

Abstract
A major obstacle to the efficient production of antibody conjugates for therapy and diagnosis is the non-ideal performance of commonly used chemical methods for the attachment of effector-molecules to the antibody of interest. Here we demonstrate that this limitation can be simply addressed using 3,4-substituted maleimides to bridge and thus functionalize disulfide bonds to generate homogeneous antibody conjugates. This one-step conjugation reaction is fast, site-specific, quantitative and generates products with full binding activity, good plasma stability and the desired functional properties. Furthermore, the rigid nature of this modification by disulfide bridging enables the successful detection of antigen with a spin labeled antibody fragment by continuous-wave electron paramagnetic resonance (cw-EPR), which we report here for the first time. Antigen detection is concentration dependent, observable in human blood and allows the discrimination of fragments with different binding affinity. We envisage broad potential for antibody based in-solution diagnostic methods by EPR or ‘spinostics'.
A substantial portion of all biotechnology products under development are antibodies and antibody fragments1. Although around 30 have obtained FDA approval for therapeutic applications to date, most of them need to be used in combination with other forms of treatment2. A major limitation is the often insufficient clinically relevant activity of this class of proteins3,4. Conjugation of the antibody to a functional molecule such as a cytotoxic drug for therapy or an imaging reagent for diagnosis is thus necessary and widely applied5. The ideal chemical coupling reaction would be fast, efficient and site-specific generating a homogeneous product which retains its antigen-binding activity. Commonly used methods do not fulfill these ideals. Lysine modification gives rise to heterogeneous mixtures of products due to the multitude of free lysines on the surface of antibodies and the result can be a narrow therapeutic window due to off-site toxicity6. In contrast, the introduction of an additional cysteine by genetic engineering, which is feasible even in a full antibody7, offers a point of attachment for site-specific modification. However an unpaired cysteine can have a detrimental effect on the production yield8 and usually results in the formation of dimers and mixed disulfides, which have to be carefully reduced before chemical coupling7. As an alternative non-natural amino acids can be used as unique chemical handles but their successful insertion is laborious and context-dependent9 and the performance of the available chemical toolbox for their modification is non-ideal10. To avoid the need for genetic engineering and obtain some site-specificity the inter-chain disulfide bonds of antibodies can be reduced, followed by reaction of the revealed cysteines with thiol-specific chemistry11. Yet the cleavage of cystines can have negative effects on the stability12 and effector-functions of antibodies13. A promising solution to these problems is the utilization of bis-reactive bridging reagents that are able to restore a covalent linkage between the two cysteines14,15.
We have recently described a new strategy for such “functionalisation by bridging” of disulfide bonds16,17,18. Substitution of the 3- and 4-positions of the widely used maleimide-motif with good leaving groups yields reagents that react rapidly and efficiently with a reduced cystine, inserting a rigid two-carbon bridge without the introduction of new chiral centers. By careful choice of the leaving groups we have synthesized reagents that can be used in tandem with reducing agents, enabling an in situ protocol in which the functionalized disulfide bonds are formed in minutes. Furthermore, the nitrogen in the maleimide ring is readily modified synthetically and various functional groups can be attached at this position, allowing diverse modification of proteins. We envisage that our methodology could be used to prepare a new generation of functionalized antibody analogues. In addition we were attracted to the possibility that the rigidity of the maleimide bridge would couple the tumbling motions of an antibody tightly to those of an attached spin label19 and thus allow the assembly of immuno-biosensors for EPR-based detection of antibody-antigen interactions or ‘spinostics' (Fig. 1). This approach would have numerous advantages over other methods for monitoring such interactions, the most prominent being its direct, and theoretically understood, connection to the molecular immobilization characteristic of binding.
Figure 1. Concept of disulfide bond-based antibody functionalisation and EPR-sensing of the antibody-antigen interaction (‘spinostics').
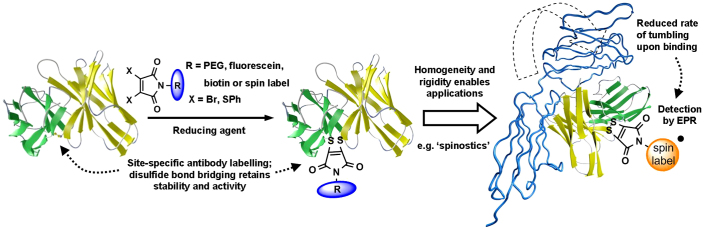
Cartoons are derived from a published model of MFE2344.
Results
Site-specific bridging of an antibody fragment disulfide bond
To test the ideas discussed above, we decided to work with a variable fragment (Fv), the smallest portion of an antibody which retains full binding activity. We chose a carcinoembryonic antigen (CEA) specific single-chain Fv fragment (scFv) shMFE20, in which the inter domain linker (Gly4Ser)3 had been changed to (Gly4Ser)4 to increase the monomer to dimer ratio in the production process (Supplementary Fig. S1). A disulfide bond had been engineered to further stabilize the monomeric form of the construct21 by performing two amino acid substitutions: Gly to Cys and Ala to Cys at Kabat positions H44 and L100 respectively, resulting in a unique cystine-stabilized single-chain Fv fragment (sscFv). The location of the artificial cystine corresponds to that identified as a general site within the scFv framework to stabilize Fvs by disulfide bonds22,23.
To modify the accessible disulfide bond we initially used a sequential protocol16: reduction of the cystine with dithiothreitol (DTT, 20 equivalents towards the antibody concentration), followed by addition of an excess of dibromomaleimide (30 equiv). Full conversion to the maleimide-bridged sscFv (Fig. 2a) was observed by LC-MS in less than 5 min. Analysis of the product revealed a small amount of higher molecular weight species (Fig. 2b and Supplementary Fig. S2) which did not appear to be formed by maleimide cross-linking as they were stable to the addition of more DTT16 (data not shown). In order to avoid this apparent multimer formation, we established the conditions for an in situ modification17. As the disulfide bond of the CEA-specific sscFv was relatively resistant to reduction by tris (carboxyethyl)phosphine (TCEP, Supplementary Methods) a mix of dithiophenolmaleimide and benzeneselenol (15 equiv each) was employed to reach full conversion (Supplementary Fig. S3). The speed of the reaction was not affected (Fig. 2c) and no higher molecular weight products were detectable (Fig. 2b and Supplementary Fig. S2), illustrating that the in situ protocol avoids the common issue of undesired scFv multimerisation24. During optimization we found that as little as 2 equiv of bridging-reagent were sufficient to yield the completely modified sscFv when two portions of the reducing agent were added or its quantity increased to 25 equiv (Supplementary Fig. S4).
Figure 2. Synthesis, activity and stability of CEA-specific sscFv analogues.

(a) Deconvoluted LC-MS spectra of unmodified and maleimide-bridged sscFv (requires 26,836 Da). Conditions a) (sequential): 20 equiv DTT for 1 h, then 30 equiv of dibromomaleimide; conditions b) (in situ): 15 equiv dithiophenolmaleimide and 15 equiv benzeneselenol; conditions c) (in situ): 2 equiv dithiophenolmaleimide and 25 equiv benzeneselenol. The additional peaks are disulfide bond containing impurities in the starting material. (b) SDS-PAGE of sequentially and in situ bridged sscFv. Processed antibody was treated with maleimides but no reducing agent. (c) Timed LC-MS data of the in situ bridging reaction of sscFv. The signal refers to the relative abundance of the mass ion of bridged sscFv. (d) ELISA of variably bridged sscFv against full length CEA. (e) ELISA of alkylated and functionalized sscFv against full length CEA. (f) ELISA of functionalized sscFv against full length CEA. (g) SDS-PAGE of bridged sscFv isolated from human plasma after incubation at 37°C and loading controls. Unmodified and alkylated sscFv were treated for 7 d and isolated. The negative control contained no antibody. (h) Stability of the bridged sscFv against a 100× excess of thiols as determined by LC-MS. The signal refers to the relative abundance of the mass ion of the un-bridged sscFv. (i) Normalized activity of the sscFv and its analogues after incubation in human plasma at 37°C as determined by ELISA against full length CEA. Full-length gels are presented in Supplementary Figure S16. ELISAs d–f and i were performed in triplicates; error bars show s.d.
When we compared the biological activity of sequentially and in situ bridged sscFv analogues with the unmodified antibody by ELISA we found that the binding activity was not merely retained but actually increased (Fig. 2d). This result was confirmed by surface plasmon resonance (SPR) measurements (Kd unmod 20.8 ± 2.9 nM, Kd bridged 6.4 ± 0.3 nM). Although the residues which had been replaced by cysteines are in the ideal position for the introduction of a disulfide bond23, its presence or geometry had possibly introduced structural strain into the sscFv which is released upon insertion of the maleimide-bridge. The likewise enhanced activity of an alkylated analogue, in which both cysteines had been reacted separately with maleimide and thus had lost the disulfide bridge completely, supports this possibility (Fig. 2e).
Functionalisation of the sscFv disulfide bond
CEA is a cell-surface glycoprotein (~200 kDa), which is overexpressed in a wide range of cancers and its blood-concentration is used as a clinical marker for post-surgical monitoring of primary colon carcinomas25. To equip the CEA-specific sscFv with imaging properties we conjugated the protein via the maleimide bridge to fluorescein, biotin and two types of nitroxide spin label. We also attached a polyethylene glycol (PEG) chain (5 kDa) as a simple model for PEGylation, a widely used process to increase the circulation time and half-life of antibody fragments26. All N-functionalized maleimides were synthesized following published protocols16,17,27 with the exception of the spin labels, for which we developed a milder one-step method of attachment to the bridging moiety (Supplementary Fig. S5 and Supplementary Methods).
The functionalisation of the sscFv via the sequential (fluorescein and biotin) or in situ protocol (spin labels and PEG) was as fast and efficient as observed for the bridging reaction with unmodified maleimides (Supplementary Fig. S6) and simple purification via a desalting column or in the case of PEGylation by nickel magnetic beads via its C-terminal his6-tag, furnished the homogeneously modified analogues in 80–90% yields. All analogues exhibited an increased activity as found for the bridged sscFv by ELISA (Fig. 2e,f) and also by SPR in the case of the PEG-sscFv (Kd PEGylated 8.7 ± 0.3 nM).
Having shown previously that an excess of thiols can cleave the maleimide from within a disulfide bond16 we tested the stability of the bridged sscFv alongside controls in human plasma, which typically contains about 18 μM of free thiol28. Relatively constant amounts of the sscFv analogue could be isolated from these samples over the time of the experiment (Fig. 2g) and the LC-MS spectra of this material showed no loss of the maleimide bridge (Supplementary Fig. S7). Notably, subjecting the modified antibody fragment to cytoplasmic concentrations of glutathione or other reducing agents (7 mM) in buffer led to the formation of un-bridged sscFv over time (Fig. 2h), implying a potential cargo-release mechanism under in vivo conditions. In addition bridging via the cystine helped to preserve the activity of the sscFv during incubation at 37°C, as did the alkylation of the cysteines (Fig. 2i), suggesting a role of the disulfide bond, once reduced, in the loss of antibody functionality.
To compare our method with the traditional method of conjugation via lysine residues, we carried out amine-based PEGylation of the sscFv with 5 kDa PEG chains activated by an N-hydroxysuccinimide ester (NHS) group. This reaction produced under varying conditions the typical mix of differentially conjugated antibodies (Fig. 3a) compared to a single product obtained by functional bridging of the disulfide bond (Fig. 3b). The mono-PEG-sscFv and a multi-PEG-sscFv species prepared with NHS-chemistry were purified (yields of 14% and 28% respectively) and their binding capacity tested by ELISA, which clearly showed the superiority of a defined and site-specific approach (Fig. 3c).
Figure 3. Functionality of the CEA-specific sscFv analogues.

(a) SDS-PAGE of variously NHS-based PEGylated sscFv. The reaction was quenched after indicated times with an excess of glycine. (b) SDS-PAGE of maleimide-based PEGylated sscFv. Processed antibody was treated with PEGylation reagent but no reducing agent. (c) ELISA of purified maleimide- and NHS-based PEGylated sscFv against full length CEA. (d) Fluorescence read-out of a dilution series of CEA expressing (CAPAN-1) and non-expressing (A375) cancer cell lines after treatment with fluorescein-sscFv (ex. 488 nm; em. 518 nm). (e) Comparison of the ELISA-activity of commonly formed reporter complexes (secondary antibody conjugated to HRP) with on-plate formation by addition of HRP-Strep conjugate to antigen-bound (full length CEA) biotin-sscFv. In the 1:4600 dilution of the HRP-Strep conjugate no non-specific background was observed. (f) One-step ELISA of preformed and purified HRP-sscFv conjugate against full length CEA. Experiments (c–f) were performed in triplicates; error bars show s.d.
The other analogues were evaluated with regard to their signaling properties: dilution series of CEA-expressing and non-expressing cell lines were settled in 96-well plates treated with the fluorescein-sscFv and analyzed with a simple ELISA reader. The signal intensity had good correspondence to the number of cells present (Fig. 3d) and the detection of as few as about 8,000 cells was possible. The biotin-sscFv analogue was coupled to horseradish peroxidase (HRP) by mixing with a commercially available streptavidin-conjugate (HRP-Strep) of the enzyme. This could be done either directly in the wells of an ELISA plate, after pre-incubation of the biotinylated antibody with the antigen, or in solution from which the final HRP-sscFv could be purified (Supplementary Fig. S8). Both antibody-enzyme conjugates produced a specific and strong signal in two- or one-step ELISAs (Fig. 3e,f and Supplementary Fig. S9).
‘Spinostics' – EPR-based immuno-sensing
To explore the idea of ‘spinostics' we conjugated two dithiophenolmaleimide-coupled nitroxide spin labels to the sscFv. They differ in that the PROXYL radical is connected either directly to the maleimide ring, yielding dithiophenolmaleimide-N-PROXYL (TPMP, Fig. 4a and Supplementary Methods) or with an additional carbon in the linkage (TPMCP, Fig. 4b and Supplementary Methods). Direct comparison of cw-EPR spectra of the free label and the spin labeled sscFv analogues confirmed successful conjugation to the antibody (Fig. 4a,b, blue lines). The antibody fragment labeled with TPMP (MP-sscFv) exhibited a more immobile nitroxide spectrum, as indicated by its broader spectral components, in comparison to the fragment labeled with TPMCP (MCP-sscFv).
Figure 4. EPR-based monitoring of antibody-antigen interactions.

(a) From top to bottom: TPMP molecule, free TPMP normalized EPR spectrum (black), MP-sscFv (blue, 10 μM) and NA1-bound state (red, 70 μM NA1) EPR spectra in human blood, MP-sscFv (blue, 250 nM) and NA1-bound state (red, 1.75 μM NA1) EPR spectra in human blood, MP-sscFv (blue, 100 nM) and NA1-bound state (red, 400 nM NA1) EPR spectra in human blood. (b) Same as (a) but for MCP-sscFv. (c) 10 μM MP-sscFv and (d) MCP-sscFv binding curves. NA1 concentrations are 0, 3, 6, 9, 12, 15, 20, 22.5, 25, 30, 35, 40, 45, 50, 60 and 70 μM. Red circles (PBS), squares (human plasma), triangles (whole human blood) represent the respective raw data points and red lines represent best fits for raw data. Blue shapes and lines represent data points for simulations and fits of simulated data, respectively. (e) 250 nM binding curves for MP-sscFv and (f) MCP-sscFv with NA1 (circles) and full length CEA (squares), respectively. Fits are as described for c and d. NA1 and full length CEA concentrations are 0, 25, 50, 75, 100, 125, 187.5, 250, 375, 500, 625, 750, 875, 1000, 1250, 1500 and 1750 nM (calculated and simulated Kd values are listed in Supplementary Table S1). (g) MP-sscFv (blue, open circles) and MP-sscFv-SM3E (red, open circles) thermal stability shift assay with (closed circles) and without NA1 (70 μM) present. Tm values were fit using Supplementary Equation S2. (h) Same as g but for MCP-sscFv and MCP-sscFv-SM3E. Tm shifts are mentioned in the main text and all other values and enthalpies of unfolding are listed in Supplementary Table S2. Raw data represent the ratio of the low field and central nitroxide peaks. Error bars shown represent the 95% confidence boundaries as calculated from the data fitting process.
Upon addition of antigen to the spin labeled sscFv analogues in the form of NA1, a 26 kDa N-terminal fragment of CEA, substantial changes in the EPR spectra were observed (Fig. 4a,b, red lines) in contrast to control experiments with BSA (Supplementary Fig. S10). To our knowledge, successful monitoring of such a binding event with EPR has not been reported before for a spin label attached to an antibody29. Similar effects were found in human plasma and also in whole blood (Supplementary Fig. S11) where the signal was surprisingly stable despite the presence of the natural reducing agent ascorbate30. We generated saturation binding curves in these media as well as in PBS, using the mean squared deviation (m.s.d.) of NA1 bound spectra from the unbound spectrum (Fig. 4c,d and Supplementary Fig. S11). The multi-component EPR spectra were also simulated and relative weightings of each of the two components accounting for approximately 95% of the spectra were extracted to monitor binding. The simulated data typically had r.m.s.d errors of < 0.06.
Fitting (Supplementary Equation S1) of MP-sscFv binding curves, obtained at 10 μM antibody concentration, yielded Kd values of: 1.91 ± 0.78 μM, 4.35 ± 1.27 μM and 6.46 ± 1.7 μM in PBS, plasma and whole blood, respectively for raw data; compared to: 1.22 ± 0.60 μM, 3.94 ± 2.06 μM and 5.63 ± 2.06 μM for data analyzed using the simulations. As expected, binding affinities are reduced as the binding matrix becomes more complex as in blood31. In contrast, when working with MCP-sscFv, the antibody concentration was constantly overestimated in comparison to its experimental value during the fitting procedure and generally predicted lower affinity values (Supplementary Table S1). The higher Kd values could be explained by the uncoupling of the binding event from the spin label reporting the protein-protein interaction, facilitated by the extra carbon atom32. This emphasizes the high degree of immobilization that is achieved by attachment of the spin label into a rigid disulfide bridge. Furthermore, by adding 30% wt/vol sucrose to both spin labeled sscFv analogues about 80% of spectral changes encountered could be replicated through the additional viscosity (Supplementary Fig. S12). These spectra illustrate the dominance of protein tumbling rates33 on sscFv EPR spectra upon binding for both labels and once again aid in proving the importance of the rigidity of the label insertion.
To simulate conditions closer to the realities of clinical settings we repeated the binding study with NA1 as well as full length CEA purified from human colon carcinomas at lower sscFv concentrations. Data – which could be successfully simulated – were acquired at antibody concentrations as low as 250 nM with a good signal to noise ratio (Fig. 4e,f and Supplementary Fig. S13). To demonstrate the potential to work at even lower concentrations we recorded spectra of bound and unbound MP- and MCP-sscFv at 100 nM respectively (Fig. 4a,b). Although relatively noisy, spectral differences that resemble those observed at higher concentrations are visible and most prominent at the low field peaks.
To further explore the utility of the ‘spinostics' platform, we investigated its application as an antibody fragment-based thermal stability shift assay (TSSA) with both labels. Here, the presence of a saturating NA1 concentration on the thermal stability of the sscFv was monitored as a function of increasing temperature. As expected, the Gibbs free energy of unfolding of the labeled sscFv was enhanced in the presence of antigen, resulting in a shift in the melting temperature (Tm) of the antibody fragment of 8.6 ± 2.7 K and 7.3 ± 1.8 K for MP- and MCP-sscFv, respectively (Fig. 4g,h, blue traces and Supplementary Fig. S14). We performed a similar experiment on sscFv-SM3E, an affinity matured variant of the CEA-specific sscFv20. sscFv-SM3E showed a further Tm shift of 13.1 ± 2.9 K and 13.8 ± 2.4 K for MP- and MCP-sscFv-SM3E, respectively (Fig. 4g,h, red traces). These values correlate well with first derivative plots of the fits (Supplementary Fig. S15).
Discussion
We were able to show that next generation maleimides allow the assembly of a range of fully active homogenous antibody conjugates in high yields and with the desired functional properties. The simple, efficient and site-specific chemistry, the plasma stability of the maleimide bridge and its potential in controllable in-cell cleavage27,34 eliminates many disadvantages of currently used conjugation chemistry and drawbacks of arduous antibody-engineering. The versatility of this chemical platform also facilitates the presented ‘spinostics' technology. The mix and measure metrology employed here for both the saturation binding curve generation and the TSSA demonstrates their potential for automation and high throughput applicability. TSSA lends itself particularly well as a complementary tool to current methods employed in screening antibody-antigen interactions, including various ligands, drug compounds and environmental targets35, with great potential for stability screening of antibody fragments. Furthermore, the matrix insensitive nature of EPR makes it attractive for ligand screening under physiological conditions, with minimal sample pre-treatment necessary.
Currently, our lower limit of detection, found in this proof-of-concept experiment using a standard commercial EPR spectrometer (Bruker EMXplus) is 100 nM. We anticipate that two to three orders of magnitude sensitivity gains would make this a clinically relevant CEA assay36. An order of magnitude in sensitivity is potentially feasible with current technologies for example through the use of improved resonators37 or rapid scan EPR38, which has future promise of decreased scan times and increased signal to noise over conventional cw-EPR. Additionally, our experiments will motivate similar work on biomarkers for other diseases and acute phase proteins with higher concentrations (e.g. C-reactive protein39), which could be detected readily with current generation EPR spectrometers. Unlike many biosensor and bio-interaction technologies, the ‘spinostics' platform described here does not require a surface and therefore is not prone to the gross overestimation of Kd typical for surface-based probes31, does not require complex surface immobilization chemistries and avoids related heterogeneity problems. Moreover, compared to conventional optical technologies, solution binding can be observed in opaque matrixes such as blood, lending itself well to point-of-care diagnostics and EPR spectra can be readily simulated to obtain more detailed information about the systems dynamics.
In conclusion, the combination of a powerful new chemical approach to site-specific antibody conjugation with an EPR-based detection method offers a platform for the development of new tools to study biological systems and for application in diagnostics and biotechnology.
Methods
Protein synthesis
We produced the sscFv variant of shMFE and of the high affinity shMFE variant SM3E using the methanol inducible Pichia pastoris production platform. In brief, a DNA fragment encoding shMFE or SM3E20 was synthesized (Genescript USA Inc.) and relative to the original sequence the following changes introduced: two substitutions at Gly'H44'Cys as well as Ala'L100'Cys (Kabat numbering), the linker was extended from (Gly4Ser)3 to (Gly4Ser)4 and sequences encoding KREAEA and His6 plus stop codon were added to the 5 and 3 prime, respectively. The sequence was digested with EcoRI and NotI, and ligated into a pPICZáB vector (Invitrogen). After amplification in E. coli TOP10 (NEB) the construct was linearized with PmeI and transferred by electroporation into Pichia pastoris X33 (Invitrogen). We used the transformant with the highest protein expression for protein production by fermentation with initial purification using radial bed adsorption IMAC as published40,41. The IMAC fraction containing the protein was concentrated with a Labscale TFF system (Millipore) using a Biomax 10 kDa ultrafiltration module and subsequently applied to a Superdex 75 size-exclusion column (500 ml bed volume, GE Healthcare) which was equilibrated with PBS (pH 7.4). The fraction containing the monomer sscFv was collected and stored at −80°C. The protein material was > 95% pure as estimated by SDS-PAGE.
We synthesized and purified the NA1 fragment of CEA as described elsewhere42.
Bridging of the sscFv
A solution of the CEA-specific sscFv in PBS (pH 7.4) was diluted in the same buffer and dimethylformamide (DMF, 10% vol/vol final amount) to yield a concentration of 70.0 μM (1.87 mg ml−1) prior to experimentation. All maleimides and benzeneselenol were prepared in DMF as highly concentrated stock solutions, while DTT was dissolved in PBS. We established three protocols: a) Sequential bridging: the antibody fragment was treated with 20 equiv DTT for 1 h at ambient temperature, then 30 equiv of dibromomaleimide were added for 10 min. b) In situ bridging: 15 equiv of dithiophenolmaleimide were added to the antibody solution followed by 15 equiv of benzeneselenol. The mixture was incubated for 20 min at ambient temperature. c) In situ bridging (alternative): 2 equiv of dithiophenolmaleimide were added to the antibody solution followed by 25 equiv of benzeneselenol. The mixture was incubated for 20 min at ambient temperature.
Successful modification was confirmed by LC-MS. Timed data was obtained by incubation of the reaction mixtures for 60 min and withdrawing and analyzing aliquots at various time points.
Functionalisation of the sscFv
We prepared all maleimides as highly concentrated stock solutions in DMF with the exception of N-PEG5000-dithiophenolmaleimide, which was prepared in PBS. The sscFv was conjugated to biotin- and fluorescein-dibromomaleimide via protocol a) and to the nitroxide spin labels as well as the PEG chain via protocol b) or c). Efficient formation of all analogues was verified by LC-MS (and SDS-PAGE in the case of PEGylated sscFv). Functionalized antibody fragments were purified with PD MiniTrap G-25 desalting columns (GE Healthcare) or PureProteome Nickel Magnetic Beads (Millipore) following manufacturers' instructions. Concentrations were determined by UV/Vis using a calculated molecular extinction coefficient of ε280 = 48,735 M−1 cm−1.
Antigen binding of the CEA-specific sscFv monitored by EPR
The concentration of spin labeled CEA-specific sscFv was held constant at 10 μM or 250 nM and the antigen concentration was varied between 0–70 μM for experiments at 10 μM and 0–1.75 μM at 250 nM. For 10 μM data, the final volumes were normalized to 15 μL with PBS. Experiments with whole human blood and human plasma were conducted under the same conditions as PBS and constituted 10 μL of the entire volume. Therefore, the only variable was the antigen in all cases. All samples were incubated for 30 min at 37°C under constant agitation. Samples were then transferred into flame sealed 1 mm (inner diameter) capillaries and briefly centrifuged. Loaded capillaries were placed into a pre-warmed (37°C) 4102ST resonator controlled by a Bruker ER4111VT temperature control unit that had been pre-calibrated with an external thermocouple. A Bruker EMXplus continuous-wave spectrometer operating at 9–10 GHz (X-band) was used as microwave source and detector. Data were recorded using the Bruker Xenon software at 63.25 mW microwave power, 70 dB gain, 2G modulation amplitude, 100 kHz modulation frequency and a typical scan time of 25–30 s. 36 scans were recorded for each spectrum using the 4102ST cavity. The same conditions were applied for the data collection at 250 nM antigen concentration with following exceptions: the number of scans was 100, sample volumes were 200 μL, microwave power was 126.2 mW and a quartz flat-cell was used as a sample holder in combination with a 4122SHQE resonator. No saturation broadening was detected when comparing low and high concentration EPR spectra. Spectra at 100 nM were acquired similarly to 250 nM data but were a total of 200 scans each. Sample holder and resonator were orientated horizontally to minimize sinking of red blood cells. The flat-cell was washed with concentrated nitric acid and ethanol between measurements. We acquired the data in random order to maintain fair testing conditions and further minimize non-specific adsorption to the flat-cell. Simulations of nitroxide EPR spectra were performed using the MATLAB plug-in Easyspin43 (v4.0.0).
Thermal stability shift assay with CEA-specific sscFv and sscFv-SM3E
The spin labelled CEA-specific sscFv or sscFv-SM3E (10 μM) alone or in its bound form (70 μM NA1) were heated within a 4102ST cavity containing an evacuated quartz dewar. Sampling temperatures were collected on heating at 293, 298, 303, 308, 313, 318, 323, 325, 327, 329, 331, 333, 335, 338, 340, 343, 345 and 348 K. A constant nitrogen gas flow was supplied at the desired temperature using a Bruker ER4111VT temperature control. The temperature offset was calibrated using an external thermocouple prior to measuring. Measuring volumes were 10 μL and were placed in a 1mm (inner diameter) glass tube and sealed at the top with clay to minimize water vapour. Microwave power was 63.25 mW, 50 dB gain and 9–16 scans were averaged for each spectrum depending on the label employed. All other parameters were the same as that for saturation binding curves.
Author Contributions
F.F.S. and J.R.B. conceived the chemical biology part of the research described, F.F.S. designed and performed the resultant experiments and wrote the manuscript together with V.A.S. and B.T. V.A.S. conceived, performed and analyzed all EPR experiments. B.T. synthesized and purified the antibody fragments and its antigen. Z.V.F.W. developed the new method for maleimide synthesis and C.P.R. synthesized the N-fluorescein- and N-biotin-maleimides. M.E.B.S., J.M.W., S.C., C.W.M.K. and G.A. provided help with the study design and access to equipment. G.A., C.W.M.K., K.A.C. and J.R.B. provided supervision and guidance for the investigation, helped with the data analysis and interpretation and edited the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
We thank K. Vigor, M. D'Alicarnasso, I. Farabella, G. Bhaysar, L. Haigh and A. E. Aliev for technical assistance. We gratefully acknowledge S. Heutz, M. Warner and A. Indermuehle for helpful discussions. We thank S. Perkins (University College London) for kindly providing the models of the bound and unbound antibody fragment. This work was supported by the Wellcome Trust (F.F.S.), EPSRC (V.A.S., C.M.W.K., G.A., K.A.C. and B.T.), BBSRC (M.E.B.S., S.C. and J.R.B.), RCUK (J.R.B.), UCLB (J.R.B. and S.C.), ECMC, CIRT, NIHR-UCLH-BRC (K.A.C. and B.T.).
Footnotes
F.F.S., M.E.B.S., S.C. and J.R.B. are founders and shareholders of Thiologics, a company developing homogeneous protein modification technology. M.E.B.S., S.C. and J.R.B. are also directors of Thiologics. The ‘spinostics' work described here is the subject of a patent application, which if licensed will afford F.F.S., V.A.S., C.W.M.K., G.A., K.A.C. and J.R.B. royalties in line with the guidelines set down by University College London.
References
- Aggarwal S. Targeted cancer therapies. Nat. Rev. Drug Discov. 9, 427–428 (2010). [DOI] [PubMed] [Google Scholar]
- Leader B., Baca Q. J. & Golan D. E. Protein therapeutics: a summary and pharmacological classification. Nat. Rev. Drug Discov. 7, 21–39 (2008). [DOI] [PubMed] [Google Scholar]
- Carter P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 1, 118–129 (2001). [DOI] [PubMed] [Google Scholar]
- Wu A. M. & Senter P. D. Arming antibodies: prospects and challenges for immunoconjugates. Nat. Biotechnol. 23, 1137–1146 (2005). [DOI] [PubMed] [Google Scholar]
- Sun M. M. et al. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 16, 1282–1290 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeger S. et al. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 49, 9995–9997 (2010). [DOI] [PubMed] [Google Scholar]
- Junutula J. R. et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 26, 925–932 (2008). [DOI] [PubMed] [Google Scholar]
- Schmiedl A., Breitling F., Winter C. H., Queitsch I. & Dubel S. Effects of unpaired cysteines on yield, solubility and activity of different recombinant antibody constructs expressed in E. coli. J. Immunol. Methods 242, 101–114 (2000). [DOI] [PubMed] [Google Scholar]
- Loscha K. V. et al. Multiple-site labeling of proteins with unnatural amino acids. Angew. Chem. Int. Ed. Engl. 51, 2243–2246 (2012). [DOI] [PubMed] [Google Scholar]
- Beal D. M. & Jones L. H. Molecular scaffolds using multiple orthogonal conjugations: applications in chemical biology and drug discovery. Angew. Chem. Int. Ed. Engl. 51, 6320–6326 (2012). [DOI] [PubMed] [Google Scholar]
- Doronina S. O. et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 21, 778–784 (2003). [DOI] [PubMed] [Google Scholar]
- Liu H., Chumsae C., Gaza-Bulseco G., Hurkmans K. & Radziejewski C. H. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal. Chem. 82, 5219–5226 (2010). [DOI] [PubMed] [Google Scholar]
- Michaelsen T. E. et al. One disulfide bond in front of the second heavy chain constant region is necessary and sufficient for effector functions of human IgG3 without a genetic hinge. Proc Natl Acad Sci U S A 91, 9243–9247 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaunak S. et al. Site-specific PEGylation of native disulfide bonds in therapeutic proteins. Nat. Chem. Biol. 2, 312–313 (2006). [DOI] [PubMed] [Google Scholar]
- Brocchini S. et al. PEGylation of native disulfide bonds in proteins. Nat. Protoc. 1, 2241–2252 (2006). [DOI] [PubMed] [Google Scholar]
- Smith M. E. et al. Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J. Am. Chem. Soc. 132, 1960–1965 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher F. F. et al. In situ maleimide bridging of disulfides and a new approach to protein PEGylation. Bioconjug. Chem. 22, 132–136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. W. et al. Polymeric dibromomaleimides as extremely efficient disulfide bridging bioconjugation and pegylation agents. J. Am. Chem. Soc. 134, 1847–1852 (2012). [DOI] [PubMed] [Google Scholar]
- Esmann M., Sar P. C., Hideg K. & Marsh D. Maleimide, iodoacetamide, indanedione, and chloromercuric spin label reagents with derivatized nitroxide rings as ESR reporter groups for protein conformation and dynamics. Anal. Biochem. 213, 336–348 (1993). [DOI] [PubMed] [Google Scholar]
- Graff C. P., Chester K., Begent R. & Wittrup K. D. Directed evolution of an anti-carcinoembryonic antigen scFv with a 4-day monovalent dissociation half-time at 37 degrees C. Protein Eng. Des. Sel. 17, 293–304 (2004). [DOI] [PubMed] [Google Scholar]
- Schmidt M. M., Thurber G. M. & Wittrup K. D. Kinetics of anti-carcinoembryonic antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer Immunol. Immunother. 57, 1879–1890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann U., Gallo M., Brinkmann E., Kunwar S. & Pastan I. A recombinant immunotoxin that is active on prostate cancer cells and that is composed of the Fv region of monoclonal antibody PR1 and a truncated form of Pseudomonas exotoxin. Proc. Natl. Acad. Sci. U. S. A. 90, 547–551 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter Y., Brinkmann U., Jung S. H., Pastan I. & Lee B. Disulfide stabilization of antibody Fv: computer predictions and experimental evaluation. Protein Eng. 8, 1323–1331 (1995). [DOI] [PubMed] [Google Scholar]
- Lee Y. C., Boehm M. K., Chester K. A., Begent R. H. & Perkins S. J. Reversible dimer formation and stability of the anti-tumour single-chain Fv antibody MFE-23 by neutron scattering, analytical ultracentrifugation, and NMR and FT-IR spectroscopy. J. Mol. Biol. 320, 107–127 (2002). [DOI] [PubMed] [Google Scholar]
- Goldstein M. J. & Mitchell E. P. Carcinoembryonic antigen in the staging and follow-up of patients with colorectal cancer. Cancer Invest. 23, 338–351 (2005). [DOI] [PubMed] [Google Scholar]
- Chapman A. P. et al. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 17, 780–783 (1999). [DOI] [PubMed] [Google Scholar]
- Ryan C. P. et al. Tunable reagents for multi-functional bioconjugation: reversible or permanent chemical modification of proteins and peptides by control of maleimide hydrolysis. Chem. Commun. (Camb.) 47, 5452–5454 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor M. A., Svardal A. M. & Ueland P. M. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal. Biochem. 200, 218–229 (1992). [DOI] [PubMed] [Google Scholar]
- Willan K. J., Golding B., Givol D. & Dwek R. A. Specific spin labelling of the Fc region of immunoglobulins. FEBS Lett. 80, 133–136 (1977). [DOI] [PubMed] [Google Scholar]
- Saphier O. et al. The reduction of a nitroxide spin label as a probe of human blood antioxidant properties. Free Radic. Res. 37, 301–308 (2003). [DOI] [PubMed] [Google Scholar]
- Wienken C. J., Baaske P., Rothbauer U., Braun D. & Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100 (2010). [DOI] [PubMed] [Google Scholar]
- Sutton B. J. et al. The gross architecture of an antibody-combining site as determined by spin-label mapping. Biochem. J. 165, 177–197 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHaourab H. S., Lietzow M. A., Hideg K. & Hubbell W. L. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry 35, 7692–7704 (1996). [DOI] [PubMed] [Google Scholar]
- Moody P. et al. Bromomaleimide-linked bioconjugates are cleavable in Mammalian cells. ChemBioChem 13, 39–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H., Berglund H. & Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 (2007). [DOI] [PubMed] [Google Scholar]
- Sharkey R. M. et al. A phase I trial combining high-dose 90Y-labeled humanized anti-CEA monoclonal antibody with doxorubicin and peripheral blood stem cell rescue in advanced medullary thyroid cancer. J. Nucl. Med. 46, 620–633 (2005). [PubMed] [Google Scholar]
- Nesmelov Y. E., Surek J. T. & Thomas D. D. Enhanced EPR sensitivity from a ferroelectric cavity insert. J. Magn. Reson. 153, 7–14 (2001). [DOI] [PubMed] [Google Scholar]
- Mitchell D. G., Quine R. W., Tseitlin M., Eaton S. S. & Eaton G. R. X-band rapid-scan EPR of nitroxyl radicals. J. Magn. Reson. 214, 221–226 (2012). [DOI] [PubMed] [Google Scholar]
- McDonnell B., Hearty S., Leonard P. & O'Kennedy R. Cardiac biomarkers and the case for point-of-care testing. Clin. Biochem. 42, 549–561 (2009). [DOI] [PubMed] [Google Scholar]
- Tolner B., Smith L., Begent R. H. & Chester K. A. Production of recombinant protein in Pichia pastoris by fermentation. Nat. Protoc. 1, 1006–1021 (2006). [DOI] [PubMed] [Google Scholar]
- Tolner B., Bhaysar G., Foster B., Vigor K. & Chester K. in Laboratory Protocols in Fungal Biology (eds V. K. Gupta et al.) Chapter 37 (Springer Science and Business Media, 2012). [Google Scholar]
- Sainz-Pastor N. et al. Deglycosylation to obtain stable and homogeneous Pichia pastoris-expressed N-A1 domains of carcinoembryonic antigen. Int. J. Biol. Macromol. 39, 141–150 (2006). [DOI] [PubMed] [Google Scholar]
- Stoll S. & Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 (2006). [DOI] [PubMed] [Google Scholar]
- Boehm M. K. & Perkins S. J. Structural models for carcinoembryonic antigen and its complex with the single-chain Fv antibody molecule MFE23. FEBS Lett. 475, 11–16 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information