

Crystal structures of three PCNA homologues from Sulfolobus solfataricus have been determined in different oligomeric states. The structural features in different assemblies present a molecular model for clamp-ring forming and opening.
Keywords: DNA replication, DNA sliding clamps, PCNA-ring opening, conformational transitions
Abstract
DNA sliding clamps form an oligomeric ring encircling DNA and serve as a moving platform for DNA-processing proteins. The opening and closing of a sliding-clamp ring is essential to load the clamp onto DNA in order to perform its functions. The molecular details of how clamp rings open and enclose DNA are still not clear. Three PCNA homologues have been found in Sulfolobus solfataricus which form a heterotrimer. Taking advantage of their hetero-oligomeric nature, the structures of the PCNAs in monomeric PCNA3, dimeric PCNA1–PCNA2 and trimeric PCNA1–PCNA2–PCNA3 forms were determined at resolutions of 2.6–1.9 Å. The distinct oligomeric structures represent different stages in ring formation, which were verified in solution by ultracentrifugation analysis. The heterodimer opens in a V-shape of 130°, while the heterotrimers form a ring with a 120° rotation between monomers. The association of a rigid PCNA3 monomer with an opened PCNA1–PCNA2 heterodimer closes the ring and introduces a spring tension in the PCNA1–PCNA2 interface, thus bending the nine-stranded intermolecular β-sheet to fit the 120° rotation. The release of the spring tension as PCNA3 dissociates from the ring may facilitate ring opening. The structural features in different assemblies present a molecular model for clamp ring assembly and opening.
1. Introduction
DNA-replication sliding clamps are essential in DNA replication and repair and are present in eukaryotes, prokaryotes and archaea (Lee & Alani, 2006 ▶; Warbrick, 2000 ▶; Tsurimoto, 1999 ▶; Hingorani & O’Donnell, 2000 ▶; Kelman, 1997 ▶; Umar et al., 1996 ▶). The sliding-clamp protein is a six-domain ring that encircles DNA and initially functions as a processivity factor for replicative DNA polymerases (Kong et al., 1992 ▶; Krishna et al., 1994 ▶; Gulbis et al., 1996 ▶; Chapados et al., 2004 ▶). The eukaryotic and archaeal sliding clamps are homologs of the proliferating cell nuclear antigen (PCNA). Interestingly, while most eucaryotic and archaeal PCNA proteins are homotrimeric rings, three PCNA homologues (PCNA1, PCNA2 and PCNA3) exist in the thermophilic archaeon Sulfolobus solfataricus (De Felice et al., 1999 ▶; Dionne et al., 2003 ▶). Although initial reports claimed that PCNA1 and PCNA3 formed functional homotrimers (De Felice et al., 1999 ▶), subsequent work revealed that individually PCNA1, PCNA2 and PCNA3 are monomers and that the functional form of Sulfolobus PCNA is a heterotrimer of PCNA1, PCNA2 and PCNA3 (hereafter called PCNA123; Dionne et al., 2003 ▶; Roberts et al., 2003 ▶; Pascal et al., 2006 ▶; Doré et al., 2006 ▶; Williams et al., 2006 ▶). Biochemical studies indicated that the heterotrimer assembles in a defined order (Dionne et al., 2003 ▶). More specifically, PCNA1 and PCNA2 first form a stable heterodimer (PCNA12) that is then capable of recruiting PCNA3 to complete the ring structure.
Sliding clamps slide along DNA through their positively charged central cavity in the direction of DNA synthesis. PCNA proteins have been found to interact with a wide variety of proteins and are involved in almost every DNA metabolic process, including replication, repair and modification (Maga & Hubscher, 2003 ▶; Majka & Burgers, 2004 ▶; Lopez de Saro & O’Donnell, 2001 ▶; Tsurimoto, 1999 ▶). The ring surface facing the primer-extension direction forms a platform that tethers DNA-modifying enzymes (PCNA-interacting proteins; PIPs) on the DNA substrate as the clamps move along the DNA (Waga & Stillman, 1998 ▶; Warbrick, 2000 ▶). The main PCNA-interacting motif, Qxx(M/L/I)xxF(Y/W), is well conserved and is called the PIP-box (Warbrick, 2000 ▶). Structural studies on PCNA–ligand complexes have shown that the PIP-box binds to the interdomain connecting loop (IDCL) of the sliding clamp and its proximal hydrophobic cavity, which is located on the front face of the ring (Gulbis et al., 1996 ▶; Sakurai et al., 2005 ▶; Bruning & Shamoo, 2004 ▶; Kontopidis et al., 2005 ▶). The PCNA123 heterotrimeric ring from S. solfataricus has been shown to stimulate the activity of flap endonuclease 1 (Fen1), DNA polymerase B1 (Pol B1) and DNA ligase 1 (Dionne et al., 2003 ▶). Fen1, Pol B1 and DNA ligase bind to PCNA1, PCNA2 and PCNA3, respectively, through their PIP-boxes (Dionne et al., 2003 ▶). A recently solved structure of PCNA12–Fen1 from S. solfataricus reveals that the PIP-box of Fen1 binds to the topologically conserved binding site of PCNA1 (Doré et al., 2006 ▶).
The sliding clamps are loaded onto double-strand/single-strand DNA junctions by ATP-driven clamp loaders (Barsky & Venclovas, 2005 ▶; Indiani & O’Donnell, 2006 ▶; Ellison & Stillman, 2001 ▶). During the clamp-loading process, homotrimeric PCNAs, with the help of replication factor C (RFC), are opened at one interface of the ring and adopt a right-handed helix structure matching the right-handed spiral arrangement of RFC (Miyata et al., 2005 ▶; Kazmirski et al., 2005 ▶). Biochemical studies of the interactions between Sulfolobus PCNA subunits revealed that the binding of PCNA3 to the PCNA12 dimer has a K d that is almost four orders of magnitude higher than that for the binding of PCNA1 to PCNA2 (Dionne et al., 2003 ▶). This strongly suggests that one or both of the PCNA1–PCNA3 or PCNA2–PCNA3 interfaces are opened during clamp loading. In support of this, recent studies using covalently fused PCNA subunits have revealed that opening of the PCNA3–PCNA1 interface is both necessary and sufficient for RFC-mediated loading of PCNA onto a DNA substrate (Dionne et al., 2008 ▶).
Structural studies of sliding clamps in different assembly intermediates will shed light on the ring opening/closing that is essential for loading onto DNA. In order to better understand the molecular mechanisms of PCNA in DNA loading, we carried out structural analyses of PCNA1, PCNA2 and PCNA3 from S. solfataricus. Although two structures of the PCNA heterotrimer have been solved in different space groups (Williams et al., 2006 ▶; Pascal et al., 2006 ▶), no other free oligomeric form has been reported to take advantage of the hetero-oligomeric system. We present here the crystal structures of the PCNA3 monomer, the PCNA12 heterodimer and the PCNA123 heterotrimer. The structures reveal a rigid PCNA3 monomer, a flexible PCNA12 dimer and a stable PCNA123 trimer. Structural comparison of PCNA in different oligomeric states provides a structural basis for the heterotrimeric ring assembly/opening. The tertiary structures of PCNA1, PCNA2 and PCNA3 also help in interpretation of their distinct binding specificities for their client proteins, such as Fen1, Pol B1 and PolIV.
2. Experimental procedures
2.1. Structure determination and analysis
Protein-sample preparation, crystallization and data collection have been reported previously (Xing et al., 2007 ▶). The structure of the SeMet-labeled PCNA12 dimer was determined at peak wavelength by single anomalous dispersion (SAD) phasing. Selenium sites were located using the program SHELXD (Sheldrick, 2008 ▶). NCS-averaged density modification and phase extension to a native resolution of 2.6 Å were performed using DM (Cowtan & Zhang, 1999 ▶). The structures of the PCNA3 monomer and the PCNA123 trimer were solved by molecular replacement with CNS (Brünger et al., 1998 ▶) and Phaser (McCoy et al., 2005 ▶) using PCNA from S. tokodaii (PDB code 1ud9) and the SAD-phased dimer as search models. Model building was performed with XtalView (McRee, 1999 ▶), O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶). The electron-density maps were improved by NCS averaging and solvent flipping with CNS (Brünger et al., 1998 ▶). The structures were refined with NCS restraints to 1.9–2.6 Å resolution with CNS (Brünger et al., 1998 ▶) and REFMAC5 (Murshudov et al., 1997 ▶). None of the protein residues are in disallowed regions of the Ramachandran plot. The refinement and data-collection statistics are summarized in Table 1 ▶. Figures were prepared using PyMOL (DeLano, 2002 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| PCNA12 dimer | ||||
|---|---|---|---|---|
| PCNA3 monomer | Native | SeMet | PCNA123 trimer | |
| Space group | P41212 | P21212 | P21212 | P21212 |
| Unit-cell parameters (Å) | a = b = 85.8, c = 264.2 | a = 105.0, b = 112.7, c = 101.9 | a = 105.0, b = 112.7, c = 101.8 | a = 148.1, b = 222.3, c = 80.2 |
| Wavelength (Å) | 1.1000 | 0.9731 | 0.9791 (peak) | 0.9195 |
| Beamline | NSLS X12B | APS 8BM | APS 8BM | APS 8BM |
| Resolution range (Å) | 30–1.90 (1.92–1.90) | 30–2.60 (2.66–2.60) | 30–3.30 (3.42–3.30) | 30–2.50 (2.59–2.50) |
| R merge | 0.110 (0.490) | 0.076 (0.555) | 0.093 (0.545) | 0.098 (0.625) |
| Unique reflections | 75802 | 37787 | 34749 | 88416 |
| Completeness (%) | 96.3 (65.6) | 99.9 (99.8) | 99.6 (100) | 95.4 (93.6) |
| I/σ(I) | 28.9 (2.2) | 8.6 (1.8) | 6.6 (2.0) | 11.7 (2.0) |
| Redundancy | 23.3 (5.8) | 5.1 (5.1) | 3.4 (3.4) | 8.0 (8.0) |
| Mosaicity (°) | 0.32 | 0.4 | 0.66 | 0.53 |
| Molecules per ASU | 4 | 2 | 2 | 3 |
| Structure solution | Molecular replacement | NA | SAD | Molecular replacement |
| Non-H atoms | 8414 | 7952 | 18472 | |
| Water molecules | 544 | 174 | 1044 | |
| R factor | 0.205 | 0.234 | 0.211 | |
| R free [No. of reflections] | 0.251 [1530] | 0.260 [1151] | 0.250 [1796] | |
| R.m.s.d. bond length (Å) | 0.017 | 0.015 | 0.011 | |
| R.m.s.d. bond angle (°) | 1.71 | 1.57 | 1.71 | |
| Average B factor (Å2) | 42.9 | 60.8 | 44.1 | |
| Wilson B factor (Å2) | 42.7 | 74.6 | 48.6 | |
2.2. Analytical ultracentrifugation
Sedimentation-equilibrium studies were conducted using a Beckman XL-A analytical ultracentrifuge. An An60Ti rotor and six channel cells with Epon charcoal centerpieces were used. Protein samples were dialyzed against a buffer containing 50 mM Tris–HCl pH 7.5 and 100 mM NaCl. Centrifugation was carried out at 293 K. PCNA subunits were analyzed at three different concentrations in the range 0.2–0.65 mg ml−1 and each concentration was sedimented to equilibrium at rotor speeds between 15 000 and 32 000 rev min−1. Data were fitted to a model using single ideal species and analyzed with the Beckman software Origin v. 6.0. Buffer density and partial specific volume were calculated from the inferred amino-acid compositions according to Cohn & Edsall (1943 ▶).
3. Results
3.1. Structures of PCNAs
The crystal structures of the PCNA3 monomer, PCNA12 dimer and PCNA123 trimer provide crystallographically independent structural information for five copies of PCNA1 and PCNA2 and seven copies of PCNA3. The structures of the PCNAs from S. solfataricus are very similar to those of PCNAs from yeast, humans and other archaea (Krishna et al., 1994 ▶; Gulbis et al., 1996 ▶; Matsumiya et al., 2001 ▶). Each PCNA monomer is composed of two topologically similar domains that are linked by an interdomain connecting loop (IDCL; Fig. 1 ▶). The three PCNAs are pairwise superimposible upon each other, although the overall sequence identity and the overall sequence similarity are only ∼8–22% among PCNA1, PCNA2 and PCNA3 (Fig. 1 ▶ b). The superposition of the three monomers in the PCNA123 heterotrimer, based on the 200 Cα atoms (excluding the loop regions), results in a root-mean-square deviation (r.m.s.d.) of 1.5 Å. PCNA2 and PCNA3 share the same number and type of secondary-structure elements, while PCNA1 has an additional β9′ strand which is located in the N-terminal domain. The β9′ strand forms four main-chain hydrogen bonds to β6 and plays an important role in the unique conformation of the N-terminal part of the PCNA1 IDCL. This part of the PCNA1 IDCL is shifted toward the back face of the ring structure by up to 10 Å compared with the corresponding Cα atoms in the IDCLs of PCNA2 or PCNA3. A similar conformation of the PCNA1 IDCL is seen in the presence of the PCNA1 client protein Fen1 (Doré et al., 2006 ▶), indicating that binding of a client protein does not significantly perturb the conformation of its docking site on a PCNA monomer. The IDCLs in PCNA2 and PCNA3 are in a similar position to those observed in their previously studied counterparts.
Figure 1.

Structures of PCNAs from S. solfataricus. (a) Ribbon diagram of PCNA1; the secondary structures are named in alphabetical or numeric order for the α-helices and β-strands, respectively, following their order in the primary sequence. (b) Superposition of the Cα traces of PCNA1 (blue), PCNA2 (red) and PCNA3 (green). The three PCNAs superimpose well with identical secondary-structure elements except for the β9′ strand in PCNA1. The additional β9′ strand moves the IDCL in PCNA1 ∼7 Å towards the back face of the ring and is shown as a thick blue arrow.
3.2. Rigid PCNA3 monomers
PCNA3 alone crystallized in space group P41212 with four monomers in the asymmetric unit. The four PCNA3 molecules in the asymmetric unit of the PCNA3 structure randomly pack against each other without forming any physiologically relevant assemblies. The apparent monomeric behavior of PCNA3 in the crystal is in agreement with our ultracentrifugation results (see below) and previous studies (Dionne et al., 2003 ▶). Pairwise superposition of these four molecules gave r.m.s.d. values ranging between 0.65 and 1.25 Å over all 244 Cα atoms. Smaller r.m.s.d. values (0.50–0.75 Å) were obtained by comparing PCNA3 monomers in the heterotrimers (see below). When the free PCNA3 monomers are compared with PCNA3 in PCNA123 heterotrimers, the r.m.s.d. values are comparable with the differences between the four free PCNA3s. The largest deviations observed in the free PCNA3s are concentrated on the N- and C-terminal end surfaces that form the interfaces contacting the neighboring monomers in the heterotrimer. Therefore, trimer formation seems to reduce the flexibility of PCNA3 at its two ends, but the overall structure of PCNA3 in different oligomeric states remains rigid.
3.3. Opened PCNA12 dimers
The PCNA12 dimer crystallized in space group P21212 with two PCNA12 dimers in the asymmetric unit. PCNA1 and PCNA2 form a V-shaped dimer with a ‘head-to-tail’ assembly, i.e. the N-terminal domain of PCNA1 interacts with the C-terminal domain of PCNA2. The two dimers in the asymmetric unit are almost identical and can be superimposed well with an r.m.s.d. of 0.60 Å on all Cα atoms. The angle between PCNA1 and PCNA2 in the two V-shaped dimers is 130°, which is 10° larger than the threefold rotation between monomers observed in the trimeric ring (Fig. 2 ▶). The two crystallographically independent dimers possess the same open conformation, which indicates that the 130° opening reflects the nature of the dimer and suggests that it is independent of the crystal-packing environment.
Figure 2.

Comparison of the PCNA12 dimer (PCNA1 is blue, PCNA2 is red) with PCNA12 from the heterotrimer (PCNA1, purple; PCNA2, green). The lateral opening in the PCNA12 dimer is about 10° wider than that in PCNA12 from the heterotrimer.
Similar to PCNA3, the PCNA1 and PCNA2 monomers in different oligomeric states are very similar. The r.m.s.d. over all Cα atoms between the two PCNA1 molecules and the two PCNA2 molecules in the PCNA12 dimers are 0.53 and 0.57 Å, respectively. Similar r.m.s.d. values (0.46–0.63 Å) were observed among the three copies of the PCNAs in the three trimers in the PCNA123 crystal. The r.m.s.d. values between both PCNA1 and PCNA2 molecules from different oligomeric states range from 0.86 to 1.36 Å. The divergence in the different oligomeric states results from changes in the N-terminal surface of PCNA1 and the C-terminal surface of PCNA2, which are exposed to solvent in the dimers and buried in the interfaces with PCNA3 in the trimers. This strongly suggests that PCNA3 binding only modifies the parts of PCNA1 and PCNA2 which participate in PCNA1–PCNA3 and PCNA2–PCNA3 interface formation.
As PCNA1 and PCNA2 seem to behave as fairly rigid molecules (r.m.s.d. < 1.5 Å), the 10° rotation difference observed between the dimer and trimer must be a rigid-body motion, which leads to a different curvature of the intermolecular nine-stranded β-sheet in PCNA1 and PCNA2. The intermolecular nine-stranded β-sheet of the dimer is less bent than that of the trimer (Fig. 3 ▶). The change in bending of the β-sheet results in a 10° lateral opening between PCNA1 and PCNA2 compared with that of PCNA12 in the heterotrimer (Fig. 2 ▶). In contrast to the wider lateral opening, ‘out-of-plane’ movement between the two PCNA molecules is insignificant, with an angular offset of less than 3° from the plane of the ring.
Figure 3.
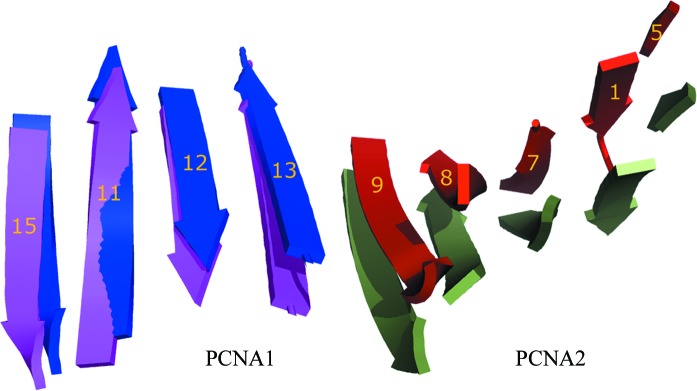
Structural changes in PCNA12 dimers. The different bending of the intermolecular nine-stranded β-sheet at the PCNA1–PCNA2 interface between the PCNA12 dimer (PCNA1, blue; PCNA2, red) and PCNA12 from the heterotrimer (PCNA1, purple; PCNA2, green).
3.4. Ring-shaped PCNA123 trimer
The PCNA123 heterotrimer crystallized in space group P21212 with three PCNA123 trimers in the asymmetric unit. The overall assembly is the same as those of the homotrimeric ring structures (Matsumiya et al., 2001 ▶; Krishna et al., 1994 ▶; Gulbis et al., 1996 ▶) and the dimeric β-subunit (Kong et al., 1992 ▶), with a pseudo-sixfold symmetry assembled by ‘head-to-tail’ interactions of N-terminal and C-terminal domains between PCNA1, PCNA2 and PCNA3 (Fig. 4 ▶). The structure of the PCNA123 heterotrimer solved in this study is almost identical to previously reported structures (Pascal et al., 2006 ▶; Williams et al., 2006 ▶).
Figure 4.

The three distinct ligand-binding sites of the PCNA123 trimer. The front face of the trimeric ring is shown as an electrostatic charged surface with the C-terminal fragment of Pol IV modeled in the three distinct ligand-binding sites. The binding sites are mapped by ribbon-styled C-termini with the side chains as white mesh surfaces. The modeling was performed by superposing the PCNA123 trimer with the β-clamp–PolIV–LF complex (PDB code 1ok7; Bunting et al., 2003 ▶).
PCNA1, PCNA2 and PCNA3 form three unique interfaces in the heterotrimer (Fig. 5 ▶). These interfaces differ from each other in amino-acid sequence composition, shape complementarity, charge distribution and number of hydrogen bonds. The PCNA1–PCNA2 interface has the largest buried surface area, 1404 Å2, and the highest shape complementarity, 0.8, of the three interfaces (Table 2 ▶). In addition to hydrophobic interactions, the PCNA1–PCNA2 interface has the most extensive charge–charge and hydrogen-bonding interactions of the three interfaces (Tables 2 ▶ and 3 ▶). The interface parameters are very similar in the three trimers observed in the asymmetric unit. Very similar interface parameters were obtained for the PCNA1–PCNA2 interface in the dimer structure, suggesting that the strength and unique features of this interface exist independently of PCNA3 binding. The strongest hydrophobic, hydrogen bonding and charge–charge interactions are observed at the PCNA1–PCNA2 interface and this is consistent with previous binding studies showing a K d of ∼10−11 M (Dionne et al., 2003 ▶).
Figure 5.

Three sets of interfaces in charged surface representation for the PCNA1–PCNA2 (a), PCNA2–PCNA3 (b) and PCNA3–PCNA1 (c) interfaces. The interfaces on the left are from the C-terminal domains which contact with the N-terminal face on the right. The side chains involved in intermolecular interactions are shown as sticks and charged residues are labeled.
Table 2. Interface parameters of PCNA.
Calculated using the PISA server (Krissinel & Henrick, 2005 ▶) and the SC program (Lawrence & Colman, 1993 ▶). The values for the heterotrimers and dimers are averaged over all of the molecules in the asymmetric units. The numbers of hydrogen bonds and salt bridges are listed for the interfaces in the three rings.
| Interface | Area (Å2) | Shape complementarity† | Hydrogen bonds | Salt bridges |
|---|---|---|---|---|
| PCNA1–PCNA2 | 1404.0 | 0.80 | 12/14/12 | 8/7/10 |
| PCNA2–PCNA3 | 1212.4 | 0.55 | 7/7/8 | 3/4/1 |
| PCNA3–PCNA1 | 1294.4 | 0.68 | 11/7/8 | 1/0/2 |
| PCNA1–PCNA2 (dimer) | 1393.0 | 0.77 | 9/13 | 11/8 |
| P. furiosus PCNA | 1438.6 | 0.68 | 14 | 14 |
| S. cerevisiae PCNA‡ | 1317.4 | 0.61 | 8 | 3 |
| Human PCNA | 1384.8 | 0.68 | 12 | 1 |
Shape complementarity (SC) is in the range 0–1. Interfaces with a SC of 1 match precisely, while interfaces with SC ≃ 0 are uncorrelated in their topography.
Krishna et al. (1994 ▶).
Table 3. Charge interactions at the interfaces.
The interactions were taken from the PISA server (Krissinel & Henrick, 2005 ▶).
| Interface | Interactions | Distance (Å) |
|---|---|---|
| PCNA1–PCNA2 | Lys175 NZ–Asp75 OD2 | 3.28 |
| Lys185 NZ–Asp103 OD1 | 3.69 | |
| Lys185 NZ–Asp103 OD2 | 3.37 | |
| Asp149 OD1–Arg82 NE | 2.51 | |
| Asp149 OD1–Arg82 NH1 | 2.74 | |
| Asp149 OD1–Arg108 NH2 | 3.13 | |
| Asp149 OD2–Arg108 NH1 | 3.01 | |
| Asp149 OD2–Arg108 NE | 3.94 | |
| PCNA2–PCNA3 | Glu146 OE1–Arg107 NE | 3.52 |
| Glu146 OE2–Arg107 NE | 2.99 | |
| Glu146 OE1–Arg107 NH1 | 3.36 | |
| PCNA3–PCNA1 | Lys110 NZ–Glu175 OE1 | 3.70 |
Several experimental observations, especially sedimentation-equilibrium analysis (see §3.5), convincingly show that there are no PCNA1, PCNA2 or PCNA3 homotrimers formed in solution. To clarify the absence of homotrimeric rings, we constructed theoretical models of PCNA1–PCNA1, PCNA2–PCNA2 and PCNA3–PCNA3 interfaces while respecting the ‘head-to-tail’ assembly. The obstacles to homotrimeric ring formation appear to be charge–charge incompatibility of each homotrimeric interface (charge–charge repulsions and close charge–hydrophobic contacts), as well as steric clashes (Fig. 5 ▶). The importance of charged interactions in PCNA123 assembly has also been shown by site-directed mutagenesis (Doré et al., 2006 ▶).
Similar to all known PCNA homotrimeric rings, the heterotrimer has an overall strong negative charge with the exception of the central channel through which DNA passes (Fig. 4 ▶). The front face contains the interdomain connecting loops which interact with most of the PCNA-binding proteins (Sakurai et al., 2005 ▶; Chapados et al., 2004 ▶; Gulbis et al., 1996 ▶; Bruning & Shamoo, 2004 ▶). In contrast to the homotrimers, the three ligand-binding sites are not identical. The sequences of the interdomain loops and the other loops at the front face are highly divergent, making the topologically equivalent binding sites vary in surface curvature and charge distribution (Fig. 4 ▶). The different physicochemical characteristics of the three distinct binding sites presumably account for the observed specificity for the distinct PCNA subunits for different binding partners such as Fen1, Pol B1, PolIV and DNA ligase 1 (Dionne et al., 2003 ▶, 2008 ▶).
3.5. Oligomeric states of the PCNAs in solution
Size-exclusion chromatography has shown distinct peaks corresponding to the individual monomers, PCNA12 dimers and PCNA123 trimers and was used to separate dimers and trimers from each other as well as from monomers (Xing et al., 2007 ▶; Dionne et al., 2003 ▶). We further studied the oligomeric states of individual PCNA subunits, the PCNA12 dimer and the PCNA123 trimer in solution by using sedimentation equilibrium to determine the molecular weight of each species. The data gave good fits for a single species as determined by the low variance and randomness of the residuals (Fig. 6 ▶). The average molecular weights for PCNA1, PCNA2, PCNA3, the PCNA12 dimer and the PCNA123 heterotrimer are in good agreement with their expected theoretical molecular weight (Table 4 ▶). The consistency in molecular weights indicates that the individual PCNAs indeed exist as monomers in solution, while PCNA1 and PCNA2 together form stable dimers and the three different PCNAs together assemble into heterotrimers. The ultracentrifugation analysis verified the structural observations in the crystal structures of monomeric, dimeric and trimeric PCNAs.
Figure 6.

Sedimentation-equilibrium analyses of PCNAs. (a) Representative scan of sedimentation-equilibrium data for PCNA1 (blue), PCNA2 (pink) and PCNA3 (black). The average molecular weights for PCNA1, PCNA2 and PCNA3 were determined as 26.2, 26.1 and 25.8 kDa, respectively. (b) Representative scan of sedimentation-equilibrium data for the PCNA12 dimer (blue) and PCNA123 trimer (pink). The average molecular weights for the PCNA12 dimer and PCNA123 trimer were determined as 54.4 and 79.2 kDa, respectively.
Table 4. Molecular weights (MW) determined by sedimentation equilibrium.
| Protein | Observed MW (Da) | Theoretical MW (Da) |
|---|---|---|
| PCNA1 | 26200 ± 1500 | 27535 |
| PCNA2 | 26100 ± 890 | 27567 |
| PCNA3 | 25800 ± 1400 | 27459 |
| PCNA12 | 54400 ± 1600 | 55102 |
| PCNA123 | 79200 ± 4800 | 82561 |
4. Discussion
PCNAs are actively loaded onto the DNA at template-primer junctions by replication factor C (RFC), which is composed of several subunits. An electron-microscopy study of a complex of homotrimeric PCNA and RFC from Pyrococcus furiosus on a DNA substrate revealed that the PCNA ring was cracked open with a gap of approximately 5 Å at a single interface (Miyata et al., 2005 ▶). Molecular-dynamics simulations have suggested that during loading, homotrimeric PCNAs are opened at one interface and adopt a right-handed helical conformation (Miyata et al., 2005 ▶; Kazmirski et al., 2005 ▶). This conformation mainly arises from a vertical twisting of the intermolecular nine-stranded antiparallel β-sheet in the homotrimeric ring (Kazmirski et al., 2005 ▶). In the present structures of the PCNA12 dimer and the PCNA123 trimer, only the horizontal curvature changes and almost no vertical twist is observed in the corresponding nine-stranded antiparallel β-sheet between the PCNA1 and the PCNA2 molecules. A similar horizontal curvature (a 130° opening) exists in the PCNA12–Fen1 structure (Doré et al., 2006 ▶). These observations strongly suggest that the individual subunits of the PCNA123 heterotrimer mainly move in the plane of the ring, which leads to the wider opening seen in the PCNA12 dimer (Figs. 2 ▶ and 7 ▶).
Figure 7.

Assembly and opening of the PCNA123 ring. (a) PCNA12 dimer and PCNA3 monomer approaching each other during ring assembly. (b) The ring of the PCNA123 trimer. (c) PCNA12 dimer with PCNA3 modeled in contact with PCNA2 with an angle of 120° as in the trimer. A gap (6–7 Å) remains in the ring-shaped molecule as the angle between PCNA1 and PCNA2 is 130° instead of 120°.
Recent studies using covalently fused dimers and trimers of Sulfolobus PCNA subunits have revealed that opening of only the PCNA3–PCNA1 interface is required for productive loading of PCNA by RFC (Dionne et al., 2008 ▶). The PCNA1–PCNA3 interface is the least complementary interface in terms of charge–charge interactions (Fig. 5 ▶), although the interface parameters in Table 2 ▶ do not show that the PCNA1–PCNA3 interface is the weakest interface. In the PCNA1–PCNA3 interface, the highly negative charged C-terminal surface of PCNA3 is buried by the hydrophobic residues clustered on the PCNA1 N-terminal surface (Fig. 5 ▶ c), which makes the PCNA1–PCNA3 interface less charge–charge compatible than the PCNA2–PCNA3 interface.
Presumably, the complex formed between RFC and PCNA favors opening of the PCNA ring and may supply the additional energy required to facilitate the proposed vertical twisting of the PCNA subunits. The tetramer of RFC small subunits contacts the PCNA12 dimer, while the RFC large subunits contact PCNA3. Previous work has revealed that ATP hydrolysis by the small subunits of RFC is required for release of RFC from PCNA after its loading onto DNA (Seybert & Wigley, 2004 ▶). It may be that this reflects conformational alterations within RFC that facilitate the closing of PCNA against the spring tension inherent in the PCNA1–PCNA2 interface. The interaction of all three PCNA subunits with RFC would also prevent dissociation of PCNA3 from PCNA2 upon opening of the PCNA3–PCNA1 interface. Correspondingly, we observe a 130° angle between PCNA1 and PCNA2 in the heterodimer and not the 120° angle found in the heterotrimer. If we model PCNA3 bound to PCNA2 in a PCNA12 heterodimer at a 120° angle, the resulting gap between PCNA1 and PCNA3 is ∼6–7 Å wide (Fig. 7 ▶ c). The shortest contacting distance is 5.8 Å, which is between the protruding side chains of Ala109 from PCNA1 and Asp179 from PCNA3. This distance is in good agreement with the ∼5 Å distance seen in the RFC–PCNA electron-microscopy structure (Miyata et al., 2005 ▶). However, the gap is not wide enough for PCNA to be loaded onto double-stranded DNA. Therefore, it is very likely that additional opening, either further lateral opening or out-of-plane twisting or a combination of both, is provided by RFC. It is also possible that the PCNA ring may be loaded onto single-stranded DNA and then translocated to the template-primer junction as noted by Miyata et al. (2005 ▶) and Kazmirski et al. (2005 ▶).
It may appear puzzling that S. solfataricus, an archaeon, has a PCNA that is organizationally more complex than that found in eukaryotes. However, in eukaryotes a second sliding clamp in addition to PCNA is found in the form of the 9-1-1 complex (a heterotrimer composed of Rad9–Hus1–Rad1), which participates in cell-cycle control and DNA repair (Kai & Wang, 2003 ▶; Parrilla-Castellar et al., 2004 ▶; Ellison & Stillman, 2003 ▶) and translesion synthesis (Paulovich et al., 1998 ▶). Rad9, Rad1 and Hus1 are broadly conserved in eukaryotes (Venclovas & Thelen, 2000 ▶; Thelen et al., 1999 ▶). A molecular-modeling study proposed that Rad9, Hus1 and Rad1 structurally resemble a PCNA structure and form a heterotrimeric ring with a head-to-tail assembly (Venclovas & Thelen, 2000 ▶). The ring structure of the human 9-1-1 heterotrimeric structure was subsequently observed by electron microscopy (Griffith et al., 2002 ▶; Shiomi et al., 2002 ▶). Thus, eukaryotic organisms possess two structurally similar ring-like proteins, homotrimeric (PCNA) and heterotrimeric (9-1-1), both of which participate in DNA replication and repair, translesion synthesis (TLS) and control of cell division. For instance, the 9-1-1 complex was found to regulate mutagenic translesion synthesis in S. cerevisiae and S. pombe (Paulovich et al., 1998 ▶; Kai & Wang, 2003 ▶; Ellison & Stillman, 2003 ▶). It may be that the heterotrimeric PCNA found in Sulfolobus represents a predecessor of its eukaryotic sliding-clamp counterparts. The selection for a heterotrimeric PCNA may in part be a consequence of the ability of the distinct PCNA subunits to have specific client proteins. This may facilitate tighter coupling of consecutive transactions on DNA than could be mediated by a homotrimer. In this regard, it may be relevant that S. solfataricus grows aerobically at 353 K and thus will be under considerable selective pressure to optimize its DNA damage-repair responses.
Supplementary Material
PDB reference: PCNA3, 2ijx, r2ijxsf
PDB reference: PCNA1–PCNA2, 2io4, r2io4sf
PDB reference: PCNA1–PCNA2–PCNA3, 2nti, r2ntisf
Acknowledgments
This work was supported by Canadian Institutes of Health Research grant MOP-67128 (HL) and funds from the Medical Research Council (SDB). The authors declare no conflict of interest.
References
- Barsky, D. & Venclovas, C. (2005). Curr. Biol. 15, R989–R992. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Bruning, J. B. & Shamoo, Y. (2004). Structure, 12, 2209–2219. [DOI] [PubMed]
- Bunting, K. A., Roe, S. M. & Pearl, L. H. (2003). EMBO J. 22, 5883–5892. [DOI] [PMC free article] [PubMed]
- Chapados, B. R., Hosfield, D. J., Han, S., Qiu, J., Yelent, B., Shen, B. & Tainer, J. A. (2004). Cell, 116, 39–50. [DOI] [PubMed]
- Cohn, E. J. & Edsall, J. T. (1943). Proteins, Amino Acids and Peptides, pp. 157–161. New York: Reinhold.
- Cowtan, K. D. & Zhang, K. Y. (1999). Prog. Biophys. Mol. Biol. 72, 245–270. [DOI] [PubMed]
- De Felice, M., Sensen, C. W., Charlebois, R. L., Rossi, M. & Pisani, F. M. (1999). J. Mol. Biol. 291, 47–57. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Dionne, I., Brown, N. J., Woodgate, R. & Bell, S. D. (2008). Mol. Microbiol. 68, 216–222. [DOI] [PubMed]
- Dionne, I., Nookala, R. K., Jackson, S. P., Doherty, A. J. & Bell, S. D. (2003). Mol. Cell, 11, 275–282. [DOI] [PubMed]
- Doré, A. S., Kilkenny, M. L., Jones, S. A., Oliver, A. W., Roe, S. M., Bell, S. D. & Pearl, L. H. (2006). Nucleic Acids Res. 34, 4515–4526. [DOI] [PMC free article] [PubMed]
- Ellison, V. & Stillman, B. (2001). Cell, 106, 655–660. [DOI] [PubMed]
- Ellison, V. & Stillman, B. (2003). PLoS Biol. 1, E33. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Griffith, J. D., Lindsey-Boltz, L. A. & Sancar, A. (2002). J. Biol. Chem. 277, 15233–15236. [DOI] [PubMed]
- Gulbis, J. M., Kelman, Z., Hurwitz, J., O’Donnell, M. & Kuriyan, J. (1996). Cell, 87, 297–306. [DOI] [PubMed]
- Hingorani, M. M. & O’Donnell, M. (2000). Curr. Biol. 10, R25–R29. [DOI] [PubMed]
- Indiani, C. & O’Donnell, M. (2006). Nature Rev. Mol. Cell Biol. 7, 751–761. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kai, M. & Wang, T. S. (2003). Genes Dev. 17, 64–76. [DOI] [PMC free article] [PubMed]
- Kazmirski, S. L., Zhao, Y., Bowman, G. D., O’Donnell, M. & Kuriyan, J. (2005). Proc. Natl Acad. Sci. USA, 102, 13801–13806. [DOI] [PMC free article] [PubMed]
- Kelman, Z. (1997). Oncogene, 14, 629–640. [DOI] [PubMed]
- Kong, X. P., Onrust, R., O’Donnell, M. & Kuriyan, J. (1992). Cell, 69, 425–437. [DOI] [PubMed]
- Kontopidis, G., Wu, S. Y., Zheleva, D. I., Taylor, P., McInnes, C., Lane, D. P., Fischer, P. M. & Walkinshaw, M. D. (2005). Proc. Natl Acad. Sci. USA, 102, 1871–1876. [DOI] [PMC free article] [PubMed]
- Krishna, T. S., Kong, X. P., Gary, S., Burgers, P. M. & Kuriyan, J. (1994). Cell, 79, 1233–1243. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2005). CompLife 2005, edited by R. B. Michael, G. Robert, K. Diederichs, O. Kohlbacher & I. Fischer, pp. 163–174. Berlin/Heidelberg: Springer.
- Lawrence, M. C. & Colman, P. M. (1993). J. Mol. Biol. 234, 946–950. [DOI] [PubMed]
- Lee, S. D. & Alani, E. (2006). J. Mol. Biol. 355, 175–184. [DOI] [PubMed]
- Lopez de Saro, F. J. & O’Donnell, M. (2001). Proc. Natl Acad. Sci. USA, 98, 8376–8380.
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Maga, G. & Hubscher, U. (2003). J. Cell Sci. 116, 3051–3060. [DOI] [PubMed]
- Majka, J. & Burgers, P. M. (2004). Prog. Nucleic Acid Res. Mol. Biol. 78, 227–260. [DOI] [PubMed]
- Matsumiya, S., Ishino, Y. & Morikawa, K. (2001). Protein Sci. 10, 17–23. [DOI] [PMC free article] [PubMed]
- Miyata, T., Suzuki, H., Oyama, T., Mayanagi, K., Ishino, Y. & Morikawa, K. (2005). Proc. Natl Acad. Sci. USA, 102, 13795–13800. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Parrilla-Castellar, E. R., Arlander, S. J. & Karnitz, L. (2004). DNA Repair (Amst.), 3, 1009–1014. [DOI] [PubMed]
- Pascal, J. M., Tsodikov, O. V., Hura, G. L., Song, W., Cotner, E. A., Classen, S., Tomkinson, A. E., Tainer, J. A. & Ellenberger, T. (2006). Mol. Cell, 24, 279–291. [DOI] [PubMed]
- Paulovich, A. G., Armour, C. D. & Hartwell, L. H. (1998). Genetics, 150, 75–93. [DOI] [PMC free article] [PubMed]
- Roberts, J. A., Bell, S. D. & White, M. F. (2003). Mol. Microbiol. 48, 361–371. [DOI] [PubMed]
- Sakurai, S., Kitano, K., Yamaguchi, H., Hamada, K., Okada, K., Fukuda, K., Uchida, M., Ohtsuka, E., Morioka, H. & Hakoshima, T. (2005). EMBO J. 24, 683–693. [DOI] [PMC free article] [PubMed]
- Seybert, A. & Wigley, D. B. (2004). EMBO J. 23, 1360–1371. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shiomi, Y., Shinozaki, A., Nakada, D., Sugimoto, K., Usukura, J., Obuse, C. & Tsurimoto, T. (2002). Genes Cells, 7, 861–868. [DOI] [PubMed]
- Thelen, M. P., Venclovas, C. & Fidelis, K. (1999). Cell, 96, 769–770. [DOI] [PubMed]
- Tsurimoto, T. (1999). Front. Biosci. 4, D849–D858. [DOI] [PubMed]
- Umar, A., Buermeyer, A. B., Simon, J. A., Thomas, D. C., Clark, A. B., Liskay, R. M. & Kunkel, T. A. (1996). Cell, 87, 65–73. [DOI] [PubMed]
- Venclovas, C. & Thelen, M. P. (2000). Nucleic Acids Res. 28, 2481–2493. [DOI] [PMC free article] [PubMed]
- Waga, S. & Stillman, B. (1998). Annu. Rev. Biochem. 67, 721–751. [DOI] [PubMed]
- Warbrick, E. (2000). Bioessays, 22, 997–1006. [DOI] [PubMed]
- Williams, G. J., Johnson, K., Rudolf, J., McMahon, S. A., Carter, L., Oke, M., Liu, H., Taylor, G. L., White, M. F. & Naismith, J. H. (2006). Acta Cryst. F62, 944–948. [DOI] [PMC free article] [PubMed]
- Xing, G., Hlinkova, V. & Ling, H. (2007). Cryst. Growth Des. 7, 2202–2205.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: PCNA3, 2ijx, r2ijxsf
PDB reference: PCNA1–PCNA2, 2io4, r2io4sf
PDB reference: PCNA1–PCNA2–PCNA3, 2nti, r2ntisf