

Abstract
Among amphitropic proteins, human glycolipid transfer protein (GLTP) forms a structurally-unique fold that translocates on/off membranes to specifically transfer glycolipids. Phosphatidylcholine (PC) bilayers with curvature-induced packing stress stimulate much faster glycolipid intervesicular transfer than nonstressed PC bilayers raising questions about planar cytosol-facing biomembranes being viable sites for GLTP interaction. Herein, GLTP-mediated desorption kinetics of fluorescent glycolipid (tetramethyl-boron dipyrromethene (BODIPY)-label) from lipid monolayers are assessed using a novel microfluidics-based surface balance that monitors lipid lateral packing while simultaneously acquiring surface fluorescence data. At biomembrane-like packing (30–35 mN/m), GLTP uptake of BODIPY-glycolipid from POPC monolayers was nearly nonexistent but could be induced by reducing surface pressure to mirror packing in curvature-stressed bilayers. In contrast, 1-palmitoyl-2-oleoyl-phosphatidylethanolamine (POPE) matrices supported robust BODIPY-glycolipid uptake by GLTP at both high and low surface pressures. Unexpectedly, negatively-charged cytosol-facing lipids, i.e., phosphatidic acid and phosphatidylserine, also supported BODIPY-glycolipid uptake by GLTP at high surface pressure. Remarkably, including both 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (5 mol%) and POPE (15 mol%) in POPC synergistically activated GLTP at high surface pressure. Our study shows that matrix lipid headgroup composition, rather than molecular packing per se, is a key regulator of GLTP-fold function while demonstrating the novel capabilities of the microfluidics-based film balance for investigating protein-membrane interfacial interactions.
Keywords: BODIPY-glycosphingolipid fluorescence, protein interaction with planar model membranes, lipid composition, microfluidic surface balance, lipid monolayer lateral packing, glycolipid transfer protein
Proteins are considered to be amphitropic when translocation to/from biomembranes: i) is required for function; ii) occurs transiently and reversibly from the cytoplasm; and iii) involves interaction with the lipid bilayer (1–3). Glycolipid transfer proteins (GLTPs) comprise a structurally-unique amphitropic protein superfamily defined by their ability to reversibly interact with membranes to achieve selective transport of glycosphingolipids (4). Human GLTP resides in the cytosol (5), but is capable of delivering glucosylceramide to the cytosolic face of the plasma membrane (6, 7) and contains a nonclassical diphenylalanine-in-an-acidic-tract targeting sequence to the endoplasmic reticulum (8). GLTPs occur widely among eukaryotes (4, 9) and GLTP-like domains serve as key functional regions in larger human proteins, i.e., FAPP2 (10, 11). The GLTP-fold is dominated by α-helices, lacks intramolecular disulfides, and uses a two-layer “sandwich” motif to form a single glycolipid binding site consisting of a surface-localized sugar headgroup recognition center and an expandable hydrophobic pocket for accommodation of the ceramide hydrocarbon chains of the glycolipid (12–17). During glycolipid transfer, GLTP interacts with membranes transiently and in a minimally perturbing manner (18, 19) via a membrane interaction domain formed by a cluster of Trp, Tyr, Lys, and nonpolar residues on the GLTP surface region adjacent to the glycolipid binding site. The spatial organization of these “membrane interaction residues” (12) differs from the membrane-docking domains of other amphitropic proteins, i.e., C1, C2, PH, PX, and FYVE domains (20).
In vitro studies of GLTP involving model membranes typically have utilized a phosphatidylcholine (PC) matrix for the glycolipid. With highly-curved PC bilayer vesicles, dramatically enhanced GLTP partitioning affinity and associated glycolipid transfer rates are observed compared with noncurvature-stressed vesicles (18, 21). Also, GLTP penetration into PC monolayers becomes undetectable above 23–26 mN/m (18, 22), i.e., packing pressures well below the 30–35 mN/m range that mimics nonstressed biomembranes. The findings raise questions about the viability of planar noncurvature-stressed membranes that typically occur in vivo for GLTP interaction. However, two factors currently limit understanding of the issues. First, the matrix lipid compositions studied to date have been rather simple, i.e., binary component, and have lacked the compositional complexity exhibited by cytosolic-facing biomembranes. So it remains unclear whether biomembrane-like lipid compositions can actually promote GLTP interaction and/or penetration into lipid monolayers at high surface pressures that mimic lipid packing in biomembranes. A second issue relates to the increase in average lipid headgroup spacing, i.e., free area, associated with positive curvature stress of the lipid matrix. Can such curvature stress alter or mask matrix lipid regulatory effects such as those of negatively-charged lipids on GLTP action? Gaining insight is important because negatively-charged phosphoglycerides typically reside or can be generated in membrane surfaces facing the cytosol (23), the cellular location where GLTP and many other amphitropic proteins localize.
Lipid monolayers provide a useful system for investigating lipid lateral interactions and protein partitioning to lipid interfaces while controlling lipid packing conditions (24). Besides serving as a model for half of a membrane bilayer, monolayers offer the advantage of maintaining the same macroscopic form over broad lipid compositional ranges and for different lipid types without transition to other mesomorphic phase structures. This is especially true for studies of lipid lateral packing involving nonbilayer-forming lipids such as phosphatidylethanolamine (PE). In the present study, we use a custom-developed microfluidics-based monolayer apparatus that simultaneously measures surface pressure and fluorescence to monitor the removal of boron dipyrromethene (BODIPY)-labeled glycolipid (1 mol%) by GLTP from monolayers comprised of different lipids and their mixtures. The ability to independently and systematically vary lipid packing and composition, parameters not easily controlled when biomolecule adsorption is studied using solid surface-supported lipids or vesicles (e.g., surface plasmon resonance), provides a new avenue for deciphering how lipid compositional changes regulate membrane interactions of amphitropic proteins. Defining the parameters that regulate amphitropic protein interaction with the lipid bilayer could also provide new ways to control their functionality.
MATERIALS AND METHODS
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), and POPC were obtained from Avanti Polar Lipids (Alabaster, AL). 1-Palmitoyl-2-[15-(4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene-8-yl)pentadecanoyl]sn-glycero-3-phosphocholine (B15-PC) and D-galactosyl-β-1,1′-N-[15-(4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene-8-yl)pentadecanoyl]D-erythro-sphingosine (B15-GalCer) were synthesized as reported previously (25–27). Use of tetramethyl BODIPY lipids (versus dimethyl BODIPY lipids) enhanced fluorophore embedding among matrix lipid chains. Lipids were spread from 1.5 mM stock solutions in hexane/ethanol (90:10, HPLC grade). Subphase buffer consisted of 50 mM sodium phosphate (pH 6.6), 100 mM NaCl, and ultrapure water (Millipore Q system).
Microfluidics-based surface balance/fluorescence platform
Figure 1A illustrates the platform components and their application to investigation of the GLTP “half-reaction” (not drawn to scale). All components, except the computer, fluorimeter, and laser power supply, are enclosed in a temperature-controlled box (not shown) maintained at 24°C. Figure 1C shows the actual surface balance which consists of a rectangular channel (0.23 × 1.5 × ∼0.015 cm) that was milled into the base (2.54 × 1.6 × 0.32 cm) cut from a 4 optical density glass absorbance filter (Edmunds Optics, Barrington, NJ). Prior to milling, the upper surface of the base was rendered hydrophobic by treating with 1H,1H,2H,2H-perfluorooctyltrichlorosilane. The base was mounted in an aluminum block recess and under a plexiglass cover to maintain a humidified argon atmosphere. Solutions were delivered/removed from the channel through PEEK tubing via three larger ports (Fig. 1C, left to right: waste, sucrose, and buffer; 0.159 cm outer diameter × 102 µm inner diameter) and six smaller sample ports (360 µm outer diameter × 150 µm inner diameter) externally manifolded together. Sample or buffer was delivered at 10 µl/min using Instech Laboratories P625/10683 peristaltic pumps (Plymouth Meeting, PA) fitted with 0.051 cm tubing. The linear flow rate in the microfluidic channel was ∼0.05 cm/s. Waste was removed using a model 625/900 pump fitted with 0.038 cm tubing. Sucrose (50% in buffer) was delivered at 2.0 µl/min using a World Precision Instruments (Sarasota, FL) SP260P syringe pump. Due to higher density, the sucrose solution moving along the bottom of the channel prevented protein adsorption. The beam from a 630 nm diode laser mounted to the aluminum block was directed across the top of the channel to a photodiode to control the waste pump. This setup enabled the channel liquid depth to be kept constant while variation of the reference voltage allowed the height of the liquid meniscus to be controlled. During normal operation the meniscus height was set to within a few microns of zero, creating an almost flat air/liquid interface. Studies of colipase and glucagon adsorption and desorption kinetics (28, 29) guided instrument design features.
Fig. 1.

A: Microfluidic flow cell monolayer apparatus configured to monitor surface pressure while simultaneously measuring fluorescence intensity (not drawn to scale; see Materials and Methods for a detailed description). B: Chemical structure of B15-GalCer fluorophore. C: Photo of microfluidic flow trough. D: Docking of GLTP containing bound 24:1-GalCer with the monolayer interface as predicted by the Orientation of Proteins in Membranes computational approach and analyzed by Trp fluorescence (19).
Air-exposed surfaces of the cell were cleaned by aspiration, followed by methanol, and then water aspiration prior to initiating buffer flow and recording surface tension/pressure. A µTrough S sensor (Kibron, Inc., Helsinki, Finland) consisting of a Wilhelmy probe, i.e., nichrome wire (28-gauge; ∼0.31 mm), was used to monitor surface tension/pressure. Buffer values in the cell agreed within 0.2 mN/m with those measured at a much larger interface. Buffer consisted of 10 mM potassium phosphate, 0.1 M NaCl buffer, pH 6.6. To set the desired surface pressure, the buffer surface was touched with a clean stainless steel wire that had been dipped into lipid solution dissolved in hexane/ethanol (9:1). Any excess lipid was removed by touching a clean wire to the surface to reduce surface pressure to the desired value. The monolayer was allowed to stabilize for ∼5 min prior to solute introduction into the flowing subphase.
During cell operation, fluorophore at the gas-liquid interface or in the subphase was excited by light from a JDSU 2212-4S 488 nm Ar laser (Santa Rosa, CA) through one arm of a bifurcated optical fiber assembly (2 × 600 µm diameter), mounted at 50° relative to the interfacial plane. The illuminated excitation spot was ∼1 mm wide. Emitted light entered the other arm of the bifurcated fiber and was passed through 490 nm cutoff and 500–570 nm bandpass filters before being projected onto a cooled two-dimensional CCD array detector (ANDOR DV420-BU2 CCD; Andor Technology, South Windsor, CT). Emission from the monolayers was measured for 0.5 s at 3 s intervals using 10% maximum laser intensity to avoid photobleaching the BODIPY fluorophore. Fluorescence data were recorded and quantified by summing the intensities of illuminated detector pixels and processed using Andor MCD v2.62. Single pixel intensity spikes >50 counts higher than adjacent pixels were presumed to originate from cosmic rays and were replaced by the adjacent pixel average. Data from the CCD camera and µTrough S sensor were analyzed on a desktop computer. Net fluorescence emission intensities were calculated by subtraction of instrumental dark spectra and the integrated intensity recorded before BODIPY-labeled lipid was introduced onto the buffer surface of the flow cell.
GLTP expression/purification
Human GLTP (GenBank AF209704) was expressed and affinity-purified (30) using pET-30 Xa/LIC vector (Novagen). 6× His-S-tag cleavage yielded native GLTP, avoiding subtle changes to protein conformation and glycolipid binding induced by N-terminal fusion tags (13). Near-UV CD confirmed native global folding (15, 16, 19). Protein purity and concentration were determined by SDS-PAGE and bicinchoninic acid assay (31).
RESULTS AND DISCUSSION
Glycolipid removal by GLTP is mitigated by PC but induced by PE
In eukaryotic membranes, PC is typically a predominant lipid constituent that drives spontaneous bilayer formation. Not surprisingly, PC is a popular model membrane matrix. We initially investigated the ability of GLTP to acquire galactosylceramide (1 mol%) labeled with tetramethyl BODIPY via a pentadecanoyl acyl linker (B15-GalCer) from POPC monolayers poised at 30 mN/m, a surface pressure that mimics packing conditions in planar biomembranes (32, 33). GLTP is known to bind and transfer without chemically altering the glycolipid itself (31). Figure 1 depicts the novel custom-designed miniature surface balance/surface fluorescence apparatus used in our study. It consists of an open microfluidic flow cell equipped with a Wilhelmy nichrome wire for measuring surface pressure and a laser-coupled fiber optic cable/diode array platform for acquiring fluorescence spectra. The fluorescence detection capability enabled direct assessment of GLTP-mediated departure of B15-GalCer from monolayers composed of different matrix lipids into a flowing subphase while controlling the lateral packing state.
Figure 2A, B shows that introduction of GLTP into the flowing subphase beneath the POPC monolayer results in both the surface pressure (upper panel) and the surface fluorescence signal of the B15-GalCer (lower panel) remaining relatively unchanged over 20 min, indicating a lack of glycolipid removal by GLTP. The finding is noteworthy because POPC remains fluid (liquid-expanded) at 30 mN/m and is packed similarly as in planar membranes. We hypothesized that the lack of B15-GalCer uptake by GLTP from the fluid PC monolayers reflects shielding by the close packed PC headgroups that limits access of GLTP to the smaller and less-hydrated galactose headgroup of GalCer (34). The phosphocholine headgroup is capable of limiting PC molecular lateral packing density in bilayers and monolayers, especially when the acyl chains contain few cis-unsaturated bonds (35–38). This “spacer effect” of the PC headgroup arises because the highly hydrated and relatively bulky PC headgroup orients almost orthogonally relative to the packed acyl chains of bilayers and monolayers rather than remaining aligned with the acyl chains. As a result, the PC headgroup can exert a shielding or “umbrella effect” over other neighboring lipids with small headgroups (e.g., GalCer, cholesterol) present as minor constituents (39). Support for this idea also comes from a recent study (40) of lectin interaction with neoglycolipid dispersed in planar PC films. The insertion of a linker to vertically displace the sugar relative to the PC headgroups was needed to ensure maximal lectin binding.
Fig. 2.
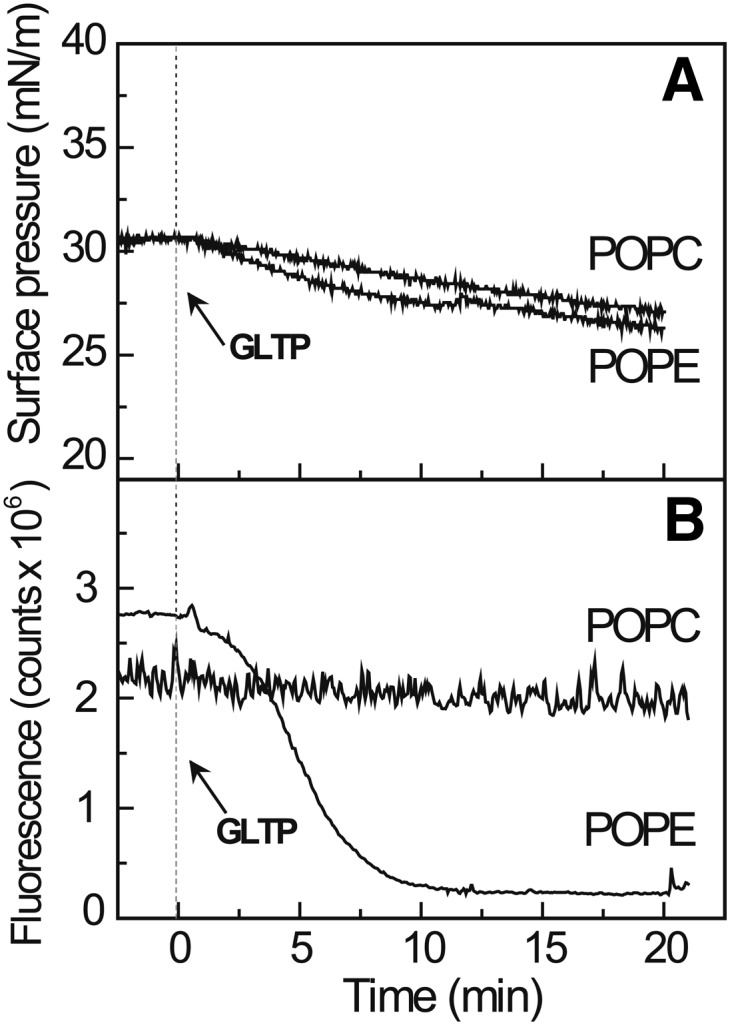
Monolayer matrices composed of POPE, but not POPC, support GalCer removal by GLTP. A: Surface pressure as a function of time for POPC or POPE monolayers, poised at 30 mN/m and containing 1 mol% B15-GalCer, prior to and after introduction of GLTP (3.0 µM) into the flowing subphase. B: Surface fluorescence changes as a function of time for POPC or POPE monolayers, under the same conditions as (A) showing the removal of B15-GalCer from the POPE, but not the POPC films, by GLTP.
To test our hypothesis, we switched the monolayer matrix to POPE because its smaller phosphoethanolamine headgroup lacks the three methyl groups that surround the terminal ammonium group of PC but still remains zwitterionic like PC. Figure 2B shows that the POPE monolayer matrix induces rapid loss of surface fluorescence, reflecting removal of B15-GalCer by GLTP, despite the initial surface pressure being 30 mN/m, i.e., packing similar to planar biomembranes (32, 33). The minimal surface pressure change observed during B15-GalCer removal by GLTP is consistent with the low surface concentration of glycolipid (1 mol%) in the POPE monolayer (Fig. 2A).
B15-GalCer removal from POPE monolayers depends on GLTP concentration and glycolipid surface concentration
To confirm that the time-dependent decrease in surface fluorescence from POPE monolayers reflects B15-GalCer removal by GLTP, the rate of B15-GalCer signal decrease from the POPE monolayer was studied as a function of GLTP concentration (0.1 to 3.0 µM) in the flowing subphase (Fig. 3A). Higher GLTP concentrations produced faster removal of B15-GalCer from the POPE monolayer. At 3.0 µM GLTP, removal of the B15-GalCer (1 mol%) begins within 30 s of protein introduction into the subphase flow and is almost complete within 5 min. At 0.6 µM GLTP, an ∼4.5 min time lag is observed prior to onset of B15-GalCer removal that takes 20 min to complete. At 0.1 µM GLTP, the removal rate of B15-GalCer is so slow that any onset time lag is obscured and the emission signal is only slightly diminished after 20 min. The lack of saturation with GLTP suggests weak binding to POPE. The linear relationship between GLTP concentration and maximum rate of B15-GalCer fluorescence signal loss from the POPE monolayer surface suggests a first-order relationship (Fig. 3B) although the precise origin of the initial lags in B15-GalCer removal at low GLTP concentrations is unclear. One possibility is diffusion-controlled time-dependent equilibration of GLTP into the unstirred layer adjacent to the lipid monolayer interface. Other studies have shown that such equilibration of protein concentration requires a few seconds following introduction into the flowing system. Figure 3C shows how different initial concentrations of B15-GalCer in POPE monolayers affect the initial fluorescence intensity and subsequent rate of decrease after introduction of GLTP into the flowing subphase beneath the POPE monolayer. Higher initial B15-GalCer surface concentrations yielded higher emission intensity and more rapidly decreasing signal upon exposure to equivalent subphase concentrations of GLTP. Analysis of highlighted data in Fig. 3C revealed a linear relationship between maximum glycolipid removal rate by GLTP and initial B15-GalCer concentration in POPE monolayers, as indicated by normalized initial fluorescence intensity (Fig. 3D). By keeping B15-GalCer concentration low (≤1 mol%), maximal emission intensity is achieved while minimizing self-quenching linked to energy transfer from excited-state monomers to ground-state dimers and complications arising from homo-Förster resonance energy transfer that occurs at high BODIPY concentrations in monolayers (25, 41, 42) and bilayers (43, 44).
Fig. 3.

Surface fluorescence rate change depends on concentration of subphase GLTP and monolayer glycolipid. A: Effect of GLTP subphase concentration (0.1, 0.3, 0.6, 1.0, and 3.0 µM) on B15-GalCer uptake from POPE monolayers as a function of time. Initial surface pressure, 25 mN/m. B: The symbol-delineated region in (A) was used to analyze the removal rate of B15-GalCer as a function of GLTP concentration. C: Effect of B15-GalCer surface concentration (0.1, 0.2, 0.5, and 1 mol%) on uptake by GLTP from POPE monolayers as a function of time. GLTP subphase concentration, 3.0 µM. No differences in the corresponding surface pressure versus time profiles for the B15-GalCer initial surface concentrations were evident (not shown). D: The symbol-delineated region in (C) was used to analyze the glycolipid removal rate as a function of B15-GalCer concentration.
B15-GalCer removal by GLTP is regulated by surface pressure but in a phospholipid composition-dependent manner
To assess the relative importance of monolayer matrix lipid composition versus lipid interfacial packing in regulating GLTP accessibility to B15-GalCer, glycolipid removal from POPC and POPE was compared at different initial surface pressures (Fig. 4). Because glycolipid removal rate depends on protein concentration, a low GLTP concentration (0.6 µM) was used in the flowing subphase. The data show that GLTP was unable to extract B15-GalCer (1 mol%) from POPC monolayers at initial surface pressures ≥24 mN/m. Below the threshold of ∼24 mN/m (Fig. 4B), glycolipid removal by GLTP commenced with the removal rate increasing as a function of decreasing initial pressure. When POPC monolayers were initially poised at surface pressures ≤20 mN/m, introduction of GLTP into the flowing subphase resulted in a sudden elevation of surface pressure (e.g., Fig. 4A, π = 15 mN/m) that preceded the typical loss of surface fluorescence associated with B15-GalCer departure (Fig. 4B). The response was consistent with deeper GLTP penetration into the monolayer, likely reflecting the increased “free area” available for GLTP adsorption (45) and the surface activity of GLTP itself. GLTP adsorption to lipid-free air/buffer interfaces results in equilibrium surface pressures of 17–19 mN/m (22).
Fig. 4.

Surface pressure regulates glycolipid removal rate from the monolayer in a phospholipid composition-dependent manner. The upper panels show the surface pressure change as a function of time when the monolayer matrix is POPC (A) or POPE (C), respectively. The initial fixed surface pressures are 15, 20, 24, 30, and 33 mN/m for POPC and 23, 25, 27, 29, 32, and 36 mN/m for POPE. Lower panels (B) and (D) show the corresponding surface fluorescence response as a function of time for the different initial surface pressures in (A) and (C). B15-GalCer uptake by GLTP from POPE occurs in a surface pressure-dependent manner but uptake from POPC ceases at surface pressures ≥24 mN/m. Additional data obtained at low initial surface pressures for POPE matrix monolayers is provided in supplementary Fig. I.
The lack of B15-GalCer departure from POPC at high surface pressures (30–35 mN/m) and stimulation by lower surface pressures (<24 mN/m) are consistent with the known ability of GLTP to transfer glycolipid rapidly when PC vesicles are small and highly curved compared with much slower transfer that occurs when PC vesicles are large and lack curvature stress (21). While surface pressures of 30–35 mN/m (or higher) generate lipid packing similar to unstressed fluid planar bilayers, surface pressures in the 20–25 mN/m range produce lipid packing that resembles the stressed environment in high-curvature PC vesicle outer surfaces (32, 33).
With POPE monolayers (Fig. 4C, D; supplementary Fig. I), B15-GalCer departure also was rapid at lower surface pressures (20–25 mN/m), but persisted at high surface pressures (30–35 mN/m) (Fig. 4D). Surface pressures >36 mN/m were needed to block glycolipid removal from POPE which remains in a fluid liquid-expanded state up to ∼40 mN/m. Above ∼40 mN/m, POPE undergoes a phase transition and both liquid-expanded and liquid-condensed phases coexist (46). Figure 5A shows that the initial removal rate of B15-GalCer is linear with respect to surface pressure regardless of whether the fluid-phase monolayer matrix is POPE or POPC, but the removal rate is much faster from POPE than from POPC and persists at higher surface pressures for POPE compared with POPC. Thus, both matrix lipid composition and lateral packing control the glycolipid removal rate. The smaller PE headgroup not only induces faster B15-GalCer removal by GLTP but also enables transfer at surface pressures producing lipid packing conditions associated with planar membranes. By contrast, the PC headgroup supports slower glycolipid removal but only when surface pressures are low enough to produce lipid packing that mimics curvature-stressed bilayers.
Fig. 5.

Effect of initial monolayer surface pressure and BODIPY-glycolipid surface density in PC or PE matrix on glycolipid uptake rate by GLTP. A: Uptake rates of B15-GalCer (1 mol%) by GLTP (0.6 µM) from either POPE or POPC monolayers poised at different initial surface pressures. GLTP uptake of B15-GalCer is ∼7-fold faster from POPE than from POPC monolayers at 24 mN/m. B: B15-GalCer surface densities in POPE and POPC monolayers and removal rates by GLTP. For each lipid matrix, higher surface density of glycolipid slows its removal rate, but much faster removal occurs from POPE than POPC at similar glycolipid surface densities. Data for (A) and (B) were derived from the symbol-delineated regions of Fig. 4C and D, respectively. Additional data showing the surface pressure versus molecular area isotherms used to calculate lipid surface densities of POPC and POPE matrices and the impact of POPC and POPE surface density on the B15-GalCer uptake rate by GLTP are provided in supplementary Fig. II.
Another consequence of the smaller PE headgroup is higher surface density of B15-GalCer in POPE compared with POPC at equivalent surface pressure. To determine if this situation accounts for the faster glycolipid extraction rate by GLTP, desorption rates were plotted as a function of B15-GalCer (pmol) per monolayer surface area (cm2) for each phosphoglyceride (Fig. 5B). Calculation of the B15-GalCer surface density is provided as supplementary information. While the B15-GalCer initial departure rate shows a linear dependence on glycolipid surface density in POPE and POPC, the departure rate actually slows as the B15-GalCer surface concentrations increase in either matrix lipid. Furthermore, the departure rates supported by POPE and POPC differ dramatically at identical B15-GalCer surface concentrations. When B15-GalCer surface densities exceed ∼2.6 pmol/cm2, departure from POPC ceases but remains fast from POPE. Supplementary Figure II shows that B15-GalCer departure exhibits similar trends when analyzed in terms of POPE and POPC surface density. These findings lead to the conclusion that the phosphoglyceride headgroup chemical composition and not the glycolipid or matrix lipid surface density per se ultimately controls whether GLTP interaction occurs when lipid packing mimics planar fluid-phase biomembranes.
GLTP removal of glycolipid from phosphatidic acid and phosphatidylserine monolayers
To determine if a PE matrix is the only phosphoglyceride matrix that supports efficient glycolipid removal by GLTP at high surface pressures, other phosphoglycerides were tested. Figure 6 shows that POPA and POPS both support fluorescent glycolipid removal by GLTP at high surface pressure despite being negatively-charged monolayer matrices. It is noteworthy that GLTP introduction into the flowing solution beneath the POPA and POPS monolayers results in sudden increases in surface pressure of 2–3 mN/m despite the relatively high initial surface pressures (∼30 mN/m) of the monolayers prior to protein addition (Fig. 6A). The sudden spikes in surface pressure are consistent with enhanced adsorption/penetration of protein into each lipid monolayer. Subsequently, the surface pressure becomes stable with the POPA surface pressure decreasing slightly over the 20 min experimental interval while the surface pressure of the POPS matrix increases slightly. The overall fluorescence response of B15-GalCer (Fig. 6B) is consistent with glycolipid removal by GLTP from the POPA matrix being rapid. Unlike a POPS matrix, which supports continuous and steady departure of B15-GalCer, GLTP interaction with a POPA monolayer matrix induces a sudden dramatic increase in the glycolipid fluorescence signal that gives way to a subsequent exponential decrease. The initial increase in fluorescence intensity suggests that POPA provides a favorable environment for GLTP-induced lateral dispersion of B15-GalCer that promotes glycolipid uptake, partially relieving the self-quenching exhibited by BODIPY lipids even at low concentrations in monolayers (35, 41, 42). In any case, the stimulation of fluorescent glycolipid desorption by phosphatidic acid (PA) and phosphatidylserine (PS) was unexpected because negatively-charged phosphoglycerides at 5 or 10 mol% in POPC small vesicles reportedly slowed GLTP transfer at low ionic strength (47). Restoration, but not stimulation, of GalCer transfer rates was observed at physiological ionic strength compared with POPC vesicles lacking negatively-charged phosphoglycerides.
Fig. 6.
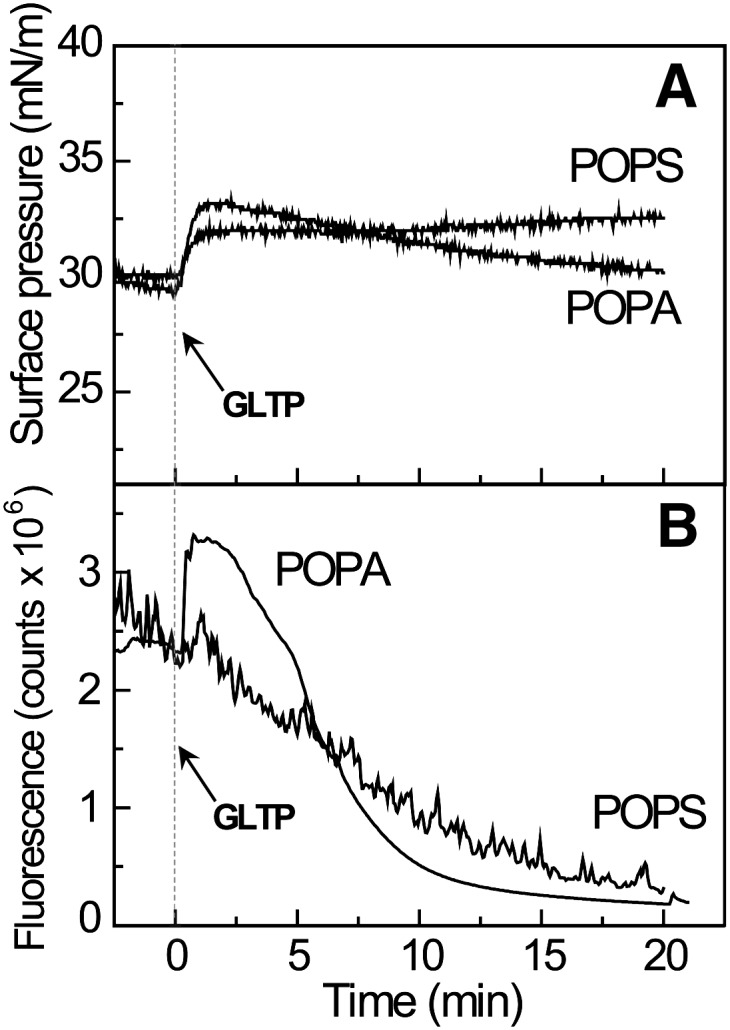
Negatively-charged matrix lipids stimulate removal of B15-GalCer by GLTP. A: Surface pressure as a function of time for POPA or POPS monolayers, poised at 30 mN/m and containing 1 mol% B15-GalCer prior to and after introduction of GLTP (3.0 µM) into the flowing subphase. B: Fluorescence changes as a function of time for POPA or POPS monolayers, under the same conditions as (A) showing the removal of B15-GalCer by GLTP.
POPE and POPA synergistically promote GLTP-mediated removal of B15-GalCer, but not B15-PC, from POPC monolayers
Biomembranes such as the plasma membrane have complex lipid compositions with different lipids distributed asymmetrically in each half of the bilayer. PC and sphingomyelin dominate the exterior-facing leaflet while PE and negatively-charged phosphoglycerides are highly enriched in the cytosol-facing leaflet (48). Because GLTP resides in the cell cytosol (5), we hypothesized that lipid compositions such as those in the cytosol-facing leaflet of the plasma membrane might enhance GLTP uptake of glycolipid compared with simple PC bilayer matrices or PC/sphingomyelin bilayers. We assessed whether PE and PA can promote GLTP action when embedded in POPC monolayers. Figure 7A shows that including 15 mol% POPE in POPC monolayers poised at high surface pressures (∼30 mN/m) only marginally stimulated removal of B15-GalCer, but not B15-PC, by GLTP (3.0 µM). In contrast, inclusion of only 5 mol% POPA in POPC monolayers moderately stimulated removal of B15-GalCer, but not B15-PC, by GLTP following an ∼4 min time lag (Fig. 7B). Notably, however, when POPE (15 mol%) and POPA (5 mol%) both were mixed with POPC, the removal of B15-GalCer, but not B15-PC, by GLTP dramatically increased (Fig. 7C) and the time lag prior to onset of B15-GalCer departure decreased nearly 10-fold (∼30 s).
Fig. 7.

POPE and POPA synergistically enhance B15-GalCer removal by GLTP from POPC monolayers. Changes in fluorescence intensity of B15-GalCer or B15-PC induced by GLTP (3.0 µM) flowing beneath POPC monolayers (∼32 mN/m) and containing: 15 mol% POPE (A); 5 mol% POPA (B); or both 15 mol% POPE and 5 mol% POPA (C). B15-PC served as control lipid.
In earlier monolayer studies involving GLTP, surface pressure change was used to detect GLTP adsorption and/or glycolipid removal (22). While a drop in surface pressure occurred with GalCer films at 36 mN/m, no change was observed with dipalmitoylphosphatidylcholine monolayers. POPC and sphingomyelin, which also have phosphocholine headgroups, did not support glycolipid uptake by GLTP. By investigating other physiologically-relevant fluid-phase phosphoglycerides, we have identified POPE and POPA as positive stimulators of GLTP uptake of glycolipids from planar monolayer matrices under lipid-packing conditions that mimic biomembranes. It is noteworthy that the stimulatory effect for POPA occurs at physiological ionic strength. This finding was unexpected because the presence of negatively-charged phosphoglycerides (5 and 10 mol%) in POPC donor vesicles previously had been shown to decrease glycolipid intervesicular transfer at low ionic strength while having minimal effect at physiological ionic strength (47). Moreover, enhanced partitioning of GLTP to donor POPC vesicles containing negatively-charged phosphoglycerides appeared to be responsible for the slowdown in glycolipid transfer (18). Fluid-phase PE, a zwitterionic lipid, had not been investigated in earlier studies involving GLTP. We suspect that the earlier use of small highly-curved vesicles masked some of the subtle packing features that exist in planar matrix environments containing PA or PC/PA mixtures, as appears to be the case for sterol (49).
From the molecular standpoint, PA, PS, and PE display similar cone-like molecular shapes that originate from the relatively small cross-sectional areas of their polar headgroups compared with their nonpolar acyl chain regions. This feature enables pure PE, PS, and PA to form inverted hexagonal II phase in excess water (50–54). The origins of the reduced headgroup size (relative to PC) can be traced to lower hydration and the ability to participate in intermolecular hydrogen bonding. It is noteworthy that small amounts of PA enhance the tendency of PE to form inverted hexagonal II phase (55). By contrast, the larger well-hydrated headgroup of PC imparts a cylindrical molecular shape that results in bilayer formation in excess water. In a POPC matrix, the molecular lateral stress profile (from “head to toe”) is less severe than in PE or PA matrices where the diglyceride regions limit close packing and leave more free area for the smaller and less hydrated headgroups (56). The diminished headgroup packing density appears to facilitate GLTP adsorption and improve access to the glycolipid headgroup. By contrast, with a PC matrix, the larger highly-hydrated and orthogonally-tilted headgroups pack more closely, and thus can exert a shielding “umbrella-like” effect over the smaller less hydrated GalCer (1 mol%). Such effects are expected to impede interaction by GLTP which binds glycolipids monovalently and shows very limited ability to penetrate into and perturb POPC vesicles during transient interaction (18). As noted earlier, despite the increased holding capacity of multivalent lectin, neoglycolipid embedded in planar PC films needs a spacer linker to improve its sugar headgroup accessibility and overcome the PC headgroup inhibitory effect (40). Also, antibody binding to sulfated GalCer (sulfatide) in PC/cholesterol liposomes reportedly is regulated by changes in hydrocarbon chain lengths of either the PC matrix or sulfated GalCer (sulfatide) ligand (57). However, the presence of other serum proteins and large amounts of cholesterol could impact the lateral distribution of the glycolipid and complicate this study.
Within a planar PC matrix, the lateral pressure profile of incorporated PE manifests itself as curvature-stress “frustration” (56, 58) which can be exacerbated by PA (55). While membrane partitioning enhancement of other peripheral amphitropic proteins by PE is known (59–61), what is noteworthy about the stimulation of the GLTP fold is the marked synergistic effect that only 5 mol% PA has on surfaces comprised of 15% POPE and 80% POPC. Because the GLTP sugar headgroup recognition center is surrounded by a membrane interaction region rich in Trp/Tyr, nonpolar (Val, Leu, Ile), and cationic (Lys, Arg) residues (12, 13), a “small-headgroup” negatively-charged lipid that can undergo hydrogen bonding (e.g., PA) might facilitate the cleft-like gating action involved in glycolipid uptake by GLTP or local conformational changes needed for the GLTP-glycolipid complex to dissociate from the matrix surface, especially in combination with PE (54). Indeed, a recent study of duramycin interaction with POPE monolayers shows that the peptide strongly alters the hydrogen bonding and hydration status of the POPE (62). An additional way that POPE could enhance GLTP uptake of glycolipids is by acting as a lateral dispersant of glycolipids. Quinn recently showed that saturated-chain PEs are immiscible with glucosylceramide, but that POPE and glucosylceramide form stable complexes above physiological temperature (36, 63).
Prior to the present study, the only membrane feature shown to markedly enhance glycolipid uptake by GLTP was high positive curvature stress in PC bilayers (18, 21, 22, 64). Findings such as these led to questions about whether cell membrane interaction sites need to be highly curved to be viable interaction sites for GLTP. Our findings show that bilayer curvature per se is not the only membrane parameter able to stimulate GLTP interaction with a fluid-phase lipid matrix. Dramatic (∼7-fold) stimulation of GLTP action can be induced from planar fluid-phase PC matrices that contain physiologically-relevant levels of PE (15 mol%) and PA (5 mol%). Thus, lipid headgroup chemical composition and the environment produced when different lipids mix can be equally important inducers of GLTP action. It is tempting to speculate that metabolic adjustment of the lipid composition in cytosol-exposed membrane surfaces could provide a new mechanism for regulating GLTP.
Our discoveries were made using a custom-built microfluidics-based surface balance that simultaneously measures surface pressure and fluorescence via a Wilhelmy nichrome wire and a laser-coupled fiber optic cable/diode array platform. This device enables lipid packing to be systematically controlled so as to reproduce conditions found in either nonstressed bilayers or high-curvature vesicles comprised of different lipids, an area of timely interest (65). The surface fluorescence detection capability enables GLTP uptake of BODIPY-labeled glycolipid from monolayers composed of different matrix lipids to be directly assessed while controlling the initial lateral packing state over subphase volumes of ∼3 μl, which is 6,000-fold lower than the subphase volume of the extra small KSV-NIMA Langmuir Trough. Compared with surface plasmon resonance approaches that rely on lipid adsorption to a solid surface, an advantage of our microfluidics-based monolayer system is control of the lipid packing state over a range that includes biomembrane-like conditions. Challenges associated with studies of peripheral amphitropic protein interactions with lipid membranes using surface plasmon resonance are well documented. For instance, uncertainties can emerge as to whether injected liposomes have formed a continuous bilayer on the surface of the chip or whether intact liposomes have been captured (66). Control of such issues usually requires specialized surface coatings. With the microfluidics-based surface balance platform, rapid and economical regeneration is achieved by simply replacing monolayers with fresh lipid, thus avoiding “fouling” artifacts that sometimes are a problem with commercial solid surface supports after multiple uses.
Supplementary Material
Acknowledgments
The authors thank Helen M. Pike for purifying the recombinant human GLTP.
Footnotes
Abbreviations:
- BODIPY
- boron dipyrromethene
- GLTP
- glycolipid transfer protein
- PA
- phosphatidic acid
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- POPA
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate
- POPE
- 1-palmitoyl-2-oleoyl-phosphatidylethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- PS
- phosphatidylserine
This work was supported by NIH/NIGMS 45928 and NIH/NHLBI 49180, Russian Foundation for Basic Research Grant 12-04-00168, and the Hormel Foundation.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of seven figures.
REFERENCES
- 1.Burn P. 1988. Amphitropic proteins: a new class of membrane proteins. Trends Biochem. Sci. 13: 79–83. [DOI] [PubMed] [Google Scholar]
- 2.Johnson J. E., Cornell R. B. 1999. Amphitropic proteins: regulation by reversible membrane interactions. Mol. Membr. Biol. 16: 217–235. [DOI] [PubMed] [Google Scholar]
- 3.Halskau Ø., Muga A., Martínez A. 2009. Linking new paradigms in protein chemistry to reversible membrane-protein interactions. Curr. Protein Pept. Sci. 10: 339–359. [DOI] [PubMed] [Google Scholar]
- 4.Brown R. E., Mattjus P. 2007. Glycolipid transfer proteins. Biochim. Biophys. Acta. 1771: 746–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuuf J., Mattjus P. 2007. Human glycolipid transfer protein—intracellular localization and effects on the sphingolipid synthesis. Biochim. Biophys. Acta. 1771: 1353–1363. [DOI] [PubMed] [Google Scholar]
- 6.Warnock D. E., Lutz M. S., Blackburn W. A., Young W. W., Jr, Baenziger J. U. 1994. Transport of newly synthesized glucosylceramide to the plasma membrane by a non-Golgi pathway. Proc. Natl. Acad. Sci. USA. 91: 2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halter D., Neumann S., van Dijk S. M., Wolthoorn J., de Maziere A. M., Vieira O. V., Mattjus P., Klumperman J., van Meer G., Sprong H. 2007. Pre- and post-Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J. Cell Biol. 179: 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuuf J., Wistbacka L., Mattjus P. 2009. The glycolipid transfer protein interacts with the vesicle-associated membrane protein-associated protein VAP-A. Biochem. Biophys. Res. Commun. 388: 395–399. [DOI] [PubMed] [Google Scholar]
- 9.Zou X., Chung T., Lin X., Malakhova M. L., Pike H. M., Brown R. E. 2008. Human glycolipid transfer protein (GLTP) genes: organization, transcriptional status, and evolution. BMC Genomics. 9: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Angelo G., Polishchuk E., Di Tullio G., Santoro M., Di Campli A., Godi A., West G., Bielawski J., Chuang C-C., van der Spoel A. C., et al. 2007. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature. 449: 62–67. [DOI] [PubMed] [Google Scholar]
- 11.Mattjus P. 2009. Glycolipid transfer proteins and membrane interaction. Biochim. Biophys. Acta. 1788: 267–272. [DOI] [PubMed] [Google Scholar]
- 12.Malinina L., Malakhova M., Teplov A., Brown R. E., Patel D. J. 2004. Structural basis for glycosphingolipid transfer specificity. Nature. 430: 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malinina L., Malakhova M., Kanack A. T., Lu M., Abagyan R., Brown R. E., Patel D. J. 2006. The liganding of glycolipid transfer protein is controlled by glycolipid acyl structure. PLoS Biol. 4: e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Airenne T. T., Kidron H., Nymalm Y., Nylund M., West G., Mattjus P., Salminen T. A. 2006. Structural evidence for adaptive ligand binding by glycolipid transfer protein. J. Mol. Biol. 355: 224–236. [DOI] [PubMed] [Google Scholar]
- 15.Kenoth R., Simanshu D. K., Kamlekar R. K., Pike H. M., Molotkovsky J. G., Benson L. M., Bergen H. R., Prendergast F. G., Malinina L., Venyaminov S. Y., et al. 2010. Structural determination and tryptophan fluorescence of heterokaryon incompatibility C2 protein (HET-C2), a fungal glycolipid transfer protein (GLTP), provide novel insights into glycolipid specificity and membrane interaction by the GLTP-fold. J. Biol. Chem. 285: 13066–13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenoth R., Kamlekar R-K., Simanshu D. K., Gao Y., Malinina L., Prendergast F. G., Molotkovsky J. G., Patel D. J., Venyaminov S. Y., Brown R. E. 2011. Conformational folding and stability of the HET-C2 glycolipid transfer protein fold: does a molten globule-like state regulate activity? Biochemistry. 50: 5163–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samygina V. R., Popov A. N., Cabo-Bilbao A., Ochoa-Lizarralde B., Goni-de-Cerio F., Zhai X., Molotkovsky J. G., Patel D. J., Brown R. E., Malinina L. 2011. Enhanced selectivity for sulfatide by engineered human glycolipid transfer protein. Structure. 19: 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao C. S., Chung T., Pike H. M., Brown R. E. 2005. Glycolipid transfer protein interaction with bilayer vesicles: modulation by changing lipid composition. Biophys. J. 89: 4017–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamlekar R-K., Gao Y., Kenoth R., Molotkovsky J. G., Prendergast F. G., Malinina L., Patel D. J., Wessels W. S., Venyaminov S. Y., Brown R. E. 2010. Human GLTP: three distinct functions for the three tryptophans in a novel peripheral amphitropic fold. Biophys. J. 99: 2626–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemmon M. A. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9: 99–111. [DOI] [PubMed] [Google Scholar]
- 21.Rao C. S., Lin X., Pike H. M., Molotkovsky J. G., Brown R. E. 2004. Glycolipid transfer protein mediated transfer of glycosphingolipids between membranes: a model for action based on kinetic and thermodynamic analyses. Biochemistry. 43: 13805–13815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nylund M., Fortelius C., Palonen E. K., Molotkovsky J. G., Mattjus P. 2007. Membrane curvature effects on glycolipid transfer protein activity. Langmuir. 23: 11726–11733. [DOI] [PubMed] [Google Scholar]
- 23.Selvy P. E., Lavieri R. R., Lindsley C. W., Brown H. A. 2011. Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111: 6064–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockman H. 1999. Lipid monolayers: why use half a membrane to characterize protein-membrane interactions? Curr. Opin. Struct. Biol. 9: 438–443. [DOI] [PubMed] [Google Scholar]
- 25.Boldyrev I. A., Zhai X., Momsen M. M., Brockman H. L., Brown R. E., Molotkovsky J. G. 2007. New BODIPY lipid probes for fluorescence studies of membranes. J. Lipid Res. 48: 1518–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boldyrev I. A., Molotkovsky J. G. 2010. New 4,4,-difluoro-3a,4a-diaza-s-indacene (BODIPY)-labeled sphingolipids for membrane studies. Russ. J. Bioorg. Chem. 36: 508–511. [DOI] [PubMed] [Google Scholar]
- 27.Sachl R., Boldyrev I., Johansson L. B. 2010. Localisation of BODIPY-labelled phosphatidylcholines in lipid bilayers. Phys. Chem. Chem. Phys. 12: 6027–6034. [DOI] [PubMed] [Google Scholar]
- 28.Momsen W. E., Mizuno N. K., Lowe M. E., Brockman H. L. 2005. Real-time measurement of solute partitioning to lipid monolayers. Anal. Biochem. 346: 139–149. [DOI] [PubMed] [Google Scholar]
- 29.Hoang K. C., Malakhov D., Momsen W. E., Brockman H. L. 2006. Open, microfluidic flow cell for studies of interfacial processes at gas-liquid interfaces. Anal. Chem. 78: 1657–1664. [DOI] [PubMed] [Google Scholar]
- 30.Malakhova M. L., Malinina L., Pike H. M., Kanack A. T., Patel D. J., Brown R. E. 2005. Point mutational analysis of the liganding site in human glycolipid transfer protein. Functionality of the complex. J. Biol. Chem. 280: 26312–26320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown R. E., Jarvis K. L., Hyland K. J. 1990. Purification and characterization of glycolipid transfer protein from bovine brain. Biochim. Biophys. Acta. 1044: 77–83. [DOI] [PubMed] [Google Scholar]
- 32.Marsh D. 1996. Lateral pressure in membranes. Biochim. Biophys. Acta. 1286: 183–223. [DOI] [PubMed] [Google Scholar]
- 33.MacDonald R. C. 1996. The relationship and interactions between lipid bilayers, vesicles, and lipid monolayers at the air/water interface. In Vesicles. M. Rosoff, editor. Marcel Dekker, NY. 3–48. [Google Scholar]
- 34.Ruocco M. J., Shipley G. G. 1983. Hydration of N-palmitoylgalactosylsphingosine compared to monosaccharide hydration. Biochim. Biophys. Acta. 735: 305–308. [DOI] [PubMed] [Google Scholar]
- 35.Nagle J. F., Stephanie Tristram-Nagle S. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469: 159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinn P. J. 2012. Lipid-lipid interactions in bilayer membranes: married couples and casual liaisons. Prog. Lipid Res. 51: 179–198. [DOI] [PubMed] [Google Scholar]
- 37.Phillips M. C., Chapman D. 1968. Monolayer characteristics of saturated 1,2-diacyl phosphatidylcholines (lecithins) and phosphatidylethanolamines at the air-water interface. Biochim. Biophys. Acta. 163: 301–313. [DOI] [PubMed] [Google Scholar]
- 38.Smaby J. M., Momsen M. M., Brockman H. L., Brown R. E. 1997. Phosphatidylcholine acyl unsaturation modulates the decrease in interfacial elasticity induced by cholesterol. Biophys. J. 73: 1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hall A., Róg T., Karttunen M., Vattulainen I. 2010. Role of glycolipids in lipid rafts: a view through atomistic molecular dynamics simulations with galactosylceramide. J. Phys. Chem. B. 114: 7797–7807. [DOI] [PubMed] [Google Scholar]
- 40.Zheng H., Du X. 2013. Reduced steric hindrance and optimized spatial arrangement of carbohydrate ligands in imprinted monolayers for enhanced protein binding. Biochim. Biophys. Acta. 1828: 792–800. [DOI] [PubMed] [Google Scholar]
- 41.Dahim M., Mizuno N. K., Li X-M., Momsen W. E., Momsen M. M., Brockman H. L. 2002. Physical and photophysical characterization of a BODIPY phosphatidylcholine as a membrane probe. Biophys. J. 83: 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugár I. P., Zhai X., Boldyrev I. A., Molotkovsky J. G., Brockman H. L., Brown R. E. 2010. Characterization of the lateral distribution of fluorescent lipid in binary-constituent lipid monolayers by principal component analysis. Int. J. Biomed. Imaging. 2010: 125850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikhalyov I., Bogen S-T., Johansson L. B-A. 2001. Donor-donor energy migration (DDEM) as a tool for studying aggregation in lipid phases. Spectrochim. Acta A Mol. Biomol. Spectrosc. 57: 1839–1845. (Pt A). [DOI] [PubMed] [Google Scholar]
- 44.Bader A. N., Hoetzl S., Hofman E. G., Voortman J., van Bergen en Henegouwen P. M., van Meer G., Gerritsen H. C. 2011. Homo-FRET imaging as a tool to quantify protein and lipid clustering. ChemPhysChem. 12: 475–483. [DOI] [PubMed] [Google Scholar]
- 45.Sugár I. P., Mizuno N. K., Brockman H. L. 2005. Peripheral protein adsorption to lipid-water interfaces: the free area theory. Biophys. J. 89: 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brockman H. L., Applegate K. R., Momsen M. M., King W. C., Glomset J. A. 2003. Packing and electrostatic behavior of sn-2-docosahexaenoyl and -arachidonoyl phosphoglycerides. Biophys. J. 85: 2384–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattjus P., Pike H. M., Molotkovsky J. G., Brown R. E. 2000. Charged membrane surfaces impede the protein-mediated transfer of glycosphingolipids between phospholipid bilayers. Biochemistry. 39: 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Meer G., Voelker D. R., Feigenson G. W. 2008. Membrane lipids: where they are and now they behave. Nat. Rev. Mol. Cell Biol. 9: 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitomo H., Chen W. H., Regen S. L. 2009. Reduced sterol-phospholipid recognition in curved fluid bilayers. Langmuir. 25: 4328–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McIntosh T. J. 1996. Hydration properties of lamellar and non-lamellar phases of phosphatidylcholine and phosphatidylethanolamine. Chem. Phys. Lipids. 81: 117–131. [DOI] [PubMed] [Google Scholar]
- 51.Fuller N., Benatti C. R., Rand R. P. 2003. Curvature and bending constants for phosphatidylserine-containing membranes. Biophys. J. 85: 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrache H. I., Tristram-Nagle S., Gawrisch K., Harries D., Parsegian V. A., Nagle J. F. 2004. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 86: 1574–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kooijman E. E., Chupin V., Fuller N. L., Kozlov M. M., de Kruijff B., Burger K. N., Rand P. R. 2005. Spontaneous curvature of phosphatidic acid and lysophosphatidic acid. Biochemistry. 44: 2097–2102. [DOI] [PubMed] [Google Scholar]
- 54.Kooijman E. E., Burger K. N. J. 2009. Biophysics and function of phosphatidic acid: A molecular perspective. Biochim. Biophys. Acta. 1791: 881–888. [DOI] [PubMed] [Google Scholar]
- 55.Li S. J., Yamazakib M. 2004. Low concentration of dioleoylphosphatidic acid induces an inverted hexagonal (HII) phase transition in dipalmitoleoylphosphatidylethanolamine membranes. Biophys. Chem. 109: 149–155. [DOI] [PubMed] [Google Scholar]
- 56.van den Brink-van der Laan E., Killian J. A., de Kruijff B. 2004. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim. Biophys. Acta. 1666: 275–288. [DOI] [PubMed] [Google Scholar]
- 57.Crook S. J., Boggs J. M., Vistnes A. I., Koshy K. M. 1986. Factors affecting surface expression of glycolipids influence of lipid environment and ceramide composition on antibody recognition of cerebroside sulfate in liposomes. Biochemistry. 25: 7488–7494. [DOI] [PubMed] [Google Scholar]
- 58.Frolov V. A., Shnyrova A. V., Zimmerberg J. 2011. Lipid polymorphisms and membrane shape. Cold Spring Harb. Perspect. Biol. 3: a004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bazzi M. D., Youakim M. A., Nelsestuen G. L. 1992. Importance of phosphatidylethanolamine for association of protein kinase C and other cytoplasmic proteins with membranes? Biochemistry. 31: 1125–1134. [DOI] [PubMed] [Google Scholar]
- 60.Heller W. T., He K., Ludtke S. J., Harroun T. A., Huang H. W. 1997. Effect of changing the size of lipid headgroup on peptide insertion into membranes. Biophys. J. 73: 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dowhan W., Bogdanov M. 2009. Lipid-dependent membrane protein topogenesis. Annu. Rev. Biochem. 78: 515–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rzeźnicka I. I., Sovago M., Backus E. H., Bonn M., Yamada T., Kobayashi T., Kawai M. 2010. Duramycin-induced destabilization of a phosphatidylethanolamine monolayer at the air-water interface observed by vibrational sum-frequency generation spectroscopy. Langmuir. 26: 16055–16062. [DOI] [PubMed] [Google Scholar]
- 63.Quinn P. J. 2011. The structure of complexes between phosphatidylethanolamine and glucosylceramide: a matrix for membrane rafts. Biochim. Biophys. Acta. 1808: 2894–2904. [DOI] [PubMed] [Google Scholar]
- 64.Brown R. E., Stephenson F. A., Markello T. M., Barenholz Y., Thompson T. E. 1985. Properties of a specific glycolipid transfer protein from bovine brain. Chem. Phys. Lipids. 38: 79–93. [DOI] [PubMed] [Google Scholar]
- 65.Huang K. C., Ramamurthi K. S. 2010. Macromolecules that prefer their membranes curvy. Mol. Microbiol. 76: 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hodnik V., Anderluh G. 2010. Capture of intact liposomes on Biacore sensor chips for protein-membrane interaction studies. Methods Mol. Biol. 627: 201–211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.