

Abstract
Motivation: Molecular simulation has historically been a low-throughput technique, but faster computers and increasing amounts of genomic and structural data are changing this by enabling large-scale automated simulation of, for instance, many conformers or mutants of biomolecules with or without a range of ligands. At the same time, advances in performance and scaling now make it possible to model complex biomolecular interaction and function in a manner directly testable by experiment. These applications share a need for fast and efficient software that can be deployed on massive scale in clusters, web servers, distributed computing or cloud resources.
Results: Here, we present a range of new simulation algorithms and features developed during the past 4 years, leading up to the GROMACS 4.5 software package. The software now automatically handles wide classes of biomolecules, such as proteins, nucleic acids and lipids, and comes with all commonly used force fields for these molecules built-in. GROMACS supports several implicit solvent models, as well as new free-energy algorithms, and the software now uses multithreading for efficient parallelization even on low-end systems, including windows-based workstations. Together with hand-tuned assembly kernels and state-of-the-art parallelization, this provides extremely high performance and cost efficiency for high-throughput as well as massively parallel simulations.
Availability: GROMACS is an open source and free software available from http://www.gromacs.org.
Contact: erik.lindahl@scilifelab.se
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Although molecular dynamics simulation of biomolecules is frequently classified as computational chemistry, the scientific roots of the technique trace back to polymer chemistry and structural biology in the 1970s, where it was used to study the physics of local molecular properties—flexibility, distortion and stabilization—and relax early X-ray structures of proteins on short time scales (Berendsen, 1976; Levitt and Lifson, 1969; Lifson and Warshel, 1968; McCammon et al., 1977). Molecular simulation in general was pioneered even earlier in physics and applied to simplified hard-sphere systems (Alder and Wainwright, 1957). The field of molecular simulation has developed tremendously since then, and simulations are now routinely performed on multi-microsecond scale where it is possible to repeatedly fold small proteins (Bowman et al., 2011; Lindorff-Larsen et al., 2011; van der Spoel and Seibert, 2006), predict interactions between receptors and ligands (Chong et al., 1999; Huang and Caflisch, 2011), predict functional properties of receptors and even capture intermediate states of complex transitions, e.g. in membrane proteins (Nury et al., 2010). This classical type of single long simulation continues to be important, as it provides ways to directly monitor molecular processes not easily observed through other means. However, many current studies increasingly rely on large sets of simulations, enabled in part by the ever-increasing number of structural models made possible by sequencing and structural genomics as well as new techniques to estimate complex molecular properties using thousands of shorter simulations (Noé and Fischer, 2008; Pande et al., 2010). Mutation studies can now easily build models and run short simulations for hundreds of mutants, model-building web servers frequently offer automated energy minimization and refinement (Zhang et al., 2011) and free-energy calculations are increasingly being used to provide better interaction energy estimates than what is possible with docking (Boyce et al., 2009; Helms and Wade, 1998). In these scenarios, classical molecular dynamics simulations based on empirical models have a significant role to play, as most properties of interest are defined by free energies, which typically require extensive sampling that traditional quantum chemistry methods can not provide for large systems.
These developments would not have been possible without significant research efforts in simulation algorithms, optimization, parallelization and not least ways to integrate simulations in modeling pipelines. The emergence of standardized packages for molecular modeling, such as CHARMM (Brooks et al., 2009), GROMOS (Christen et al., 2005), Amber (Case et al., 2005), NAMD (Phillips et al., 2005) and GROMACS (Hess et al., 2008), has been important, as these have helped commoditize simulation and molecular modeling research and made the techniques available to life science application researchers, who are not specialists in simulation development.
All these packages have complementary strengths and profiles; for the GROMACS molecular simulation toolkit, one of our primary long-term development goals has been to achieve the highest possible simulation efficiency for the small- to medium-size clusters that were present in our own research laboratories. As computational resources are typically limited in those settings, it is sometimes preferable to use throughput approaches with moderate parallelization that yield whole sets of simulations rather than maximizing performance in a single long simulation. However, in recent years, we have combined this with optimizing parallel scaling to enable long simulations when dedicated clusters or supercomputers are available for select critical problems.
During the past 4 years since GROMACS 4, we have developed a number of new features and improvements that have led up to release 4.5 of the software and significantly improved both performance and efficiency for throughput as well as massively parallel applications. Many tasks that only a decade ago required exceptionally large dedicated supercomputing resources are now universally accessible, and sometimes they can even be run efficiently on a single workstation or laptop. However, contemporary ‘low-end’ machines are now parallel computers ranging from 2 to 4 cores on a laptop and up to 16–32 cores on workstations and require parallel programs to use all resources in a single job. Here, we present the work and features that have gone into GROMACS 4.5, including development to make the code fully portable and multithreaded on a wide range of platforms, features to facilitate high-throughput simulation and not least more efficient tools to help automate complex simulations, such as free-energy calculations, with another long-term goal of commoditizing affinity prediction as well. High-end performance in GROMACS has also been improved with new decomposition techniques in both direct and reciprocal space that push parallelization further and that have made microsecond simulation timescales reachable in a week or two even for large systems using only modest computational resources.
2 RESULTS
2.1 An open source molecular simulation framework
The development of GROMACS was originally largely driven by our own needs for efficient modeling. However, in hindsight, the decision to release the package as both open source and free software was a significant advance for the project. The codebase has become a shared infrastructure with contributions from several research laboratories worldwide, where every single patch and all code review are public as soon as they are committed to the repository. We explicitly encourage extensions and re-use of the code; as examples, GROMACS is used as a module to perform energy minimization in other structural bioinformatics packages (including commercial ones); it is available as a component from many vendors that provide access to cloud computing resources; and some of the optimized mathematical functions (such as inverse square roots) have been reused in other codes. Many Linux distributions also provide pre-compiled or contributed binaries of the package. These features per se do not necessarily say anything about scientific qualities, but we believe this open development platform ensures (i) intensive code scrutiny, (ii) several state-of-the art implementations of algorithms and (iii) immediate availability of research work to end users. Compared with only 10 years ago, the project is now used everywhere from the smallest embedded processors to the largest supercomputers in the world, with applications ranging from genome-scale refinement of coarse-grained models to multi-microsecond simulations of membrane proteins or vesicle fusion.
2.2 Enabling efficient simulation on desktop resources
Supercomputers are still important for the largest molecular simulations, but many users rely on modest systems for their computational needs. For many applications, one can even argue this is the most important target: researchers often use interactive tools, companies are hesitant to invest in expensive computational infrastructure and there is an increasing focus on high-throughput studies, where a single calculation cannot use 50% of a cluster. Historically, this low-end regime has been the primary goal for GROMACS [whereas NAMD (Phillips et al., 2005) has focused on parallel scaling], and we have specifically focused efforts on achieving the highest possible efficiency on single nodes. GROMACS is designed for maximum portability, with external dependencies kept to a minimum and fall-back internal libraries provided whenever possible. It is possible to build GROMACS on almost any Unix-based system (including many embedded architectures). In GROMACS 4.5, we have extended this further, making Microsoft Windows a fully supported platform. This is obviously relevant for many researchers’ desktops, but it is also critical for distributed computing projects where the software runs on participant-controlled computers, e.g. in the Folding@Home project. One of the main challenges in the past few years has been the emergence of multicore machines. Although GROMACS runs in parallel, it was designed to use message-passing interface (MPI) communication libraries present on supercomputers rather than automatically using multiple cores. In release 4.5, we have solved this by designing a new internal ‘thread_MPI’ interface layer that implements the MPI communication calls using multithreading and automatically uses every core available on a laptop or desktop for increased performance.
2.3 High-throughput simulation and modeling
As simulation software and computer performance has improved, biomolecular dynamics has increasingly been used for structure equilibration, sampling of models or to test what effects mutations might have on structure and dynamics by introducing many different mutations and perform comparatively short simulations on multiple structures. Although this type of short simulation might not look as technically impressive as long trajectories, we strongly feel it is a much more powerful approach for many applications. As simulations build on statistical mechanics, a result seen merely in one long trajectory might as well be a statistical fluctuation that would never be accepted as significant in an experimental setting. In contrast, by choosing to perform 50 100-ns simulations instead of a single 5 μs one, it is suddenly possible to provide standard error estimates and quantitative instead of qualitative results from simulations (Lange et al., 2010). In addition, the same toolbox can be applied to liquid simulations, where the need for sampling is limited, but where one often needs to study a range of systems under different conditions (e.g. temperature) to extract data that can be compared with experiments. (Caleman et al., 2012). As discussed earlier in the text, GROMACS has always been optimized to achieve the best possible efficiency using scarce resources (which we believe is the norm for most users), and version 4.5 has introduced several additional features to aid high-throughput simulation. All GROMACS runs are now automatically checkpointed and can be interrupted and continued as frequently as required, and optional flags have been added to enable binary reproducibility of trajectories. As GROMACS is successfully used in a number of distributed computing projects where both CPU and storage hardware might be less controlled, GROMACS’s main simulation executable mdrun now flushes all pending buffers after each file-writing step and tries to flush file system cache when writing checkpoints. As users often work with many datasets at once, we have implemented MD5 hashes on checkpoint continuation files to guarantee their integrity and to make sure the user does not append to the wrong file by mistake. These additional checks have allowed us to enable file appending on job continuation: repeated short jobs that continue from checkpoints will yield a single set of output just as from a single long job. Hundreds or even thousands of smaller simulations can be started with a single GROMACS execution command to optimize use on supercomputers that favor large jobs, and each of these can be parallel themselves if advantageous. GROMACS also supports simulations running in several modern cloud computing environments where virtual server instances can be started on demand. As cloud computing usage is also billed by the hour, we believe the most instructive metric for performance and efficiency is to actually measure simulation performance in terms of the cost to complete a given simulation—for an example, see the performance Section 2.4.
2.4 Implicit solvent and knowledge-based modeling
In addition to the high-throughput execution model, there are a number of new code features developed to support modeling and rapid screening of structures. In previous versions, GROMACS has not supported implicit solvent, as it seemed of little use when it was slower than explicit water. This has changed with version 4.5, and the code now comes with efficient implementations of the Still (Qui et al., 1997), HCT (Hawkins–Cramer–Truhlar) (Hawkins et al., 1996) and OBC (Onufriev–Bashford–Case) (Onufriev et al., 2004) models for generalized born interactions based on tabulated interaction rescaling (Larsson and Lindahl, 2010). Together with manually tuned assembly kernels, implicit solvent simulations can reach performance in excess of a microsecond per day for small proteins even on standard CPUs. The neighbor-searching code has been updated to support grid-based algorithms even in vacuo—including support for atoms diffusing away towards infinity with maintained performance—and there are now also highly optimized kernels to compute all–versus–all interactions without cut-offs both for standard and generalized born interactions. The program now also supports arbitrary knowledge-based statistical interactions through atom-group–specific tables both for bonded and non-bonded interactions. Constraints such as those used in refinement can be applied either to positions, atomic distances or torsions, and there are several options for ensemble weighting of contributions from multiple constraints.
2.5 Strong scaling on massively parallel clusters
Despite the rapid emergence of high-throughput computing, the usage of massively parallel resources continues to be a cornerstone of high-end molecular simulation. Absolute performance is the goal for this usage too, but here, it is typically limited by the scalability of the software. GROMACS 4.0 introduced a number of new features, including new neutral territory domain decomposition algorithm that is also used in the Desmond software (Bowers et al., 2006), but the performance was still limited by the scaling of the particle-mesh Ewald (Essmann et al., 1995) (PME) implementation, in particular the single-dimensional decomposition of the fast Fourier transform (FFT) grids. For GROMACS 4.5, this has been solved with a new implementation of 2D or ‘pencil’ decomposition of reciprocal space. A subset of nodes are dedicated to the PME calculation, and at the beginning of each step, the direct-space nodes send coordinate and charge data to them. As direct space can be composed in all three dimensions, a number of direct-space nodes (typically 3–4) map onto a single reciprocal-space node (Fig. 1). Limiting the computation of the 3D–FFT to a smaller number of nodes improves parallel scaling significantly (Hess et al., 2008) and is now also used by NWChem (Valiev et al., 2010). The new pencil decomposition makes it much easier to automatically determine both real- and reciprocal-space decompositions of arbitrary systems to fit a given number of nodes. The automatic load balancing step of the domain decomposition has also been improved; domain decomposition now works without periodic boundary conditions (important for implicit solvent); and GROMACS now includes tools to automatically tune the balance between direct and reciprocal-space work. In particular when running in parallel over large numbers of nodes, it is advantageous to move more work to real space (which scales near-linearly) and decrease the reciprocal-space load to reduce the dimensions of the 3D–FFT grid (where the number of communication messages scales with the square of the number of nodes involved). The latest version of GROMACS also supports many types of multilevel parallelism; in addition to coding-level optimizations, such as single-instruction multiple-data instructions and the multithreaded execution, GROMACS supports replica-exchange ensemble simulations where a single simulation can use hundreds of replicas that only communicate every couple of seconds, which makes it possible to scale even fairly small systems (e.g. a protein) to thousands of nodes for these kinds of calculations. Finally, for the largest systems comprising hundreds of millions of particles, we now achieve true linear weak scaling for reaction-field and other non–lattice-summation methods (Schulz et al., 2009). A lot of recent work has been invested in reducing memory needs and enabling parallel IO, and the code has been shown to successfully scale to >150 000 cores.
Fig. 1.

3D Domain decomposition in real space combined with 2D pencil domain decomposition in reciprocal space. The scaling in previous versions of GROMACS was limited by the reciprocal space PME set-up, and in particular the 1D decomposition of FFTs along the x-axis. The pencil grid decomposition improves reciprocal space scaling considerably and makes it easier to use arbitrary numbers of nodes. Colors in the plot refer to a hypothetical system with four cores per node, where three are used for direct-space and one for reciprocal-space calculations
2.6 Automated topology generation for wide classes of molecules and force fields
It was clear that the automated tools to generate input files were somewhat limited in earlier releases of GROMACS; few molecules apart from single-chain proteins worked perfectly. For version 4.5, the pdb2gmx tool has been reworked, and we now support automatic topology generation for proteins, DNA, RNA and many small molecules. Any number of chains and different molecule classes can be mixed, and they are automatically detected. The program provides several different options for how to handle termini and HETATM records in structures, and residue names and numbering from the input files are now maintained throughout the main simulation and analysis tools. With the most recent version, the package now comes with standard support for virtually all major point-charge force fields: GROMOS43a1, GROMOS43a2, GROMOS45a3, GROMOS53a5, GROMOS53a6, Encad, OPLS, OPLS-AA/L, CHARMM19, CHARMM27, Amber94, Amber96, Amber99, Amber99SB, AmberGS, Amber03 and Amber99SB-ILDN. To the best of our knowledge, this range of forcefield support is currently unique among packages and makes it straightforward to systematically compare the influence of the parameter approximations in biomolecular modeling. The code also provides name translation files to support all the conventions used in the different force fields. In addition, a number of databases provide topologies for small molecules for use in GROMACS (Malde et al., 2011; van der Spoel et al., 2012), whereas small molecule topologies for the generalized Amber force field (Wang et al., 2005), as well as topologies using the Charm general force field (Vanommeslaeghe et al., 2010), can be readily converted to GROMACS format, further increasing the applicability of the software.
2.7 Accurate and flexible integration
GROMACS 4.5 also includes a number of additional integrators of the equations of motion. Originally, the code only supported leapfrog verlet, which keeps track of the positions at the full step, whereas the velocities are offset by half a time step. The velocity Verlet algorithm (Swope et al., 1982) is now also fully supported. In velocity Verlet, positions and velocities are both at the same time point. For constant energy simulations, both algorithms give the same trajectories, but for constant temperature or constant pressure simulations, velocity Verlet integration provides many additional features. A number of pressure control or temperature control algorithms are only possible with velocity Verlet integrators because these algorithms require both the pressure and temperature to be specified at the same time. Additionally, velocity Verlet is more common in other simulation packages (Amber, NAMD and Desmond); therefore, it makes it easier to perform detailed comparisons between GROMACS and other molecular simulation engines. Good temperature algorithms already exist for leapfrog algorithm; however, velocity Verlet is, hence, not generally necessary in such cases.
For pressure control, the existing algorithm by Parrinello and Rahman (Nosé and Klein, 1983; Parrinello and Rahman, 1981) is not correct using leapfrog, although it has been verified to give a correct distribution of volumes within statistical noise in many situations. Slight errors arise because of the time step mismatch between the components of the pressure calculation involving kinetic energy and potential energy. The introduction of velocity Verlet allows the use of additional, more rigorous pressure control algorithms, such as that of Martyna, Tuckerman, Tobias, and Klein (Martyna et al., 1996), which can be useful for accurate lipid bilayer simulations in GROMACS 4.5.
The leapfrog Verlet and velocity Verlet are both implemented as specific instances of a method called Trotter factorization, a general technique for decomposition of the equations of motion, which makes it possible to write out different symplectic integrators based on different ordering of the integration of different degrees of freedom. This Trotter factorization approach will make it possible to eventually support a large range of multistep and other accelerated sampling integrators in the future, and it is already used for more efficient temperature and pressure scaling. Historically, GROMACS has relied on virtual interaction sites and all-bond constraints to extend the shortest time step in integration (in contrast to NAMD that uses multiple time step integration), but this approach will make it possible to support both alternatives in future versions.
2.8 A state-of-the-art free-energy calculation toolbox
Simulation-based free-energy calculations provide a way to accurately include effects both of interactions and entropy, and accurately predict solvation and binding properties of molecules. It is one of the most direct ways that simulations can provide specific predictions of properties that can also be measured experimentally. GROMACS as well as other packages have long supported free-energy perturbation and slow-growth methods to calculate free-energy differences when gradually changing the properties of molecules. The present release of the code provides an extensive new free-energy framework based on Bennett Acceptance Ratios (BAR). As a free-energy perturbation technique, this allows the calculation of a free-energy difference along an arbitrary coupling parameter λ, typically in multiple steps to provide sufficient phase space overlap between each step. The total energy (or actually, Hamiltonian) is then defined as 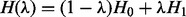 , where H0 and H1 are the Hamiltonians for the two end states. BAR uses differences in Hamiltonian as the basis for calculating the free-energy difference, and it has been shown to be both the most efficient free-energy perturbation method for extracting free-energy differences (Bennett, 1976), and a statistically unbiased estimator for this free-energy difference (Shirts and Pande, 2005).
, where H0 and H1 are the Hamiltonians for the two end states. BAR uses differences in Hamiltonian as the basis for calculating the free-energy difference, and it has been shown to be both the most efficient free-energy perturbation method for extracting free-energy differences (Bennett, 1976), and a statistically unbiased estimator for this free-energy difference (Shirts and Pande, 2005).
The Hamiltonian differences needed for BAR are now calculated automatically on the fly in simulations, rather than as a post-processing step using large full-precision trajectories, which makes it possible to use distributed computing or cloud resources where the available storage and bandwidth are limited. Rather than manually defining how to modify each molecule, the user can now simply specify that they want to calculate the free energy of decoupling a particular molecule or group of atoms from the system as a simulation parameter. Given the set of output files from such a project, the code also comes with a new tool g_bar that automatically calculates the free energy and its uncertainty at each step, and it provides a finished estimate of the free energy required to change from one end state to the other, including estimates of the standard error and measures for the phase space overlap (Fig. 2).
Fig. 2.

Free-energy calculations using BAR. GROMACS 4.5 provides significantly enhanced tools to automatically create topologies describing decoupling of molecules from the system to calculate binding or hydration free energies. The Hamiltonian of the system is defined as H(λ) = (1−λ)H0 + λ H1, where H0 and H1 are the Hamiltonians for the two end states. The user specifies a sequence of lambda points and runs simulations where the phase space overlaps and Hamiltonian differences are calculated on the fly. Finally, all these files are provided to the new g_bar tool that automatically analyses the results and provides free energies as well as standard error estimates for the system change
2.9 Other features
In addition to the larger development concepts covered here, several additional parts of GROMACS have been improved and extended for version 4.5.
We now support symplectic leapfrog and velocity Verlet integrators for fully reversible temperature and pressure coupling, with several new barostats and thermostats, including Nose–Hoover chains for ergodic temperature control and Martyna-Tuckerman-Tobias-Klein (MTTK) pressure control integrators. These are important for calculating accurate free energies, in particular for smaller systems or cases where pressure will affect the result.
A new file-format plugin has been designed to allow GROMACS to directly read any trajectory or coordinate format supported by the optional VMD libraries (Humphrey et al., 1996).
The previously labor-intensive task of embedding and equilibrating membrane proteins in lipid bilayers has been automated with the new tool g_membed (Wold et al., 2010). Given a membrane protein structure and an arbitrary bilayer (including lipid mixtures or proteins), this tool virtually shrinks the membrane protein to a small axis and then gently ‘grows’ it in over a few thousand steps. Lipids are removed based on overlap, and the tool has full support for asymmetrically shaped proteins.
Non-equilibrium simulation: it is now possible to pull any number of groups in arbitrary directions and to apply torques in addition to forces. The corresponding g_wham tool for analysis of non-equilibrium simulations has been updated extensively to allow robust error estimates using Bayesian bootstrapping (Hub et al., 2010).
The multi-scale modeling now includes a Quantum mechanics (QM)/Molecular mechanics (MM) interface to a number of common quantum chemistry programs and algorithms, coarse-grained (CG) modeling with force fields, such as MARTINI (Marrink et al., 2007), and a highly efficient parallel implicit solvent algorithm that can all be used in combination.
Normal-mode analysis can now be performed for extremely large systems through a new sparse-matrix diagonalization engine that also works in parallel, and even for PME simulations, it is possible to perform the traditional non-sparse (computationally costly) diagonalization in parallel.
3 PERFORMANCE
3.1 Scaling
For systems where absolute speed matters, the final simulation performance can be expressed as speed_per_core * ncores * scaling_efficiency. Rather than optimizing only for scaling efficiency, we have aimed to improve both absolute performance per core and the scaling efficiency at the same time. Recent enhancements in this respect include the better PME parallel decomposition described earlier in the text. The choice of method for calculating long-range electrostatics can greatly affect simulation performance, and rather than simply optimizing the method that scales best (reaction-field), we have worked to optimize the method that is currently viewed as best practice in the field (van der Spoel and van Maaren, 2006). By implementing 2D pencil node decomposition for PME and improving the dynamic load-balancing algorithms, we obtain close-to-linear scaling over large numbers of nodes for a set of benchmark systems that were selected as real world applications from our and others recent work. Scaling results are plotted in Figure 3 for a ligand-gated ion channel (Murail et al., 2011) (a typical membrane protein in a bilayer), a massive vesicle fusion simulation (Kasson et al., 2010), a virus capsid (Larsson et al., 2012) and a large methanol–water mixture. To estimate real-world performance, we report scaling and performance results on two clusters: a Cray XE6 with a Gemini interconnect and a more commodity cluster with Quadruple data rate (QDR) Infiniband and significantly less than full bisectional bandwidth. Both machines were equipped with AMD Magny-cours processors clocked at 1.9–2.1 GHz. For all simulations except the smaller ion channel (130 000 atoms), we obtain strong linear scaling well >1000 cores; for the ion channel, the linear scaling regime still extends <500 atoms/core. All of these benchmark simulations use PME long-range electrostatics where the lattice component is evaluated every single step; our tests with reaction field electrostatics show virtually perfect linear scaling for any number of cores as long as the system is large enough (>250 atoms/core).
Fig. 3.

Strong scaling of medium-to-large systems. Simulation performance is plotted as a function of number of cores for a series of simulation systems. Performance data were obtained on two clusters: one that is thinly connected using QDR Infiniband but not full bisectional bandwidth and a more expensive Cray XE6 with a Gemini interconnect. In increasing order of molecular size: the ion channel with virtual sites had 129 692 atoms, the ion channel without virtual sites had 141 677 atoms, this virus capsid had 1 091 164 atoms, the vesicle fusion system had 2 511 403 atoms and the methanol system had 7 680 000 atoms. See Supplementary Data for details
3.2 Single-node parallelization
GROMACS 4.5 implements parallelization at a low level through single-instruction multiple-data (SIMD) operations, and at a high level through the MPI, and ongoing efforts add an intermediate level of OpenMP parallelization. This provides good scaling at high core counts but adds complexity to code deployment for small installations. We have, therefore, written a threads-only implementation of MPI calls that allows single-node parallelization of GROMACS using either POSIX or Windows threads without additional dependencies, using hardware-supported atomic and lock-free synchronization, with non-blocking communication when the MPI specification allows it. Scaling of the thread_MPI implementation is plotted in Figure 4 for two different systems. The Villin headpiece is a small protein in water (7300 atoms) simulated here using short 8 Å cut-offs, whereas the 2-oleoyl-1-pamlitoyl-sn-glyecro-3-phosphocholine (POPC) bilayer is a membrane/water system with 17 400 atoms using PME electrostatics and 10 Å cut-offs. The scaling behavior is near-identical to OpenMPI (an open source MPI library) on a single node, which is gratifying, as OpenMPI is pretty much a state-of-the-art implementation. The advantage of the GROMACS thread_MPI implementation is that it is lightweight, reduces build complexity and works on a wide variety of systems, including Linux, OS/X, Windows and most embedded systems. This development greatly facilitated large-scale deployment of parallel GROMACS simulations on architectures such as Folding@Home where a user cannot install an MPI library without administrator privileges.
Fig. 4.
Efficient and portable parallel execution using POSIX or Windows threads. Performance is plotted as a function of number of cores using the thread\_MPI library and compared with using OpenMPI. Simulations were run on a single node with 24 AMD 8425HE cores running at 2.66 GHz. Performance is nearly identical between the two parallel implementations. Data are plotted for the Villin and POPC bilayer benchmark systems
3.3 Throughput simulations
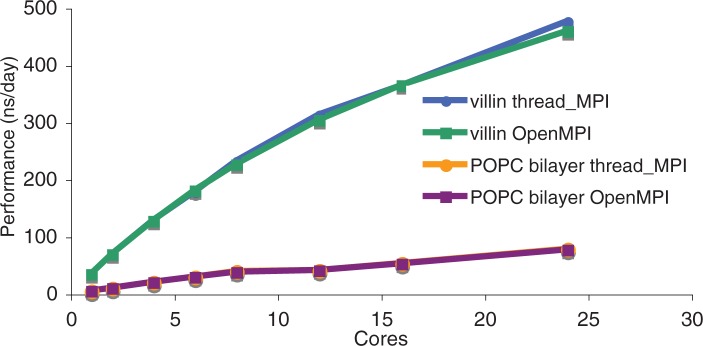
As modern computers have increased in processing power, simulations that used to require supercomputers become tractable on small clusters and even single machines. This has two important consequences: moderate-size simulations become accessible to non-specialists without major allocations of supercomputing resources, and it becomes possible to run many simulations at once to perform moderate-throughput computation on different conditions, mutants of a protein or small-molecule ligands. To illustrate both of these, and to show that it is not always necessary to invest in clusters to perform efficient simulations, we have benchmarked GROMACS running on instances at one of the current major cloud providers. The cloud-computing market gives access to relatively capable machines and good burst capacity to thousands of cores or more. With the thread_MPI parallelization in GROMACS, simulation performance is good on single nodes, and the installation is trivial. To emphasize the general accessibility of performing these simulations, we have selected a number of small-to-medium size proteins and other common biological systems, such as membranes, hydrocarbons and water, and illustrate throughput as the cost for completing a microsecond of simulation at a major cloud provider in October 2012 (Fig. 5). The competitive marked means pricing is relatively similar from all providers. As an example, the 1998 1-µs Villin simulation by Duan and Kollman was a landmark computational achievement at the time (Duan and Kollman, 1998). That simulation required months of supercomputer time using hundreds of nodes on one of the fastest machines in USA. Today, anybody can repeat the same simulation in under a week on a single cloud node at a cost of $11, bringing this well within range of a student project (and soon a class laboratory exercise). Equivalently, screening of hundreds of mutants becomes feasible even without large dedicated resources. The systems and settings have been selected to illustrate a range of different settings, which are described in more detail in the Supplementary Data.
Fig. 5.
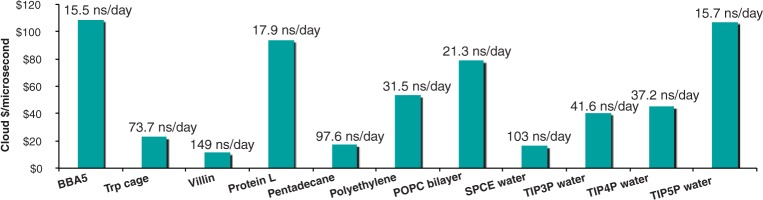
Cost efficiency of GROMACS on small systems. Cost per microsecond is plotted for a series of small systems running on a single eight-core node at a major cloud provider. Simulation details are available in the Supplementary Data. The label on each bar indicates the performance (inversely proportional to cost). The ready availability of cloud compute instances enables extremely cost-efficient high-throughput simulation using individual nodes
4 CONCLUSIONS AND OUTLOOK
Improvements in processor power, simulation algorithms and new computing paradigms are opening a new frontier for molecular dynamics where many simulations of a moderate-sized system are now tractable. The amount of applicable methods and levels of accuracy available in molecular simulation packages is likewise expected to increase, with both more accurate polarizable models and less-detailed coarse-grained force fields gaining popularity (van der Spoel and Hess, 2011). This enables a fundamental change in the way we approach molecular simulation as a tool. The traditional use of molecular dynamics can be thought of as probing the physical consequences of a given starting protein sequence, ligand and structure. Now, given an ensemble of 50 candidate models (as might be generated from nuclear magnetic resonance of a flexible or underdetermined complex), we can evaluate the relative probability of the models and define the accessible conformation space. We can also do mutant scans—the computational equivalent of combinatorial mutagenesis. Soon, approaches such as random walks in sequence space or ligand scaffold space will become routinely tractable. These new capabilities will demand a different approach to simulation—in addition to the underlying physics- and chemistry-based methodology, scientists will need to devote more attention to statistical sampling and leverage benefits of classical informatics techniques, such as randomized search algorithms and network flow theory.
For the next couple of years, we expect the high-throughput trend to become increasingly accentuated: despite massively increased computational power, researchers have been reluctant to merely push longer simulations. Instead of extending membrane proteins simulations to a single 5–10 µs trajectory, most current publications rather use the same amount of total computing time for a whole set of shorter simulations to provide statistics. Fundamentally, we believe this is a scientifically sound development, and one that is likely to move biomolecular simulation and modeling from compute-centric to data-centric approaches more similar to other methods used in bioinformatics.
Supplementary Material
ACKNOWLEDGEMENTS
GROMACS would not have been possible without a large number of users contributing ideas, suggestions and patches—thank you.
Funding: European Research Council [209825 to E.L., 258980 to B.H.]; Swedish research council (to E.L., D.V.D.S.); Foundation for Strategic Research (to E.L.); Swedish e-Science Research Center (to E.L., B.H.); eSSENCE (to D.V.D.S.); National Institute of Health [R01GM098304 to P.K.]; ScalaLife & CRESTA EU FP7 projects (to E.L., B.H.). Computing resources have been provided by the Swedish National Infrastructure for Computing.
Conflict of Interest: none declared.
REFERENCES
- Alder B, Wainwright T. Phase transition for a hard sphere system. J. Chem. Phys. 1957;27:1208–1209. [Google Scholar]
- Bennett CH. Efficient estimation of free energy differences from Monte Carlo data. J. Comp. Phys. 1976;22:245. [Google Scholar]
- Berendsen H. Technical report. CECAM, Lyon: 1976. Report of CECAM workshop: models for protein dynamics. [Google Scholar]
- Bowers KJ, et al. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) ACM, New York, NY: 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. [Google Scholar]
- Bowman G, et al. Atomistic folding simulations of the five helix bundle protein 6-85. J. Am. Chem. Soc. 2011;133:664–667. doi: 10.1021/ja106936n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce S, et al. Predicting ligand binding affinity with alchemical free energy methods in a polar model binding site. J. Mol. Biol. 2009;394:747–763. doi: 10.1016/j.jmb.2009.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B, et al. Charmm: The biomolecular simulation program. J. Comp. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleman C, et al. Force field benchmark of organic liquids: density, enthalpy of vaporization, heat capacities, surface tension, compressibility, expansion coefficient and dielectric constant. J. Chem. Theor. Comput. 2012;8:61–74. doi: 10.1021/ct200731v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D, et al. The amer biomolecular simulation programs. J. Comp. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong L, et al. Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48g7. Proc. Natl Acad. Sci. USA. 1999;96:14330–14335. doi: 10.1073/pnas.96.25.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M, et al. The gromos software for biomolecular simulation: Gromos05. J. Comp. Chem. 2005;26:1719–1951. doi: 10.1002/jcc.20303. [DOI] [PubMed] [Google Scholar]
- Duan Y, Kollman P. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- Essmann U, et al. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8592. [Google Scholar]
- Hawkins D, et al. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. A. 1996;100:19824–19839. [Google Scholar]
- Helms V, Wade R. Computational alchemy to calculate absolute protein-ligand binding free energy. J. Am. Chem. Soc. 1998;120:2710–2713. [Google Scholar]
- Hess B, et al. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theor. Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Huang D, Caflisch A. The free energy landscape of small molecule unbinding. PLoS Comput. Biol. 2011;7:e1002002. doi: 10.1371/journal.pcbi.1002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hub JS, et al. g_wham-a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theor. Comput. 2010;6:3713–3720. [Google Scholar]
- Humphrey W, et al. VMD—Visual Molecular Dynamics. J. Molec. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kasson PM, et al. Atomic-resolution simulations predict a transition state for vesicle fusion defined by contact of a few lipid tails. PLoS Comput. Biol. 2010;6:e1000829. doi: 10.1371/journal.pcbi.1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange OF, et al. Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data. Biophys. J. 2010;99:647–655. doi: 10.1016/j.bpj.2010.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson D, et al. Virus capsid dissolution studied by microsecond molecular dynamics simulations. PLoS Comput. Biol. 2012;8:e1002502. doi: 10.1371/journal.pcbi.1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson P, Lindahl E. A high-performance parallel-generalized born implementation enabled by tabulated interaction rescaling. J. Comp. Chem. 2010;31:2593–2600. doi: 10.1002/jcc.21552. [DOI] [PubMed] [Google Scholar]
- Levitt M, Lifson S. Refinement of protein conformations using a macromolecular energy minimization procedure. J. Mol. Biol. 1969;46:269–279. doi: 10.1016/0022-2836(69)90421-5. [DOI] [PubMed] [Google Scholar]
- Lifson S, Warshel A. A consistent force field for calculation on conformations, vibrational spectra and enthalpies of cycloalkanes and n-alkane molecules. J. Phys. Chem. 1968;49:5116–5130. [Google Scholar]
- Lindorff-Larsen K, et al. How fast-folding proteins fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- Malde AK, et al. An automated force field topology builder (ATB) and repository: version 1.0. J. Chem. Theor. Comput. 2011;7:4026–4037. doi: 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, et al. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Martyna GJ, et al. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996;87:1117–1157. [Google Scholar]
- McCammon J, et al. Dynamics of folded proteins. Nature. 1977;267:585–590. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- Murail S, et al. Microsecond simulations indicate that ethanol binds between subunits and could stabilize an open-state model of a glycine receptor. Biophys J. 2011;100:1642–1650. doi: 10.1016/j.bpj.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noé F, Fischer S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008;18:154–162. doi: 10.1016/j.sbi.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Nosé S, Klein ML. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983;50:1055–1076. [Google Scholar]
- Nury H, et al. One-microsecond molecular dynamics simulation of channel gating in a nicotinic receptor homologue. Proc. Natl Acad. Sci. USA. 2010;107:6275–6280. doi: 10.1073/pnas.1001832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev A, et al. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- Pande V, et al. Everything you wanted to know about Markov state models but were afraid to ask. Methods. 2010;52:99–105. doi: 10.1016/j.ymeth.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- Phillips J, et al. Scalable molecular dynamics with namd. J. Comp. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qui D, et al. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate born radii. J. Phys. Chem. A. 1997;101:3005–3014. [Google Scholar]
- Schulz R, et al. Scaling of multimillion-atom biological molecular dynamics simulation on a petascale supercomputer. J. Chem. Theor. Comput. 2009;5:2798–2808. doi: 10.1021/ct900292r. [DOI] [PubMed] [Google Scholar]
- Shirts MR, Pande VS. Comparison of efficiency and bias of free energies computed by exponential averaging, the Bennett acceptance ratio, and thermodynamic integration. J. Chem. Phys. 2005;122:144107. doi: 10.1063/1.1873592. [DOI] [PubMed] [Google Scholar]
- Swope WC, et al. A computer-simulation method for the calculation of equilibrium-constants for the formation of physical clusters of molecules: application to small water clusters. J. Chem. Phys. 1982;76:637–649. [Google Scholar]
- Valiev M, et al. NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010;181:1477–1489. [Google Scholar]
- van der Spoel D, Hess B. Gromacs—the road ahead. WIREs Comput. Mol. Sci. 2011;1:710–715. [Google Scholar]
- van der Spoel D, Seibert M. Protein folding kinetics and thermodynamics from atomistic simulations. Phys. Rev. Lett. 2006;96:238102–238106. doi: 10.1103/PhysRevLett.96.238102. [DOI] [PubMed] [Google Scholar]
- van der Spoel D, van Maaren PJ. The origin of layer structure artifacts in simulations of liquid water. J. Chem. Theor. Comput. 2006;2:1–11. doi: 10.1021/ct0502256. [DOI] [PubMed] [Google Scholar]
- van der Spoel D, et al. GROMACS molecule and liquid database. Bioinformatics. 2012;28:752–753. doi: 10.1093/bioinformatics/bts020. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. Antechamber, an accessory software package for molecular mechanical calculations. J. Comput. Chem. 2005;25:1157–1174. [Google Scholar]
- Wold M, et al. g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comp. Chem. 2010;31:2169–2174. doi: 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- Zhang J, et al. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011;19:1784–1795. doi: 10.1016/j.str.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.