3 (Cbz-aryl-C-4), 128.6 (Cbz-aryl-C-3, Cbz-aryl-C-5), 136.0 (Cbz-aryl-C-1), 153.5 (Cbz-C=O), 162.7 (C-1). MS (ESI+): m/z = 715.3 [2 M + Na]+. HRMS (ESI): calcd. for C17H22N4O4 ([M + Na]+) 369.1533, found 369.1535. IR (ATR): ν = 2972, 2095, 1697, 1509, 1363, 1215, 1151, 1046, 846, 696. UV (MeCN): λmax (log ε) = 238 (3.85). TLC: Rf = 0.78 (petroleum ether–EtOAc 1:1).
N2-Cbz-capreomycidine tert-butyl ester ((rac)-23) and N2-Cbz-epicapreomycidine tert-butyl ester ((rac)-24)
General procedure B with (Z)-tert-butyl N2-Cbz-2-amino-5-azido-pent-2-enoate (Z)-21 (1.05 g, 3.04 mmol), PPh3 (2.41 g, 9.18 mmol), water (1.8 mL) and THF (46 mL) for the Staudinger reduction as well as guanidinylation reagent 13 (433 mg, 2.96 mmol), NEt3 (326 mg, 446 μL, 3.23 mmol) and DMF (14 mL) for the domino reaction. The reaction mixture was stirred at 80 °C for 19 h. Purification by column chromatography (CH2Cl2–MeOH gradient (8–11 %)) gave a mixture of (rac)-23 and (rac)-24 (ratio 1.0 : 1.0) as a brownish foam (683 mg, 62 % over 2 steps from (Z)-21). 1H NMR (300 MHz, CDCl3): δ = 1.43 (s, 2 × 9 H, t-Bu-CH3), 1.65–1.99 (m, 2 × 2 H, H-4), 3.13–3.26 (m, 1 × 2 H, H-5), 3.26–3.42 (m, 1 × 2 H, H-5), 3.70–3.80 (m, 1 × 1 H, H-3), 3.81–3.91 (m, 1 × 1 H, H-3), 4.29–4.40 (m, 2 × 1 H, H-2), 5.02–5.14 (m, 2 × 2 H, Cbz-CH2), 6.18 (dbr, J = 8.5 Hz, 2 × 1 H, Cbz-NH), 7.03 (sbr, 1 × 2 H, guanidine-NH), 7.13 (sbr, 1 × 2 H, guanidine-NH), 7.22–7.35 (m, 2 × 5 H, aryl-H), 7.89 (sbr, 1 × 1 H, guanidine-NH), 8.02 (sbr, 1 × 1 H, guanidine-NH), 8.10 (sbr, 1 × 1 H, guanidine-NH), 8.13 (sbr, 1 × 1 H, guanidine-NH). 13C NMR (76 MHz, CDCl3): δ = 21.8 (1 × C-4), 23.0 (1 × C-4), 27.9 (2 × t-Bu-CH3), 36.6 (1 × C-5), 36.7 (1 × C-5), 50.2 (1 × C-3), 51.4 (1 × C-3), 56.9 (1 × C-2), 57.4 (1 × C-2), 67.3 (2 × Cbz-CH2), 83.8 (1 × t-Bu-C), 83.9 (1 × t-Bu-C), 128.0 (2 × Cbz-aryl-C-2, 2 × Cbz-aryl-C-6), 128.1 (1 × Cbz-aryl-C-4), 128.2 (1 × Cbz-aryl-C-4), 128.5 (2 × Cbz-aryl-C-3, 2 × Cbz-aryl-C-5), 136.0 (1 × Cbz-aryl-C-1), 136.1 (1 × Cbz-aryl-C-1), 154.9 (1 × Cbz-C=O), 155.0 (1 × Cbz-C=O), 156.3 (1 × guanidine-C), 156.6 (1 × guanidine-C), 167.6 (1 × C-1), 168.4 (1 × C-1). MS (ESI+): m/z = 363.2 [M + H]+. HRMS (ESI+): calcd. for C18H26N4O4 ([M + H]+) 363.2027, found 363.2029. TLC: Rf = 0.33 (CH2Cl2–MeOH 4:1).
N2-Acetyl-capreomycidine hydrochloride ((rac)-27) and N2-acetyl-epicapreomycidine hydrochloride ((rac)-28)
A mixture of N2-Cbz-capreomycidine tert-butyl ester (rac)-23 and N2-Cbz-epicapreomycidine tert-butyl ester (rac)-24 (174 mg, 0.481 mmol), acetic anhydride (123 mg, 113 μL, 1.20 mmol) and Pd/C (10 %, 13 mg, 12 μmol) in anhydrous MeOH (4.8 mL) was stirred under a hydrogen atmosphere (1 bar, balloon) at rt for 22 h. After filtration through Celite and washing the Celite with MeOH (2 ×), the filtrate was evaporated under reduced pressure to give a mixture of (rac)-25 and (rac)-26 containing acetic anhydride as a yellow oil (170 mg). This mixture was then dissolved in hydrochloric acid (6 M, 4.0 mL, 24 mmol), and the solution was stirred at rt for 3 h. The solvent was evaporated under reduced pressure to give a mixture of (rac)-27 and (rac)-28 (ratio 1.2 : 1.0) as an orange foam (93 mg, 77 % over 2 steps from (rac)-23 and (rac)-24). 1H NMR (300 MHz, D2O): δ = 1.88–2.20 (m, 2 × 2 H, H-4), 2.11 (s, 1 × 3 H, Ac-CH3), 2.13 (s, 1 × 3 H, Ac-CH3), 3.36–3.46 (m, 2 × 2 H, H-5), 4.02–4.11 (m, 2 × 1 H, H-3), 4.65–4.82 (m, 2 × 1 H, H-2). 13C NMR (76 MHz, D2O): δ = 20.7 (1 × C-4), 21.7 (1 × Ac-CH3), 21.8 (1 × Ac-CH3), 22.2 (1 × C-4), 36.1 (1 × C-5), 36.2 (1 × C-5), 49.3 (2 × C-3), 54.7 (1 × C-2), 55.2 (1 × C-2), 154.1 (1 × guanidine-C), 154.4 (1 × guanidine-C), 172.4 (2 × C-1), 174.6 (2 × Ac-C=O). MS (ESI+): m/z = 237.1 [M + Na]+. HRMS (ESI+): calcd. for C8H14N4O3 ([M + Na]+) 237.0958, found 237.0958.
X-ray structure determination of (Z)-11a
Single crystals of (Z)-11a were selected and covered with perfluorinated polyether oil on a microscope slide, which was cooled with a nitrogen gas flow using the X-TEMP2 (Kottke and Stalke 1993; Kottke et al. 1996; Stalke 1998). An appropriate crystal was selected using a polarize microscope, mounted on the tip of a MiTeGen©-MicroMount, fixed to a goniometer head and shock-cooled by the crystal cooling device.
The data for (Z)-11a were collected from shock-cooled crystals at 100(2) K (Kottke and Stalke 1993; Kottke et al. 1996; Stalke 1998). The data of (Z)-11a were collected on a Incoatec Mo Microsource (Schulz et al. 2009) with mirror optics and APEX II detector with a D8 goniometer. The diffractometer was equipped with a low-temperature device and used MoKα radiation, λ = 71.073 pm. The crystal of (Z)-11a used for data collection was a non-merohedral twin with two twin domains. The two domains were separated using RLATT. Two orientation matrices were used for the integration with Saint (Bruker AXS Inst. Inc. 2011) and a semi-empirical absorption correction with Twinabs (Sheldrick 2012) was applied. The structure was solved by direct methods (Shelxs-97) (Sheldrick 1990) using untwinned data (HKLF 4) and refined by full-matrix least-squares methods against F2 (Shelxl-97) (Sheldrick 2008; Müller et al. 2006) using the twinned data (HKLF 5) within the ShelXle GUI (Hübschle et al. 2011). The fractional distribution of the second domain refines to 0.4669 (9). All non-hydrogen-atoms were refined with anisotropic displacement parameters. The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to equal to 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Only the hydrogen atoms of the nitrogen atoms (H1 and H8) were found from difference density map and restrained to same distance within the esd (0.02 Å). Disordered moieties were refined using bond lengths and angles restraints and anisotropic displacement parameter restraints.
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. The CCDC number, crystal data and further details for the X-ray measurement are listed in the Supplementary Material. Copies of the data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif or from the corresponding author.
Results and discussion
For the synthesis of potential cyclization precursors 8a–h (Table 1), a general synthetic approach was developed. In order to achieve diversity with respect to the protecting group pattern of the guanidine moiety, it was envisaged to introduce this functionality at a late stage. Guanidinylation of primary or secondary amines is a common way to prepare guanidines (for the terminology of guanidine formation, see Jones 2002). However, since δ-amino-didehydro α-amino acid esters (i.e., didehydro-ornithine derivatives) with an unprotected δ-amino group display limited stability, the corresponding δ-azido analogues were synthesized first. Reduction of the azide moiety would then provide the amines suitable for guanidinylation reactions. This approach has previously been used in the synthesis of a protected didehydro-arginine derivative (Yonezawa et al. 2000). However, to the best of our knowledge, the literature precedent for the synthesis and further transformation of a δ-azido-didehydro α-amino acid ester is limited to this one example.
Thus, rapid access to protected δ-azido-didehydro α-amino acids was an essential requirement for the described synthetic strategy. All δ-azido-didehydro α-amino acids used in this study were prepared by Wittig-Horner reactions of amino acid phosphonates 9a–c (Ducho et al. 2009; Schmidt et al. 1984) with 3-azido-propionaldehyde 10 (Davies et al. 1967) (Fig. 3). This approach proved to be significantly more efficient than the previously used lengthy transformation of protected didehydro-glutamate into the respective δ-azido compound (Yonezawa et al. 2000). Wittig-Horner reactions of amino acid phosphonates with aldehydes are known to furnish the according (Z)-didehydro amino acids with excellent diastereoselectivities (Schmidt et al. 1992; for further examples, see Ducho et al. 2009; Spork and Ducho 2010; Spork et al. 2011). Accordingly, the (Z)-configured products (Z)-11a–c were obtained in isolated yields of 52–77 %. The synthesis of cyclization precursors 8b and 8g,h required the presence of a second Boc group at the α-amino moiety. Thus, (Z)-11a was treated with di-tert-butyl dicarbonate ((Boc)2O) to afford (Z)-11d in 92 % yield. For the stereochemical assignment of (Z)-11a, the according (E)-isomer was needed as a reference compound. Following careful column chromatography, the (E)-configured congener (E)-11a was isolated as a byproduct from the Wittig-Horner transformation of 9a with 10 in 4 % yield. Application of the established 1H NMR criteria for the distinction of (Z)- and (E)-didehydro amino acids (Mazurkiewicz et al. 2005) on both isomers of 11a clearly revealed the major product to display (Z)-configuration (for details see Supplementary Material). For a completely unambiguous assignment of the double-bond geometry, single crystals of (Z)-11a suitable for X-ray crystallography could be obtained by slow evaporation of a solution of the compound in diethyl ether. The elucidated structure confirmed the (Z)-configuration of the double bond (Fig. 4). The crystals of (Z)-11a grew as non-merohedral twins in the space group P21/c with two hydrogen-bonded (N–H···O d = 206(2) pm) molecules within the asymmetric unit (see Supplementary Material). The azide groups of both molecules are disordered on two positions with an occupancy of 50 %.
Fig. 3.

Synthesis of protected δ-azido-didehydro α-amino acids 11
Fig. 4.

Molecular structure of protected δ-azido-didehydro α-amino acid (Z)-11a. Ellipsoids are shown at 50 % probability level. Carbon atoms are shown in black, nitrogen in blue, oxygen red and hydrogen white. The other hydrogen atoms and the disorder of the azide-group are omitted for clarity
The first attempt to achieve the desired biomimetic cyclization was to synthesize precursor 8a without protecting groups at the guanidine moiety (Fig. 5). Azido derivative (Z)-11a was therefore reduced under Staudinger conditions to give amino intermediate 12, which was not purified with respect to anticipated stability issues (see Yonezawa et al. 2000). Guanidinylation of 12 was carried out using commercially available reagent 13 in the presence of triethylamine to liberate the reactive agent from the employed hydrochloride. However, instead of the expected product (Z)-8a, a mixture of protected capreomycidine (rac)-14 and epicapreomycidine (rac)-15 (diastereomeric ratio (d.r.) ca. 1 : 1) was isolated in 54 % yield over two steps from (Z)-11a. Thus, after formation of intermediate (Z)-8a, an immediate aza-Michael-addition occurred in a domino fashion (for the concept of domino reactions, see Tietze and Beifuss 1993; Tietze 1996; Tietze et al. 2006). Similar results were obtained with precursor (Z)-11d, which was reduced to ornithine derivative 16. Intermediate 16 was then used in the domino reaction to give a mixture of protected capreomycidine (rac)-17 and epicapreomycidine (rac)-18 (d.r.~1.8 : 1.0) in 49 % yield over two steps from (Z)-11d. Hence, the higher electrophilicity of the precursor 16 bearing two Boc groups did not result in higher isolated yields of the domino product (Fig. 5). Interestingly, when isomer (E)-11a was used in the sequence of Staudinger reduction and subsequent domino-guanidinylation-aza-Michael-addition, no product could be isolated. It was therefore concluded that either the (E)-configured ornithine derivative was more instable than the (Z)-isomer or that the (E)-didehydro-arginine underwent decomposition under the reaction conditions.
Fig. 5.
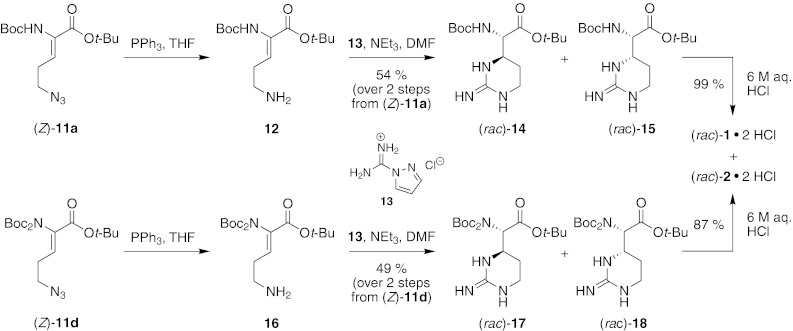
Synthesis of capreomycidine (rac)-1 and epicapreomycidine (rac)-2 using a biomimetic domino reaction
The thus synthesized mixtures of protected capreomycidine (rac)-14 and epicapreomycidine (rac)-15 as well as (rac)-17 and (rac)-18, respectively, were deprotected under acidic conditions to furnish a mixture of target compounds capreomycidine (rac)-1 and epicapreomycidine (rac)-2 as their dihydrochlorides in yields of 99 and 87 %, respectively (Fig. 5). A procedure for the separation of 1 and 2 by crystallization of the picrates is established (Bycroft et al. 1971b). Our novel biomimetic synthesis can therefore be considered a very convenient and rapid access to pure 1 and/or 2 in racemic form.
It was envisaged that a stereocontrolled version of the domino reaction would be difficult to achieve. First attempts to employ chiral bases such as (−)−quinine or (−)−sparteine instead of triethylamine resulted in low conversions and side reactions. Hence, it was decided to prepare N2-acetyl derivatives of 1 and 2 using the biomimetic route to enable acylase-mediated resolution of the product mixtures for separation of the l- from the d-isomers. Didehydro amino acids (Z)-11b and (Z)-11c were therefore used in the newly established protocol of Staudinger reduction (products 19a and 19b, not purified) and subsequent biomimetic domino transformation (Fig. 6). Surprisingly, the anticipated domino process stopped at the didehydro-arginine stage after the reduction and guanidinylation steps for both precursors (Z)-11b and (Z)-11c. Thus, compounds 8c and 8d were obtained in impure form and did not undergo cyclization towards the capreomycidine scaffold even at elevated temperatures up to 100 °C. This indicates that the didehydro amino acid moiety is required to display a certain electrophilicity to enable the aza-Michael addition and that even slight changes, such as the shift from carbamate to amide protection, can lead to a complete loss of reactivity.
Fig. 6.

Synthesis of Nα-acetylated capreomycidines for enzymatic resolution: first attempt with the domino reaction stopping after the guanidinylation step
N-Acetylated capreomycidin derivatives for enzymatic resolution therefore had to be prepared via a different route. An attempted direct acetylation of target structures (rac)-1 and (rac)-2 did not provide the desired Nα-acetylated products. It was therefore envisaged to synthesize the according N-Cbz-protected congeners first and then to exchange the N-Cbz for an N-acetyl moiety after the biomimetic cyclization process, followed by acidic cleavage of the tert-butyl ester (Fig. 7). When Cbz-protected phosphonate 20 (Schmidt et al. 1982, 1984; Schmidt and Wild 1985; Hamzavi et al. 2003) was employed in the Wittig-Horner reaction with aldehyde 10, Cbz-protected δ-azido-didehydro α-amino acid ester (Z)-21 was obtained in 81 % yield (calculated as traces of aldehyde 10 could not completely be removed). Stereoisomer (E)-21 could also be isolated in 3 % yield, and again, application of the established 1H NMR criteria for the distinction of (Z)- and (E)-didehydro amino acids (Mazurkiewicz et al. 2005) confirmed the stereochemical assignment. Subsequent Staudinger reduction of (Z)-21 furnished ornithine derivative 22, which was directly treated with guanidinylation reagent 13 to initiate the domino reaction. Thus, formation of the respective arginine derivative and domino-type ring closure provided a mixture of protected capreomycidine (rac)-23 and epicapreomycidine (rac)-24 (d.r.~1 : 1) in 62 % yield over two steps. The mixture of (rac)-23 and (rac)-24 was then subjected to a sequence of hydrogenolysis with in situ-acetylation (products (rac)-25 and (rac)-26) and acidic cleavage of the tert-butyl ester to afford a mixture of Nα-acetylated products (rac)-27 and (rac)-28 in 77 % yield over two steps from the mixture of (rac)-23 and (rac)-24 (Fig. 7). However, attempts to subject this mixture of Nα-acetylated capreomycidines to acylase-mediated deacetylation to separate the l- from the d-isomers failed as no conversion could be detected. Apparently, Nα-acetylated capreomycidines are no substrates for standard acylases. This might result either from steric hindrance due to the adjacent six-membered ring or from the positively charged guanidine group being fixed in a position close to the reaction site.
Fig. 7.
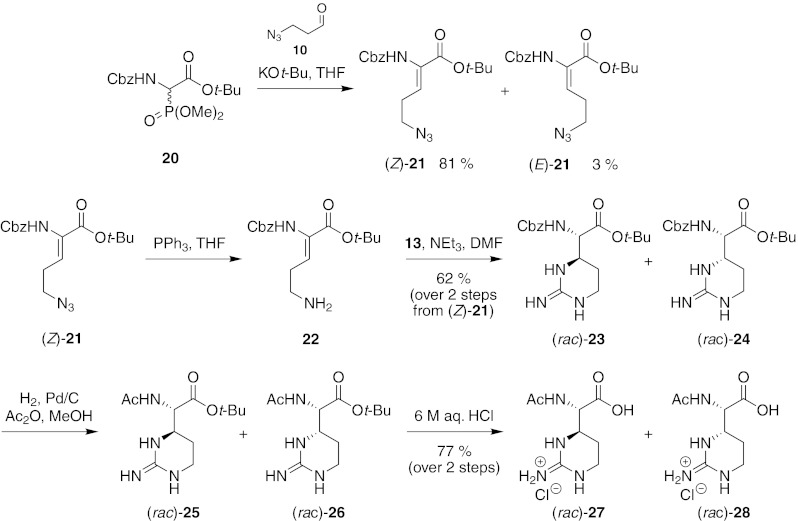
Synthesis of Nα-acetylated capreomycidines for enzymatic resolution: successful route via Nα-Cbz-protected derivatives
The synthesis of guanidine-protected cyclization precursors 8e–h (see Table 1) was then attempted. It was anticipated that these compounds should not spontaneously undergo the aza-Michael addition as a result from the reduced nucleophilicity of the guanidine group. In contrast, one should be able to isolate fully protected didehydro-arginine derivatives 8e–h and to subject them to aza-Michael cyclization in a separate step. This second reaction would then require activation either with a Lewis acid (in order to enhance the electrophilicity of the didehydro amino acid moiety) or a base (to deprotonate the urethane-protected guanidine and make it nucleophilic). Thus, the domino reaction would be dissected into two separate transformations, and a chiral activating agent for the cyclization step might potentially provide stereoinduction and enable a stereoselective biomimetic synthesis of capreomycidines. In principle, the acidic properties of urethane-protected guanidines are established (Feichtinger et al. 1998). However, it was unclear if the guanidine moiety had to be fully protected in order to allow for sufficient deprotonation of the urethane moiety, and therefore, the additional N-benzyl group was introduced into potential cyclization precursors 8f and 8h.
For an efficient synthesis of 8e–h, δ-azido-didehydro α-amino acids (Z)-11a and (Z)-11d were used and again reduced to moderately stable ornithine derivatives 12 and 16 (vide supra, Fig. 8). For the introduction of the benzyl group, 12 and 16 were subjected to reductive amination reactions with benzaldehyde to furnish Nδ-benzylated ornithines 29 and 30, which could not be obtained in pure form. All didehydro-ornithines 12, 16, 29 and 30 were transformed into the respective arginine derivatives by guanidinylation with reagent 31 (Tian et al. 1992) in the presence of silver(I) triflate. Thus, target structures 8e–h were obtained in yields of 42–71 % (over two or three steps from the respective δ-azido-didehydro α-amino acid).
Fig. 8.

Synthesis of potential cyclization precursors 8e–h with protected guanidine moieties
Guanidine-protected cyclization precursors 8e–h were then subjected to numerous attempts to activate them for the aza-Michael addition. Initially, 8e was treated with a series of bases (K2CO3 in MeCN, NaH, KHMDS or DBU in THF) for urethane deprotonation or with iron(III) chloride as a Lewis acid in dichloromethane for activation of the Michael acceptor system (reactions not displayed). No conversion towards the cyclic capreomycidine system was detected. As the bis-Boc-protected congener 8g was anticipated to be more reactive, its possible cyclization in the presence of DBU in THF was investigated next, but no conversion was observed even at elevated temperatures. It was therefore decided to skip cyclization studies on 8f and to conduct more detailed investigations regarding the aza-Michael cyclization of what should be the most reactive precursor both in terms of guanidine-urethane acidity and Michael acceptor activity, i.e. didehydro-arginine 8h. Hence, compound 8h was treated with a series of bases (K2CO3 in DMSO, basic Al2O3 in MeCN, KOt-Bu or NaH in THF) as well as a series of Lewis acids (FeCl3, SnCl4, Me2AlCl, EtAlCl2 or Yb(OTf)3 in dichloromethane, reactions not displayed). Furthermore, Takemoto’s bifunctional chiral organocatalyst (Inokuma et al. 2006) was added to a solution of 8h in toluene to study if the combination of thiourea activation and basic moiety might be able to trigger the cyclization reaction. However, no conversion towards the cyclic capreomycidine system was detected in any of the listed cases. It was therefore concluded that the reactivity of the didehydro amino acid moiety was just not sufficient for aza-Michael additions if the nucleophilicity of the guanidine group is too low resulting from its derivatization. This again demonstrated that the biomimetic cyclization reaction could only work within a certain frame of reactivity of both moieties involved (vide supra). Thus, a dissection of the domino reaction into two separate transformations was impossible, preventing further attempts to conduct the biomimetic cyclization in a stereocontrolled fashion.
Conclusion
In summary, we have developed an unprecedented biomimetic pathway for the concise synthesis of the non-proteinogenic amino acids capreomycidine 1 and epicapreomycidine 2 in racemic form. The two key steps of this efficient synthetic route were (1) the preparation of protected δ-azido-didehydro α-amino acid precursors using Wittig-Horner reactions and (2) a sequence of Staudinger reduction and a novel domino reaction for the construction of the capreomycidine scaffold. This domino-guanidinylation-aza-Michael-addition sequence mimics the biosynthetic ring closure of didehydro-arginine towards capreomycidine. Attempts to subject the furnished racemic mixtures of l- and d-amino acids to acylase-mediated enzymatic resolution were unsuccessful as Nα-acetylated capreomycidines were apparently no substrates for acylases. Investigations on didehydro-arginine cyclization precursors with different protecting group patterns revealed that the domino reaction would only work within a certain frame of reactivity, i.e. both sufficient electrophilicity of the Michael acceptor and nucleophilicity of the guanidine moiety were essential. Fully protected didehydro-arginine derivatives 8e–h therefore did not undergo the aza-Michael addition anymore, even in the presence of highly reactive activating agents. However, both compounds 8e–h and their respective azide precursors might be useful synthetic intermediates for the preparation of isotope-labelled ornithine or arginine derivatives (also see Baldwin et al. 1993a, b). Overall, the obtained results demonstrate that a biomimetic 1,4-addition of a guanidine moiety to a didehydro amino acid unit is synthetically feasible, but that a fine-tuning of the reactivity is essential.
Electronic supplementary material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, SFB 803 “Functionality controlled by organization in and between membranes”) and the Fonds der Chemischen Industrie (FCI, Sachkostenzuschuss). MG and DS thank the DFG Priority Programme 1178 and the Danish National Research Foundation (DNRF) funded Center for Materials Crystallography (CMC).
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Ando T, Matsuura K, Izumi R, Noda T, Take T, Nagata A, Abe J. Studies on tuberactinomycin. II. Isolation and properties of tuberactinomycin-N, a new tuberactinomycin group antibiotic. J Antibiot. 1971;24:680–686. doi: 10.7164/antibiotics.24.680. [DOI] [PubMed] [Google Scholar]
- Baldwin JE, Merritt KD, Schofield CJ. Synthesis of l-ornithines stereospecifically deuterated at C-3. Tetrahedron Lett. 1993;34:3919–3920. doi: 10.1016/S0040-4039(00)79263-X. [DOI] [Google Scholar]
- Baldwin JE, Merritt KD, Schofield CJ, Elson SW, Baggaley KH. Studies on the stereospecificity of the Clavaminic acid synthase catalysed hydroxylation reaction. J Chem Soc Chem Commun. 1993;1993:1301–1302. doi: 10.1039/c39930001301. [DOI] [Google Scholar]
- Bartz QR, Ehrlich J, Mold JD, Penner MA, Smith RM. Viomycin, a new tuberculostatic antibiotic. Amer Rev Tuberculosis. 1951;63:4–6. doi: 10.1164/art.1951.63.1.4. [DOI] [PubMed] [Google Scholar]
- Bruker AXS Inst. Inc. (2011) SAINT v7.68A in Bruker APEX v2011.9, Bruker AXS Inst. Inc., Madison, USA
- Bycroft BW, Croft LR, Johnson AW, Webb T. Structure, stereochemistry, and reactions of the guanidine moiety of viomycin. J Antibiot. 1969;22:133–134. doi: 10.7164/antibiotics.22.133. [DOI] [PubMed] [Google Scholar]
- Bycroft BW, Cameron D, Croft LR, Hassanali-Walji A, Johnson AW, Webb T. Total structure of capreomycin IB, a tuberculostatic peptide antibiotic. Nature. 1971;231:301–302. doi: 10.1038/231301a0. [DOI] [PubMed] [Google Scholar]
- Bycroft BW, Cameron D, Johnson AW. Synthesis of capreomycidine and epicapreomycidine, the epimers of α-(2-iminohexahydro-4-pyrimidyl)glycine. J Chem Soc C. 1971;1971:3040–3047. doi: 10.1039/j39710003040. [DOI] [Google Scholar]
- Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. Identification of the gene cluster involved in muraymycin biosynthesis from Streptomyces sp. NRRL 30471. Mol BioSyst. 2011;7:920–927. doi: 10.1039/c0mb00237b. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Shlaes D. Fix the antibiotics pipeline. Nature. 2011;472:32. doi: 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- Davies AJ, Donald ASR, Marks RE. The acid-catalysed decomposition of some β-azido-carbonyl compounds. J Chem Soc C. 1967;1967:2109–2112. doi: 10.1039/j39670002109. [DOI] [Google Scholar]
- DeMong DE, Williams RM. The asymmetric synthesis of (2S,3R)-capreomycidine. Tetrahedron Lett. 2001;42:3529–3532. doi: 10.1016/S0040-4039(01)00487-7. [DOI] [Google Scholar]
- DeMong DE, Williams RM. Asymmetric synthesis of (2S,3R)-capreomycidine and the total synthesis of capreomycin IB. J Am Chem Soc. 2003;125:8561–8565. doi: 10.1021/ja0351241. [DOI] [PubMed] [Google Scholar]
- Ducho C, Hamed RB, Batchelar ET, Sorensen JL, Odell B, Schofield CJ. Synthesis of regio- and stereoselectively deuterium-labelled derivatives of l-glutamate semialdehyde for studies on carbapenem biosynthesis. Org Biomol Chem. 2009;7:2770–2779. doi: 10.1039/b903312b. [DOI] [PubMed] [Google Scholar]
- Dyer JR, Kellogg CK, Nassar RF, Streetman WE. Viomycin II. Structure of viomycin. Tetrahedron Lett. 1965;6:585–592. doi: 10.1016/S0040-4039(00)90001-7. [DOI] [PubMed] [Google Scholar]
- Feichtinger K, Sings HL, Baker TJ, Matthews K, Goodman M. Triurethane-protected guanidines and triflyldiurethane-protected guanidines: new reagents for guanidinylation reactions. J Org Chem. 1998;63:8432–8439. doi: 10.1021/jo9814344. [DOI] [Google Scholar]
- Finlay AC, Hobby GL, Hochstein F, Lees TM, Lenert TF, Means JA, P’An SY, Regna PP, Routien JB, Sobin BA, Tate KB, Kane JH. Viomycin, a new antibiotic active against mycobacteria. Amer Rev Tuberculosis. 1951;63:1–3. doi: 10.1164/art.1951.63.1.1. [DOI] [PubMed] [Google Scholar]
- Hamzavi R, Dolle F, Tavitian B, Dahl O, Nielsen PE. Modulation of the pharmacokinetic properties of PNA: preparation of galactosyl, mannosyl, fucosyl, n-acetylgalactosaminyl, and n-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers. Bioconjugate Chem. 2003;14:941–954. doi: 10.1021/bc034022x. [DOI] [PubMed] [Google Scholar]
- He H, Williamson RT, Shen B, Graziani EI, Yang HY, Sakya SM, Petersen PJ, Carter GT. Mannopeptimycins, novel antibacterial glycopeptides from Streptomyces hygroscopicus. J Am Chem Soc. 2002;124:9729–9736. doi: 10.1021/ja020257s. [DOI] [PubMed] [Google Scholar]
- Herr EB, Haney ME, Pittenger GE, Higgens CE. Isolation and characterization of a new peptide antibiotic. Proc Ind Acad Sci. 1960;69:134. [Google Scholar]
- Hübschle CB, Sheldrick GM, Dittrich B (2011) ShelXle: a Qt graphical user interface for SHELXL. J Appl Cryst 44:1281–1284 [DOI] [PMC free article] [PubMed]
- Inokuma T, Hoashi Y, Takemoto Y. Thiourea-catalyzed asymmetric michael addition of activated methylene compounds to α,β-unsaturated imides: dual activation of imide by intra- and intermolecular hydrogen bonding. J Am Chem Soc. 2006;128:9413–9419. doi: 10.1021/ja061364f. [DOI] [PubMed] [Google Scholar]
- Izumi R, Noda T, Ando T, Take T, Nagata A. Studies on tuberactinomycin. 3. Isolation and characterization of two minor components, tuberactinomycin B and tuberactinomycin O. J Antibiot. 1972;25:201–207. doi: 10.7164/antibiotics.25.201. [DOI] [PubMed] [Google Scholar]
- Jackson MD, Gould SJ, Zabriskie TM. Studies on the formation and incorporation of streptolidine in the biosynthesis of the peptidyl nucleoside antibiotic streptothricin F. J Org Chem. 2002;67:2934–3941. doi: 10.1021/jo016182c. [DOI] [PubMed] [Google Scholar]
- Jones JH. The terminology of guanidine formation. J Pept Sci. 2002;8:285–287. doi: 10.1002/psc.408. [DOI] [PubMed] [Google Scholar]
- Ju J, Ozanick SG, Shen B, Thomas MG. Conversion of (2S)-arginine to (2S,3R)-capreomycidine by VioC and VioD from the viomycin biosynthetic pathway of Streptomyces sp. strain ATCC11861. ChemBioChem. 2004;5:1281–1285. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- Koehn FE. New strategies and methods in the discovery of natural product anti-infective agents: the mannopeptimycins. J Med Chem. 2008;51:2613–2617. doi: 10.1021/jm070432l. [DOI] [PubMed] [Google Scholar]
- Kottke T, Stalke D. Crystal handling at low temperature. J Appl Crystallogr. 1993;26:615–619. doi: 10.1107/S0021889893002018. [DOI] [Google Scholar]
- Kottke T, Lagow RJ, Stalke D. Low cost conversion of a coaxial nozzle arrangement into a stationary low temperature attachment. J Appl Crystallogr. 1996;29:465–468. doi: 10.1107/S0021889896003172. [DOI] [Google Scholar]
- Lemke A, Büschleb M, Ducho C. Concise synthesis of both diastereomers of 3-hydroxy-l-arginine. Tetrahedron. 2010;66:208–214. doi: 10.1016/j.tet.2009.10.102. [DOI] [Google Scholar]
- Mahady GB, Huang Y, Doyle BJ, Locklear T. Natural products as antibacterial agents. Stud Nat Prod Chem. 2008;35:423–444. doi: 10.1016/S1572-5995(08)80011-7. [DOI] [Google Scholar]
- Mazurkiewicz R, Kuźnik A, Grymel M, Kuźnik N. 1H NMR spectroscopic criteria for the configuration of N-acyl-α, β-dehydro-α-amino acid esters. Magn Reson Chem. 2005;43:36–40. doi: 10.1002/mrc.1496. [DOI] [PubMed] [Google Scholar]
- McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. Structures of the muraymycins, novel peptidoglycan biosynthesis inhibitors. J Am Chem Soc. 2002;124:10260–10261. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]
- Müller P, Herbst-Irmer R, Spek AL, Schneider TR, Sawaya MR. In: Crystal structure refinement: a crystallographer’s guide to SHELXL, IUCr texts on crystallography. Müller P, editor. Oxford: Oxford University Press; 2006. [Google Scholar]
- Nagata A, Ando T, Izumi R, Sakakibara H, Take T, Hayano K, Abe J. Studies on tuberactinomycin (tuberactin), a new antibiotic. I. Taxonomy of producing strain, isolation and characterization. J Antibiot. 1968;21:681–687. doi: 10.7164/antibiotics.21.681. [DOI] [PubMed] [Google Scholar]
- Noda T, Take T, Nagata A, Wakamiya T, Shiba T. Chemical studies on tuberactinomycin. III. Chemical structure of viomycin (tuberactinomycin B) J Antibiot. 1972;25:427–428. doi: 10.7164/antibiotics.25.427. [DOI] [PubMed] [Google Scholar]
- Nomoto S, Teshima T, Wakamiya T, Shiba T. The revised structure of capreomycin. J Antibiot. 1977;30:955–959. doi: 10.7164/antibiotics.30.955. [DOI] [PubMed] [Google Scholar]
- Nomoto S, Teshima T, Wakamiya T, Shiba T. Total synthesis of capreomycin. Tetrahedron. 1978;34:921–927. doi: 10.1016/0040-4020(78)88140-X. [DOI] [Google Scholar]
- Okura A, Morishima H, Takita T, Aoyagi T, Takeuchi T, Umezawa H. The structure of elastatinal, an elastase inhibitor of microbial origin. J Antibiot. 1975;28:337–339. doi: 10.7164/antibiotics.28.337. [DOI] [PubMed] [Google Scholar]
- Razzak M, De Brabander JK. Lessons and revelations from biomimetic syntheses. Nat Chem Biol. 2011;7:865–875. doi: 10.1038/nchembio.709. [DOI] [PubMed] [Google Scholar]
- Schmidt U, Wild J. Synthesen von Peptidalkaloiden, 11. Über Aminosäuren und Peptide, 51—Dehydroaminosäuren, 19. Totalsynthese von Hexaacetylcelenamid A. Liebigs Ann Chem. 1985;1985:1882–1894. doi: 10.1002/jlac.198519850915. [DOI] [Google Scholar]
- Schmidt U, Lieberknecht A, Schanbacher U, Beuttler T, Wild J. Facile preparation of N-acyl-2-(diethoxyphosphoryl) glycine esters and their use in the synthesis of dehydroamino acid esters. Angew Chem Int Ed. 1982;21:776–777. doi: 10.1002/anie.198207761. [DOI] [Google Scholar]
- Schmidt U, Lieberknecht A, Wild J. Amino acids and peptides; XLIII. dehydroamino acids; XVIII. Synthesis of dehydroamino acids and amino acids from N-acyl-2-(dialkyloxyphosphinyl)-glycin esters; II. Synthesis. 1984;1984:53–60. doi: 10.1055/s-1984-30730. [DOI] [Google Scholar]
- Schmidt U, Griesser H, Leitenberger V, Lieberknecht A, Mangold R, Meyer R, Riedl B (1992) Diastereoselective formation of (Z)-didehydroamino acid esters. Synthesis 1992:487–450
- Schulz T, Meindl K, Leusser D, Stern D, Graf J, Michaelsen C, Ruf M, Sheldrick GM, Stalke D. A comparison of a microfocus X-ray source and a conventional sealed tube for crystal structure determination. J Appl Crystallogr. 2009;42:885–891. doi: 10.1107/S0021889809030921. [DOI] [Google Scholar]
- Schwörer CJ, Oberthür M. Synthesis of highly functionalized amino acids: an expedient access to l- and d-β-hydroxyenduracididine derivatives. Eur J Org Chem. 2009;2009:6129–6139. doi: 10.1002/ejoc.200900971. [DOI] [Google Scholar]
- Seiple IB, Su S, Young IS, Nakamura A, Yamaguchi J, Jorgensen L, Rodriguez RA, O’Malley DP, Gaich T, Kock M, Baran PS. Enantioselective total syntheses of (-)-palau’amine, (-)-axinellamines, and (-)-massadines. J Am Chem Soc. 2011;133:14710–14726. doi: 10.1021/ja2047232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM. Phase annealing in SHELX-90: direct methods for larger structures. Acta Crystallogr Sect A. 1990;46:467–473. doi: 10.1107/S0108767390000277. [DOI] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2012) TWINABS 2012/1, Georg-August-University Göttingen, Göttingen, Germany
- Shiba T, Nomoto S, Teshima T, Wakamiya T. Revised structure and total synthesis of capreomycin. Tetrahedron Lett. 1976;17:3907–3910. doi: 10.1016/0040-4039(76)80179-7. [DOI] [Google Scholar]
- Shiba T, Ukita T, Mizuno K, Teshima T, Wakamiya T. Total synthesis of l-capreomycidine. Tetrahedron Lett. 1977;18:2681–2684. doi: 10.1016/S0040-4039(01)83045-8. [DOI] [Google Scholar]
- Singh MP, Petersen PJ, Weiss WJ, Janso JE, Luckman SW, Lenoy EB, Bradford PA, Testa RT, Greenstein M. Mannopeptimycins, new cyclic glycopeptide antibiotics produced by Streptomyces hygroscopicus LL-AC98: antibacterial and mechanistic activities. Antimicrob Agents Chemother. 2003;47:62–69. doi: 10.1128/AAC.47.1.62-69.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spork AP, Ducho C. Novel 5’-deoxy nucleosyl amino acid scaffolds for the synthesis of muraymycin analogues. Org Biomol Chem. 2010;8:2323–2326. doi: 10.1039/c003092a. [DOI] [PubMed] [Google Scholar]
- Spork AP, Wiegmann D, Granitzka M, Stalke D, Ducho C. Stereoselective synthesis of uridine-derived nucleosyl amino acids. J Org Chem. 2011;76:10083–10098. doi: 10.1021/jo201935w. [DOI] [PubMed] [Google Scholar]
- Stalke D. Cryo crystal structure determination and application to intermediates. Chem Soc Rev. 1998;27:171–178. doi: 10.1039/a827171z. [DOI] [Google Scholar]
- Tanino T, Hirano S, Ichikawa S, Matsuda A. Synthetic study of muraymycins using Ugi-four component reaction. Nucleic Acids Symp Series. 2008;52:557–558. doi: 10.1093/nass/nrn282. [DOI] [PubMed] [Google Scholar]
- Tanino T, Ichikawa S, Shiro M, Matsuda A. Total synthesis of (-)-muraymycin D2 and its epimer. J Org Chem. 2010;75:1366–1377. doi: 10.1021/jo9027193. [DOI] [PubMed] [Google Scholar]
- Tanino T, Ichikawa S, Matsuda A. Synthesis of l-epi-capreomycidine derivatives via C-H amination. Org Lett. 2011;13:4028–4031. doi: 10.1021/ol201527k. [DOI] [PubMed] [Google Scholar]
- Tatsuta K, Mikami N, Fujimoto K, Umezawa S, Umezawa H, Aoyagi T. The structure of chymostatin, a chymotrypsin inhibitor. J Antibiot. 1973;26:625–646. doi: 10.7164/antibiotics.26.625. [DOI] [PubMed] [Google Scholar]
- Taubes G. The bacteria fight back. Science. 2008;321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- Teshima T, Konishi K, Shiba T. Synthesis of l-epicapreomycidine. Bull Chem Soc Jpn. 1980;53:508–511. doi: 10.1246/bcsj.53.508. [DOI] [Google Scholar]
- Tian Z, Edwards P, Roeske RW. Synthesis of optically pure Cα-methyl-arginine. Int J Pept Protein Res. 1992;40:119–126. doi: 10.1111/j.1399-3011.1992.tb01459.x. [DOI] [PubMed] [Google Scholar]
- Tietze LF. Domino reactions in organic synthesis. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- Tietze LF, Beifuss U. Sequential transformations in organic chemistry: a synthetic strategy with a future. Angew Chem Int Ed. 1993;32:131–163. doi: 10.1002/anie.199301313. [DOI] [Google Scholar]
- Tietze LF, Brasche G, Gericke K. Domino reactions in organic synthesis. Weinheim: Wiley-VCH; 2006. [Google Scholar]
- Umezawa H, Aoyagi T, Morishima H, Kunimoto S, Matsuzaki M, Hamada M, Takeuchi T. Chymostatin, a new chymotrypsin inhibitor produced by actinomycetes. J Antibiot. 1970;23:425–427. doi: 10.7164/antibiotics.23.425. [DOI] [PubMed] [Google Scholar]
- Umezawa H, Aoyagi T, Okura A, Morishima H, Takeuchi T, Okami Y. Elastatinal, a new elastase inhibitor produced by actinomycetes. J Antibiot. 1973;26:787–789. doi: 10.7164/antibiotics.26.787. [DOI] [PubMed] [Google Scholar]
- Wakamiya T, Shiba T, Kaneko T, Sakakibara H, Take T, Abe J. Chemical studies on tuberactinomycin. I. The structure of tuberactidine, guanidino amino acid component. Tetrahedron Lett. 1970;11:3497–3500. doi: 10.1016/S0040-4039(01)98511-9. [DOI] [PubMed] [Google Scholar]
- Wakamiya T, Mizuno K, Ukita T, Teshima T, Shiba T. Chemical studies on tuberactinomycin. XIV. Novel synthesis of dl-capreomycidine. Bull Chem Soc Jpn. 1978;51:850–854. doi: 10.1246/bcsj.51.850. [DOI] [Google Scholar]
- Walsh C. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Norton EB, Mansour TS. Improved procedures for preparation of racemic capreomycidine. Synth Commun. 2004;5:795–803. doi: 10.1081/SCC-120028353. [DOI] [Google Scholar]
- Yin X, Zabriskie TM. VioC is a non-heme iron, α-ketoglutarate-dependent oxygenase that catalyzes the formation of 3S-hydroxy-l-arginine during viomycin biosynthesis. ChemBioChem. 2004;5:1274–1277. doi: 10.1002/cbic.200400082. [DOI] [PubMed] [Google Scholar]
- Yin X, McPhail KL, Kim K, Zabriskie TM. Formation of the nonproteinogenic amino acid 2S,3R-capreomycidine by VioD from the viomycin biosynthesis pathway. ChemBioChem. 2004;5:1278–1281. doi: 10.1002/cbic.200400187. [DOI] [PubMed] [Google Scholar]
- Yonezawa Y, Hirosaki T, Hayashi T, Shin CG. Convenient synthesis and conversion of a (Z)-α,β-didehydroornithine derivative to α,β-didehydrokyotorphin. Synthesis. 2000;2000:144–148. doi: 10.1055/s-2000-6219. [DOI] [Google Scholar]
- Yoshioka H, Aoki T, Goko H, Nakatsu K, Noda T, Sakakibara H, Take T, Nagata A, Abe J, Wakamiya T, Shiba T, Kaneko T. Chemical studies on tuberactinomycin. II Structure of tuberactinomycin O. Tetrahedron Lett. 1971;12:2043–2046. doi: 10.1016/S0040-4039(01)96775-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.