

Abstract
Like many coactivators, the GACKIX domain of the master coactivator CBP/p300 recognizes transcriptional activators of diverse sequence composition via dynamic binding surfaces. The conformational dynamics of GACKIX that underlie its function also render it especially challenging for structural characterization. We find that the ligand discovery strategy of Tethering is an effective method for identifying small molecule fragments that stabilize the GACKIX domain and enables, for the first time, the crystallographic characterization of this important motif. The 2.0 Å resolution structure of GACKIX complexed to a small molecule was further analyzed by molecular dynamics simulations, revealing the importance of specific side chain motions that remodel the activator binding site in order to accommodate binding partners of distinct sequence and size. More broadly, these results suggest that Tethering can be a powerful strategy for identifying small molecule stabilizers of conformationally malleable proteins, thus facilitating their structural characterization and accelerating the discovery of small molecule modulators.
Transcriptional coactivators are among the most conformationally malleable of proteins and contain binding surfaces that undergo rapid remodeling as complexes are formed with their cognate ligands.1,2 This plasticity is essential to their function, enabling recognition of an often diverse array of transcriptional activator sequences.3,4 Perhaps the best-studied example of this is the GACKIX domain of the coactivator CBP/p300, a small (90 amino acid) domain that is known to interact with >10 distinct amphipathic sequences at two distinct binding sites (Figure 1a) in order to stimulate transcription at hundreds of genes,5–9 including those regulating hematopoiesis, memory formation and the inflammatory response.10–12 Not surprisingly, the malleability of this class of proteins renders them especially intractable to crystallographic characterization, either alone or in complex with their binding partners. In the case of the GACKIX domain, there are no crystal structures of either free protein or any complexed form. Here we demonstrate that a covalently linked small-molecule ligand of this conformationally dynamic protein enables, for the first time, a high resolution snapshot of the coactivator interacting with a ligand. This first crystal structure of GACKIX provides important insight to the side chain orientations of this domain in the context of ligand recognition, particularly with regard to small molecules. Furthermore, these results show that the ligand discovery strategy of Tethering13–16 can be expanded to targeting conformationally dynamic proteins and enable their structural characterization.
Figure 1.

a) The GACKIX domain is in the N-terminal region of CBP/p300. GACKIX interacts with >10 amphipathic transcriptional activators using two distinct sites.5–9 MLL, HBZ and c-Jun target a smaller, deeper site while the activation domains of cMyb and CREB (pKID) utilize a second, broader site. b) Schematic of the Tethering screen used to identify small molecule fragments (1–10 and 2–64) that form a disulfide bond with a cysteine introduced at position 664 (L664C) within GACKIX. See Supporting Information for details.
We screened for small molecules that interact with the GACKIX domain using the Tethering approach,16 a strategy that provides a mechanism for the rapid discovery of covalent ligands (Figure 1b). Attention was focused on the binding site that is targeted by the transcriptional activation domains of proteins such as the Mixed Lineage Leukemia (MLL) activator and c-Jun; the Tethering approach is a fragment discovery method and the smaller, deeper MLL/c-Jun binding site appeared the more targetable by low molecular weight compounds.17,18 Towards this end, a residue at the rim of the binding surface, L664, was mutated to a cysteine and the resulting GACKIX L664C mutant fully characterized (see SI for details). Small molecule fragments containing a disulfide motif were then screened for the ability to form a disulfide bond with GACKIX L664C in the presence of a competitor, β-mercaptoethanol. Two fragment ligands emerged from the screen with high Tethering efficiency to GACKIX L664C as quantified by DR(Dose Response)50 values (2–8 μM), fragments 1–10 and 2–64 (Figure 1b).
To assess the effect of tethered 1–10 or 2–64 on the binding properties of GACKIX, fluorescent anisotropy binding assays were used to measure the binding affinity of wildtype GACKIX, GACKIX L664C and fragment-tethered GACKIX L664C complexes to native transcriptional activator ligands that target the two different binding sites (Figure 2a). Consistent with the screen design, the presence of 1–10 or 2–64 decreased MLL binding to GACKIX L664C by ~22 to 33-fold. Also, while tethered 2–64 does not affect GACKIX’s binding affinity for pKID, the transcriptional activation domain of CREB that interacts with the distal binding site,19 GACKIX tethered to fragment 1–10 does exhibit attenuated binding to pKID (~2-fold). This suggests that 1–10 engages the amino acid side chains comprising the allosteric network connecting the two binding sites.6,20,21
Figure 2.

a) KDs for GACKIX constructs interacting with fluorescein-labeled MLL and pKID peptides were determined by fluorescent anisotropy. Each KD is a fitted result of experiments performed in triplicate with the indicated error (SD). b) Bar graph depicts the percent increase in melting temperature (TM) upon tethering to either 1–10 or 2–64 as monitored by circular dichroism (blue bars) and the percent of backbone amides protected from H-D exchange upon attachment of the small molecules (red bars). The green bars represent the fold-increase in resistance to thermolysin degradation of the GACKIX mutants when tethered to 1–10 and 2–64. Data is normalized to GACKIX L664C.
The tethered fragments significantly altered the stability of the GACKIX domain. This was assessed for each of the fragment-protein pairs by measuring changes in CD-monitored thermal melting temperature, amide hydrogen-deuterium (H-D) exchange and thermolysin-mediated proteolysis (Figure 2b). For example, the 1–10—GACKIX L664C and the 2–64—GACKIX L664C complexes exhibit a 15–18 °C (≥20%) increase in melting temperature. In the H-D exchange experiment, the mass of free GACKIX L664C shifted 29 Da upon exposure to D2O for 1 min as monitored by mass spectrometry,13 whereas the mass shift was 17 and 13 Da when 1–10 and 2–64 were tethered to GACKIX L664C, respectively, showing that 40%–55% of the exchangeable amides were protected from H-D exchange compared to the free protein. The proteolytic stability (half life) of the tethered complex increased 5–37 fold compared to the untethered protein, (for 1–10 for example: T½ of 10 minutes versus 2.1 minutes).22,23 These findings encouraged pursuit of crystallization of fragment—GACKIX L664C complexes.
Of the various fragment—protein complexes and conditions that were screened, the best results were obtained with 1–10—GACKIX L664C under the crystallizing condition of 1.8 M ammonium sulfate and 0.1 M Tris, pH 7.0 at 25° C, leading to crystals amenable for diffraction. However, only microcrystals of 2–64 tethered to GACKIX L664C were obtained and were of too poor quality to solve. Initially, molecular replacement strategies using the NMR structures of GACKIX bound to native transcriptional activation domains were used but did not lead to the 1–10—GACKIX structure.19,24 Therefore, a selenomethionine-incorporated GACKIX L664C tethered to 1–10 was prepared and the X-ray structure was solved. Using these data, the structure of 1–10—GACKIX L664C was determined to 2.0 Å resolution.
As illustrated in Figure 3a, the small molecule 1–10 sits within the MLL/c-Jun binding site of GACKIX, and is oriented toward the core of the protein between helices α3(residues 646–664) and α2(residues 623–638). Notably, the aromatic ring of 1–10 is positioned relatively deep in a hydrophobic pocket lined by the side chains of Ile611, Leu628, Leu607, Val635, and Tyr631 (Figure 3b); Leu628 and Tyr631 have previously been shown to be key residues involved in GACKIX interacting with MLL.24,25 Tyr631 in particular, closely contacts the aromatic ring of 1–10 (~4Å), illustrated by the above 2σ deviation of the Tyr631 ϕ and ψ angles.26 Consistent with these data, chemical shift perturbation experiments with 15N-labeled KIX L664C free and covalently tethered to 1–10 (Figure 4a) revealed significant changes in the backbone amide shifts of the residues lining the hydrophobic binding surface for 1–10 (Ile611, Leu628, Leu607, Val635 and Tyr631).
Figure 3.
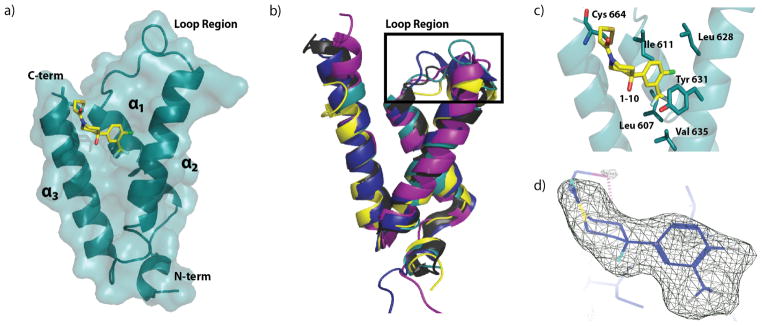
a) Refined crystal structure of GACKIX L664C covalently tethered to fragment 1–10. Refined resolution = 2.0 Å, Rwork/Rfree= 0.2064/0.2329. b) Crystal structure of GACKIX L664C tethered to 1–10 (teal) superimposed using Coot on the NMR solution structures of GACKIX in complex with cognate transcriptional activation domains: pKID (yellow, PDB ID 1KDX, R.M.S.D.=1.40 Å); with MLL and c-Myb (deep blue, PDB ID 2AGH, R.M.S.D.=1.80 Å); with PCET (purple, PDB ID 2KWF, R.M.S.D.=1.81 Å); and with FOXO3A (black, PDB ID 2LQH, R.M.S.D.=1.07 Å). c) Interactions between 1–10 (yellow) and residue side chains of GACKIX L664C (blue) at the binding surface. d) 3σ electron density map (Fo-Fc) of 1–10 illustrates the fit of the small molecule.
Figure 4.

a) Results from chemical shift perturbation experiment (1H,15N-HSQC) with 1–10-tethered GACKIX L664C. Residues that shifted more than 1 SD upon 1–10 tethering are in yellow and include Ile611, Leu628, Leu607, Val635 and Tyr631, and Ile660; b) The difference in the average SASA (Solvent Accessible Surface Area) calculated by residue between simulations of GACKIX L664C untethered and tethered to 1–10 in units of Å2. A residue colored red is less solvent-exposed in the 1–10-tethered structure, with color intensity indicating the extent of the change; blue resides are more solvent-exposed in the 1–10-tethered structure.
The prevailing structural model of the amphipathic class of activator-coactivator complexes is that the activator forms an amphipathic helix upon binding to the surface of the coactivator.27–29 Although only a limited suite of surfaces have been characterized, the available data suggest that the binding surfaces are often broad,2,30 making them particularly challenging to target with small molecules that have far less volume and surface area than the typical helix of a transcriptional activator.31 Overlay of the 1–10—GACKIX L664C structure with the averages of the previously reported NMR structures of GACKIX-ligand complexes24,32,33 yields R.M.S.D. values between 1.07–1.81 Å, demonstrating the overall similarities in the backbone structure. The exception to this similarity is in the loop region (residues 612–622) between helices α1 and α2, which deviates significantly with R.M.S.D. values between 2.73–3.11 Å. This difference is not surprising, as conformational changes in the loop regions are thought to be integral to the ability of GACKIX to accommodate diverse native ligands.5,19,21,32
To dissect in more detail how the GACKIX surface remodels itself to recognize fragment 1–10 we carried out 40 ns molecular dynamics simulations of the GACKIX crystal structure with or without ligand 1–10. A gross comparison of the backbone reveals that a change in the loop conformation is the most significant, as shown in the root mean square fluctuations (Figure S6) and in the average structure overlay (Figure S7). These changes are often difficult to visualize by solution methods because the loop region contains several proline residues, but mutagenesis and NMR methods have suggested that conformational plasticity in this region underlies the ability of GACKIX to recognize diverse amphipathic sequences.20,21,24 It is this movement of the loop and a rotation of helix α1 that enable the formation of a narrower binding surface to accommodate a molecule that is considerably smaller than a peptidic helix (~77% smaller volume)(Figure S7). The binding surface that is targeted by 1–10 is also significantly different, both as a result of loop conformational changes and because of side chain motions as demonstrated by the change in solvent accessible surface area of the residues when the fragment is tethered (Figure 4b). For example, the liganded GACKIX shows a population shift in the Tyr 631 side chain χ angles relative to the untethered protein, leading to a hydrophobic binding surface for deeper interactions (Movie S1). Simulations of 2–64 tethered to GACKIX L664C suggest that the binding mode of this ligand is similar to that of 1–10 and further demonstrates the ability of this protein to adapt to different binding partners (Figure S8). The helices α3 and α2 must open to accommodate this larger ligand and corresponding changes in the chemical shifts of residues involved in this opening are observed by NMR (Figures S5 and S7).
In conclusion, we have obtained a 2Å-resolution snapshot of the conformationally dynamic coactivator GACKIX domain complexed with a small molecule. This will significantly facilitate using rational structure-based approaches to design more potent analogs; for example, current efforts include extending the molecule 1–10 at the C4 position of the aromatic ring in order to more effectively engage with the hydrophobic space within the GACKIX site. From a broader perspective, these results in combination with recent studies showing noncovalent small molecules stabilizing conformationally dynamic proteins34,35 suggest that Tethering may be an exceptionally enabling approach to obtain long-sought x-ray crystallography data of conformationally dynamic proteins. This includes transcriptional coactivators such as CBP/p300 targeted here, but also members of other cellular machines that rely upon conformationally dynamic interfaces to recognize binding partners.36,37
Supplementary Material
Acknowledgments
AKM is grateful for support of this work from NIH 2RO1 GM65330 and RO1 CA140667. W.C.P. acknowledges NIH F32 GM090550 and N.W. is a fellow of the UM PSTP (GM07767 NIGMS). CLB acknowledges GM037554. J.A.W. is grateful for support of this work from NIH R01 AI070292. J.D.S. is a fellow of California TRDRP (award #110385).
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). We thank Dr. David Smith of LS-CAT for help with remote data collection. Coordinates for the 1-10—GACKIX L664C complex have been deposited in the PDB (4I9O).
Footnotes
The authors declare no competing financial interests.
Details of the Tethering screen and additional structural details can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sugase K, Dyson HJ, Wright PE. Nature. 2007;447:1021–5. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 2.Thompson AD, Dugan A, Gestwicki JE, Mapp AK. ACS Chem Biol. 2012;7:1311–20. doi: 10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh GP, Ganapathi M, Dash D. Proteins. 2007;66:761–5. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 4.Boehr DD, Nussinov R, Wright PE. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzman RNDe, Goto NK, Dyson HJ, Wright PE. J Mol Biol. 2006;355:1005–13. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 6.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. J Biol Chem. 2002;277:43168–74. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 7.Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. Mol Cell Biol. 2001;21:2249. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cook PR, Polakowski N, Lemasson I. J Mol Biol. 2011;409:384–98. doi: 10.1016/j.jmb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vendel AC, McBryant SJ, Lumb KJ. Biochemistry. 2003;42:12481–7. doi: 10.1021/bi0353023. [DOI] [PubMed] [Google Scholar]
- 10.Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, Brindle PK. Nature. 2002;419:738–43. doi: 10.1038/nature01062. [DOI] [PubMed] [Google Scholar]
- 11.Wood MA, Attner MA, Oliveira AMM, Brindle PK, Abel T. Learning & Memory. 2006;13:609–17. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu GH, Qu J, Shen X. Biochim Biophys Acta. 2008;1783:713–27. doi: 10.1016/j.bbamcr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Proc Natl Acad Sci U S A. 2011;108:6056–61. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA. Proc Natl Acad Sci U S A. 2004;101:12461–6. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheer JM, Romanowski MJ, Wells JA. Proc Natl Acad Sci U S A. 2006;103:7595–600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Eric M, Gordon A, Wells JA. Proc Natl Acad Sci U S A. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Two natural products that target the MLL site have recently been identified.: Majmudar CY, Højfeldt JW, Arevang CJ, Pomerantz WC, Gagnon JK, Schultz PJ, Cesa LC, Doss CH, Rowe SP, Vasquez V, Tamayo-Castillo G, Cierpicki T, Brooks CLI, Sherman DH, Mapp AK. Angew Chem, Int Ed Engl. 2012;51:11258–11262. doi: 10.1002/anie.201206815.
- 18.A small pilot screen using protein observed 19F-NMR identifed a small molecule fragment that indicates with the MLL sites: Pomerantz WC, Wang N, Lipinski AK, Wang R, Cierpicki T, Mapp AK. ACS Chem Biol. 2012;7:1345–50. doi: 10.1021/cb3002733.
- 19.Radhakrishnan I, Perezalvarado G, Parker D, Dyson H, Montminy M, Wright P. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 20.Brüschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, Tollinger M. J Am Chem Soc. 2009;131:3063–8. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- 21.Korkmaz EN, Nussinov R, Haliloğlu T. PLoS Comput Biol. 2012;8:e1002420. doi: 10.1371/journal.pcbi.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park C, Marqusee S. Nature Methods. 2005;2:207–12. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 23.Hoofnagle AN, Resing KA, Ahn NG. Annu Rev Biophys Biomol Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- 24.De Guzman RN, Goto NK, Dyson HJ, Wright PE. J Mol Biol. 2006;355:1005–13. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 25.Arai M, Dyson HJ, Wright PE. FEBS Lett. 2010:4500–4504. doi: 10.1016/j.febslet.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.This is consistent with data from a solution binding study of untethered 1–10 interacting with GACKIX containing 19F-labeled Tyr631 that showed a dose-dependent change in the 19F chemical shift (see ref 18).
- 27.Sugase K, Dyson HJ, Wright PE. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 28.Uesugi M, Nyanguile O, Lu H, Levine aJ, Verdine GL. Science. 1997;277:1310–3. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- 29.Thoden JB, Ryan LA, Reece RJ, Holden HM. J Biol Chem. 2008;283:30266–30272. doi: 10.1074/jbc.M805200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuller JC, Burgoyne NJ, Jackson RM. Drug Discovery Today. 2009;14:155–61. doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Fry D. Peptide Science. 2006;84:535–552. [Google Scholar]
- 32.Wang F, Marshall C. Proc Natl Acad Sci U S A. 2012;109:6078–83. doi: 10.1073/pnas.1119073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zor T, De Guzman RN, Dyson HJ, Wright PE. J Mol Biol. 2004;337:521–34. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 34.Saalau-Bethell SM, Woodhead AJ, Chessari G, Carr MG, Coyle J, Graham B, Hiscock SD, Murray CW, Pathuri P, Rich SJ, Richardson CJ, Williams PA, Jhoti H. Nat Chem Biol. 2012;8:920–5. doi: 10.1038/nchembio.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basse N, Kaar JL, Settanni G, Joerger AC, Rutherford TJ, Fersht AR. Chemistry & Biology. 2010;17:46–56. doi: 10.1016/j.chembiol.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Dunker AK, Silman I, Uversky VN, Sussman JL. Curr Opin Struct Biol. 2008;18:756–64. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Dyson HJ, Wright PE. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.