

Background: Thrombin utilizes active site and anion-binding exosites (ABE) to fulfill diverse roles.
Results: ABE I ligands PAR3(44–56), PAR1(49–62), and Hirudin(54–65) exert different effects on thrombin exosite-active site and exosite-exosite interactions. More consequences occur with added PPACK and ABE II ligands.
Conclusion: Thrombin employs ligands to transmit unique events across the enzyme.
Significance: Ligand binding may be tailored to therapeutically regulate multifunctional thrombin.
Keywords: Blood Coagulation Factors, Mass Spectrometry (MS), Protein Complexes, Protein Conformation, Thrombin
Abstract
Thrombin participates in coagulation, anticoagulation, and initiation of platelet activation. To fulfill its diverse roles and maintain hemostasis, this serine protease is regulated via the extended active site region and anion-binding exosites (ABEs) I and II. For the current project, amide proton hydrogen-deuterium exchange coupled with MALDI-TOF mass spectrometry was used to characterize ligand binding to individual exosites and to investigate the presence of exosite-active site and exosite-exosite interactions. PAR3(44–56) and PAR1(49–62) were observed to bind to thrombin ABE I and then to exhibit long range effects over to ABE II. By contrast, Hirudin(54–65) focused more on ABE I and did not transmit influences over to ABE II. Although these three ligands were each directed to ABE I, they did not promote the same conformational consequences. d-Phe-Pro-Arg-chloromethyl ketone inhibition at the thrombin active site led to further local and long range consequences to thrombin-ABE I ligand complexes with the autolysis loop often most affected. When Hirudin(54–65) was bound to ABE I, it was still possible to bind GpIbα(269–286) or fibrinogen γ′(410–427) to ABE II. Each ligand exerted its predominant influences on thrombin and also allowed interexosite communication. The results obtained support the proposal that thrombin is a highly dynamic protein. The transmission of ligand-specific local and long range conformational events is proposed to help regulate this multifunctional enzyme.
Introduction
Thrombin is a serine protease whose functions are quite diverse. This multifaceted enzyme is involved in procoagulation, anticoagulation, and platelet activation. To maintain hemostasis and prevent thrombosis, the actions of thrombin must be carefully regulated (1–3).
To achieve this balance, thrombin contains insertion loops and two anion-binding exosites (ABE)3 that cause it to be a more selective enzyme than other members of the serine protease family (1, 2). The 60-insertion (or β-insertion) loop functions like a lid regulating entrance into the active site, and the autolysis (or γ) loop participates in controlling substrate specificity. Removal of the autolysis loop leads to a thrombin mutant with greatly hindered ability to convert fibrinogen to fibrin (4). In addition to the β- and γ-loops, thrombin exosites ABE I and ABE II also play key roles in regulating substrate specificity (Fig. 1). Exosite binding has been shown to direct substrates to the active site, contribute to opening of the active site region, and help in attracting other regulatory molecules to thrombin. Targeting ligands to these ABEs promotes local and/or long range conformational changes to thrombin. Together, these different actions allow thrombin to fulfill its array of protease functions (3).
FIGURE 1.
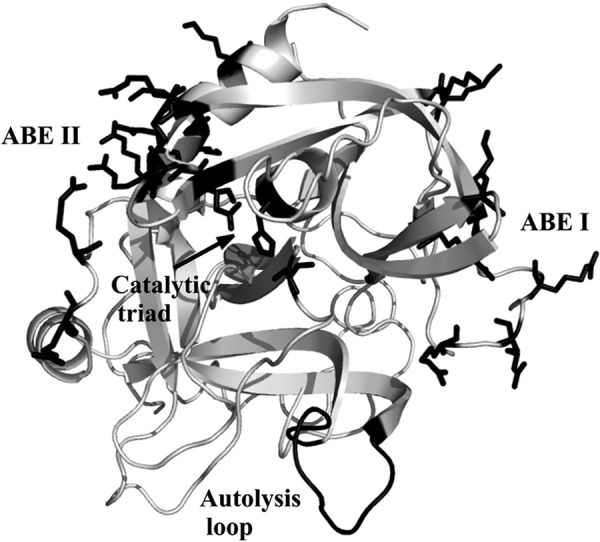
Highlighting key sites on the serine protease thrombin. The catalytic triad, the anion-binding exosites, and the autolysis loop are marked in black. The catalytic triad is represented by Ser195, His57, and Asp102 and shown as sticks. The anion-binding exosites of thrombin (ABE I and ABE II) are located at opposite sides of the active site and are shown as sticks. The Protein Data Bank code was 1PPB. The figure was prepared with PyMOL.
Both thrombin ABEs are highly positively charged and are located on opposite sides of the serine protease active site (Fig. 1) (5). ABE I contains some additional hydrophobic character relative to ABE II. ABE I is utilized by fibrinogen, thrombomodulin (TM), protease-activated receptors PAR1 and PAR3, and the leech-derived thrombin inhibitor Hirudin. The opposite exosite ABE II accommodates heparin, sulfated benzofurans, fibrinogen γ′, Factor VIII, and the platelet receptor GpIbα (1–3, 6).
The ability of exosite interactions to influence events at the thrombin active site is well established (7–9). The presence and consequence of long range communication between exosites, however, is not fully defined. Studies in this research area have often focused on fluorescence and/or surface plasmon resonance effects. Some reports indicate that thrombin can readily accommodate ligands at both exosites (7, 10, 11). With these examples, the conversion from a binary (enzyme-ligand) to a ternary (ligand 1-enzyme-ligand 2) complex does not lead to major changes in the KD value of either ligand. By contrast, other investigations suggest that binding at one exosite can greatly hinder binding events at the opposite exosite (12, 13). As a result, ternary complexes are challenging to maintain. The discrepancies in these different results may be due to the individual ligands tested and/or the environments in which the complexes are examined.
An alternative approach utilizes amide proton hydrogen-deuterium exchange (HDX) coupled with mass spectrometry to characterize ligand binding to thrombin exosites in solution (14–19). The HDX method focuses on documenting the exchange of solvent-accessible backbone amide protons with heavier deuterons. Unlike the fluorescence and surface plasmon resonances studies, no bulky spectroscopic probes are introduced, and no player becomes immobilized.
With the HDX technique, it is possible to assess which regions of the protein become more or less exposed to solvent in the presence versus the absence of ligand. Our previous HDX data demonstrated that binding of either GpIbα(269–286) or fibrinogen γ′(410–427) to ABE II led to distinctive interexosite communication over to ABE I (15, 16). By contrast, other HDX studies showed that binding of Hirudin (54–65) to ABE I did not cause the same kind of extensive long range effect to ABE II (16). Interestingly, recent NMR studies (20, 21) have further documented that thrombin is a highly flexible molecule in solution whose conformations can be modulated upon introduction of ligands.
In light of these HDX-MS and NMR results, it is very intriguing to further characterize interexosite communication in thrombin and the contributions made by individual ligands. To achieve this goal, we employed HDX coupled with MALDI-TOF mass spectrometry to explore three important ideas. First, we examined the ability of peptides derived from PAR1 and PAR3 to target ABE I and then influence the solvent accessibility of thrombin both locally and long range. Both PAR1(49–62) and PAR3(44–56) promoted interexosite communication between ABE I and II. Next, we examined the consequences of active site occupation on the ABE I-dependent communication events. Results revealed that the conformational dynamics of thrombin are further affected when ABE I ligands are added to d-Phe-Pro-Arg-chloromethyl ketone (PPACK)-thrombin. Finally, we tested whether thrombin could accommodate ligands at both exosites and evaluated the influences on thrombin solvent accessibility. The HDX results supported the proposal of allowing ligand binding at both thrombin exosites. Moreover, each ligand is able to maintain its predominant influences on thrombin.
With these findings, we provide further evidence that thrombin is a highly dynamic protein. Upon ABE binding, information is transmitted across the protease, thus assisting in regulation of this multifaceted enzyme. A better understanding of these effects may aid in design of new therapeutics to control this vital coagulation enzyme.
EXPERIMENTAL PROCEDURES
Materials
The peptides Hirudin(54–65) 54GDFEEIPEEsYLQ65, GpIbα(269-286) DEGDTDLpYPDpYPpYPPEEDTEG, and PAR3(44–56) 44QNTFEEFPLSDIE56 were synthesized by Bachem Bioscience Inc. PAR1(49–62) 49NDKYEPFWEDEEKN62 was prepared by New England Peptide. Fibrinogen γ′(410–427) PEHPAETEpYPDSLpYPPEDDL was synthesized by SynPep (Dublin, CA). PPACK was purchased from Calbiochem. Peptide m/z values were verified by MALDI-TOF MS on an Applied Biosystems Voyager DE-Pro MS. The concentrations of peptides in solution were determined by quantitative amino acid analysis (AAA Service Laboratory, Boring, OR).
When generating thrombin-ligand complexes, the following KD values were used. For Hirudin(54–65), we employed a KD of 225 nm for bovine thrombin (22, 23). For PAR3(44–56), we estimated a KD of 1.8 μm based on PAR3(31–60) and for PAR1(49–62) and a KD of 500 nm based on PAR1(33–62) (24). Other values that were utilized include the following: GpIbα 269–286 (KD of 5.9 nm) (25) and Fbg γ′ 410–427 (KD of 680 nm) (26).
Thrombin Preparation
Thrombin was purified from bovine barium sulfate eluate (Sigma) on a Waters 600 HPLC system as described by Trumbo and Maurer (27) The purified thrombin was concentrated with Amicon Ultra-15 centrifugal filter units (Millipore) on an Allegra 21R centrifuge (Beckman Coulter). Aliquots of thrombin were frozen at −70 °C for future use. The binding patterns of Hirudin(54–65), PAR3(44–56), PAR1(49–62), GpIbα(269–286), and Fbg γ′(410–427) to bovine thrombin are anticipated to be comparable with those with human thrombin. Bovine thrombin exhibits an 87% sequence similarity with human thrombin (5). Furthermore, key regions of thrombin such as the active site, Na+-binding site, ABE I, and ABE II remain conserved among these species. Biophysical studies that probe the conformational features of thrombin-ligand interactions have yielded similar results for both bovine and human thrombins. Ligands that have been examined include PPACK (28), the thrombomodulin EGF domains (14, 17, 19), fibrinopeptide A (29), PAR1(30), and fibrinogen γ′ (15). There are some sequence differences between bovine and human thrombin within the autolysis loop region. These differences result in some changes in proteolytic cleavage sites. However, peptic digests involving the autolysis region do yield overlapping segments (bovine residues 135–149D versus human residues 131–144 and 145–155; chymotrypsin numbering). As a result, information can still be collected on the full loop region. Prior hydrogen deuterium exchange studies demonstrated that the same conformational conclusions could be made with both thrombin forms (17, 28). Moreover, the autolysis loops reported on similar changes in solvent accessibility. The bovine-derived autolysis segment has the added benefit of being easier to detect and quantitate by MALDI-TOF mass spectrometry than the human segments.
HDX Sample Preparation
Thrombin was buffer exchanged into 75 mm NaCl, 12.5 mm NaH2PO4, pH 6.5. The enzyme solution was then concentrated to 50 μm, a value verified by UV measurements on a Cary 100 spectrophotometer. The more physiological buffer concentrations of 150 mm NaCl and 25 mm NaH2PO4 would later be achieved by drying down thrombin aliquots and resuspending them in a smaller volume of D2O. The resolubilized thrombin maintained 80% of its original activity toward the chromogenic substrate Pefachrome TH (H-d-CHG-Ala-Arg-p-nitroanilide). Moreover, this thrombin sample could still readily clot fibrinogen as measured by a turbidity assay (see supplemental Fig. S1). A key cleavage site used to generate ligand-impaired γ-thrombin occurs at the Arg75-Tyr76 site (31). With the resolubilized thrombin samples, the ABE I segment containing residues 65–84 was still intact and could be isolated from thrombin following a peptic digest. The thrombin used in the current HDX studies must therefore still have the ABE I residues 65–84 available for ligand binding.
Solution aliquots of buffer-exchanged thrombin (50 μm) were incubated with ligands of interest for 10 min at room temperature. Hirudin(54–65), PAR1(49–62), and GpIbα(269–286) peptides were added to thrombin at 20:1 ratios, whereas PAR3(44–56) and Fbg γ′(410–427) were added at a 40:1 ratio.
The enzyme to ligand ratios that were chosen would help assure full occupancy of a thrombin exosite during the course of the HDX experiment. Under these conditions, a maximal amount of solvent protection at the targeted exosite could be observed after 1 min of deuteration. With the PAR peptides, 99.9% occupancy should be achieved with a 40:1 PAR3(44–56) to thrombin ratio and a 20:1 PAR1(49–62) to thrombin ratio (14). A considerable deterioration in mass spectral quality occurred when a 40:1 PAR1(49–62) to thrombin ratio was used. The ratios used in this project should allow both PAR peptides to exhibit similar contact times with the thrombin surface as part of an enzyme-ligand complex.
With high ratios of ligand to enzyme, there may be concerns that nonspecific effects are being promoted. Titration control studies were carried out with the PAR peptides. As shown in supplemental Fig. S2, the overall conformational events were still preserved when the PAR1(49–62) to thrombin ratios were lowered from 20:1 to 10:1. Nonspecific binding effects that are only present at high ligand concentrations have not been observed. With higher affinity ligands such as our previous work with GpIbα (1–290), a lower ligand to thrombin ratio of 2:1 can be monitored. The GpIbα domain could still maintain the same local and long range HDX effects as observed with the GpIbα(269–286) peptide model (16).
Control studies to assess whether an ABE I-directed ligand could also bind to another site are also valuable. A strategy to alleviate this concern would be to work with a thrombin species where the ABE I site is no longer available for ligand binding. This condition may be achieved by using γ-thrombin, a proteolytically derived variant of this serine protease that contains a disrupted ABE I region (31). With this variant, the ABE II region would be intact. Control HDX experiments were thus performed with human γ-thrombin (Hematologic Technologies, Inc.) and a series of the peptide ligands. The ligand to thrombin ratios were the same as those used for α-thrombin. The fragments resulting from peptic digest of human γ-thrombin were identified by MS/MS analysis on an Applied Biosystems 4700 mass spectrometer.
An alternative strategy to monitor for nonspecific binding to ABE II would be to employ a very high affinity ligand that targets ABE I. A candidate has not yet been found that fully blocks ABE I, does not risk interfering with ABE II regions probed by HDX, and does not promote long range effects. For HDX studies, γ-thrombin was found to serve as the best control for blocking access to ABE I.
After the single ligand complexes were examined, a series of double ligand complexes were probed in the presence of α-thrombin. Hirudin(54–65) was added first to the thrombin solution, and the mixture was incubated for 10 min at room temperature. The second ligand was then introduced, and the resultant ternary complex was incubated for an additional 10 min. For studies on active site-inhibited thrombin, PPACK was added to thrombin at a ratio of 4:1, and the mixture was incubated at 37 °C for 30 min. Inhibition of thrombin activity was verified by kinetic assay using the chromogenic substrate Pefachrome TH. The PPACK-inhibited thrombin was then subjected to buffer exchange and concentration. Later, exosite-directed ligands could be introduced.
After the different thrombin-ligand incubation periods, all complexes were dried as aliquots using a CentriVap concentrator and CentriVap cold trap (Labconco). These dried samples were stored at −70 °C for future use.
HDX Experiments
Dry aliquots were defrosted and allowed to come to room temperature. 12 μl of 99.996% D2O (Cambridge Isotope Laboratories) were added, yielding final concentrations of 100 μm thrombin, 150 mm NaCl, and 25 mm NaH2PO4, pH 6.5. The samples were incubated in a desiccator at room temperature. After 1 min of incubation, 6 μl of sample were added to 57 μl of 0.1% TFA, pH 2.5 (on ice), to quench the exchange. The 63 μl of the quenched solution were transferred to a tube of activated pepsin bound to agarose. The remaining 6 μl of sample were quenched after 10 min of HDX and added to another tube of pepsin. Digestion occurred on ice for 10 min. The enzyme digest was separated from the pepsin-immobilized agarose by performing a centrifugation step in a mini-centrifuge kept on ice. Aliquots of 8.2 μl were immediately frozen in liquid nitrogen.
HDX Analysis
Each frozen aliquot of thrombin that had been subjected to HDX was thawed and immediately mixed with 8 μl of matrix (10 mg/ml α-cyano-hydroxycinnamic acid (Aldrich) in 1:1:1 ethanol:acetonitrile:0.1% TFA), pH 2.2. 0.5 μl of the resultant mixture were spotted onto a chilled stainless steel MALDI plate, dried with a SpeedVac unit (Savant), and inserted into the MALDI-TOF MS. This procedure was completed within a 5-min time frame, limiting the amount of hydrogen back exchange.
All of the spectra were collected in reflector mode with 256 shots/spectrum and a 800–3500 m/z range. At least three spectra were collected for each trial, and each trial was repeated at least three times. Time points for 1 and 10 min of deuteration were examined.
Data Explorer software (Applied Biosystems) was used to analyze the obtained spectra. All of the bovine thrombin peptides in the peptic digest were previously identified by Croy et al. (28), and the identities were verified in our laboratory by using post-source decay sequencing (15). All of the spectra were calibrated using two reference peptides, including the singly protonated monoisotopic mass 888.4943 Da (46–52 residues) and singly protonated pentaisotopic mass 2106.1505 Da (85–99 residues). The amount of deuterium uptake by each peptide was quantified as described by Sabo et al. (15, 16).
The number of deuterons (D) and the accompanying standard deviations were calculated for free thrombin, PPACK-inhibited thrombin, and the single versus double-ligand bound thrombin. Next, two-tailed t tests were performed on the D ± S.D. series, and the p values were calculated. As part of this work, pooled standard deviations (SAB) were determined, and tcalc values were tabulated. Four degrees of freedom were employed. For the single ligand studies, comparisons were made relative to free thrombin. Select comparisons were also made relative to PPACK-thrombin or to one of the ligand-thrombin complexes. The data were then ranked as p ≤ 0.05 (*, 95% confidence; 2.78 ≤ tcal ≤ 4.58), p ≤ 0.01 (**, 99% confidence; 4.60 ≤ tcalc ≤ 8.57), p ≤ 0.001 (***, 99.9% confidence; tcalc ≥ 8.61).
RESULTS
Monitoring Shifts in HDX Centroids in Presence of Ligand
HDX coupled with mass spectrometry provides an opportunity to further examine the conformational dynamics associated with the binding of ligands to thrombin. In this HDX technique, the exchange primarily of solvent-accessible backbone amide protons with heavier deuterons is detected as an increase in centroid mass (in terms of m/z) for a protein fragment (32). Upon complex formation, some regions of the intact protein can become protected from deuteration as a result of direct interaction with the ligand. The affected region becomes less accessible to the heavier deuterium and is recorded by mass spectrometry as a decrease in centroid mass compared with the same region of a deuterated ligand-free protein. In addition to detecting binding interfaces, HDX protection can be due to ligand-induced conformational changes that are propagated to distant sites (14, 18, 32).
The quenching conditions for the HDX experiments (pH 2.5 and low temperature) are well suited for pepsin-dependent cleavage of thrombin. The resultant sequence coverage allows several key thrombin regions to be probed by the HDX method. For the current work, we quantified deuteration profiles for ABE I fragment 65–84; three ABE II fragments 85–99, 173–180, and 173–181; autolysis loop fragment 135–149D; and the fragments −13 to −4, 46–52, 106–113, 106–116, and 202–207. Chymotrypsin numbering is employed throughout. Residues −13 to −4 are part of the thrombin A-chain, and residues 202–207 are positioned in a hinge region near the A-chain. Residues 46–52 are located near ABE I 65–84. Finally, residues 106–116 are part of a β-strand located antiparallel to a strand near the N terminus of ABE II residues 85–99. The various pepsin derived fragments that are mentioned above are color-coded in Fig. 2.
FIGURE 2.

Thrombin-ligand complexes. Ligands bound to thrombin such as Hirudin(54–65), PAR3(44–56), PAR1(49–62), PPACK, fibrinogen γ′(410–427), and GpIbα(269–286) are depicted as black sticks. Thrombin fragments obtained by peptic digest that represent ABE I, ABE II, autolysis loop, 106–116 and 202–207 regions are colored in red, dark blue, green, cyan, and orange, respectively. The catalytic triad is shown as sticks in yellow (for the IIa-PPACK complex). Protein Data Bank codes include 1HAH for Hirudin(54–65), 2PUX for PAR3(44–56), 1PPB for PPACK, 2HWL for fibrinogen γ′(410–427), and 1P8V for GpIbα(269–286). The figure was prepared with PyMOL.
Following HDX Centroids for Different ABE-directed Peptides
In agreement with previous thrombin-ligand crystal structures (Fig. 2), binding of the individual ligands Hirudin(54–65), PAR3(44–56), and PAR1(49–62) to thrombin each caused the centroid for the ABE I 65–84 segment to shift to lower m/z at 1 min of deuteration because of direct solvent protection at this thrombin exosite (Fig. 3). At the same time, the ABE II(85–99) was not solvent protected by Hirudin(54–65). By contrast, PAR3(44–56) and PAR1(49–62) caused definite centroid shifts for both ABE I(65–84) and the more distant ABE II(85–99) at 1 min of deuteration (Fig. 3). The current HDX results thus suggest that the PAR3 and PAR1 segments induce both local and longer range conformational effects to thrombin.
FIGURE 3.
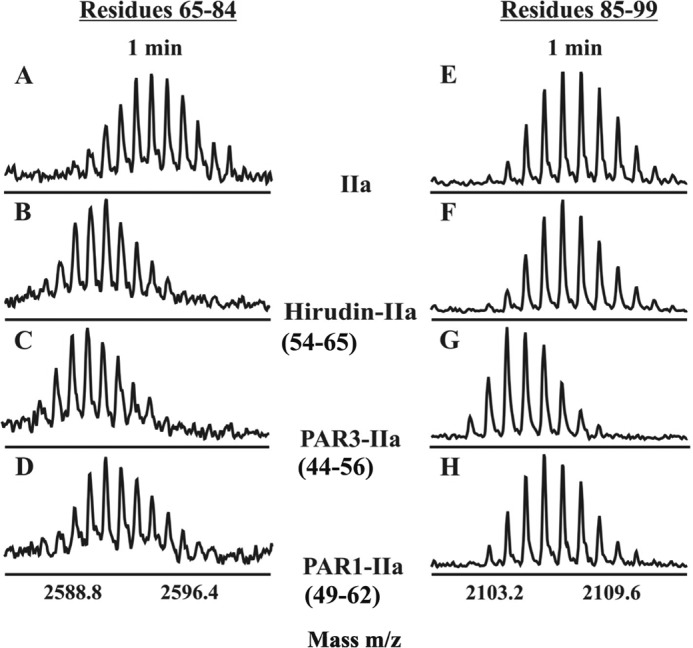
HDX centroids shifts because of exosite ligand binding. Centroids for the ABE I segment 65–84 (A–D) and the ABE II segment 85–99 (E–H) following 1 min of deuteration in the following environments: free thrombin (A and E), Hirudin(54–65)-thrombin (B and F), PAR3(44–56)-thrombin (C and G), and PAR1(49–62)-thrombin (D and H).
A control experiment was then carried out using ABE I impaired γ-thrombin and a set of peptide ligands. The disrupted ABE I region could not be monitored, but any solvent protection at the key ABE II regions could be documented. Fig. 4 shows 1-min HDX centroids for γ-thrombin complexes involving Hirudin(54–65), PAR3(44–56), or GpIbα(269–286). GpIbα(269–286), an ABE II ligand, was the only peptide that could target residues 85–99 and protect this thrombin segment from deutertation. Such studies helped verify that PAR3 was not switching its dominant solvent protection effect from ABE I to ABE II when the impaired γ-thrombin was employed. For HDX studies, γ-thrombin was found to serve as the best control for blocking access to ABE I.
FIGURE 4.
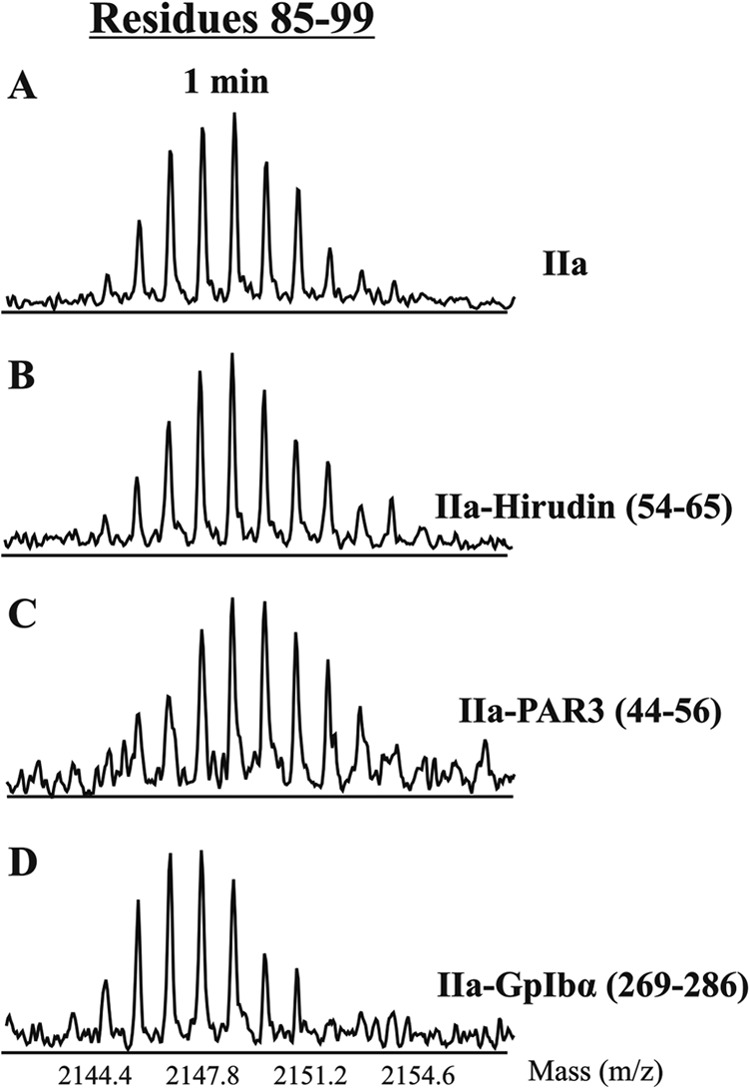
HDX centroid shifts examining ability of γ-thrombin to accommodate exosite ligands. Shown are centroids for the ABE II segment 85–99 following 1 min of deuteration in the following environments: free γ-thrombin (A), γ-thrombin-Hirudin(54–65) (B), γ-thrombin-PAR3(44–56) (C), and γ-thrombin-GpIbα(269–286) (D).
Based upon the γ-thrombin control studies described above, the influences of PAR3(44–56) on the native α-thrombin are proposed to be due to PAR3(44–56) targeting ABE I and then exhibiting some long range effects over to ABE II and other regions. The same results are anticipated for PAR1(49–62). Although comparing HDX centroid shifts is highly valuable, determination of the actual deuterium uptake yields more quantitative results.
Deuteration Values for ABE I Ligands
In the presence of Hirudin(54–65), the number of deuterons incorporated into the thrombin ABE I 65–84 region was indeed less than in free thrombin. Deuteration (D) values for residues 65–84 at 1 min included 6.81 ± 0.29 D for free thrombin versus 3.36 ± 0.17 D for Hirudin(54–65)-thrombin and then 7.80 ± 0.50 D versus 4.43 ± 0.13 D, respectively, at 10 min (p values of ≤ 0.001) (Fig. 5 and supplemental Table S1). The other regions of thrombin displayed modest, if any, differences in D across the 1–10-min time course.
FIGURE 5.

Deuterium incorporation for the Hirudin(54–65)-thrombin complex in the presence and absence of PPACK. The bars in the graphs correspond to deuterium incorporation of free thrombin (white), Hirudin(54–65)-bound thrombin (white striped), PPACK-inhibited thrombin (gray), and PPACK-inhibited thrombin in the presence of Hirudin(54–65) (gray striped). Values for both 1 and 10 min of deuteration are displayed. The error bars correspond to standard deviations for three independent trials.
PAR3(44–56) exhibited a quite different binding pattern from Hirudin(54–65). As expected, there was protection observed for ABE I 65–84 (7.63 ± 0.41 D for free thrombin versus 2.74 ± 0.25 D for thrombin-PAR3 at 1 min and then 7.97 ± 0.45 D versus 3.98 ± 0.26 D at 10 min). Interestingly, the ABE II segments 85–99, 173–180, and 173–181 were also significantly protected by the PAR3 segment. Long range solvent protection was maintained after 10 min of deuteration for 85–99, 173–180, and 173–181 (Fig. 6 and supplemental Table S2). The p values for these four thrombin segments ranged from p < 0.01 to 0.001. Other regions of thrombin that underwent substantial solvent protection in the presence of PAR3(44–56) included thrombin segments −13 to −4, 106–113, 106–116, and 202–207. The autolysis loop segment 135–149D experienced a modest decrease in D across the 1–10-min deuteration period, p < 0.05 (Fig. 6 and supplemental Table S2).
FIGURE 6.

Deuterium incorporation for the PAR3(44–56)-thrombin complex in the presence and absence of PPACK. The bars in the graphs correspond to deuterium incorporation of free thrombin (white), PAR3(44–56)-bound (striped white), PPACK-inhibited thrombin (gray), and the ternary IIa-PPACK-PAR3(44–56) complex (gray striped). Values for both 1 and 10 min of deuteration are displayed. The error bars correspond to standard deviations for three independent trials.
The binding patterns of PAR1(49–62) were similar to PAR3(44–56). Introduction of PAR1(49–62) led to a significant decrease in deuteration for the ABE I fragment 65–84 (6.25 ± 0.27 D versus 4.37 ± 0.19 D at 1 min, p < 0.001; Fig. 7 and supplemental Table S3). With the PAR1 complex, solvent protection was also observed with thrombin ABE II residues 85–99, 173–180, and 173–181. More modest decreases in deuteration were observed for the 106–113 fragment of thrombin.
FIGURE 7.

Deuterium incorporation for the PAR1(49–62)-thrombin complex in the presence and absence of PPACK. The bars in the graphs correspond to deuterium incorporation of free thrombin (white), PAR1(49–62)-bound thrombin (white striped), PPACK-inhibited thrombin (gray), and the ternary IIa-PPACK-PAR1(49–62) complex (gray striped). Values for both 1 and 10 min of deuteration are displayed. The error bars correspond to standard deviations for three independent trials.
Evaluating the Influence of Active Site Binding
To investigate whether occupation of the active site would influence the binding pattern of the ABE I ligands, PPACK, an irreversible active site inhibitor of thrombin, was introduced prior to the addition of the ligands. By 10 min of deuteration, the presence of PPACK had consistently protected the ABE II fragment 173–181 from solvent exchange (p values < 0.05 to 0.01; see Figs. 5–7 and supplemental Tables S1–S3). The N-terminal portion of PPACK is directed toward this area that borders the thrombin active site region. In addition, modest protection could at times be documented for the thrombin fragments 85–99 and 135–149D. Previous HDX studies also reported effects on these three thrombin fragments (15, 28).
The solvent protection observed for thrombin fragments 173–180 and 173–181 in the presence of PPACK was further maintained upon the addition of Hirudin(54–65). Interestingly, the autolysis loop fragment 135–149D appeared to exhibit a more than additive protection in the ternary thrombin-PPACK-Hirudin(54–65) complex following 10 min of deuteration, p < 0.01 (Fig. 5 and supplemental Table S1). A comparison of the IIa-PPACK versus IIa-PPACK-Hirudin(54–65) data helped to confirm that Hirudin(54–65) introduces protective effects on 65–84 and 135–149D in the ternary complex. A comparison of IIa-Hirudin(54–65) versus IIa-PPACK-Hirudin(54–65) emphasized the contributions that PPACK makes toward protecting the solvent accessibility of thrombin 85–99, 135–149D, and 173–181.
For the PAR3(44–56) investigations (Fig. 6 and supplemental Table S2), PPACK inhibition appeared again to exhibit a more than additive protection on the 135–149D segment in the ternary complex following 10 min of deuteration, p < 0.001. Other thrombin segments that were protected within the thrombin-PPACK-PAR3(44–56) complex included 65–84, 85–99, 106–116, 173–181, and 202–207 (p values ranged from 0.05 to 0.001). A comparison of the deuteration values of IIa-PPACK versus IIa-PPACK-PAR3(44–56) further highlights the contributions that PAR3(44–56) makes on several local and long range regions on thrombin. PAR3(44–56) introduces its own effects even in the presence of the active site inhibitor PPACK. For the PAR1(49–62) studies, the protective effects in the presence of PPACK-thrombin focused on 65–84, 85–99, 135–149D, and 173–181 (Fig. 7 and supplemental Table S3).
Characterizing Thrombin Complexes Involving Both Anion-binding Exosites
To probe dual occupation of the exosites, HDX experiments were carried out where Hirudin(54–65) is directed to ABE I, and then GpIbα(269–286) or fibrinogen γ′(410–427) is directed to ABE II. First, interactions only involving the ABE II directed ligands were examined. As expected from prior crystal structures (Fig. 2), the ligands GpIbα(269–286) and γ′(410–427) targeted ABE II 85–99, 173–180, and 173–181 (p < 0.01 to 0.001). Similar to prior HDX studies, the other thrombin regions experiencing solvent protection included 106–113, 202–207, and −13 to −4 (p < 0.05 to 0.001) (Fig. 8 and supplemental Tables S4 and S5). Solvent protection was also observed at ABE I 65–84 after 1 min of deuteration (p < 0.01) but was diminished by 10 min.
FIGURE 8.

Deuterium incorporation when both exosites are occupied. The bars in the graphs correspond to deuterium incorporation of free thrombin (white), binary thrombin-Hirudin(54–65) complex (gray), binary thrombin-GpIbα(269–286) complex (striped white), binary thrombin-γ′(410–427) complex (dotted white), ternary thrombin-Hirudin(54–65)-GpIbα(269–286) complex (striped gray), and ternary thrombin-Hirudin(54–65)-γ′(410–427) complex (dotted gray). Values for both 1 and 10 min of deuteration are displayed. The error bars correspond to standard deviations for three independent trials.
HDX control studies with γ-thrombin (Fig. 4) verified that ligand binding to ABE II is not dependent on the presence of an intact ABE I. Prior NMR studies on the binary fibrinogen γ′(410–427) to γ-thrombin complex could only demonstrate that thrombin contact sites were maintained in the presence of a species with a disrupted ABE I (15). The current HDX studies on γ-thrombin revealed that the GpIbα and fibrinogen γ′ ligands were still protecting ABE II from hydrogen-deuterium exchange (see Fig. 4 for centroid shifts involving GpIbα(269–286) in the presence of γ-thrombin). By contrast, when both exosites are intact as in α-thrombin, long range influences from ABE II to ABE I are possible.
For the ternary complexes (GpIbα-IIa-Hirudin(54–65) and Fbg γ′-IIa-Hirudin(54–65)), solvent protection at ABE I and ABE II was strongly maintained for the full 10 min of deuteration. Such results are consistent with each exosite being occupied by its preferred ligand. With these thrombin systems, protection was also observed for the thrombin fragments 106–113, 106–116, 173–180, 173–181, and 202–207 at 1 min and/or 10 min of deuteration (p values range from <0.05 to <0.001). For the current ternary complexes, the ABE II ligands appeared to exert a greater influence on the conformational dynamics of thrombin than the ABE I ligand Hirudin(54–65). As a result, the ABE II-directed ligands dominated the HDX effects observed.
DISCUSSION
Probing Conformational Events after ABE I Binding
X-ray crystallography and site-directed mutagenesis studies have indicated that Hirudin, PAR3, and PAR1 all target similar regions of thrombin ABE I (33–42). Our HDX studies effectively complement this analysis by showing solvent protection for the thrombin segment 65–84 in the presence of Hirudin(54–65), PAR3(44–56), and PAR1(49–62).
Distinct characteristics for the different ABE I ligands become apparent when evaluating the HDX results for the presence of local and long range interactions. Hirudin(54–65) is a tight binding ligand, and HDX studies confirm that it targets ABE I (Fig. 9). Some modest HDX protection at the autolysis loop and/or near the active site could, at times, be observed in the presence of Hirudin(54–65) (16). Earlier fluorescence studies also documented Hirudin(54–65) influences to the thrombin active site and to the autolysis loop (43). Mutagenesis studies revealed thrombin residues involved in establishing such networks (38).
FIGURE 9.

The HDX binding patterns of various thrombin ligands. Thrombin segments that were protected from solvent exchange following 10 min of deuteration are colored in red. The observed HDX effects were statistically different from the values obtained with free thrombin at a level of p ≤ 0.001. The individual ligands bound to thrombin including GpIbα(269–286) (Protein Data Bank code 1P8V), Hirudin(54–65) (Protein Data Bank code 1HAH), and PAR3(44–56) (Protein Data Bank code 2PUX) are depicted as black sticks. The figure was prepared with PyMOL.
An important finding from the current HDX studies was that Hirudin(54–65) and the PAR family could both target ABE I; however, the PARs could introduce more extensive long range effects across the thrombin surface. The PAR3(44–56) and PAR1(49–62) sequences were shown previously by x-ray crystallography to interact with thrombin ABE I (35, 36). In our HDX control studies, PAR3(44–56) has not been observed to bypass ABE I and move on to bind ABE II.
The long range ABE I to ABE II influences observed with the PAR peptides are complementary to the ABE II to ABE I effects observed with either Fbg γ′ or GpIbα peptides (15, 16). The PAR3(44–56) and PAR1(49–62) peptides induced solvent protection that extended from thrombin ABE I over to positions 85–99 (ABE II), 106–116 (segment between ABE I and ABE II), and 173–181 (ABE II near the active site). PAR3(44–56) promoted greater effects over to positions 135–149D (autolysis loop) and 202–207 (segment in the B chain linking over toward the A chain; Fig. 9).
For ABE I ligands, the position 106–116 HDX effects appear to be most dominant for PAR3(44–56). The 106–116 segment is part of a β-strand located antiparallel to a strand near the N terminus of ABE II residues 85–99. Intriguingly, the ABE II ligands Fbg γ′ and GpIbα also take advantage of 106–116 (15, 16). The β-strand may serve as a pathway to transmit information between the two thrombin exosites.
The reasons why PAR1 and PAR3 segments induce greater long range conformational effects on thrombin than Hirudin(54–65) are valuable to consider. Prior studies demonstrated that the presence of GpIbα at thrombin ABE II leads to enhanced cleavage of PAR 1 (44, 45). PAR1 hydrolysis within the thrombin active site region benefits from anchoring a distal portion of PAR1 to ABE I. To achieve appropriate regulation of this GpIbα-IIa-PAR1 system on platelets, it may be advantageous to have communication between the two exosites and also with the active site. Hirudin, by contrast, is an effective inhibitor, and the 54–65 segment may predominantly focus on targeting ABE I and influencing the active site. Less is known about the effects of PAR3 on thrombin conformation. Interestingly, PAR3 has been reported to serve as a cofactor in promoting interactions between murine thrombin and PAR4 (46). X-ray crystallography has documented that PAR3 peptide binding at thrombin ABE I influences the orientation of the 60s insertion loop and thus PAR 4 access to the catalytic site (35).
Introducing a Covalently Bound Inhibitor
Placing an inhibitor at the thrombin active site also has distinct consequences. Solution NMR studies by Lechtenberg et al. (20) suggest that PPACK helps to establish the functional core of thrombin and aids in reducing the solution dynamics of this serine protease. Bringing in Hirudin(54–65) at ABE I introduces further conformational changes thus preparing thrombin for utilizing its substrates and ligands (20). The first backbone dynamics study on PPACK-thrombin was recently reported by Fuglestad et al. (21) and involved a combination of NMR relaxation rate measurements and accelerated molecular dynamics simulations. Their research confirmed that the functional core of the serine protease is stabilized upon PPACK binding; however, the surface loops remain surprisingly dynamic. Moreover, these loops exhibit motions on multiple time scales. The thrombin autolysis loop is a clear participant in this phenomenon. Other regions include the 90s and 60s loops, the loops involved in the Na+ site, and the thrombin A-chain. Such a dynamic thrombin environment may be crucial for promoting communication between the active site and the anion-binding exosites (21).
HDX coupled with mass spectrometry provides an opportunity to further monitor the solvent accessibility changes that occur with a series of thrombin-ligand complexes in the presence of PPACK. The HDX studies carried out here and in earlier studies (15, 28) reveal that the presence of PPACK leads primarily to solvent protection in ABE II regions near the active site and also modest effects at the autolysis loop (15, 28). The current HDX studies indicate that the ABE I ligands PAR3(44–56) and PAR1(49–62) can then introduce their own further effects at local and more distant sites on thrombin. The highly flexible autolysis loop is an example of a region that responds to dual ligand occupation at the thrombin active site and at ABE I. Earlier HDX work indicated that ABE II ligands could also be accommodated by PPACK thrombin (15). The effects were often larger than observed with ABE I ligands. The more closely located ABE II-based regions may have a more direct and substantial influence on the thrombin active site.
Examining the Influences of Dual Occupancy
Our studies provide the first examples of probing ligand binding to both anion-binding exosites using HDX. In complementary fluorescence-based approaches, dual occupancies with Fbg γ′ peptide or GpIbα at ABE II and then Hirudin(54–65) or DNA aptamer at ABE I have been observed (10, 11). The HDX studies provide the advantage of demonstrating that Hirudin(54–65) focuses its effects on ABE I and then allowing Fbg γ′ or GpIbα peptides to target specific ABE II segments and influence other more distant regions (Fig. 9).
Interestingly, the Fbg γ′ peptide has been shown by De Cristofaro and co-workers (11) to inhibit thrombin cleavage of PAR1(38–60), leading to a decrease in kcat value. This PAR1 sequence extends from the active site to ABE I. Hydrolysis of full-length PAR1 on intact platelets is also hindered by increasing concentrations of Fbg γ′ peptide. By contrast, GpIbα binding can promote hydrolysis of PAR1 on platelets (44). Similar effects were not observed with a comparable PAR4 peptide substrate sequence (11). The Fbg γ′ peptide may be propagating an effect over to the active site region that has greater influence on the PAR1 LDPR sequence than the PAR4 PAPR sequence. PAR domains with affinities more closely approaching that of Hirudin would allow other dual occupancy complexes to be probed by HDX approaches.
Comparing and Contrasting Ligands That Target Anion-binding Exosites
To better understand the characteristics and functions of thrombin exosites, it is valuable to also consider other ligands that have been studied. Hirudin(54–65), a monoclonal antibody against human thrombin (47), TMEGF45 (thrombomodulin fourth and fifth EGF domains) (17), TMEGF56 (thrombomodulin fifth and sixth EGF domains) (17), PAR3(44–56), and PAR1(49–62) all interact at the same region of thrombin ABE I, but they do not all exhibit the same influences on the solvent accessibility properties of thrombin (Table 1). Hirudin(54–65) focuses on ABE I and then has only a very modest influence over to the autolysis loop and ABE II(173–181). TMEGF45 and TMEGF56 both bind to ABE I via the fifth EGF domain and affect the solvent accessibility of the autolysis loop (17, 37).
TABLE 1.
Summary of target sites and long range HDX effects on thrombin
| Ligand | Target site | Long range influences |
|---|---|---|
| TMEGF45a | ABE I | ABE II (90s loop, 85–99), autolysis loop |
| PAR3(44–56) | ABE I | ABE II (90s loop, 85–99), autolysis loop |
| ABEII (near the active site, 173–181) | ||
| 106 to 116, 202 to 207, −13 to −4 | ||
| PAR1(49–62) | ABE I | ABE II (90s loop, 85–99) |
| ABEII (near the active site, 173–181) | ||
| 106–116 | ||
| Hirudin(54–65) | ABE I | ABE II (near the active site, 173–181) |
| Autolysis loop | ||
| TMEGF56a | ABE I | Autolysis loop |
| Thrombin mAbb | ABE I | 108–116 |
| GpIbα(269–286)c | ABE II | 106 to 116, 202 to 207, −13 to −4 |
| ABE I(65–84) | ||
| Fbg γ′(410–427)d | ABE II | 106 to 116, 202 to 207, −13 to −4 |
| ABE I(65–84) |
TMEGF45 has the added benefit of also promoting long range influences over to the 90s loop (within ABE II residues 85–99) (14, 17, 18). Interestingly, TMEGF45 supports thrombin activation of protein C, and residues in the 90s region (Arg93, Arg97, and Arg101) are important in the activation process (48). This benefit occurs even though TMEGF45 and TMEGF56 bind with similar affinity to thrombin. These HDX data suggest that the presence of the EGF4 domain contributes to the conformational events involving the 90s loop (17). In support of these HDX observations, conventional and accelerated molecular dynamics studies have recently revealed an allosteric pathway involving key components of thrombin that is present only in the slow time scale of tens of microseconds (49). This pathway contains a network of correlated motions linking the fourth and fifth EGF domains and surface loops near the thrombin active site region.
The current HDX studies suggest that PAR3(44–56) and PAR1(49–62) exhibit some conformational properties similar to those of TMEGF45. Both PAR peptides promote long range influences over to the 90s loop (within ABE II residues 85–99), and PAR3(44–56) has a greater influence on the autolysis-loop. PAR3(44–56) and PAR1(49–62) also influence other thrombin segments including residues 106–116 and 173–181. The 106–116 segment is particularly affected. Further studies are needed to evaluate the extent to which these different segments may influence PAR binding, ABE II ligand binding, and/or functional roles in thrombin. The focus of the HDX approach is to identify regions that undergo conformational events reflected in terms of solvent accessibility changes.
A final class of ABE I targeted ligands that has been probed includes a monoclonal antibody against human thrombin (47). HDX studies reveal that the epitope for this antibody involves ABE I and the segment 108–116. Curiously, there are no other long range effects.
In contrast to the ABE I-directed ligands mentioned above, Fbg γ′ and GpIbα generated similar local and long range effects following binding to ABE II (15, 16). A review of different thrombin-ligand complexes reveals that the ABE II binding surface is somewhat broader than that of ABE I (3). Whether other ABE II ligands can achieve a different set of long range consequences is still to be seen (3, 10).
Thrombin Is Highly Dynamic
Solution-based evidence continues to accumulate supporting the proposal that free thrombin is highly dynamic and thus may adopt multiple conformations. Thrombin may have properties similar to that of ubiquitin, a protein recently shown to contain a rich ground state ensemble (50, 51). The ubiquitin ensemble has an array of conformers that interconvert with each other on the low microsecond time scale (51). Fuglestad et al. (21) already have evidence that thrombin contains amino acids that exhibit motions in this supra τc time scale. For the thrombin system, substrate or cofactors may seek out select conformations and further promote thrombin to adopt particular structural and dynamical features. Some of these free thrombin conformations may be trapped in the thrombin-bound x-ray crystal structures already being reported. Furthermore, key conformational states of thrombin have been identified in solution (52–55).
NMR studies have revealed that the functional core of thrombin is stabilized upon PPACK binding. Intriguingly, the surface loops remain dynamic with motions on multiple time scales (21). Additional ligand binding has the opportunity to further influence the conformational and functional properties of thrombin (20). Some of the different interpretations about the presence and consequences of interexosite communication may well be related to the ligands examined. Hydrogen-deuterium exchange, NMR ligand titrations, relaxation rate studies on amino acid backbone events, and molecular dynamics methods all contribute additional strategies for probing the conformational properties of thrombin (15–17, 20, 21, 49).
Conclusions
The HDX studies carried out in the current project indicate that ligands that target a particular thrombin exosite do not all promote identical conformational consequences. Similar and distinct patterns of local and long range effects were observed in the presence of the ABE I ligands Hirudin(54–65), PAR3(44–56), and PAR1(49–62). Occupation of the active site with the inhibitor PPACK may further influence the HDX effects observed. Moreover, it is possible to occupy ABE I with Hirudin(54–65) and still get effective binding of GpIbα and Fbg γ′ peptides to ABE II. As with the single ligands, individual regions of thrombin that are affected can be mapped out. The local and long range effects of these ternary complexes are maintained, thus permitting the utilization of interexosite communication. The knowledge gained may later be used to design new ligand-based therapeutics that exert an effect at a specific thrombin site or that could also influence a distant region of the enzyme.
Acknowledgments
We are thankful for the guidance and assistance of M. Merchant and D. Wilkie (University of Louisville Clinical Proteomics Center) in performing the MS/MS analysis on an ABI 4700 MALDI-TOF/TOF mass spectrometer. We appreciate helpful discussions and critical evaluations of this work from Donghan Lee, M. C. Yappert, R. T. Woofter, P. G. Doiphode, and M. A. Jadhav.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL68440. This work was also supported by Grant W81XWH from the Telemedicine and Advanced Technology Research Center of the United States Army.

This article contains supplemental Tables S1–S5 and Figs. S1 and S2.
- ABE
- anion-binding exosite
- IIa
- thrombin
- PAR
- protease-activated receptor
- TM
- thrombomodulin
- EGF45
- epidermal growth factor domains 4 and 5
- EGF56
- epidermal growth factor domains 5 and 6
- Fbg
- fibrinogen
- γ-thrombin
- thrombin with hydrolysis at ABE I and at the autolysis loop
- HDX
- hydrogen-deuterium exchange
- PPACK
- d-Phe Pro Arg chloromethyl ketone
- sY
- sulfotyrosine
- pY
- phosphotyrosine
- 60s
- thrombin insertion loop involving residues 60A-60I (extends beyond chymotrypsin numbering)
- 90s
- thrombin surface loop region 90–99
- 149D
- fourth residue within the 149 thrombin insertion loop.
REFERENCES
- 1. Di Cera E. (2008) Thrombin. Mol. Aspects Med. 29, 203–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lane D. A., Philippou H., Huntington J. A. (2005) Directing thrombin. Blood 106, 2605–2612 [DOI] [PubMed] [Google Scholar]
- 3. Bock P. E., Panizzi P., Verhamme I. M. (2007) Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 5, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dang Q. D., Sabetta M., Di Cera E. (1997) Selective loss of fibrinogen clotting in a loop-less thrombin. J. Biol. Chem. 272, 19649–19651 [DOI] [PubMed] [Google Scholar]
- 5. Bode W., Turk D., Karshikov A. (1992) The refined 1.9-A x-ray crystal structure of d-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin. Structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1, 426–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sidhu P. S., Liang A., Mehta A. Y., Abdel Aziz M. H., Zhou Q., Desai U. R. (2011) Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J. Med. Chem. 54, 5522–5531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verhamme I. M., Olson S. T., Tollefsen D. M., Bock P. E. (2002) Binding of exosite ligands to human thrombin. Re-evaluation of allosteric linkage between thrombin exosites I and II. J. Biol. Chem. 277, 6788–6798 [DOI] [PubMed] [Google Scholar]
- 8. Ng N. M., Quinsey N. S., Matthews A. Y., Kaiserman D., Wijeyewickrema L. C., Bird P. I., Thompson P. E., Pike R. N. (2009) The effects of exosite occupancy on the substrate specificity of thrombin. Arch. Biochem. Biophys. 489, 48–54 [DOI] [PubMed] [Google Scholar]
- 9. Ye J., Liu L. W., Esmon C. T., Johnson A. E. (1992) The fifth and sixth growth factor-like domains of thrombomodulin bind to the anion-binding exosite of thrombin and alter its specificity. J. Biol. Chem. 267, 11023–11028 [PubMed] [Google Scholar]
- 10. Li W., Johnson D. J., Adams T. E., Pozzi N., De Filippis V., Huntington J. A. (2010) Thrombin inhibition by serpins disrupts exosite II. J. Biol. Chem. 285, 38621–38629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lancellotti S., Rutella S., De Filippis V., Pozzi N., Rocca B., De Cristofaro R. (2008) Fibrinogen-elongated γ chain inhibits thrombin-induced platelet response, hindering the interaction with different receptors. J. Biol. Chem. 283, 30193–30204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fredenburgh J. C., Stafford A. R., Weitz J. I. (1997) Evidence for allosteric linkage between exosites 1 and 2 of thrombin. J. Biol. Chem. 272, 25493–25499 [DOI] [PubMed] [Google Scholar]
- 13. Petrera N. S., Stafford A. R., Leslie B. A., Kretz C. A., Fredenburgh J. C., Weitz J. I. (2009) Long range communication between exosites 1 and 2 modulates thrombin function. J. Biol. Chem. 284, 25620–25629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mandell J. G., Falick A. M., Komives E. A. (1998) Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. U.S.A. 95, 14705–14710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sabo T. M., Farrell D. H., Maurer M. C. (2006) Conformational analysis of γ′ peptide (410–427) interactions with thrombin anion binding exosite II. Biochemistry 45, 7434–7445 [DOI] [PubMed] [Google Scholar]
- 16. Sabo T. M., Maurer M. C. (2009) Biophysical investigation of GpIbα binding to thrombin anion binding exosite II. Biochemistry 48, 7110–7122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koeppe J. R., Seitova A., Mather T., Komives E. A. (2005) Thrombomodulin tightens the thrombin active site loops to promote protein C activation. Biochemistry 44, 14784–14791 [DOI] [PubMed] [Google Scholar]
- 18. Mandell J. G., Falick A. M., Komives E. A. (1998) Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal. Chem. 70, 3987–3995 [DOI] [PubMed] [Google Scholar]
- 19. Mandell J. G., Baerga-Ortiz A., Akashi S., Takio K., Komives E. A. (2001) Solvent accessibility of the thrombin-thrombomodulin interface. J. Mol. Biol. 306, 575–589 [DOI] [PubMed] [Google Scholar]
- 20. Lechtenberg B. C., Johnson D. J., Freund S. M., Huntington J. A. (2010) NMR resonance assignments of thrombin reveal the conformational and dynamic effects of ligation. Proc. Natl. Acad. Sci. U.S.A. 107, 14087–14092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fuglestad B., Gasper P. M., Tonelli M., McCammon J. A., Markwick P. R., Komives E. A. (2012) The dynamic structure of thrombin in solution. Biophys. J. 103, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kroh H. K., Tans G., Nicolaes G. A., Rosing J., Bock P. E. (2007) Expression of allosteric linkage between the sodium ion binding site and exosite I of thrombin during prothrombin activation. J. Biol. Chem. 282, 16095–16104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anderson P. J., Nesset A., Dharmawardana K. R., Bock P. E. (2000) Characterization of proexosite I on prothrombin. J. Biol. Chem. 275, 16428–16434 [DOI] [PubMed] [Google Scholar]
- 24. Krem M. M., Di Cera E. (2003) Dissecting substrate recognition by thrombin using the inactive mutant S195A. Biophys. Chem. 100, 315–323 [DOI] [PubMed] [Google Scholar]
- 25. De Marco L., Mazzucato M., Masotti A., Ruggeri Z. M. (1994) Localization and characterization of an α-thrombin-binding site on platelet glycoprotein Ib α. J. Biol. Chem. 269, 6478–6484 [PubMed] [Google Scholar]
- 26. Lovely R. S., Moaddel M., Farrell D. H. (2003) Fibrinogen γ′ chain binds thrombin exosite II. J. Thromb. Haemost 1, 124–131 [DOI] [PubMed] [Google Scholar]
- 27. Trumbo T. A., Maurer M. C. (2000) Examining thrombin hydrolysis of the factor XIII activation peptide segment leads to a proposal for explaining the cardioprotective effects observed with the factor XIII V34L mutation. J. Biol. Chem. 275, 20627–20631 [DOI] [PubMed] [Google Scholar]
- 28. Croy C. H., Koeppe J. R., Bergqvist S., Komives E. A. (2004) Allosteric changes in solvent accessibility observed in thrombin upon active site occupation. Biochemistry 43, 5246–5255 [DOI] [PubMed] [Google Scholar]
- 29. Ni F., Meinwald Y. C., Vásquez M., Scheraga H. A. (1989) High-resolution NMR studies of fibrinogen-like peptides in solution. Structure of a thrombin-bound peptide corresponding to residues 7–16 of the Aα chain of human fibrinogen. Biochemistry 28, 3094–3105 [DOI] [PubMed] [Google Scholar]
- 30. Ni F., Ripoll D. R., Martin P. D., Edwards B. F. (1992) Solution structure of a platelet receptor peptide bound to bovine α-thrombin. Biochemistry 31, 11551–11557 [DOI] [PubMed] [Google Scholar]
- 31. Hofsteenge J., Braun P. J., Stone S. R. (1988) Enzymatic properties of proteolytic derivatives of human α-thrombin. Biochemistry 27, 2144–2151 [DOI] [PubMed] [Google Scholar]
- 32. Hoofnagle A. N., Resing K. A., Ahn N. G. (2003) Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32, 1–25 [DOI] [PubMed] [Google Scholar]
- 33. Xu H., Bush L. A., Pineda A. O., Caccia S., Di Cera E. (2005) Thrombomodulin changes the molecular surface of interaction and the rate of complex formation between thrombin and protein C. J. Biol. Chem. 280, 7956–7961 [DOI] [PubMed] [Google Scholar]
- 34. Rydel T. J., Ravichandran K. G., Tulinsky A., Bode W., Huber R., Roitsch C., Fenton J. W., 2nd. (1990) The structure of a complex of recombinant hirudin and human α-thrombin. Science 249, 277–280 [DOI] [PubMed] [Google Scholar]
- 35. Bah A., Chen Z., Bush-Pelc L. A., Mathews F. S., Di Cera E. (2007) Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc. Natl. Acad. Sci. U.S.A. 104, 11603–11608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gandhi P. S., Chen Z., Di Cera E. (2010) Crystal structure of thrombin bound to the uncleaved extracellular fragment of PAR1. J. Biol. Chem. 285, 15393–15398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fuentes-Prior P., Iwanaga Y., Huber R., Pagila R., Rumennik G., Seto M., Morser J., Light D. R., Bode W. (2000) Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature 404, 518–525 [DOI] [PubMed] [Google Scholar]
- 38. Mengwasser K. E., Bush L. A., Shih P., Cantwell A. M., Di Cera E. (2005) Hirudin binding reveals key determinants of thrombin allostery. J. Biol. Chem. 280, 26997–27003 [DOI] [PubMed] [Google Scholar]
- 39. Ayala Y. M., Cantwell A. M., Rose T., Bush L. A., Arosio D., Di Cera E. (2001) Molecular mapping of thrombin-receptor interactions. Proteins 45, 107–116 [DOI] [PubMed] [Google Scholar]
- 40. Pineda A. O., Cantwell A. M., Bush L. A., Rose T., Di Cera E. (2002) The thrombin epitope recognizing thrombomodulin is a highly cooperative hot spot in exosite I. J. Biol. Chem. 277, 32015–32019 [DOI] [PubMed] [Google Scholar]
- 41. Hall S. W., Nagashima M., Zhao L., Morser J., Leung L. L. (1999) Thrombin interacts with thrombomodulin, protein C, and thrombin-activatable fibrinolysis inhibitor via specific and distinct domains. J. Biol. Chem. 274, 25510–25516 [DOI] [PubMed] [Google Scholar]
- 42. Adams T. E., Li W., Huntington J. A. (2009) Molecular basis of thrombomodulin activation of slow thrombin. J. Thromb. Haemost. 7, 1688–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parry M. A., Stone S. R., Hofsteenge J., Jackman M. P. (1993) Evidence for common structural changes in thrombin induced by active-site or exosite binding. Biochem. J. 290, 665–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Candia E., Hall S. W., Rutella S., Landolfi R., Andrews R. K., De Cristofaro R. (2001) Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J. Biol. Chem. 276, 4692–4698 [DOI] [PubMed] [Google Scholar]
- 45. De Cristofaro R., De Candia E., Landolfi R., Rutella S., Hall S. W. (2001) Structural and functional mapping of the thrombin domain involved in the binding to the platelet glycoprotein Ib. Biochemistry 40, 13268–13273 [DOI] [PubMed] [Google Scholar]
- 46. Nakanishi-Matsui M., Zheng Y. W., Sulciner D. J., Weiss E. J., Ludeman M. J., Coughlin S. R. (2000) PAR3 is a cofactor for PAR4 activation by thrombin. Nature 404, 609–613 [DOI] [PubMed] [Google Scholar]
- 47. Baerga-Ortiz A., Hughes C. A., Mandell J. G., Komives E. A. (2002) Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci. 11, 1300–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He X., Ye J., Esmon C. T., Rezaie A. R. (1997) Influence of arginines 93, 97, and 101 of thrombin to its functional specificity. Biochemistry 36, 8969–8976 [DOI] [PubMed] [Google Scholar]
- 49. Gasper P. M., Fuglestad B., Komives E. A., Markwick P. R., McCammon J. A. (2012) Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. U.S.A., 109, 21216–21222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lange O. F., Lakomek N. A., Farès C., Schröder G. F., Walter K. F., Becker S., Meiler J., Grubmüller H., Griesinger C., de Groot B. L. (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320, 1471–1475 [DOI] [PubMed] [Google Scholar]
- 51. Ban D., Funk M., Gulich R., Egger D., Sabo T. M., Walter K. F., Fenwick R. B., Giller K., Pichierri F., de Groot B. L., Lange O. F., Grubmüller H., Salvatella X., Wolf M., Loidl A., Kree R., Becker S., Lakomek N. A., Lee D., Lunkenheimer P., Griesinger C. (2011) Kinetics of conformational sampling in ubiquitin. Angew. Chem. Int. Ed. Engl. 50, 11437–11440 [DOI] [PubMed] [Google Scholar]
- 52. Bradford H. N., Krishnaswamy S. (2012) Meizothrombin is an unexpectedly zymogen-like variant of thrombin. J. Biol. Chem. 287, 30414–30425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pozzi N., Vogt A. D., Gohara D. W., Di Cera E. (2012) Conformational selection in trypsin-like proteases. Curr. Opin. Struct. Biol. 22, 421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vogt A. D., Di Cera E. (2012) Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894–5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kamath P., Huntington J. A., Krishnaswamy S. (2010) Ligand binding shuttles thrombin along a continuum of zymogen-like and proteinase-like states. J. Biol. Chem. 285, 28651–28658 [DOI] [PMC free article] [PubMed] [Google Scholar]