

Abstract
A new withanolide (1) named withawrightolide, and four known withanolides (2–5) were isolated from the aerial parts of Datura wrightii (Solanaceae). The structure of compound 1 was elucidated through 2D NMR and other spectroscopic techniques. In addition, the structure of withametelin L (2) was confirmed by X-ray crystallographic analysis. Using MTS viability assays, withanolides 1–5 showed antiproliferative activities against human glioblastoma (U251 and U87), head and neck squamous cell carcinoma (MDA-1986), and normal fetal lung fibroblast (MRC-5) cells with IC50 values in the range between 0.56 and 5.6 μM.
Withanolides are a group of modified, highly-oxygenated C28 ergostane-type steroids, present primarily in several genera of the Solanaceae which include Acnistus, Datura, Dunalia, Jaborosa, Physalis, and Withania. In recent years these compounds have gained significant scientific interest due to their structural and biological diversity, in conjunction with their antitumor capacities.1–3 Recently we reported, as part of an ongoing study, the isolation and antiproliferative activities of a series of withanolides from Physalis longifolia, Vassobia brevifolia, and Withania somnifera with oxygenation at C-1, 3, 4, 5, 6, 7, 11, 17, 19, 20, 22, 27 and 28.4–7
The Datura genus is a rich source of oxygen substituted C-21 withanolides2 yet there have been limited biological activity studies. We therefore chose to investigate the native Kansas plant Datura wrightii Regel to continue our Solanaceae derived withanolide work, and to further probe withanolide structure-activity relationships.3,4
We report herein the first phytochemical and bioactivity study of withanolides from D. wrightii including all details pertaining to the isolation, structure elucidation, and cytotoxicity (using MTS viability assays) of the new withanolide withawrightolide (1) and four known withanolides (2–5).
Compounds 1–5 were isolated from a CH2Cl2–MeOH (1:1) extract of the aerial parts of the title plant (see Experimental Section). The molecular formula of the minor component 1 was determined to be C28H38O5 by HRESIMS and NMR experiments, equating to ten double-bond equivalents. The IR absorptions of 1 indicated the presence of OH (3435 cm−1), keto and ester (1740 and 1705 cm−1) groups. The 1H NMR spectrum (Table 1) showed signals of three methyl groups at δ 0.72 (3H, s), 1.12 (3H, s), 1.42 (3H, s); four protons attached to oxygenated carbons at δ 3.31 (1H, t, J = 2.9 Hz), 3.71 (1H, dd, J = 3.3, 13.3 Hz), 3.87 (1H, d, J = 13.3 Hz), 4.64 (1H, brs); and two olefinic methine groups at δ 6.00 (1H, s) and 6.75 (1H, s). The 13C NMR (APT) and HSQC spectra for 1 (Table 1) displayed 28 carbon signals differentiated as three CH3, ten CH2 (including an olefinic at δ 130.3 and an oxygenated at δ 60.7), eight CH (including two oxygenated at δ 75.7 and 73.1), and seven C (including a keto carbonyl at δ 217.7, an ester carbonyl at δ 165.4, an olefinic at δ 139.0, and an oxygenated at δ 69.5), corresponding to C28H37. The remaining hydrogen atom was therefore assigned as an OH group, indicating that seven rings must be present in the structure.
Table 1.
1H (500 MHz) and 13C (125 MHz) NMR Data for Withawrightolide 1 in CDCl3
| Position | δc, type | δH (J in Hz) | Position | δc, type | δH (J in Hz) | |
|---|---|---|---|---|---|---|
|
|
|
|||||
| 1 | 217.7, C | 15 | 24.1, CH2 | 1.73, m; 1.26, m | ||
| 2 | 39.6, CH2 | 2.81, ddd (1.7, 5.4, 18.1) 2.14, d (18.1) |
16 | 26.7, CH2 | 1.74, m; 1.42, m | |
| 3 | 16.0, CH | 1.39, m | 17 | 47.7, CH | 1.75, m | |
| 4 | 17.2, CH2 | 0.065, dd (3.8, 5.8) 0.78, ddd (1.7, 5.8, 5.9) |
18 | 13.0, CH3 | 0.72, s | |
| 5 | 36.0, C | 19 | 15.0, CH3 | 1.12, s | ||
| 6 | 73.1, CH | 3.31, t (2.9) | 20 | 39.8, CH | 1.84, m | |
| 7 | 37.4, CH2 | 1.96, m; 1.27, m | 21 | 60.7, CH2 | 3.87, d (13.3); 3.71, dd (3.3, 13.3) | |
| 8 | 29.3, CH | 1.86, m | 22 | 75.7, CH | 4.64, brs | |
| 9 | 47.4, CH | 0.97, ddd (3.4, 10.7, 12.2) | 23 | 33.5, CH2 | 2.0, dd (1.7, 9.0); 1.90, m | |
| 10 | 52.6, C | 24 | 69.5, C | |||
| 11 | 22.2, CH2 | 1.42, m; 1.34, m | 25 | 139.0, C | ||
| 12 | 39.5, CH2 | 1.89, m; 1.31, m | 26 | 165.4, C | ||
| 13 | 43.3, C | 27 | 130.3, CH2 | 6.75, s; 6.00, s | ||
| 14 | 55.8, CH | 1.15, m | 28 | 25.9. CH3 | 1.42, s | |
The NMR data of 1 were closely related to, a major isolate of our investigation, the six-ringed withanolide withametelin L (2) [(20R,22R,24R)-21,24-epoxy-12β-hydroxy-1-oxowitha-2,25,25(27)-trienolide].8 The structure of 2 was confirmed by X-ray crystallographic studies as shown in Figure 1. Compounds 1 and 2 were found to contain identical bicyclic side chain moieties; an exocyclic double bond [an olefinic methylene C-27 at δC 130.3 and two singlet olefinic protons at δH 6.00 and 6.75 (each 1H, s, H2-27)] conjugated with the lactone carbonyl (C-26: δC 165.4); characteristic signals for oxygenated C-21 [a methylene with δC 60.7 and two protons at δH 3.87 (1H, d, J = 13.3 Hz), 3.71 (1H, dd, J = 13.3, 3.3 Hz)] and C-22 [a methine with δC 75.7 and δH 4.64 (1H, brs)], and a tertiary methyl group C-28 with δC 25.9 and δH 1.42 (3H, s) adjacent to the oxygenated quaternary C-24 with δC 69.5.
Figure 1.

X-ray ORTEP drawing of withametelin L (2)
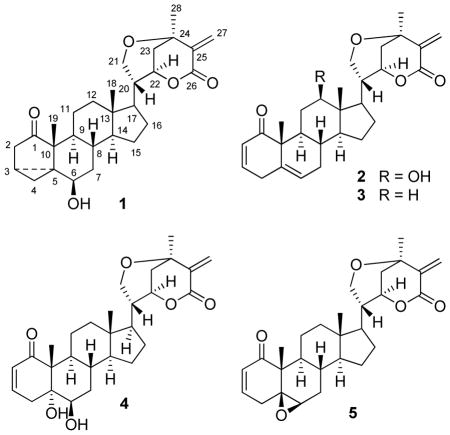
The differences between 1 and 2 were observed within the steroid nucleus moieties. Compound 1 showed unusual NMR signals for a keto group (δ 217.7) and a methylene group [δC 17.2, δH 0.78 (1H, ddd, J = 1.7, 5.8, 5.9 Hz) and 0.065 (1H, dd, J = 3.8, 5.8 Hz)], implying the presence of a five-membered keto ring (the chemical shift value of a keto group in a six-membered-ring is less than 210 ppm) and a cyclopropane ring (when considering the markedly low chemical shift value at δH 0.065 and small geminal coupling constant 5.8 Hz), respectively. The presence of 1-oxo-3,5-cyclo-6-hydroxy in the A/B rings of the steroid nucleus was deduced by HSQC and 1H-1H COSY fragment of -C(2)H2-C(3)H-C(4)H2-; HMBC correlations between H3-19 (δ 1.12, 3H, s) and C-1 (δ 217.7), C-5 (δ 36.0), C-9 (δ 47.4), and C-10 (δ 52.6); between H-3 (δ 1.39, 1H, m) and C-1, C-5, and C-6 (δ 73.1). The presence of a three-membered ring formed by C-3, C-4, C-5 was also supported by the chemical shift values of C-3 (CH, δ 16.0) C-4 (CH2, δ 17.2), and C-5 (C, δ 36.0). Thus, the planar structure of 1 was represented as shown.
As for the stereochemistry, β orientation of the H-3 proton was assigned based on the ROESY correlation between H-3 and H3-19. Axial β orientation of the OH group at C-6 was established by the small coupling constant between H-6 (δ 3.31, t, J = 2.9 Hz) and H2-7, and the ROESY correlation between H-6 and H-4α (δ 0.78, 1H, ddd, J = 1.7, 5.8, 5.9 Hz). These NMR data and assignments were in good agreement with those withanolides reported in the literature with a 1-oxo-3,5-cyclo-6-hydroxy functionality in the A/B rings of the steroid nucleus.9,10 Accordingly, the structure of 1 was elucidated as (20R,22R,24R)-21,24-epoxy-3α,5α-cyclo-1-oxowitha-25(27)-enolide on the basis of biogenetic grounds, and subsequently named withawrightolide. Detailed 1H and 13C NMR spectrometric analysis utilizing 2D correlational techniques were undertaken and summarized in Table 1.
The four other withanolides were identified through data comparisons with those published in the literature, as withametelin L (2),8 withametelin also known as daturilin (3),11,12 withametelin O (4),8 and withametelin F (5).13
Withawrightolide 1 contains a cyclopropane ring, a rarity in withanolides. A literature investigation showed that only five of the 820 withanolides reported to date contain a cyclopropane ring: physalin S isolated from Physalis alkekengi var. francheti,9 cilistols p, pm, p1 and u from Solanum cilistum (Solanaceae).10 This is the first report of such a structural type3 present in Datura species.
All the five withanolides (1–5) isolated were tested for cytotoxicity against human glioblastoma (U251 and U87), head and neck squamous cell carcinoma (MDA-1986), and normal fetal lung fibroblast (MRC-5) cells, the results of which were summarized in Table 2. Withanolides 1–5 demonstrated cytotoxicity with IC50 values ranging from 0.56–3.6 μM in the cancer cells and 3.3–5.6 μM in the normal fibroblasts. Specifically, concentrations required to obtain cytotoxicity in the normal MRC-5 cells were 3.8–7.4 fold higher than those in U87 cells. Our previously published work on a commonly known withanolide, withaferin A,4,13 is provided as a control for comparison. The potencies of 1–5 are on average slightly lower than withaferin A, suggesting that oxygenation of C-21 did not contribute toward the observed antiproliferative activities. Furthermore, the comparison of withanolides 2 (12β-hydroxy 3) and 3 revealed that oxygenation of C-12 is also not a contributor for antiproliferative activities. These observations are in good agreement to those reported in the literature.3 On the other hand, withanolides 1–5 appear to have greater selectivity than withaferin A for cancer cells (especially in the glioma and head and neck squamous cancer cells tested) compared to normal cells such as fibroblasts with average selectivity of between 4 and 7 fold. Such a level of selectivity may provide for a larger therapeutic window for treatment in vivo.
Table 2.
Cytotoxicity (IC50) of Isolated Withanolides (μM) against Four Cell Linesa
| Compound | U87 | U251 | MDA-1986 | MRC-5 | MRC-5:U87 |
|---|---|---|---|---|---|
| 1 | 1.5 | 3.6 | 3.1 | 5.6 | 3.8 |
| 2 | 0.57 | 1.3 | 2.3 | 4.2 | 7.4 |
| 3 | 1.0 | 2.2 | 2.7 | 5.1 | 5.1 |
| 4 | 1.1 | 2.8 | 3.0 | 4.6 | 4.1 |
| 5 | 0.56 | 1.4 | 1.5 | 3.3 | 6.0 |
| Withaferin Ab | 1.1 | 0.69 | 0.80 | 0.20 | 0.19 |
For cell lines used, see text.
Withaferin A was used as control.
Due to the observed proliferation inhibition of the withanolides 1–5, further U251cell-based western blot analysis was performed. Total cell expression of heat shock protein 70 (HSP70) and HSP32, proteins associated with cellular stress and withaferin A activity,13 were monitored 24 hours post-treatment (Figure 2). Post-treatment HSP70 upregulation was observed for 5 μM concentrations of withanolides 2, 3, and 5. Withanolides, 1–5, elevated HSP32 levels in a concentration-dependent manner.
Figure 2.
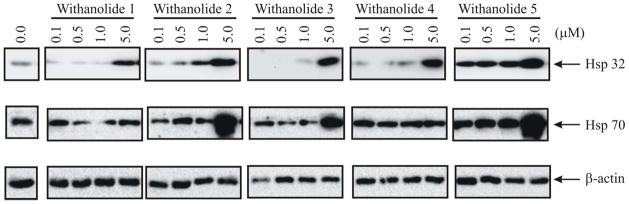
Western blot analysis of glioblastoma cell line U251 following treatment with withanolides 1–5. Total levels of HSP32 and HSP70, biomarkers of cellular stress and withanolide activity, were screened at 0, 0.1, 0.5, 1, and 5 μM concentrations of each compound for 24h. HSP70 was markedly upregulated at 5 μM with withanolides 2, 3, and 5, while HSP32 levels were elevated in a concentration-dependent manner with all compounds 1–5 tested. The β-actin protein was used as a loading control marker.
In this report, the heat shock effect and oxidative stress effect was abrogated following administration of N-acetyl-cysteine to the cells. This stress response effect was only observed with compounds 2, 3, and 5. It is likely that the epoxide at C5–6 in compound 5 may lead to free radical or reactive-oxygen species formation that could explain an oxidative stress on the cells. Additionally the double bond at C5–6 (compounds 2 and 3) may also convey some of the biologic activity seen with these molecules as compounds 1 and 4 lack either an epoxy group or a double bond at C5–6 and do not exhibit this stress response following treatment. Further studies, however, are warranted to validate this observation and more completely evaluate the mechanism of antiproferative action of these withanolides.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured with a Rudolph RS Autopol IV automatic polarimeter. IR data were obtained with a Thermo Nicolet Avatar 360 FT-IR spectrometer. NMR spectra were recorded with a Bruker AV-400 or AV-500 instrument with a cryoprobe for 1H, APT, COSY/DQF-COSY, HSQC, HMBC, and NOESY/ROESY. Chemical shift values are given in δ (ppm) using the peak signals of the solvent CDCl3 (δH 7.26 and δC 77.23) as references and coupling constants were reported in Hz. ESIMS data were measured with an Agilent 1200 Series LC-MS/MS ion trap 6300 mass spectrometer. HRESIMS data were collected with a LCT Premier time of flight mass spectrometer (Waters Corp., Milford, MA). Column chromatography was performed on silica gel (particle size 12–25 μm) (Sorbent Technologies, Atlanta, GA), or MCI CHP20P (particle size 75–150 μm) (Sigma-Aldrich, St. Louis, MO), or Sephadex LH-20 (GE Healthcare, Piscataway, NJ), or C18 reversed-phase silica gel (particle size 40–65 μm) (Sigma-Aldrich, St. Louis, MO). Normal-phase silica gel G TLC plates (w/UV 254) and reversed-phase C18 TLC plates (w/UV 254) (Sorbent Technologies, Atlanta, GA) were used for fraction and compound detection. The spots were visualized using UV light at 254 nm and spraying with 10% EtOH-sulfuric acid reagent. Semi-preparative HPLC was performed on an Agilent 1200 unit equipped with a DAD detector, utilizing a Lichrospher RP-18 column (250 × 10 mm, 5 μm).
Plant Material
The fresh aerial parts of D. wrightii were collected in July 2010 from the Quivira National Wildlife Refuge (latitude: 38.10165°; longitude: 98.45496°), Stafford County, Kansas and authenticated by Dr. Kelly Kindscher of the Kansas Biological Survey, University of Kansas, Lawrence, Kansas, United States. A voucher specimen, 4091, was deposited in the R.L. McGregor Herbarium of the University of Kansas.
Extraction and Isolation
The collected biomass was air dried, ground to a coarse powder (2.1 Kg), and extracted three times with CH2Cl2–MeOH (50:50, 6.0 L) at room temperature. After removing the solvents under vacuum, the extract (220 g) was suspended in 400 mL H2O, followed by successive partitions with n-hexane, ethyl acetate and n-butanol (3 × 500 mL). The resulting n-BuOH fraction (52 g) collected was applied to a MCI CHP20P column (2.0 Kg) and eluted subsequently with mixtures of H2O–MeOH (100:0, 80:20, 60:40, 40:60, 85:15, 0:100), in order of increasing concentrations of MeOH. The 85% MeOH fraction (12.0 g) was subjected to silica gel CC, eluted with CH2Cl2-CH3COCH3 with increasing amounts of acetone to afford compounds withametelin L 2 (340 mg), withametelin also known as daturilin 3 (150 mg), withametelin O 4 (12 mg), and withametelin F 5 (7 mg). The fractions eluted by CH2Cl2-CH3COCH3 (6:1) were subjected to semi-preparative HPLC, with the mobile phase CH3CN-H2O (44:56), to afford 1 (8 mg).
Withawrightolide (1): amorphous powder; [α]25D +19.2 (c 0.18, CHCl3); UV (MeOH) λmax (log ε) 226 (3.05) nm; IR (neat) νmax 3435 (br), 2922, 1740, 1705 cm−1; 1H NMR and 13C NMR, see Table 1; ESIMS (positive-ion mode) m/z 455 [M + H]+; HRESIMS m/z 477.2610 [M + Na]+ (calcd for C28H38O5Na, 477.2617).
Single-Crystal X-ray Structure Determination of withametelin L (2)
Crystal analysis was performed with a colorless cubic crystal (dimensions 0.48 × 0.12 × 0.05 mm3) obtained from CH2Cl2–CH3CN (1:1) using Cu Kα radiation (λ = 1.54178 Å) on a Bruker APEX2 diffractometer equipped with a Bruker MicroStar microfocus rotating anode X-ray source and Helios multilayer optics. Crystal data for 2: C28H36O5 (formula weight 452.57), monoclinic, space group P21, T = 100(2) K, crystal cell parameters a = 11.7590(3) Å, b = 6.5766(2) Å, c = 15.0921(4) Å, β = 96.0790(10)°, V = 1160.57(10) Å3, Dc = 1.295 Mg/m3, Z = 2, F(000) = 488, absorption coefficient μ = 0.700 mm−1. A total of 10166 reflections were collected in the range 2.94 < θ < 69.26°, with 2971 independent reflections [R(int) = 0.0155], completeness to θ = 66° was 96.5%. Multi-scan absorption correction applied; full-matrix least-squares refinement on F2, the number of data/restraints/parameters were 2971/1/443; goodness-of-fit on F2 = 1.060; final R indices [I > 2σ(I)], R1 = 0.0249, ωR2 = 0.0646; R indices (all data), R1 = 0.0249, ωR2 = 0.0646; largest difference peak and hole, 0.169 and −0.126 e/Å−3.
Cell Culture and Cell Proliferation and Viability Assay
Two human glioblastoma cell lines, U251 and U87, were generously provided by Dr. Jann Sarkaria (Mayo Clinic, Rochester, MN). The head and neck squamous cell carcinoma (HNSCC) cell line MDA-1986 was a gift from Dr. Jeffrey Myers (University of Texas, M.D. Anderson Cancer Center, Houston, TX). The fetal lung fibroblast cell line MRC-5 was obtained from ATCC. Cell maintenance, experimental procedures, and data presentation were as similar as previously described.4,14 The U87 and U251 cell lines were grown in DMEM (#6429; Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) and 1% penicillin/streptomycin (100 IU/mL/100 μg/mL; Sigma-Aldrich). The MDA-1986 and MRC-5 cell lines were grown in similar media but also supplemented with 1% L-glutamine (200 mM; Sigma-Aldrich), 1% MEM-vitamin (100x; Hyclone, Logan, UT), and 1% MEM-nonessential amino acids (Sigma Aldrich). The cells were grown in a monolayer and were incubated at 37 °C in a humidified atmosphere containing 5% CO2 until they reached 75–90% confluence level. The viability of cells after treatment with withanolides (1–5) were determined using an MTS assay. Cells were seeded in 96-well plates at 2,500 cells/well in 90 μL of media. Following an approximate 6 h incubation period, 10 μL of drug-containing media in various concentrations was added to each well, and the cells were incubated for an additional 72 h. The number of viable cells was quantified by the colorometric CellTiter96 Aqueous MTS assay (Promega, Fitchburg, WI) at 490 nm on a BioTek Synergy 2 plate reader (BioTek, Winooski, VT) as per the manufacturer’s instructions. All experiments were carried out in triplicate on two separate occasions, and GraphPad (GraphPad Inc., San Diego, CA) was used to generate best-fit sigmoidal dose response curves for IC50 determination.
Western Blotting Analysis
Cells were plated at an amount previously determined, based on the growth characteristics of the cells, to have a high enough yield without achieving complete confluency upon harvest and were allowed to grow overnight. Cells were treated with appropriate concentrations of the withanolides. Upon completion of treatment, proteins were collected, quantified, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and electrotransfered onto a Hybond nitrocellulose membrane as previously described.4 Actin levels were assessed to ensure equal loading and transfer of proteins. Western analysis was completed in the human U251 cell line. Primary antibodies were utilized against HSP70 (Enzo Life Sciences, Farmingdale, NY; ADI-SPA-810; 1:1000), HSP32/heme oxygenase 1 (Life Sciences, Farmingdale, NY; SPA-894; 1:1000), and total actin (EMD Millipore, Billerica, MA; MAB1501; 1:50,000). Donkey anti-rabbit IgG HRP (sc-2313; 1:5000) and goat anti-mouse IgG HRP (sc-2005; 1:5000) secondary antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, CA).
Supplementary Material
Acknowledgments
This study was supported, in part, by grant IND 0061464 (awarded to B.N.T. and K.K.) from the Kansas Bioscience Authority (KBA) and Center for Heartland Plant Innovations (HPI). The authors also acknowledge partial financial assistance from grant NFP0066367 from the Institute for Advancing Medical Innovation (IAMI) (awarded to M.S.C. and to B.N.T.). Partial support of the in vitro experiments was provided by the University of Kansas Center for Cancer Experimental Therapeutics NIH-COBRE P20 RR015563 (PI: B.N.T., project award PI: M.S.C.). The authors are grateful to NSF-MRI grant CHE-0923449 that was used to purchase a Bruker APEX2 X-ray diffractometer and the software. The authors thank Q. Long, H. Loring, G. Beverlin and M. Ferreira, for assistance in plant collection and identification and R. Gollapudi with initial extraction and partition of biomass.
Footnotes
Dedicated to Dr. Lester A. Mitscher of the University of Kansas for his pioneering work on the discovery of bioactive natural products and their derivatives.
Supporting Information. 1H and 13C NMR spectra of withanolide 1 are available free of charge via http://pubs.acs.org. Crystallographic data for the structure of 2 as reported in this paper were deposited with the Cambridge Crystallographic Data Centre, under reference number CCDC 907707. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033 or deposit@ccdc.cam.ac.uk.
References
- 1.Chen LX, Hao H, Qiu F. Nat Prod Rep. 2011;28:705–740. doi: 10.1039/c0np00045k. [DOI] [PubMed] [Google Scholar]
- 2.Misico RI, Nicotra VE, Oberti JC, Barboza G, Gil RR, Burton G. Prog Chem Org Nat Prod. 2011;94:127–229. doi: 10.1007/978-3-7091-0748-5_3. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Samadi AK, Cohen MS, Timmermann BN. Pure Appl Chem. 2012;84:1353–1367. doi: 10.1351/PAC-CON-11-10-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samadi AK, Tong XQ, Mukerji R, Zhang HP, Timmermann BN, Cohen MS. J Nat Prod. 2010;73:1476–1481. doi: 10.1021/np100112p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong X, Zhang H, Timmermann BN. Phytochemistry Lett. 2011;4:411–414. doi: 10.1016/j.phytol.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Samadi AK, Gallagher RJ, Araya JJ, Tong X, Day VW, Cohen MS, Kindscher K, Gollapudi R, Timmermann BN. J Nat Prod. 2011;74:2532–2544. doi: 10.1021/np200635r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H, Motiwala H, Samadi A, Day V, Aubé J, Cohen M, Kindscher K, Gollapudi R, Timmermann B. Chem Pharm Bull. 2012;60:1234–1239. doi: 10.1248/cpb.c12-00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan Y, Wang X, Hu X. J Nat Prod. 2007;70:1127–1132. doi: 10.1021/np070096b. [DOI] [PubMed] [Google Scholar]
- 9.Makino B, Kawai M, Kito K, Yamamura H, Butsugan Y. Tetrahedron. 1995;51:12529–12538. [Google Scholar]
- 10.Zhu XH, Ando J, Takagi M, Ikeda T, Yoshimitsu A, Nohara T. Chem Pharm Bull. 2001;49:1440–1443. doi: 10.1248/cpb.49.1440. [DOI] [PubMed] [Google Scholar]
- 11.Oshima Y, Bagchi A, Hikino H, Sinha SC, Sahai M, Ray AB. Tetrahedron Lett. 1987;28:2025–2028. [Google Scholar]
- 12.Siddoqio S, Sultana N, Ahmad SS, Haider SI. Phytochemistry. 1987;26:2641–2643. [Google Scholar]
- 13.Jahromi MAF, Manickam M, Gupta M, Oshima Y, Hatakeyama S, Ray AB. J Chem Research (S) 1993:234–235. [Google Scholar]
- 14.Grogan PT, Sleder KD, Samadi AK, Zhang H, Timmermann BN, Cohen MS. Invest New Drugs. 2012;30 doi: 10.1007/s10637-012-9888-5. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.