

Background: TGFβ induces a Smad3-Smad4 complex and PKA-R interaction.
Results: We define interaction domains between Smad4 and PKA-R required for TGFβ-mediated cell growth and EMT regulation.
Conclusion: An interaction between Smad4 and PKA-R regulates TGFβ signaling.
Significance: A novel cross-talk mechanism between TGFβ and PKA signaling is identified that is critical for execution of TGFβ-mediated cellular responses.
Keywords: Cell Growth, EMT, Protein Kinase A (PKA), SMAD Transcription Factor, Transforming Growth Factor α (TGFα)
Abstract
Transforming growth factor β (TGFβ) signaling normally functions to regulate embryonic development and cellular homeostasis. It is increasingly recognized that TGFβ signaling is regulated by cross-talk with other signaling pathways. We previously reported that TGFβ activates protein kinase A (PKA) independent of cAMP through an interaction of an activated Smad3-Smad4 complex and the regulatory subunit of the PKA holoenzyme (PKA-R). Here we define the interaction domains of Smad4 and PKA-R and the functional consequences of this interaction. Using a series of Smad4 and PKA-R truncation mutants, we identified amino acids 290–300 of the Smad4 linker region as critical for the specific interaction of Smad4 and PKA-R. Co-immunoprecipitation assays showed that the B cAMP binding domain of PKA-R was sufficient for interaction with Smad4. Targeting of B domain regions conserved among all PKA-R isoforms and exposed on the molecular surface demonstrated that amino acids 281–285 and 320–329 were required for complex formation with Smad4. Interactions of these specific regions of Smad4 and PKA-R were necessary for TGFβ-mediated increases in PKA activity, CREB (cAMP-response element-binding protein) phosphorylation, induction of p21, and growth inhibition. Moreover, this Smad4-PKA interaction was required for TGFβ-induced epithelial mesenchymal transition, invasion of pancreatic tumor cells, and regulation of tumor growth in vivo.
Introduction
Transforming growth factor β (TGFβ) is a member of a large family of structurally related proteins that normally functions to regulate embryonic development and cellular homeostasis, including regulation of proliferation, differentiation, matrix production, and apoptosis in a cell and context-specific manner (1–4). Alterations in the TGFβ signaling pathway may result in human diseases such as developmental disorders, vascular diseases, and cancer. Although TGFβ acts as a tumor suppressor in the early stage of epithelial carcinogenesis, TGFβ promotes tumor progression in advanced stages by inducing tumor growth, epithelial mesenchymal transition (EMT),2 invasion, evasion of immune surveillance, and metastasis.
TGFβ signaling is mediated by two types of transmembrane serine/threonine kinase receptors (type I and II) that bind ligand and transmit signals from the cell surface to the cell interior by activating a signaling cascade that involves Smad proteins, a family of highly conserved intracellular proteins. Smads can be subdivided into three classes based on their functional properties: receptor-regulated Smads (Smad1, -2, -3, -5, -8), a common mediator Smad (Smad4), and the inhibitory Smads (Smad6 and -7) (5–7). Although each Smad has a distinct function, all are composed of highly conserved N-terminal (MH1) and C-terminal (MH2) domains with a variable proline-rich linker region, with exception of the inhibitory Smads, which lack a MH1 domain. The MH1 domain is involved in DNA binding and maintaining the Smad in an inactive conformation. The linker region that connects the MH1 and MH2 domains contains important peptide motifs with binding sites for Smurf (Smad ubiquitination-related factor) ubiquitin ligases, phosphorylation sites for several classes of protein kinases, and specific to Smad4, a nuclear export signal. The MH2 domain participates in the formation of homo- and hetero-oligomers and mediates interactions with other proteins during transcriptional regulation (8–11).
Among the R-Smads, Smad2 and Smad3 respond specifically to TGFβ and activin (12). Upon ligand binding, Smad2 and Smad3 bind directly to the type I receptor and are phosphorylated by TGFβ receptor complexes at the SSXS motif at the C terminus of the proteins, which results in release from the receptor. The phosphorylated Smads form heteromeric complexes with Smad4 and translocate into the nucleus to regulate gene expression (13, 14). Inside the nucleus, the activated R-Smad-Smad4 complex can either bind with recruited co-activator CBP/p300 to target-specific promoters or engage in protein-protein interactions with other functional molecules to induce or suppress gene transcription (15, 16).
It is increasingly recognized that the strength and duration of the TGFβ signal is regulated by cross-talk with other signaling pathways. One potentially important interaction was suggested in a report that TGFβ could activate cAMP-dependent protein kinase (also known as protein kinase A (PKA)) through an unknown mechanism (17). PKA is a cytosolic, tetrameric holoenzyme that is composed of two regulatory (R) subunits associated with two catalytic (C) subunits (18, 19). Elevation of intracellular cAMP level causes binding of cAMP to the regulatory subunits and leads to a dissociation of the tetrameric complex, thus allowing the free catalytic subunit to be active as a serine-threonine kinase in the cytoplasm and nucleus. The dissociated, active C subunits can then affect cell physiology via phosphorylation of a wide variety of protein substrates. PKA signaling has been shown to play a role in a wide range of physiological processes, including growth, differentiation, extracellular matrix production, and apoptosis (18, 20). Many of these cellular effects are similar to those elicited by TGFβ.
In support for the role of PKA in mediating TGFβ-induced responses, Sharma et al. (21) demonstrated that TGFβ-induced phosphorylation of the type I 1,4,5 triphosphate receptor in mesangial cells is mediated by PKA. Inhibition of PKA has also been previously found to attenuate TGFβ-induced stimulation of CREB phosphorylation and fibronectin gene expression (17, 22). Recently, cross-talk between the TGFβ and PKA signaling pathways has been found to be important in colon cancer cell survival and metastasis through regulation of survivin and XIAP signaling (23). We previously identified a novel mechanism of PKA activation by TGFβ via the formation of a trimeric complex composed of activated Smad3, Smad4, and the regulatory subunit of PKA, with the resulting release of the catalytic subunit from the PKA holoenzyme (24). Smad2 did not participate in complex formation. This effect was not observed in Smad3- or Smad4-deficient cells but did occur in the absence of an increase in intracellular cAMP, the major known activator of PKA. We found that the activation of PKA was required for TGFβ-mediated activation of the transcription factor CREB, induction of the cell cycle regulatory protein p21Cip1, and inhibition of cell growth. These results indicate an important cross-talk mechanism between the TGFβ/Smad and PKA signaling pathways. However, the molecular mechanism by which this cross-talk occurs is not known.
In this study we define the interaction domains of the Smad4 protein and the regulatory subunit of PKA that form the trimeric complex with Smad3 responsible for TGFβ-induced PKA activation. We demonstrate that the interaction of these defined regions of Smad4 and the regulatory subunit of PKA was responsible for TGFβ-mediated increases in PKA activity, CREB phosphorylation, induction of the cell cycle regulatory protein p21Cip1, growth inhibition, EMT, and pancreatic cancer cell invasion. Furthermore, this interaction mediated tumor growth in vivo. These findings have significant implications for understanding how TGFβ signaling may be regulated during normal cellular function and in disease states.
EXPERIMENTAL PROCEDURES
Cell Culture
MvlLu cells, a well studied TGFβ responsive mink lung epithelial cell line, were a gift from L. Mathews (University of Michigan). Panc1 and CFPAC1 pancreatic cancer cell lines were purchased from American Type Culture Collection (ATCC). Both Mv1Lu and Panc1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM), CFPAC1 cells were cultured in Iscove's modified Dulbecco's medium (ATCC), and the UM2 primary pancreatic cancer cell line generated in our laboratory using Institutional Review Board (IRB) guidelines from an invasive pancreatic adenocarcinoma of a 76-year-old female patient who underwent surgical resection at University of Michigan Hospital was cultured in RPMI1640 supplemented with 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (Invitrogen) in 95% air and 5% carbon dioxide at 37 °C.
Mutant-expressing Plasmids
A series of Smad4 and PKA-RIIα deletion mutants were created using a TOPO TA Cloning vector (Invitrogen) according to the manufacturer's instructions. The plasmid constructs expressing full-length Smad4 (residues 1–552) and the Smad4 deletion mutants Smad4Δ142 (residues 143–552), Smad4Δ250 (residues 251–552), Smad4Δ259 (residues 260–552), Smad4Δ267 (residues 268–552), Smad4Δ281 (residues 282–552), Smad4Δ290 (residues 291–552), Smad4Δ300 (residues 301–552), and Smad4Δ320 (residues 321–552) were prepared by cloning into an N-terminal 3× FLAG-tagged expression vector (Sigma). The directional TOPO cloning vector (Invitrogen) for PKA-RIIα contains a 14-amino acid V5 epitope and a polyhistidine (His6) tag at the C-terminal region to aid in detection of the recombinant proteins. To make interstitial deletion mutants of the B domain and Smad4, we used a PCR-based, site-directed mutagenesis kit (Stratagene, La Jolla, CA). Pairs of primers were designed to reproduce the whole plasmid DNA, placing the regions to be deleted on the 3′ end of the PCR products. A Dpn1 restriction enzyme was used to digest parental and hybrid plasmid DNAs, and the remaining new plasmid DNAs were ligated at room temperature for 1 h with T4 DNA ligase. The ligated DNA was then transformed into XL1-Blue supercompetent cells (Stratagene, La Jolla, CA). Colonies were selected to grow in LB medium, and plasmid DNA was isolated. All mutants were verified by DNA sequencing.
Transient Transfection
Mv1Lu cells or CFPAC1 cells cultured in 6-well plates or 100-mm culture dishes at 80% confluency were transiently transfected with 2 μg/well or 10 μg/dish of plasmids using the lipofectamine2000 reagent (Invitrogen). Forty-eight hours after transfection, cells were serum-starved for 18 h and then left untreated or treated with TGFβ (R&D System, Minneapolis, MN) or forskolin at described concentrations.
Western Blot Analysis
Cells were lysed in cell lysis buffer (50 mm Tris-HCl, pH 8.0, containing 1% Triton X-100, 25 μg/ml aprotinin and leupeptin, 1 mm PMSF, and 10% glycerol). The cell lysates were then centrifuged at 10,000 × g for 10 min to remove debris. Protein concentrations were measured using the Bradford method (Bio-Rad). Proteins separated by SDS-PAGE were transferred onto nitrocellulose membranes. After blocking with blocking buffer (Tris-buffered saline (TBS), pH 7.4, with 0.1% Tween 20 containing 5% skim milk powder) for 1 h at room temperature, the membranes were incubated for 1 h at room temperature with the following primary antibodies in blocking buffer: anti-FLAG, 1:2000 dilution (Sigma); anti-CREB, anti-phospho-CREB (Ser 133), anti-Smad3 (Cell Signaling Technology, Beverly, MA), anti-Smad4, anti-His, anti-PKA-RIα, anti-RIβ, anti-RIIα, anti-RIIβ, anti-MMP-2, anti-MMP-9, anti-COL1A1, anti-COL1A2, anti-PAI, (Santa Cruz Biotechnology) at dilutions of 1:1000; anti-V5 conjugated with HRP (Invitrogen) at a 1:5000 dilution. After incubation with secondary antibodies conjugated with HRP, the proteins were visualized using an ECL detection kit (Pierce) according to the manufacturer's instructions.
Co-immunoprecipitation Assays
Mv1Lu cells were transiently transfected with FLAG-tagged Smad4, V5-tagged PKA-RIIα, or their various deletion mutants. After serum starvation, the cells were treated with 100 pm TGFβ for 15 min. Cells were lysed in ice-cold lysis buffer (50 mm Tris, 150 mm NaCl, 0.5% Igepal, pH 7.4) containing freshly added protease inhibitors (Roche Applied Science). The lysates were centrifuged for 15 min at 10,000 × g to remove debris. PKA-RII or Smad4 was immunoprecipitated by incubating with anti-PKA-RIIα or anti-Smad4 antibodies (Santa Cruz, CA) and protein G-agarose beads (Invitrogen) at 4 °C overnight. Immunoprecipitates were washed 4 times with ice-cold lysis buffer and resolved by reducing SDS-PAGE. Co-immunoprecipitated Smad4 and its mutants were detected with anti-FLAG antibody (Sigma). Co-immunoprecipitated full-length and deletion mutants of PKA-RIIα were detected using an anti-V5 antibody.
Peptide Synthesis and Peptide Competition Experiments
Four small specific peptides, amino acids 281–285 (P1) and 320–329 (P2) of the PKA-RIIα B domain, amino acids 290–300 (PS4) of Smad4, and one mutant peptide with scrambled amino acid sequence of peptide P2 as control peptide (Pc) were synthesized in the University of Michigan Protein Structure Core Facility (Ann Arbor, MI). The peptides were rendered cell-permeant by fusing each to the cell membrane transduction domain of the human immunodeficiency virus-type 1 (HIV-1) Tat protein (Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg) to obtain Tat peptides (25) preceded by an N-terminal fluorescein isothiocyanate. In experiments utilizing the peptides, cell lysates were incubated and mixed either with P1, P2, or the control peptide (Pc) at 4 °C. After 3 h, co-immunoprecipitation assays were conducted using an anti-Smad4 antibody and protein G-agarose beads. The co-immunoprecipitates were then subjected to SDS-PAGE. Immunoblotting was performed using an HRP-conjugated anti-V5 antibody. For in vivo competition experiments, Mv1Lu cells were incubated either with P1, P2, or a combination of P1 and P2 at different concentrations for 30 min and then treated with 100 pm TGFβ for 30 min. Cell lysates were subjected to PKA activity assays according to the manufacturer's instructions (Promega, Madison, WI) and Western blotting to assess CREB phosphorylation.
Preparation of GST-tagged Wild-type B Domain and Mutant Proteins
The cDNAs for wild-type and mutant B domain of PKA-RIIα were subcloned into the glutathione S-transferase (GST) expression plasmid, pGEX4T1 (GE Healthcare). These plasmids were individually transformed into the Escherichia coli strain BL21(DE3) (Invitrogen). Bacterial growth was initiated at 37 °C until an A600 nm of 0.6–0.8 optical density was reached, and protein expression was induced by the addition of 1.0 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 37 °C. The cells were harvested by centrifugation at 5000 × g, 4 °C for 10 min, then resuspended in lysis buffer (50 mm Tris-HCl, pH 8.5, 300 mm NaCl, 5 mm EDTA) and lysed by adding 0.5% Triton X-100 after sonication. The soluble fraction was retained after centrifugation at 10,000 × g at 4 °C for 30 min. GST fusion proteins were isolated by glutathione-Sepharose affinity chromatography. Glutathione-Sepharose affinity columns were pre-equilibrated with 20 column volumes of binding buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl) at room temperature. Soluble protein fractions were allowed to bind to the column for 4 h at 4 °C. Columns were then drained and washed with 3 × 50 ml of binding buffer. To remove the tightly bound cAMP from the recombinant wild-type B domain and mutant proteins, GST proteins in dialysis bags were incubated with 8 m urea in a dialysis buffer (5 mm MOPS, 0.5 mm EDTA, 100 mm KCl, and 5 mm β-mercaptoethanol, pH 7.0) overnight at 4 °C, then the dialysis process was continued using serial 6, 4, 2, 1, and 0.5 m urea in the dialysis buffer for 1 h each with agitation and finally equilibrated in the urea-free dialysis buffer.
GST Pulldown Assays
10 μg of GST-Smad4 proteins were incubated with 10 μl of glutathione-Sepharose beads and 400 μl of wild-type PKA-RIIα B domain-transfected Mv1lu cell lysates without or with 100 μm peptide P1, P2, and Pc at 4 °C overnight. The beads were washed 4 times with a washing buffer (PBS, 0.05% Triton X-100, 2 mm EDTA) and analyzed by Western blot analysis using anti-Smad4 and anti-V5 antibodies.
Cell Proliferation Assays
Cell proliferation was measured by using a CellTiter 96 AQ nonradioactive cell proliferation assay (Promega) as described (24).
cAMP Binding Assays
200 nm GST fusion proteins, either wild-type PKA RIIα B domain or the B domain mutants Δ281–285, Δ320–329, and Δ380–383 conjugated to glutathione beads, were mixed with 600 nm [3H]cAMP in cAMP binding buffer (20 mm MOPS, pH 7.0, 150 mm KCl, 1 mm DTT) for 2 h at room temperature. After washing with PBS-T solution three times, the GST beads were mixed with scintillation fluid, and binding of cAMP to the B domain or its mutants was determined by counting bound [3H]cAMP in a scintillation counter. The percentage of cAMP bound to wild-type B domain and its mutants was obtained by dividing bound [3H]cAMP by the total radioactive input of [3H]cAMP.
Real-time Quantitative Polymerase Chain Reaction
Purification of total RNA and cDNA synthesis were performed using the RNeasy Mini kit (Qiagen, Valencia, CA) and High Capacity cDNA RT kit (Applied Biosystems, Foster City, CA), respectively. Real-time PCR experiments were conducted using the SYBER Green PCR system (Applied Biosystems) on a Rotor-gene Q cycler (Qiagen). Cycling temperatures were as follows: denaturing, 95 °C; annealing, 60 °C; extension, 72 °C. The following primers were used: snail, forward (5′-ACCACTATGCCGCGCTCTT-3′) and reverse (5′-GGTCGTAGGGCTGCTGGAA-3′); vimentin, forward (5′-TCTACGAGGAGGAGATGCGG-3′) and reverse (5′-GGTCAAGACGTGCCAGAGAC-3′); E-cadherin, forward (5′GTCAGTTCAGACTCCAGCCC-3′) and reverse (5′-AAATTCACTCTGCCCAGGACG-3′).
In Vitro Extracellular Matrix Invasion Assays
Cell invasion was assayed using an extracellular matrix invasion assay kit (ECMatrix Cell Invasion Assay (catalogue #ECM550), Millipore, Billerica, MA) according to the manufacturer's protocol. In brief, 3 × 105 cells incubated for 1 h in serum-free media were resuspended in 300 μl of serum-free media and plated on the top of an ECM-coated membrane insert. The insert was then incubated for 72 h in serum-containing media with 15 μm H89, 5 μm P1/P2, and 5 μm PS4 peptides either alone or combination with 20 ng/ml TGFβ1. Invading cells were quantified using the manufacturer provided cell dye and digital imaging software.
In Vivo Tumorigenicity Studies
Six-week-old male NOD/SCID mice were housed under pathogen-free conditions. Animal experiments were approved by the University of Michigan Animal Care and Use Committee and were performed in accordance with established guidelines. Mice were anesthetized with an intraperitoneal injection of xylazine (9 mg/kg) and ketamine (100 mg/kg). 1 × 106 CFPAC1 cells with stable expression of vector alone or FLAG-tagged Smad4 or FLAG-Smad4(Δ290–300) were suspended in phosphate-buffered saline/Matrigel mixture (1:1 mixture, total volume of 100 μl) and were injected into the subcutaneous tissue of the abdomen using a 30-gauge needle. Four weeks after cancer cell injection, mice were euthanized with carbon dioxide inhalation, and necropsies were performed to assess the extent of primary tumor growth as described (25). Paraffin-embedded pancreatic tissue sections (4 μm thick) were cut, and hematoxylin and eosin staining of cut sections was performed.
Statistical Analysis
Data are represented as the mean ± S.E. from at least three independent experiments. Significance of differences between groups was evaluated by Student's t test or analysis of variance. p < 0.05 was considered significant.
RESULTS
Identification of the Smad4 Region That Interacts with PKA-R
We previously reported that TGFβ activates PKA through a direct interaction between an activated Smad3-Smad4 complex and the PKA regulatory subunit (PKA-R). Using in vitro binding assays, we previously found that constitutively active Smad3 (Smad3D) could not bind to PKA-RIIα, yet Smad3D could form a complex with Smad4 that was then able to bind PKA-RIIα (24). Therefore, we reasoned Smad4 was likely the component of the activated Smad3-Smad4 complex responsible for PKA-R binding and sought to determine which region of Smad4 was responsible for this interaction. To test this hypothesis, a series of N-terminal FLAG-tagged Smad4 truncation mutants were created and transfected into Mv1Lu cells, a mink epithelial cell line commonly used for studies of TGFβ signaling (Fig. 1A). Co-immunoprecipitation experiments showed that Smad4 was able to interact with PKA-RIIα in a TGFβ dependent manner (Fig. 1B). Like wild-type Smad4, Smad4Δ142 (deletion of MH1 domain) was capable of binding PKA-R when cells were treated with TGFβ (Fig. 1C). Analysis of additional deletion mutants showed that whereas further deletion of amino acids 1–250 did not affect binding, deletion of amino acids 1–320 prevented Smad4 binding to PKA-R, suggesting that the linker C-terminal 70 amino acids of Smad4 contained the binding site for PKA-R (Fig. 1D). Consecutive serial deletions of Smad4 within this portion of the linker region indicated that amino acids 290–300 were involved in the interaction between Smad4 and PKA-R (Fig. 1E). To further refine the significance of this region of Smad4 in binding to PKA-R, we created the Smad4 interstitial deletion mutant Smad4(Δ290–300) and found that unlike wild-type Smad4, Smad4(Δ290–300) was not able to bind to PKA-R after TGFβ stimulation (Fig. 1F), highlighting the importance of amino acids 290–300 of Smad4 for this interaction.
FIGURE 1.

Amino acids 290–300 of Smad4 interact with the regulatory subunit of PKA. A, shown is a schematic of Smad4 and truncation and interstitial deletion mutants used for interaction assays with PKA-R. B–F, shown is the determination of PKA-R binding site of Smad4. Mv1Lu cells were transfected with N-terminal FLAG-tagged wild-type Smad4 or Smad4 mutant-expressing plasmids and then treated with 100 pm TGFβ for 30 min. Cell lysates were subjected to coimmunoprecipitation (IP) assays. The regulatory subunit of PKA was immunoprecipitated by an anti-PKA-RIIα antibody followed by immunoblotting (IB) of FLAG-Smad4 using an anti-FLAG antibody. 10% of cell lysates were loaded and immunoblotted for input control. The results are the representative of three independent experiments.
The Smad4(Δ290–300) Deletion Mutant Is Unable to Rescue TGFβ-induced PKA Activation and CREB Phosphorylation in Smad4-deficient Cells
To further demonstrate that amino acids 290–300 of Smad4 represent a functional PKA-R binding site, wild-type Smad4 and its deletion mutants were transiently transfected into CFPAC1 cells, a pancreatic cancer cell line that lacks endogenous Smad4. Forty-eight hours post transfection, cells were treated with 100 pm TGFβ for 30 min. Control vector transfected cells did not demonstrate enhanced PKA activity (Fig. 2A). However, cells expressing Smad4 and several of its mutants, including Smad4Δ290 but not Smad4Δ300 and Smad4(Δ290–300), were able to rescue TGFβ-induced PKA activation. Similar results were obtained when examining phosphorylation of the transcription factor CREB (Fig. 2, B and C), a known downstream target of PKA signaling (27). Unlike Smad4, Smad4(Δ290–300), due to lack of PKA-R binding site, was not able to rescue TGFβ-induced CREB phosphorylation (Fig. 2C). We also observed that TGFβ induced a comparable increase in Smad3 phosphorylation both in Smad4- and Smad4(Δ290–300)-transfected CFPAC1 cells (Fig. 2C), demonstrating that these cells did respond to TGFβ as previously described (28). To test whether Smad4Δ300 and Smad4(Δ290–300) retain their ability to interact with Smad3 in a TGFβ-dependent manner, FLAG-tagged full-length Smad4 and Smad4 mutants were transfected into Mv1Lu cells, and the ability of the FLAG-tagged Smad4 proteins to bind to Smad3 in response to 100 pm TGFβ was assessed. The Smad4Δ300 and Smad4(Δ290–300) mutants displayed similar levels of binding to Smad3 as was observed with full-length Smad4, suggesting that the inability of Smad4Δ300 and Smad4(Δ290–300) to activate PKA and phosphorylate CREB in Smad4-deficient CFPAC1 cells was not due to the inability to bind to Smad3 (supplemental Fig. 1). Thus, we identified a domain of Smad4 that is required for PKA interaction but not for interaction with Smad3. This also demonstrates that the Smad4 deletion mutants are not grossly misfolded proteins.
FIGURE 2.

The effect of TGF-β-induced PKA activation and CREB phosphorylation in CFPAC1 cells transfected with Smad4 and its deletion mutants. Smad4-deficient CFPAC1 cells were transiently transfected with Smad4 and its deletion mutants. Transfected cells were treated with TGF-β (100 pm) for 30 min, and cell lysates were prepared for PKA activity assays (A) and Western blotting to measure CREB phosphorylation (B and C). The results are from three separate experiments. **, p < 0.01 versus vector control.
Amino Acids 281–285 and 320–329 of the PKA RIIα B Domain Are Necessary for the Interaction between the PKA-RIIα B Domain and Smad4
All PKA regulatory subunit isoforms have a well conserved and defined domain structure that consists of a dimerization domain, a linker region, and cAMP binding domains A and B (18). Each domain has its own function and also communicates with the other domains as the regulatory subunit undergoes conformational changes that are induced by cAMP binding. To identify which region(s) of the PKA regulatory subunit interacts with Smad4, a series of PKA-R truncation mutants was generated using PCR-based mutagenesis (Fig. 3A). Mutants of PKA-RIIα were constructed lacking the 1) dimerization and linker domains and 2) dimerization, linker, and A domains (thus containing only the B domain). Western blot analysis of MvlLu cell lysates transfected with full-length PKA-R or its deletion mutants demonstrated comparable levels of expression (Fig. 3B). In co-immunoprecipitation experiments we found that the full-length PKA regulatory subunit, the A+B domains, and the B domain alone all interacted with Smad4 in a TGFβ-dependent manner (Fig. 3B), demonstrating that the B domain alone was sufficient for the interaction between the PKA regulatory subunit and Smad4.
FIGURE 3.

The B domain of the regulatory subunit of PKA-RIIα interacts with Smad4. Shown is a schematic of the regulatory subunit of PKA and its truncation mutants (A). Mv1Lu cells were transiently transfected with PKA-RIIα full-length or deletion mutant-expression plasmids in the presence or absence of 100 pm TGF-β for 30 min. Co-immunoprecipitation (IP) assays were performed using anti-Smad4 and V5 antibodies (B). The data shown are representative of three independent experiments with similar results. IB, immunoblot.
Because the B domain alone was sufficient for the molecular interaction between PKA-RIIα and Smad4, we next focused on identifying the region(s) of the B domain responsible for this interaction. We determined that in addition to RIIα, the remaining three isoforms of PKA regulatory subunit (RIα, RIβ, and RIIβ) also interacted with Smad4 in response to TGFβ (Fig. 4A), suggesting that the site of interaction(s) in the B domain was within the conserved regions of all four PKA regulatory subunit isoforms. The cAMP binding site (amino acids 330–350) within the B domain was excluded from consideration because it does not lie on the surface of the molecule (29). We then defined highly conserved regions among the human PKA-R isoforms based on sequence analysis (30) that had a high surface probability based on the crystal structure of human RIIβ (29) utilizing the three-dimensional computer modeling program Cn3D4.1 (NCBI, Bethesda, MD).
FIGURE 4.
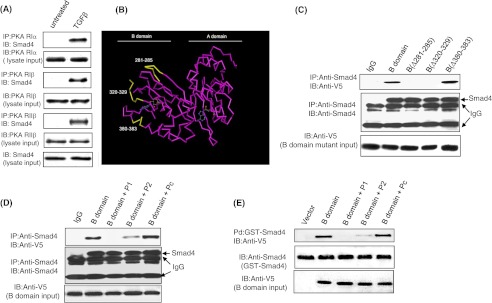
Defining sites of Smad4 interaction with the B domain of PKA-R subunit. Mv1Lu cells were treated in the presence or absence of 100 pm TGF-β for 15 min, then co-immunoprecipitation (IP) experiments were performed using anti-RIα, anti-RIβ, and anti-RIIβ antibodies and an anti-Smad4 antibody (A). Results are representative of three separate experiments. Protein structure of the PKA-R subunit demonstrating three regions within the B domain that are highly conserved among PKA regulatory isoforms and located on the surface of the molecules (B) are: amino acids 281–285 (YKDGERI), 320–320(QEVEIARC), and 380–383 (ISHY) (shown in yellow). cAMP binding sites are depicted in blue. PKA-RII B domain mutants with interstitial deletions of amino acids 281–285 and 320–329 are unable to bind to Smad4 (C). Peptides P1 or P2 but not control peptide (Pc) blocked the ability of the PKA-RIIα B domain to interact with Smad4 in both by co-immunoprecipitation and GST pulldown assays (D and E). Mv1Lu cells transfected with PKA-RIIα B domain and its interstitial deletion mutants were treated with 100 pm TGF-β for 15 min, and co-immunoprecipitation assays were done with anti-Smad4 and anti-V5 antibodies. In peptide blocking experiments, lysates were preincubated with 10 μm P1, P2, or Pc for 30 min and then subjected to co-immunoprecipitation and GST pulldown assays. Co-precipitated B domain and its deletion mutants were detected with an anti-V5 antibody by Western blotting. Lysates (10% of immunoprecipitates) were also immunoblotted (IB) for input control. Data is representative of three independent experiments.
Three candidate Smad4 binding regions located on the surface of the B domain of the RIIα molecule were conserved among all four PKA-R isoforms: amino acids 281–285, 320–329, and 380–383 (Fig. 4B, highlighted in yellow). To determine whether these regions were involved in the interaction with Smad4, three interstitial deletion mutants, B(Δ281–285), B(Δ320–329), and B(Δ380–383), were generated using PCR-based site-directed mutagenesis. Western blot analysis of lysates from TGFβ-treated cells confirmed that the B domain and its deletion mutants were expressed at comparable levels (Fig. 4C). Co-immunoprecipitation experiments in TGFβ-treated cells revealed that deletion of either amino acids 281–285 or amino acids 320–329 prevented complex formation between Smad4 and the B domain, whereas deletion of amino acids 380–383 had no effect on this interaction (Fig. 4C). To further verify the importance of these specific sites within the B domain in Smad4 interaction, we utilized two specific peptides, amino acids 281–285 (YKDGERI, P1) and amino acids 320–329 (QEVEIARC, P2), and a control peptide with scrambled amino acid sequence of peptide P2 (Pc). Co-immunoprecipitation experiments were conducted as described above, except that the small peptides (100 μm) were added to the cell lysates 1 h before the addition of an anti-Smad4 antibody and agarose protein G beads. We found that peptides P1 and P2 competed with the wild-type B domain of PKA-R for interaction with Smad4 in TGFβ-treated cells, resulting in a significant decrease in binding (Fig. 4D), whereas the control peptide had no effect. Similar results were obtained when using an anti-His antibody to immunoprecipitate B domain and its mutants and anti-Smad4 to identify co-precipitated Smad4 (data not shown). In a GST pulldown assay using purified GST-Smad4 protein and B domain-transfected Mv1Lu cell lysates, peptides P1 and P2 were able to significantly block the interaction between Smad4 and B domain (Fig. 4E). These experiments demonstrate that amino acids 281–285 and 320–329 within the B domain of the regulatory subunit of PKA are necessary for complex formation with Smad4.
Amino Acids 281–285 and 320–329 of PKA-R Block TGFβ-induced PKA Activation and CREB Phosphorylation
To determine whether amino acids 281–285 and 320–329 of PKA-R are critical sites of the B domain not only for Smad4 interaction but also downstream signaling events, Mv1Lu cells were exposed to the blocking peptides P1, P2, or the control peptide Pc for 1 h and then treated with 100 pm TGFβ for 30 min. Cell lysates were prepared and subjected to PKA activity assays and Western blotting to measure changes in CREB phosphorylation. All of the peptides demonstrated equivalent uptake by these cells (supplemental Fig. 2). TGFβ-induced PKA activation (Fig. 5A) and CREB phosphorylation (Fig. 5B) were completely blocked in both cells treated with PS4, comprised of amino acids 290–300 of Smad4, and the combination of P1 and P2 peptides but not blocked in cells treated with the control peptide Pc. P1 or P2 peptides alone showed a partial blocking effect on TGFβ-induced PKA activation. P1 and P2 peptides had no effect on forskolin-induced PKA activation and CREB phosphorylation (Fig. 5, A and B), indicating that they are specific for TGFβ/Smad-induced PKA signaling. These results together with our above binding experiments suggest that the amino acids 281–285 and 320–329 of PKA-R are necessary for Smad4 binding and PKA signaling and suggest that amino acids 281–285 and 320–329 may form a Smad4 binding pocket to exert cross-talk between these two pathways.
FIGURE 5.

TGF-β-induced PKA activation and CREB phosphorylation were inhibited by specific blocking peptides in Mv1Lu cells. Mv1Lu cells were incubated with control Pc or P1 and P2 given alone or in combination for 30 min followed by treatment with 100 pm TGFβ or forskolin at indicated concentrations for 30 min. Cell lysates were prepared for PKA activity measurement using a nonradioactive PepTag Assay kit (Promega) (A). CREB phosphorylation in the lysates was measured by Western blotting using anti-p-CREB antibodies (B). Results shown are representative of three independent experiments.
cAMP Binding Ability Was Not Affected by the Deletion Mutations of the PKA Regulatory Subunit B Domain
Although the deletions made in the PKA regulatory subunit B domain did not overlap with the cAMP binding site, it is possible that the interstitial deletions may have altered the conformation of the B domain, affecting its ability to bind Smads or cAMP. To examine this possibility, cAMP binding assays were conducted to compare the ability of the wild-type B domain and its interstitial deletion mutants to bind cAMP. We found that the wild-type B domain and its deletion mutants B(Δ281–285), B(Δ320–329), and B(Δ380–383) bound cAMP comparably, with a binding percentage at 3.3% for control, 14.25% for wild-type B domain, 16.45% for B(Δ281–285), 12.5% for B(Δ320–329), and 16% for B(Δ380–383) (supplemental Fig. 3A). Competition with 10-fold excess nonradioactive cAMP decreased [3H]cAMP binding to control levels, demonstrating that cAMP binding was specific (supplemental Fig. 3B). These results show that B domain mutants retained their ability to bind cAMP at levels comparable to the wild-type B domain, suggesting that the structural integrity of the B domain mutants was maintained. These findings support our data identifying amino acids 281–285 and 320–329 as critical sites within the B domain for the interaction of PKA-R with Smad4.
TGFβ-induced p21 Expression and Growth Inhibition Require Smad4 and PKA-R Interaction
To determine whether the interaction between Smad4 and PKA-R might affect cellular responses of TGFβ beyond PKA activation and CREB phosphorylation, Mv1Lu cells were treated with blocking peptides P1 and P2, peptide PS4, or control peptide Pc, each for 1 h, and then cells were treated with 100 pm TGFβ. Induction of the cell cycle regulatory protein p21 and growth inhibition were assessed. Both combination of P1 and P2 peptides and the PS4-(290–300) peptide were able to block the ability of TGFβ to induce p21 expression (Fig. 6A) and growth inhibition (Fig. 6B), whereas the control peptide had no effect, suggesting that the interaction of amino acids 290–300 of Smad4 and the “Smad4 binding pocket” created by amino acids 281–285 and 320–329 of the B domain of PKA-R are required for TGFβ to elicit these cellular responses. The importance of the 290–300 amino acid region of Smad4 in regulating p21 expression and growth inhibition by TGFβ in cells was also examined in the Smad-4 deficient CFPAC1 cells. In CFPAC1 cells, transfection with wild-type Smad4 allowed TGFβ to restore p21 induction (Fig. 6C) and growth inhibition (Fig. 6D), whereas transfection with the Smad4(Δ290–300) mutant did not. Thus, binding of the 290–300 amino acid region of Smad4 to PKA-R is required for TGFβ-induced p21 induction and growth inhibition. To further investigate the functional relevance of the 290–300 amino acid region of Smad4 in TGFβ mediated responses, we examined five proteins COL1A1, COL1A2, MMP-2, MMP-9, and plasminogen activator inhibitor-1 (PAI-1), previously described as being up-regulated in response to TGFβ in a Smad4-dependent manner (31–33). We found that TGFβ treatment (100 pm, 4 h) in CFPAC1 cells stably expressing Smad4 showed increased expression of all 5 genes (Fig. 6, E and F), whereas CFPAC1 cells without Smad4 showed no increases in response to TGFβ, highlighting the Smad4 dependence of these responses. TGFβ-mediated increases in COL1A1, COL1A2, and PAI-1 protein expression observed in CFPAC1 cells expressing Smad4 were not observed in Smad4(Δ290–300)-expressing CFPAC1 cells, demonstrating that this region of Smad4 was required for these changes. In contrast, TGFβ treatment increased MMP-2 (pro and active forms) and MMP-9 protein expression in both the CFPAC1 cells expressing Smad4 and the Smad4(Δ290–300) mutant, demonstrating that for these two proteins the 290–300 region of Smad4 is not required for the TGFβ-mediated increased in protein expression (Fig. 6, E and F).
FIGURE 6.

TGFβ-induced p21 expression and growth inhibition were blocked by specific peptides in Mv1Lu cells or by Smad4(Δ290–300) transfection in CFPAC1 cells. Mv1Lu cells were incubated with P1, P2, PS4, or control peptide Pc (5 μm each) given alone or in combination for 30 min followed by treatment with 100 pm TGFβ for 4 or 72 h. After a 4-h TGFβ treatment, p21 was detected by Western blotting (A). Cell proliferation was assessed after 72 h of TGFβ treatment (B). To further validate the importance of amino acids 290–300 of Smad4 in TGF-β-induced Smad4 target gene expression and cell growth regulation, CFPAC1 cells were either stably expressed with wild-type Smad4 or the Smad4(Δ290–300) mutant. Protein expression was measured by immunoblotting after 4 h of TGFβ treatment (C and E), and cell proliferation assays was performed after 3 days of TGFβ treatment (D), (*, p < 0.05 versus control). Relative protein expression from immunoblot was determined by densitometry from three independent experiments (F).
TGFβ-induced EMT and Invasion of Pancreatic Tumor Cells Were Blocked by Inhibition of PKA
In addition to the known effects of TGFβ on cell growth regulation, in advanced cancers TGFβ elicits tumor-promoting effects through its ability to induce EMT, which enhances cellular invasiveness and metastasis. To assay whether inhibition of PKA could suppress TGFβ-induced EMT and invasion in pancreatic tumor cells, Panc1 cells were treated with TGFβ (20 ng/ml) alone or in the presence of the PKA inhibitor H89 (15 μm) or the Smad4-PKA interaction blocking peptides (5 μm each) for 72 h. Cells treated with TGFβ underwent a morphologic change from a round into a spindle-like phenotype, characteristic of EMT. Interestingly, TGFβ-induced EMT was strongly blocked by H89, blocking peptides P1/P2 and PS4, respectively (Fig. 7A). We next examined the changes of the well described epithelial markers Zo-1 and E-cadherin and the mesenchymal markers vimentin and snail by Western blotting and real-time quantitative PCR. Both Zo-1 and E-cadherin were significantly down-regulated, and the EMT-associated proteins vimentin and snail were up-regulated after treatment of Panc1 cells with TGFβ (Fig. 7, B and C). TGFβ-induced down-regulation of Zo-1 and E-cadherin and up-regulation of vimentin and snail was significantly blocked both by H89 and the blocking peptides P1P2 and PS4 (Fig. 7, B and C). Similar results were obtained with the primary pancreatic tumor UM2 cell line (supplemental Fig. 4). To test whether the Smad4-PKA-R interaction was required for TGFβ-mediated cellular invasion, cell invasion assays were performed and revealed that TGFβ promoted Pancl tumor cell invasion, which was strongly inhibited by H89 and the blocking peptides P1/P2 or PS4 (Fig. 7D). Similar results were obtained from UM2 cells (supplemental Fig. 4C). Together these results demonstrate that a Smad4-PKA regulatory subunit interaction is needed for several key TGFβ-mediated cellular responses, including cell invasion and induction of an EMT phenotype.
FIGURE 7.
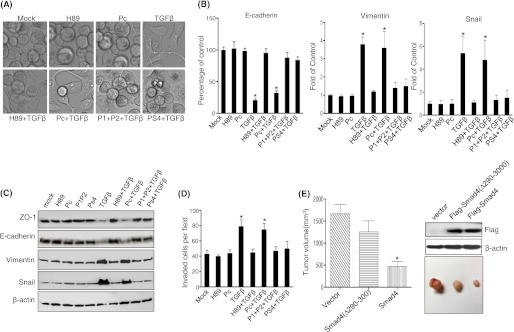
TGFβ-induced EMT, invasion, and tumor growth are dependent on the 290–300-amino acid region of Smad4. Panc1 cells were treated with TGFβ (20 ng/ml) in the presence of PKA inhibitor H89 (15 μm), synthesized peptides P1/P2 (each 5 μm), or PS4 (5 μm) for 3 days (A). RNA and total protein were isolated and subjected to real-time quantitative PCR and Western blotting to measure Zo-1, E-cadherin, vimentin, and snail expression (B and C). Cell invasion assays was performed, and invading cells were counted as cells per field under an inverted Nikon microscope (D). Comparison of tumor growth at 4 weeks between CFPAC1 cells expressing vector only, Smad4, and the Smad4(Δ290–300) mutant (n = 5 animals per group; *, p < 0.01 versus control (E).
Smad4-dependent Tumor Growth Inhibition Is Dependent on Amino Acids 290–300 of Smad4
To investigate the effect of TGFβ/Smad and PKA cross-talk on pancreatic tumor cell growth in vivo, a xenograft tumorigenicity assay was carried out in NOD/SCID mice. CFPAC1 cells expressing vector control, Smad4, and Smad4(Δ290–390) were injected subcutaneously (1 × 106 cells, n = 5 animals per group), and the tumor volumes were measured weekly for a total of 4 weeks. Expression of Smad4 in CFPAC1 cells significantly inhibited tumor growth compared with control cells (Fig. 7E). In contrast, tumor growth inhibition of CFPAC1 cells stably expressing Smad4(Δ290–300) was significantly reduced compared with CFPAC1 cells expressing Smad4. Histological analysis of the tumors in each group showed no visible differences in the appearance of the tumors (data not shown). Overall, these data demonstrate that the portion of Smad4 that binds PKA-R is important in mediating the inhibitory effects of Smad4 on tumor growth in vivo (Fig. 7E).
DISCUSSION
The dominant model of signaling induced by TGFβ family members is a linear signaling pathway from receptor activation to Smad activation, resulting in ligand-induced transcriptional regulation (34). It is becoming increasingly clear that other signaling pathways also define cellular responses to TGFβ signaling and that TGFβ family members activate not only Smads but other signaling pathways. We have previously reported that TGFβ activates PKA in a cAMP-independent manner by direct binding of an activated Smad3-Smad4 complex to the regulatory subunit of the PKA holoenzyme, forming a trimeric complex and resulting in activation of PKA (24). Because TGFβ signaling is critically important in a wide range of physiological and pathological processes, we investigated the molecular interactions between Smad4 and the regulatory subunit of PKA to determine the mechanism by which the Smad4-PKA-R interaction regulated TGFβ signaling. We defined the specific regions of Smad4 and PKA-R necessary for this interaction and demonstrated that this interaction was functionally significant in PKA activity and CREB activation as well as TGFβ-mediated p21 induction, growth regulation, invasion, and induction of an EMT phenotype.
Using a series of N-terminal FLAG-tagged Smad4 deletion mutants, we first determined that the linker C-terminal 70 amino acids of Smad4 contained an interaction domain for the regulatory subunit of PKA. Further consecutive deletions showed that amino acids 290–300 of the linker region were essential for binding of Smad4 to PKA-RIIα. It is well known that the MH2 domain of Smads is responsible for their interaction with other proteins, including transcription factors, co-activators, and co-repressors (35, 36). For example, a recent study demonstrated that Smad3 and protein kinase B (PKB/Akt) interacted with each other in human hepatoma Hep3B cells, with this interaction induced by insulin and inhibited by TGFβ (37). Complex formation between Smad3 and protein kinase B occurred specifically between the MH2 domain of Smad3 and the C-terminal domain of protein kinase B. The transcriptional co-repressor Ski/Sno has been shown to interact with Smad3-Smad4 and negatively regulate TGFβ signaling (38). Ski/Sno binds only to the C terminus of Smad4. The Ski binding surface on Smad4 significantly overlaps with the surface required for binding the phosphorylated tail of the R-Smads, thus disrupting the active heteromeric complex formed between the co-Smads and the R-Smads (39).
The function of the linker region of Smad4 is less well defined. de Caestecker et al. (40) reported that a 47-amino acid region from amino acids 274 to 321 within the proline-rich middle linker domain of Smad4 served as the Smad4 activation domain (SAD), which was essential for TGFβ signaling activities. They later reported that the functional activity of the Smad4 SAD domain is p300-dependent due to a physical interaction between the SAD domain and the N terminus of p300 (41). In the present study we have defined an additional functional role for the SAD domain. We have shown that the SAD region of Smad4 contains a novel binding site for the PKA regulatory subunit, which is the molecular basis of cross-talk between the TGFβ/Smad and PKA signal pathways, and that amino acid residues 290–300 of Smad4 are indispensable not only for binding of Smad4 to PKA but also for PKA signal pathway activation by TGFβ. This 11-amino acid sequence of Smad4 is a proline- and histidine-rich region that contains the amino acids (N) HHPPMPPHPGH (C). This region is unique for Smad4 among the Smads and does not share homology with Smad2 or Smad3. Of note, proline-rich motifs are well documented to be critical in protein-protein interactions (42).
The regulatory subunits of PKA are modular proteins that regulate PKA holoenzyme activation in a ligand-dependent, location-specific manner. The PKA-R subunits are composed of several distinct, well defined domains. Each domain has its own function and also communicates with other regions of the molecule as the holoenzyme undergoes conformational changes induced by cAMP binding (29). At the N terminus of the PKA-R subunit is a dimerization domain that functions to maintain the R-subunits as a stable dimer and provides a docking site for a variety of A kinase anchoring proteins (AKAPs), thereby localizing PKA to specific subcellular locations (43–47). After a variable, intervening linker domain at the C terminus of the PKA-R subunits are two tandem cAMP binding domains (A and B). The cAMP binding domains, presumably resulting from gene duplication, have extensive sequence similarity and bind cAMP cooperatively (47, 48). Although each domain has a functional cAMP binding site, the domains serve different functions. In the holoenzyme, cAMP binds first to domain B, inducing a conformational change and thus allowing access of cAMP to domain A. Subsequent binding of cAMP to domain A causes dissociation of the PKA catalytic subunit and activation of the holoenzyme (49).
In this study we found that the B domain of PKA-R alone was sufficient for interaction between PKA-R and Smad4 after TGFβ stimulation. The ability of an isolated B domain to function independently of other domain structures in the R subunit is supported by work by Shabb et al. (50) where they found that the B domain of the PKA-RIα subunit was able to function independently as a high affinity cAMP- binding protein. In further experiments using B domain interstitial deletion mutants, we identified that amino acids 281–285 and 320–329 of the B domain of PKA-R were critical for the TGFβ-induced interaction between the PKA regulatory subunit and Smad4 and for TGFβ-induced PKA activation and CREB phosphorylation. Interaction of Smad4 with these regions of the B domain of PKA-R were also essential for TGFβ-mediated induction of p21 and growth inhibition. Both of these regions, located in physical proximity on the surface of the molecule, were required to interact with Smad4, suggesting that they may form a “pocket” for Smad4 binding. The ability of a bound Smad complex to induce a conformational change in the PKA holoenzyme is supported by our previous work demonstrating that an activated Smad3-Smad4 complex is able to directly cause dissociation of the PKA holoenzyme and resultant PKA activity in vitro (24).
Local invasion can be considered an initial and essential step in the malignant process and often leads to the development of distant metastasis. TGFβ is a potent inducer of cellular invasion and EMT (51, 52). EMT is a process whereby cells lose cell-cell interactions and other epithelial properties while acquiring a more migratory and mesenchymal phenotype. EMT occurs at several stages of early development, and its exploitation during cancer progression is thought to contribute to tumor invasion and metastasis (53, 54). A hallmark of EMT is the functional loss of E-cadherin which mediates cell-cell interaction. The loss of E-cadherin expression has been shown to coincide with the transition from well differentiated tumor tissue to invasive carcinoma in a transgenic mouse model of pancreatic β-cell carcinogenesis, giving further support for a role of E-cadherin as a suppressor of invasion in pancreatic tumorigenesis (55). Smad4 has been shown to be essential for down-regulation of E-cadherin induced by TGFβ in the pancreatic cancer cell line Panc1 (56). Snail is one of a family of transcription factors shown to repress transcription of the E-cadherin gene (57). PKA has been shown to phosphorylate Snail at serine 11, with mutation of serine 11 abrogating the EMT-inducing activity of Snail, indicating that intact serine 11 is required for Snail to drive the EMT program (58). Previously, we have shown that the TGFβ ability to inhibit cell growth and induce p21 expression was dependent on a Smad complex-PKA interaction, and here we show that it requires a specific interaction of the 290–300 amino acid region of Smad4 and the B domain of the regulatory subunit of PKA. Furthermore, we show that this specific interaction is also required for the TGFβ ability to promote invasion of pancreatic cancer cells, induce an EMT phenotype, and regulate Smad4-mediated growth inhibition in vivo, highlighting the important role of this molecular interaction in pancreatic cancer tumorigenesis.
Interestingly, we found that the expression of the proteins COL1A1, COL1A2, PAI-1, and MMP-2 and MMP-9, all reported to be induced by TGFβ and Smad4-dependent (31–33), had differential requirements for amino acids 290–300 of Smad4. Although COL1A1, COL1A2, and PAI-1 were dependent on the Smad4-PKA-R interaction, MMP-2 and MMP-9 did not require this interaction of Smad4 and PKA-R for TGFβ to increase their expression. These results are consistent with a previous study that showed that TGFβ induced COL1A1 and PAI-1 expression and that overexpression of a Smad4 mutant, Smad4ΔM, a mutant of Smad4 lacking amino acids 275–322, inhibited TGFβ-induced COL1A1 and PAI-1 mRNA expression (59). The specific domain(s) of Smad4 responsible for regulation of MMP-2 and MMP-9 expression have not been previously defined, but our data would suggest that regulation of this subset of genes is not dependent on an interaction between Smad4 and PKA-R, at least in this cell line studied.
In summary, we have defined the specific interaction domains of Smad4 and the R subunit of the PKA holoenzyme that form a functional complex in vivo in response to TGFβ. Our results suggest that when cells are treated with TGFβ, an activated Smad3/Smad complex translocates into the nucleus, and the 290–300-amino acid sequence in the SAD domain of Smad4 binds to a pocket on the surface of the PKA-R subunit, distinct from the cAMP binding site, and that binding of an activated Smad3-Smad4 complex to this pocket results in PKA activation, p21, COL1A, COL1A2, PAI-1 protein expression, and TGFβ-mediated growth regulation, pancreatic tumor cell EMT, and invasion. In addition, this interaction was required for Smad4-dependent growth inhibition in a pancreatic cancer cell line in vivo. This study provides new insight into the molecular basis of Smad-PKA-R subunit complex formation and discloses a novel mechanism of cross-talk signaling that regulates PKA and TGFβ-induced cellular responses.
Acknowledgments
We thank the Proteomics and Peptide Synthesis Core at University of Michigan for synthesis and purification of the synthetic peptides used in this study.
This work was supported, in whole or in part, by National Institutes of Health Grant DK-061507 (to D. M. S.).

This article contains supplemental Figs. S1–S4.
- EMT
- epithelial mesenchymal transition
- PKA
- protein kinase A
- CREB
- cAMP-response element-binding protein
- PAI-1
- plasminogen activator inhibitor-1
- SAD
- Smad4 activation domain.
REFERENCES
- 1. Blobe G. C., Schiemann W. P., Lodish H. F. (2000) Role of transforming growth factor β in human disease. N. Engl. J. Med. 342, 1350–1358 [DOI] [PubMed] [Google Scholar]
- 2. Massagué J. (1990) The transforming growth factor-β family. Annu. Rev. Cell Biol. 6, 597–641 [DOI] [PubMed] [Google Scholar]
- 3. Massagué J. (2008) TGFβ in Cancer. Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asiedu M. K., Ingle J. N., Behrens M. D., Radisky D. C., Knutson K. L. (2011) TGFβ/TNFα-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 71, 4707–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Massagué J., Chen Y. G. (2000) Controlling of TGF-β signaling. Genes Dev. 14, 627–644 [PubMed] [Google Scholar]
- 6. Huminiecki L., Goldovsky L., Freilich S., Moustakas A., Ouzounis C., Heldin C. H. (2009) Emergence, development and diversification of the TGF-β signalling pathway within the animal kingdom. BMC Evol. Biol. 9, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ross S., Hill C. S. (2008) How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 40, 383–408 [DOI] [PubMed] [Google Scholar]
- 8. Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem. 67, 753–791 [DOI] [PubMed] [Google Scholar]
- 9. Massagué J., Seoane J., Wotton D. (2005) Smad transcription factors. Genes Dev. 19, 2783–2810 [DOI] [PubMed] [Google Scholar]
- 10. Attisano L., Lee-Hoeflich S. T. (2001) The Smads. Genome Biology 2, reviews 3010.1–reviews 3010.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Makkar P., Metpally R. P., Sangadala S., Reddy B. V. (2009) Modeling and analysis of MH1 domain of Smads and their interaction with promoter DNA sequence motif. J. Mol. Graph. Model. 27, 803–812 [DOI] [PubMed] [Google Scholar]
- 12. Wrana J. L., Attisano L. (2000) The Smad pathway. Cytokine Growth Factor Rev. 11, 5–13 [DOI] [PubMed] [Google Scholar]
- 13. Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hana J., Heldin C. H., Miyazono K., ten Dijke P. (1997) TGF-β receptor-mediated signalling through Smad2, Smad3, and Smad4. EMBO J. 17, 5353–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 15. Derynck R., Zhang Y., Feng X. H. (1998) Smads. Transcriptional activators of TGF-β responses. Cell 95, 737–740 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y., Feng X., We R., Derynck R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 383, 168–172 [DOI] [PubMed] [Google Scholar]
- 17. Wang L., Zhu Y., Sharma K. (1998) Transforming growth factor-β1 stimulates protein kinase A in mesangial cells. J. Biol. Chem. 273, 8522–8527 [DOI] [PubMed] [Google Scholar]
- 18. Taylor S. S., Buechler J A., Yonemoto W. (1990) cAMP-dependent protein kinase. Framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 59, 971–1005 [DOI] [PubMed] [Google Scholar]
- 19. Uhler M. D., Carmichael D. F., Lee D. C., Chrivia J. C., Krebs E. G., McKnight G.S. (1986) Isolation of cDNA clones coding for the catalytic subunit of mouse cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 83, 1300–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walsh D. A., Van Patten S. M. (1994) Multiple pathway signal transduction by the cAMP-dependent protein kinase. FASEB J. 8, 1227–1236 [DOI] [PubMed] [Google Scholar]
- 21. Sharma K., Wang L., Zhu Y., Bokkala S., Joseph S. K. (1997) Transforming growth factor-β1 inhibits type I inositol 1,4,5-trisphosphate receptor expression and enhances its phosphorylation in mesangial cells. J. Biol. Chem. 272, 14617–14623 [DOI] [PubMed] [Google Scholar]
- 22. Peng F., Zhang B., Wu D., Ingram A. J., Gao B., Krepinsky J. C. (2008) TGFβ-induced RhoA activation and fibronectin production in mesangial cells require caveolae. Am. J. Physiol. Renal Physiol. 295, F153–F164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chowdhury S., Howell G. M., Rajput A., Teggart C. A., Brattain L. E., Weber H. R., Chowdhury A., Brattain M. G. (2011) Identification of a novel TGFβ/PKA signaling transduceome in mediating control of cell survival and metastasis in colon cancer. PLoS One 6, e19335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang L., Duan C. J., Binkley C., Li G., Uhler M. D., Logsdon C. D., Simeone D. M. (2004) A transforming growth factor β-induced Smad3-Smad4 complex directly activates protein kinase A. Mol. Cell. Biol. 24, 2169–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li C., Wu J. J., Hynes M., Dosch J., Sarkar B., Welling T. H., Pasca di Magliano M., Simeone D. M. (2011) c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 141, 2218–2227 [DOI] [PubMed] [Google Scholar]
- 26. Deleted in proof.
- 27. Montminy M. R., Bilezikjian L. M. (1987) Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature 328, 175–178 [DOI] [PubMed] [Google Scholar]
- 28. Subramanian G., Schwarz R. E., Higgins L., McEnroe G., Chakravarty S., Dugar S., Reiss M. (2004) Targeting endogenous transforming growth factor β receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 64, 5200–5211 [DOI] [PubMed] [Google Scholar]
- 29. Diller T. C., Madhusudan, Xuong N. H., Taylor S. S. (2001) Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase. Crystal structure of the type II β regulatory subunit. Structure 9, 73–82 [DOI] [PubMed] [Google Scholar]
- 30. Canaves J. M., Taylor S. S. (2002) Classification and phylogenetic analysis of the cAMP-dependent protein kinase regulatory subunit family. J. Mol. Evol. 54, 17–29 [DOI] [PubMed] [Google Scholar]
- 31. Holmes A., Abraham D. J., Sa S., Shiwen X., Black C. M. (2001) CTGF and SMADS, maintenance of scleroderma phenotype is independent of SMAD signaling. J. Biol. Chem. 276, 10594–10601 [DOI] [PubMed] [Google Scholar]
- 32. Tsuchida K., Zhu Y., Siva S., Dunn S. R., Sharma K. (2003) Role of Smad4 on TGFβ induced extracellular matrix stimulation in mesangial cells. Kidney Int. 63, 2000–2009 [DOI] [PubMed] [Google Scholar]
- 33. Wiercinska E., Naber H. P., Pardali E., van der Pluijm G., van Dam H., ten Dijke P. (2011) The TGFβ/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res. Treat. 128, 657–666 [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Pan X., Lei W., Wang J., Shi J., Li F., Song J. (2006) Regulation of transforming growth factor-β1-induced apoptosis and epithelial-to-mesenchymal transition by protein kinase A and signal transducers and activators of transcription 3. Cancer Res. 66, 8617–8624 [DOI] [PubMed] [Google Scholar]
- 35. Derynck R., Zhang Y. E. (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425, 577–584 [DOI] [PubMed] [Google Scholar]
- 36. Heldin C. H., Miyazono K., ten Dijke P. (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- 37. Remy I., Montmarquette A., Michnick S. W. (2004) PKB/Akt modulates TGF-β signalling through a direct interaction with Smad3. Nat. Cell Biol. 6, 358–365 [DOI] [PubMed] [Google Scholar]
- 38. Liu X., Sun Y., Weinberg R. A., Lodish H. F. (2001) Ski/Sno and TGF-β signaling. Cytokine Growth Factor Rev. 12, 1–8 [DOI] [PubMed] [Google Scholar]
- 39. Luo K. (2004) Ski and SnoN. Negative regulators of TGF-β signaling. Curr. Opin. Genet. Dev. 14, 65–70 [DOI] [PubMed] [Google Scholar]
- 40. de Caestecker M. P., Hemmati P., Larisch-Bloch S., Ajmera R., Roberts A. B., Lechleider R. J. (1997) Characterization of functional domains within Smad4/DPC4. J. Biol. Chem. 272, 13690–13696 [DOI] [PubMed] [Google Scholar]
- 41. de Caestecker M. P., Yahata T., Wang D., Parks W. T., Huang S., Hill C. S., Shioda T., Roberts A. B., Lechleider R. J. (2000) The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. J. Biol. Chem. 275, 2115–2122 [DOI] [PubMed] [Google Scholar]
- 42. Kay B. K., Williamson M. P., Sudol M. (2000) The importance of being proline. The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14, 231–241 [PubMed] [Google Scholar]
- 43. Huang L. J., Taylor S. S. (1998) Dissecting cAMP binding domain A in the RIα subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J. Biol. Chem. 273, 26739–26746 [DOI] [PubMed] [Google Scholar]
- 44. Banky P., Roy M., Newlon M. G., Morikis D., Haste N. M., Taylor S. S., Jennings P. A. (2003) Related protein-protein interaction modules present drastically different surface topographies despite a conserved helical platform. J. Mol. Biol. 330, 1117–1129 [DOI] [PubMed] [Google Scholar]
- 45. Newlon M. G., Roy M., Morikis D., Carr D. W., Westphal R., Scott J. D., Jennings P. A. (2001) A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 20, 1651–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Newlon M. G., Roy M., Morikis D., Hausken Z. E., Coghlan V., Scott J. D., Jennings P. A. (1999) The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat. Struct. Biol. 6, 222–227 [DOI] [PubMed] [Google Scholar]
- 47. Døskeland S. O., Ogreid D. (1984) Characterization of the interchain and intrachain interactions between the binding sites of the free regulatory moiety of protein kinase I. J. Biol. Chem. 259, 2291–2301 [PubMed] [Google Scholar]
- 48. Robinson-Steiner A. M., Corbin J. D. (1983) Probable involvement of both intrachain cAMP binding sites in activation of protein kinase. J. Biol. Chem. 258, 1032–1040 [PubMed] [Google Scholar]
- 49. Herberg F. W., Taylor S. S., Dostmann W. R. (1996) Active site mutations define the pathway for the cooperative activation of cAMP-dependent protein kinase. Biochemistry 35, 2934–2942 [DOI] [PubMed] [Google Scholar]
- 50. Shabb J. B., Poteet C. E., Kapphahn M. A., Muhonen W. M., Baker N. E., Corbin J. D. (1995) Characterization of the isolated cAMP-binding B domain of cAMP-dependent protein kinase. Protein Sci. 4, 2100–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heldin C. H., Landström M., Moustakas A. (2009) Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 21, 166–176 [DOI] [PubMed] [Google Scholar]
- 52. Moustakas A., Heldin C. H. (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 98, 1512–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moreno-Bueno G., Portillo F., Cano A. (2008) Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 27, 6958–6069 [DOI] [PubMed] [Google Scholar]
- 54. Yang J., Weinberg R. A. (2008) Epithelial-mesenchymal transition. At the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829 [DOI] [PubMed] [Google Scholar]
- 55. Perl A. K., Wilgenbus P., Dahl U., Semb H., Christofori G. (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392, 190–193 [DOI] [PubMed] [Google Scholar]
- 56. Takano S., Kanai F., Jazag A., Ijichi H., Yao J., Ogawa H., Enomoto N., Omata M., Nakao A. (2007) Smad4 is essential for down-regulation of E-cadherin induced by TGF-β in pancreatic cancer cell line PANC-1. J. Biochem. 141, 345–351 [DOI] [PubMed] [Google Scholar]
- 57. Cano A., Pérez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., Nieto M. A. (2000) The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherinexpression. Nat. Cell Biol. 2, 76–83 [DOI] [PubMed] [Google Scholar]
- 58. MacPherson M. R., Molina P., Souchelnytskyi S., Wernstedt C., Martin-Pérez J., Portillo F., Cano A. (2010) Phosphorylation of serine 11 and serine 92 as new positive regulators of human Snail 1 function. Potential involvement of casein kinase-2 and the cAMP-activated kinase protein kinase A. Mol. Biol. Cell 21, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Durand M. K. V., Bodker J. S., Christensen A., Dupont D. M., Hansen M., Jensen J/K/., Kjelgaard S., Mathiasen L., Pedersen K. E., Skeldal S., Wind T., Andreasen P. A. (2004) Plasminogen activator inhibitor-1 and tumor growth, invasion, and metastasis. Thromb. Haemostasis 91, 428–449 [DOI] [PubMed] [Google Scholar]