

Abstract
The cell microenvironment has a profound influence on the behaviour, growth and survival of cells. The extracellular matrix (ECM) provides not only mechanical and structural support to cells and tissues but also binds soluble ligands and transmembrane receptors to provide spatial coordination of signalling processes. The ability of cells to sense the chemical, mechanical and topographical features of the ECM enables them to integrate complex, multiparametric information into a coherent response to the surrounding microenvironment. Consequently, dysregulation or mutation of ECM components results in a broad range of pathological conditions. Characterization of the composition of ECM derived from various cells has begun to reveal insights into ECM structure and function, and mechanisms of disease. Proteomic methodologies permit the global analysis of subcellular systems, but extracellular and transmembrane proteins present analytical difficulties to proteomic strategies owing to the particular biochemical properties of these molecules. Here, we review advances in proteomic approaches that have been applied to furthering our understanding of the ECM microenvironment. We survey recent studies that have addressed challenges in the analysis of ECM and discuss major outcomes in the context of health and disease. In addition, we summarize efforts to progress towards a systems-level understanding of ECM biology.
Keywords: cell adhesion, extracellular matrix, mass spectrometry, proteomics
Intercellular cohesion and communication are fundamental requirements of multicellular organisms. Indeed, the selective adhesion of cells to one another and to the extracellular matrix (ECM) within which they reside is necessary for much of metazoan anatomy and development (Özbek et al. 2010; Hynes 2012). The ECM provides a structural framework for cell binding, and this enables tissues and organs to form. In addition, the composition and organization of the ECM play key roles in determining how cells interact with and respond to their microenvironment (Frantz et al. 2010). Thus, the ECM defines the physical and chemical interactions that control cellular physiology and fate.
The identification and quantification of the components of distinct ECMs, the spatial and temporal dynamics of ECM molecules and the interactions underpinning ECM protein networks represent key steps towards understanding the role of the ECM in health and disease. Proteomics, the study of all proteins in a given system, offers an opportunity to address these challenges in a global manner, without the need for investigations based on predetermined molecular candidates. Here, we review recent progress in the isolation and proteomic analysis of extracellular molecules, with a focus on mass spectrometry (MS)–based proteomic strategies aimed at the non-specialist. A complete overview of ECM composition is beyond the scope of this review; rather, we highlight the technical and biological aspects of the latest ECM proteomic studies and discuss implications for future research on the regulation and dysregulation of ECM.
Ethical approval
For work involving the authors, ethical approval was obtained for investigations using human tissue, and animal experiments were carried out under the authority of the UK Home Office.
Roles of the ECM in health and disease
The ECM is composed of a complex network of proteins, glycoproteins and proteoglycans, which together provide tissue-specific biophysical and biochemical properties (Hynes 2009). Extracellular matrix molecules form basement membranes (which separate epithelium or endothelium from stroma and consist primarily of collagen IV, laminins, nidogens and perlecan) and interstitial ECM structures (Table 1), which support cell migration and endow tissue architecture and integrity. To become functional components of ECM, many ECM molecules require complex levels of transcriptional, translational and post-translational control (Table 1). For example, collagen synthesis involves several post-translational modifications, such as hydroxylation of proline and lysine residues, pro-peptide cleavage and covalent crosslinking by lysyl oxidases (Myllyharju & Kivirikko 2004). A characteristic feature of many ECM molecules is their modular structure. Exon replication, rearrangement and diversification and alternative splicing permit a limited number of modules to be combined into large multidomain molecules (Table 1) that have specific structural and functional characteristics. Fibronectin, for example, consists of a series of repeating modules (type I, II and III fibronectin repeats), several of which serve as binding sites for other ECM components, such as heparin, fibrin and collagens (Singh et al. 2010). Indeed, polymerization of fibronectin is necessary for the incorporation of collagen, fibrillin and thrombospondin into the ECM (Sottile & Hocking 2002; Kinsey et al. 2008). As is also the case for many proteoglycans, fibronectin presents soluble factors, such as bone morphogenetic proteins and fibroblast growth factors, to cells (Hynes 2009). Thus, the ECM acts as an accessible reservoir for signalling proteins, which enables local triggering of cellular signal transduction.
Table 1.
Major ECM components and their key domains, post-translational modifications and tissue distributions
| Molecule name | Gene name(s)* | Domains† | Post-translational modification(s)†,‡ | Examples of tissue distribution† |
|---|---|---|---|---|
| Fibril-forming collagens | ||||
| Collagen I | COL1A1, COL1A2 | Fibrillar collagen NC1; VWFC | Glyco; Hyl; Hyp; pro; X | Bone, cornea, skin, tendon |
| Collagen II | COL2A1 | Fibrillar collagen NC1; VWFC | Glyco; Hyl; Hyp; X | Cartilage, vitreous body |
| Collagen III | COL3A1 | Fibrillar collagen NC1; VWFC | Glyco; Hyl; Hyp; phospho; S–S; X | Blood vessels, bone, skin |
| Collagen V | COL5A1–COL5A3 | Collagen-like; fibrillar collagen NC1; laminin G-like/TSP N-terminal; VWFC | Glyco; Hyl; Hyp; phospho; sulf; S–S; X | Blood vessels, bone, cornea, placenta, skin, tendon |
| Collagen XI | COL11A1, COL11A2 | Collagen-like; fibrillar collagen NC1; laminin G-like/TSP N-terminal | Glyco; Hyp; pro; S–S; X | Cartilage, placenta, tendon |
| Collagen XXIV | COL24A1 | Collagen-like; fibrillar collagen NC1; laminin G-like/TSP N-terminal | Glyco | Bone, cornea |
| Collagen XXVII | COL27A1 | Collagen-like; fibrillar collagen NC1; laminin G-like/TSP N-terminal | Glyco; phospho | Cartilage |
| Network-forming collagens | ||||
| Collagen IV | COL4A1–COL4A6 | Collagen IV NC1 | Glyco; Hyl; Hyp; phospho; pro; S–S; Ubl; X | Basement membranes |
| Collagen VIII | COL8A1, COL8A2 | C1q | Hyp; pro | Basement membranes, blood vessels, connective tissues |
| Collagen X | COL10A1 | C1q | Hyp | Calcifying cartilage |
| Beaded filament–forming collagens | ||||
| Collagen VI | COL6A1–COL6A3, COL6A5, COL6A6 | BPTI/Kunitz inhibitor; collagen-like; FNIII; VWFA | Glyco; Hyl; Hyp; phospho; S–S | Bone, cartilage, cornea, skin |
| Collagen XXVI | EMID2 | Collagen-like; EMI | Glyco; Hyp; S–S | Ovary, testis |
| Collagen XXVIII | COL28A1 | BPTI/Kunitz inhibitor; collagen-like; VWFA | S–S | Basement membranes |
| Anchoring fibrils | ||||
| Collagen VII | COL7A1 | BPTI/Kunitz inhibitor; collagen-like; FNIII; VWFA | Glyco; Hyl; Hyp; S–S | Anchoring fibrils, basement membranes |
| Fibril-associated collagens with interrupted triple helices | ||||
| Collagen IX | COL9A1–COL9A3 | Collagen-like; laminin G-like/TSP N-terminal | Glyco; Hyp; S–S; X | Cartilage, vitreous body |
| Collagen XII | COL12A1 | Collagen-like; FNIII; laminin G-like/TSP N-terminal; VWFA | Glyco; Hyp; S–S | Connective tissues |
| Collagen XIV | COL14A1 | Collagen-like; FNIII; laminin G-like/TSP N-terminal; VWFA | Glyco; Hyl; Hyp; S–S | Connective tissues |
| Collagen XVI | COL16A1 | Collagen-like; laminin G-like/TSP N-terminal | Glyco; Hyp; S–S | Cartilage, papillary dermis, placenta |
| Collagen XIX | COL19A1 | Collagen-like; laminin G-like/TSP N-terminal | Hyp; S–S | Basement membranes |
| Collagen XX | COL20A1 | Collagen-like; FNIII; laminin G-like/TSP N-terminal; VWFA | Glyco | Widespread |
| Collagen XXI | COL21A1 | Collagen-like; laminin G-like/TSP N-terminal; VWFA | Glyco | Widespread |
| Collagen XXII | COL22A1 | Collagen-like; laminin G-like/TSP N-terminal; VWFA | Glyco | Tissue junctions |
| Transmembrane collagens and collagen-like proteins | ||||
| Collagen XIII | COL13A1 | Collagen-like; helical TM | S–S | Cell junctions |
| Collagen XVII | COL17A1 | Collagen-like; helical TM | Glyco; Hyp; phospho; pro; S–S | Hemidesmosomes |
| Collagen XXIII | COL23A1 | Collagen-like; helical TM | Pro | Brain, cornea, kidney, lung, skin, tendon |
| Collagen XXV | COL25A1 | Collagen-like; helical TM | Glyco; Hyl; Hyp; pro | Brain |
| Ectodysplasin A | EDA | Collagen-like; helical TM | Glyco; pro | Hair follicles, keratinocytes, sweat glands |
| EMID1 | EMID1 | Collagen-like; EMI | Glyco; S–S | |
| Gliomedin | GLDN | Collagen-like; helical TM; olfactomedin-like | Glyco; pro | Brain, placenta, sciatic nerve, spinal cord |
| MARCO | MARCO | Collagen-like; helical TM; SRCR | Glyco; S–S | Macrophages |
| MSR1 | MSR1 | Collagen-like; helical TM; SRCR | Glyco; phospho; S–S | Macrophages |
| PSPs | SFTPA1, SFTPA2, SFTPD | C-type lectin; collagen-like | Acetylation; glyco; Hyl; Hyp; S–S | Lung |
| Multiplexin collagens | ||||
| Collagen XV | COL15A1 | Collagen-like; laminin G-like/TSP N-terminal | Glyco; Hyp; S–S | Basement membranes |
| Collagen XVIII | COL18A1 | Frizzled; laminin G-like/TSP N-terminal | Glyco; Hyp; pro; S–S | Basement membranes |
| Elastin and microfibrillar proteins | ||||
| Elastin | ELN | Hyp; S–S; X | Elastic fibres | |
| Emilins | EMILIN1–EMILIN3 | C1q; collagen-like; EMI | Glyco; phospho; S–S | Elastic fibres |
| Fibrillins | FBN1–FBN3 | EGF-like; TGF-BP | Glyco; phospho; pro; S–S; X | Microfibrils |
| Fibulins | EFEMP1, EFEMP2, FBLN1, FBLN2, FBLN5, FBLN7, HMCN1 | Anaphylatoxin-like; EGF-like; Ig-like C2-type; nidogen G2 β-barrel; sushi; TSP type 1; VWFA | Glyco; phospho; S–S | Basement membranes, connective tissues, retina (EFEMP1), teeth (FBLN7) |
| MAGPs | MFAP1, MFAP2, MFAP4, MFAP5 | Fibrinogen C-terminal; SXC | Acetylation; glyco; phospho; S–S; sulf; X | Microfibrils |
| Non-collagenous glycoproteins | ||||
| Fibronectin | FN1 | FNI; FNII; FNIII | Glyco; phospho; pro; S–S; sulf; X | Widespread |
| Laminins | LAMA1–LAMA5, LAMB1–LAMB4, LAMC1–LAMC3 | Laminin EGF-like; laminin G-like/TSP N-terminal; laminin IV type A; laminin N-terminal | Glyco; phospho; pro; S–S | Basement membranes |
| Nidogens | NID1, NID2 | EGF-like; NIDO; nidogen G2 β-barrel; thyroglobulin type 1 | Glyco; S–S; sulf | Basement membranes |
| SPARC | SPARC | EF-hand; follistatin-like; Kazal-like | Glyco; S–S | Basement membranes |
| Tenascins | TNC, TNN (tenascin-W), TNR, TNXB | EGF-like; fibronectin C-terminal; FNIII | Glyco; phospho; S–S | Nervous system, skeletal tissues, vasculature |
| Thrombospodins | COMP, THBS1–THBS4 | COMP N-terminal (COMP); EGF-like; laminin G-like/TSP N-terminal; TSP C-terminal; TSP type 1; TSP type 3; VWFC | Glyco; phospho; S–S | Cartilage (COMP), widespread |
| Matrilins | ||||
| Matrilins | MATN1–MATN4 | EGF-like; VWFA | Glyco; phospho; S–S | Cartilage (MATN1, MATN3), connective tissues (MATN2, MATN4) |
| Basement membrane proteoglycans | ||||
| Agrin | AGRN | EGF-like; Kazal-like; laminin EGF-like; laminin G-like; NtA; SEA | Glyco; S–S | Basement membranes |
| Bamacan | SMC3 | Acetylation; glyco; phospho | Basement membranes | |
| Perlecan | HSPG2 | EGF-like; Ig-like C2-type; laminin EGF-like; laminin G-like; laminin IV type A; LDL-receptor class A; SEA | Glyco; pro; S–S | Basement membranes |
| Hyalectans (lecticans) | ||||
| Aggrecan | ACAN | C-type lectin; EGF-like; Ig-like V-type; link; sushi | Glyco; pro; S–S | Cartilage |
| Brevican | BCAN | C-type lectin; EGF-like; Ig-like V-type; link; sushi | Glyco; S–S | Nervous system |
| Neurocan | NCAN | C-type lectin; EGF-like; Ig-like V-type; link; sushi | Glyco; S–S | Nervous system |
| Versican | VCAN | C-type lectin; EGF-like; Ig-like V-type; link; sushi | Glyco; phospho; S–S | Widespread |
| Small leucine-rich proteoglycans | ||||
| Asporin | ASPN | LRRNT | Glyco; S–S | Cartilage |
| Biglycan | BGN | Glyco; pro; S–S | Connective tissues | |
| Chondroadherin | CHAD | LRRNT | Glyco; S–S | Chondrocytes |
| Decorin | DCN | Glyco; pro; S–S | Connective tissues | |
| Epiphycan | EPYC | LRRNT | Glyco; S–S | Cartilage, ligament, placenta |
| Fibromodulin | FMOD | LRRNT | Glyco; S–S; sulf | Connective tissues |
| Keratocan | KERA | LRRNT | Glyco; S–S | Cornea, trachea |
| Lumican | LUM | LRRNT | Glyco; S–S; sulf | Cartilage, cornea |
| Mimecan | OGN | Glyco; S–S | Bone, cornea | |
| Nyctalopin§ | NYX | LRRNT | Glyco; S–S | Kidney, retina |
| Opticin§ | OPTC | LRRNT | Glyco; S–S; sulf | Brain, cartilage, iris, retina |
| Osteomodulin | OMD | LRRNT | Glyco; S–S; sulf | Bone |
| Podocan§ | PODN | LRRNT | Glyco | Heart, kidney, liver, pancreas, smooth muscle |
| Podocan-like protein–1§ | PODNL1 | LRRNT | Glyco | Bone |
| Prolargin | PRELP | Glyco; S–S | Basement membranes, connective tissues | |
| Tsukushin§ | TSKU | LRRNT | Glyco | Brain, iris |
| Testicans | ||||
| Testicans | SPOCK1–SPOCK3 | Kazal-like; thyroglobulin type 1 | Glyco; phospho; S–S | Cornea, nervous system |
BPTI, bovine pancreatic trypsin inhibitor; COMP, cartilage oligomeric matrix protein; ECM, extracellular matrix; EGF, epidermal growth factor; EMI, EMILIN domain; EMID, EMI domain–containing protein; FNI, fibronectin, type I; FNII, fibronectin, type II; FNIII, fibronectin, type III; G-like, globular-like; Ig, immunoglobulin; LDL, low-density lipoprotein; LRRNT, leucine-rich repeat N-terminal domain; MAGP, microfibril-associated glycoprotein; MARCO, macrophage receptor with collagenous structure; MSR1, macrophage scavenger receptor types I and II; NC1, non-collagenous-1; NtA, N-terminal agrin; PSP, pulmonary surfactant-associated protein; SEA, sea urchin sperm protein, enterokinase, agrin; SPARC, secreted protein, acidic and rich in cysteine; SRCR, scavenger receptor cysteine-rich; SXC, six-cysteine; TGF-BP, transforming growth factor–β–binding protein-like; TM, transmembrane; TSP, thrombospondin; VWFA, von Willebrand factor, type A; VWFC, von Willebrand factor, type C.
Human gene names are listed.
Data were extracted from the UniProt Knowledgebase (http://uniprot.org) (release 2012_07) (UniProt Consortium 2012).
Modifications (including inferred): glyco, glycosylation; Hyl, lysine hydroxylation; Hyp, proline hydroxylation; phospho, phosphorylation; pro, proteolytic processing; S–S, disulphide bonding; sulph, sulphation; Ubl, ubiquitin-like conjugation; X, covalent crosslinking.
Small leucine-rich proteoglycan family member that is not considered a proteoglycan due to lack of glycosaminoglycan chains.
Certain ECM molecules, such as fibronectin, can extend to reveal cryptic binding sites that promote fibre formation and cell adhesion. The orientation of ECM fibres is important to direct effective cell migration (Doyle et al. 2009), and the stiffness of the ECM plays a key role in regulating sites of ECM attachment (Pelham & Wang 1997), including multimolecular signalling complexes generically termed cell adhesions (Byron et al. 2010). Cells on soft substrates exhibit smaller and more dynamic adhesions, whereas cells on stiff substrates have larger and more stable adhesions. By determining ECM compliance and applied force, the composition and organization of the ECM therefore plays an important role in regulating mechanosensitive cell adhesion complexes (DuFort et al. 2011). Furthermore, cell adhesion can actively remodel the ECM, highlighting the bidirectional interplay between the cell and its microenvironment. Importantly, the biomechanical properties of ECM regulate fundamental cellular properties, such as differentiation and stem cell pluripotency (Engler et al. 2006; Gilbert et al. 2010). Recent work has demonstrated a role for the transcription regulators Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) as mediators of mechanical signals exerted by ECM stiffness (Dupont et al. 2011), although a complete understanding of the molecular interplay between ECM properties and cellular function is lacking. It is noteworthy that changes in ECM stiffness, as a consequence of altered ECM composition and organization, are often associated with ageing, injury or pathological conditions (Frantz et al. 2010; DuFort et al. 2011; Lu et al. 2012). Abnormal ECM dynamics are linked to tissue fibrosis of many organs (Frantz et al. 2010), chronic inflammation (Sorokin 2010) and are a hallmark of cancer (Levental et al. 2009; Barker et al. 2012; Lu et al. 2012). The considerable evidence implicating ECM dysregulation and remodelling with disease progression renders ECM molecules attractive targets for biomarker studies and therapeutic development. Indeed, although beyond the scope of this review, extracellular candidates have recently been identified as predictive markers for several diseases using proteomics (Lindsey et al. 2012; Shang et al. 2012).
In addition to dysregulation of global ECM dynamics, mutations in individual ECM genes cause a range of pathological conditions (Table 2). For example, numerous forms of chondrodysplasia, a disorder that disrupts cartilage development with a spectrum of severities, result from mutations in collagens, thrombospondins and matrilins (Table 2) (Piróg-Garcia et al. 2007; Fresquet et al. 2008). The most common collagen mutations are glycine substitutions, which disrupt collagen triple helix folding (Bateman et al. 2009). Mutations causing misfolding of collagen I in patients with the bone disorder osteogenesis imperfecta or with chondrodysplasias have been shown to result in procollagen aggregation in the endoplasmic reticulum, highlighting the importance of correct extracellular molecule biosynthesis in the regulation of healthy ECM (Lamandé et al. 1995; Rajpar et al. 2009; Nundlall et al. 2010). Epidermolysis bullosa, a group of heritable blistering disorders presenting with fragile skin and mucous membranes, is caused by several mutations in genes expressed within the cutaneous basement membrane zone (Table 2), such as genes encoding laminins and collagens, the laminin receptor integrin α6β4 and cytoskeletal proteins (for example, keratins and plectin). Dystrophic epidermolysis bullosa results from mutations in collagen VII, which has led to attempts to address the resultant abnormal collagen VII expression, including cell therapy approaches that are providing promising outcomes in clinical trials (Uitto et al. 2012). Mutations in fibrillin-1 reduce its levels in extracellular microfibrils, depleting a sink for transforming growth factor–β (TGF-β), which results in Marfan syndrome, a multisystemic disorder that manifests with long bone overgrowth and heart defects. Antagonism of TGF-β signalling using an angiotensin II type 1 receptor blocker in Marfan syndrome mouse models and patients has proved effective at reducing aortic defects (Habashi et al. 2006; Brooke et al. 2008), and multiple trials are currently underway to assess the efficacy of angiotensin II blockade on aortic properties in Marfan syndrome (Hartog et al. 2012). Indeed, mouse models have been valuable in understanding the molecular mechanisms of several pathological ECM abnormalities, as many models recapitulate the clinical features observed in diseased patients (Aszódi et al. 2006; Bateman et al. 2009). Thus, such models provide important preclinical systems for the interrogation of ECM defects and the development of molecular therapies for diseases of the ECM.
Table 2.
Major ECM components involved in genetic disorders
| Molecule name | Gene name(s)* | Phenotype(s)†,‡ |
|---|---|---|
| Fibril-forming collagens | ||
| Collagen I | COL1A1, COL1A2 | Caffey disease (COL1A1); Ehlers-Danlos syndrome; osteogenesis imperfecta |
| Collagen II | COL2A1 | Achondrogenesis; avascular necrosis of the femoral head; Czech dysplasia; Kniest dysplasia; Legg-Calve-Perthes disease; multiple epiphyseal dysplasia with myopia and deafness; osteoarthritis with mild chondrodysplasia; otospondylomegaepiphyseal dysplasia; platyspondylic skeletal dysplasia; spondyloepimetaphyseal dysplasia; spondyloepiphyseal dysplasia; spondyloperipheral dysplasia; Stickler syndrome; vitreoretinopathy with phalangeal epiphyseal dysplasia |
| Collagen III | COL3A1 | Ehlers-Danlos syndrome |
| Collagen V | COL5A1, COL5A2 | Ehlers-Danlos syndrome |
| Collagen XI | COL11A1, COL11A2 | Fibrochondrogenesis; Marshall syndrome (COL11A1); non-syndromic hearing loss (COL11A2); otospondylomegaepiphyseal dysplasia (COL11A2); Stickler syndrome; Weissenbacher-Zweymueller syndrome (COL11A2) |
| Network-forming collagens | ||
| Collagen IV | COL4A1–COL4A6 | Alport syndrome (COL4A3–COL4A5); benign familial hematuria (COL4A3, COL4A4); brain small vessel disease with Axenfeld-Rieger anomaly (COL4A1); brain small vessel disease with haemorrhage (COL4A1); diffuse leiomyomatosis with Alport syndrome (COL4A6); hereditary angiopathy with nephropathy, aneurysms and muscle cramps (COL4A1); porencephaly (COL4A1, COL4A2) |
| Collagen VIII | COL8A2 | Fuchs endothelial corneal dystrophy; posterior polymorphous corneal dystrophy |
| Collagen X | COL10A1 | Schmid-type metaphyseal chondrodysplasia |
| Beaded filament–forming collagens | ||
| Collagen VI | COL6A1–COL6A3 | Bethlem myopathy; myosclerosis (COL6A2); Ullrich congenital muscular dystrophy |
| Anchoring fibrils | ||
| Collagen VII | COL7A1 | Epidermolysis bullosa dystrophica; toenail dystrophy; transient bullous of the newborn |
| Fibril-associated collagens with interrupted triple helices | ||
| Collagen IX | COL9A1–COL9A3 | Multiple epiphyseal dysplasia; Stickler syndrome (COL9A1, COL9A2) |
| Transmembrane collagens and collagen-like proteins | ||
| Collagen XVII | COL17A1 | Junctional epidermolysis bullosa |
| Ectodysplasin A | EDA | Hypohidrotic ectodermal dysplasia; selective tooth agenesis |
| MSR1 | MSR1 | Barrett oesophagus; hereditary prostate cancer |
| PSPs | SFTPA2 | Idiopathic pulmonary fibrosis |
| Endostatin-producing collagens | ||
| Collagen XVIII | COL18A1 | Knobloch syndrome |
| Elastin and microfibrillar proteins | ||
| Elastin | ELN | Cutis laxa; supravalvular aortic stenosis |
| Fibrillins | FBN1, FBN2 | Acromicric dysplasia (FBN1); distal arthrogryposis (FBN2); geleophysic dysplasia (FBN1); isolated ectopia lentis (FBN1); Marfan syndrome (FBN1); MASS syndrome (FBN1); Shprintzen-Goldberg syndrome (FBN1); stiff skin syndrome (FBN1); Weill-Marchesani syndrome (FBN1) |
| Fibulins | EFEMP1, EFEMP2, FBLN1, FBLN5 | Age-related macular degeneration (FBLN5); cutis laxa (EFEMP2, FBLN5); Doyne honeycomb retinal dystrophy (EFEMP1); synpolydactyly (FBLN1) |
| Non-collagenous glycoproteins | ||
| Fibronectin | FN1 | Glomerulopathy with fibronectin deposits; plasma fibronectin deficiency |
| Laminins | LAMA2, LAMA3, LAMB2, LAMB3, LAMC2, LAMC3 | Cortical malformations, occipital (LAMC3); generalized atrophic benign epidermolysis bullosa (LAMA3); junctional epidermolysis bullosa (LAMA3, LAMB3, LAMC2); laryngoonychocutaneous syndrome (LAMA3); merosin-deficient muscular dystrophy (LAMA2); nephrotic syndrome (LAMB2); Pierson syndrome (LAMB2) |
| Tenascins | TNXB | Ehlers-Danlos syndrome, hypermobility type; Ehlers-Danlos-like syndrome due to tenascin-X deficiency |
| Thrombospodins | COMP | Multiple epiphyseal dysplasia; pseudoachondroplasia |
| Matrilins | ||
| Matrilins | MATN3 | Multiple epiphyseal dysplasia; spondyloepimetaphyseal dysplasia |
| Basement membrane proteoglycans | ||
| Bamacan | SMC3 | Cornelia de Lange syndrome |
| Perlecan | HSPG2 | Dyssegmental dysplasia; Schwartz-Jampel syndrome |
| Hyalectans (lecticans) | ||
| Aggrecan | ACAN | Osteochondritis dissecans, short stature and early-onset osteoarthritis; spondyloepimetaphyseal dysplasia; spondyloepiphyseal dysplasia |
| Versican | VCAN | Wagner syndrome |
| Small leucine-rich proteoglycans | ||
| Decorin | DCN | Stromal corneal dystrophy |
| Keratocan | KERA | Cornea plana |
| Nyctalopin | NYX | Stationary night blindness |
ECM, extracellular matrix.
Human gene names are listed.
Mutations that contribute to susceptibility to multifactorial diseases or infection or to ‘non-diseases’ are excluded.
Data were extracted from the Online Mendelian Inheritance in Man resource (http://omim.org) (accessed 18 August 2012) (Hamosh et al. 2005).
Isolation and analysis of ECM
In recent years, the quest for a comprehensive understanding of the molecular mechanisms of ECM regulation in health and disease, and for markers and therapies for ECM dysfunction, has exploited global analytical approaches, such as proteomics. Mass spectrometry–based proteomic technologies, in particular, provide the opportunity for sensitive and large-scale analyses of biological systems, and rapid developments in instrumentation continue to increase the accuracy, sensitivity and robustness of MS (Box 1). The ability of MS to elucidate sites of post-translational modification makes it especially useful for the analysis of the heavily modified ECM (Table 1). Despite its advantages, there are many challenges associated with MS-based analysis of extracellular molecules (Wilson 2010). The complex, multimolecular nature of the ECM, as with all cellular organelles, renders its proteome intractable to in-depth analysis because of the large number and large dynamic range of analytes. Thus, many strategies incorporate various isolation, enrichment or fractionation methodologies to increase the signal over the noise and enable the detection of a large dynamic range of molecule concentrations (Box 1, Figure 1) (Righetti et al. 2005). Extracellular matrix components interact with many other components and often undergo covalent crosslinking, resulting in low solubility, which requires careful dissociation and extraction to avoid sample loss or bias (Wilson 2010). The source of ECM and its purity therefore place specific constraints on the methods chosen for proteomic analysis, and below, we outline recent MS-based proteomic studies that analysed extracellular components derived from tissues, cell culture models and purification in vitro.
Box 1. MS-based proteomic technologies
MS as a tool in life science
Most proteomic studies rely on MS as the central analytical technology by which measurements are acquired. Mass spectrometry methods are sensitive (permitting routine detection of femtomole amounts of peptides), rapid (sequencing thousands of peptides in a single run) and readily automated (permitting high-throughput strategies). Thus, MS provides an attractive methodology for the investigation of complex protein mixtures isolated from cells (Aebersold & Mann 2003). Although it remains challenging to achieve a comprehensive analysis of a proteome, rapid developments in MS-based proteomic technologies have enabled increasingly detailed coverage of analysed proteomes (Mallick & Kuster 2010).
MS data acquisition and analysis
The instrument of MS, a mass spectrometer, ionizes sample molecules, separates the ions according to their mass-to-charge ratio and measures the signal intensity of the ions. Tandem MS results in the fragmentation of peptide ions to provide structural information about the ions, which enables the determination of their amino acid sequence and the location of post-translational modifications (Aebersold & Mann 2003; Han et al. 2008). Tandem MS is the core technology used in ‘bottom-up’ proteomics, in which protein samples are enzymatically digested into peptides prior to analysis. With the availability of completed genome sequences, protein sequence databases (digested in silico) can be queried with MS data using database search tools to infer the identity of peptides, and thus proteins, present in the sample. The matching of theoretical mass spectra to observed mass spectra and the validation of assigned peptide and protein inferences represent complex computational and statistical challenges, to address which a large number of scoring and analysis strategies have been developed (Nesvizhskii et al. 2007).
Quantitative MS analysis
Quantification of peptides and proteins using MS provides additional insights to protein identification results, which are especially relevant to the investigation of dynamic biological systems. Several techniques for the quantification of relative or absolute amounts of proteins have been developed in recent years. Incorporation of stable isotope labels into different samples enables identical peptides from different samples to be distinguished within a single MS analysis. Such a method forms the basis of several quantification strategies, e.g. SILAC, which uses metabolic labelling (Ong et al. 2002), and iTRAQ, which yields isobarically tagged peptides (Ross et al. 2004). Label-free quantitative strategies, which compare two or more MS analyses in the absence of a label, include spectral counting, which correlates the number of observed mass spectra matched to peptides from a protein to the abundance of that protein (Liu et al. 2004), peptide counting (Ishihama et al. 2005), peptide ion signal measurements (Silva et al. 2005) and a combination of these parameters (Griffin et al. 2010). As well as enabling the study of protein dynamics, quantitative proteomics plays a crucial role in the identification of non-specific proteins that can contaminate biochemical samples. A protein with similar abundance in both an experimental sample and a control suggests that the protein is a contaminant, which can be filtered out of the data set (Rinner et al. 2007; Trinkle-Mulcahy et al. 2008).
Overcoming biological complexity
Separation of complex samples is a useful approach to address the challenges presented by proteomic examination of cells or tissues using current MS-based technologies, such as the limited sequencing speed of mass spectrometers (Righetti et al. 2005). In addition to first-level purification or enrichment of proteins from cells or tissues, resolution by one- or two-dimensional gel electrophoresis is often used to reduce further sample complexity (although two-dimensional gel electrophoresis is generally less favoured due to its reduced depth of proteome coverage). Following proteolytic digestion, peptides are usually fractionated based on the hydrophobicity by reversed-phase liquid chromatography or on the basis of multiple properties, such as by strong cation exchange and reversed-phase chromatographies for multidimensional separation. Such ‘shotgun’ analyses of peptides can be successful even in the absence of prior protein fractionation (Han et al. 2008). To overcome the biological and technical variability inherent in the analysis of complex samples, multiple replicate analyses (particularly biological replicates) are critical to provide the statistical power required to identify significant results (Karp et al. 2005; Gan et al. 2007; Dephoure & Gygi 2012). Furthermore, multiple testing is a common feature of proteomic experiments, which usually compare numerous peptides or proteins using a statistical test, and the resulting inflation of false positive results should be corrected by recalculating the obtained probabilities, for example, using false discovery rates. Developments in separation and MS technologies continue to provide increased performance in terms of sensitivity, accuracy and analytical robustness, which brings us closer to comprehensive analysis of complex biological systems.
Figure 1.
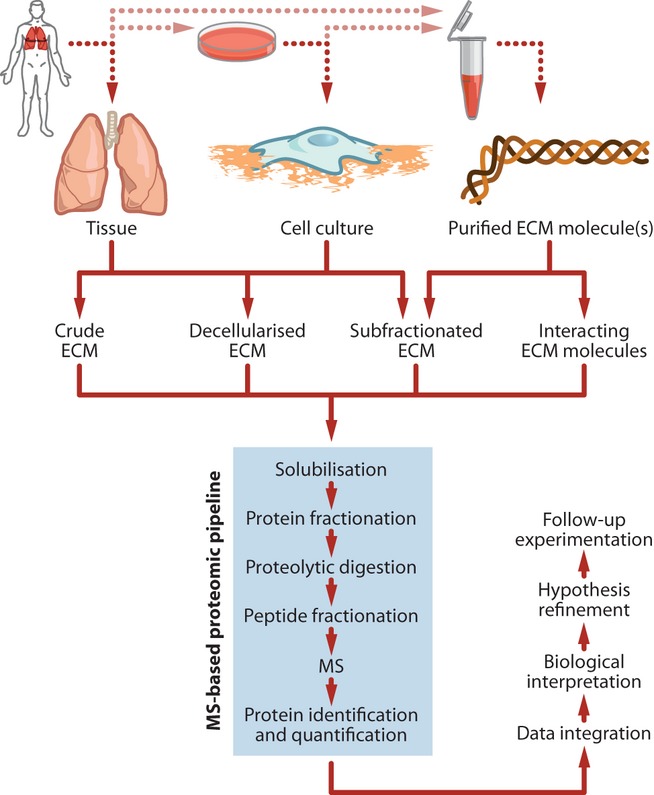
Approaches for mass spectrometry (MS)–based proteomic analysis of the extracellular matrix (ECM). The schematic depicts multiple workflows for the isolation and proteomic analysis of extracellular molecules. For the derivation of ECM, tissue could be of human or animal origin from any part of healthy or diseased sources. Human tissue could be acquired from biopsies or biofluids. Cell culture models could be based on primary cells or cell lines or used as xenografts in animal models. Individual or groups of ECM molecules could be recombinantly expressed or purified from tissue or cells. Molecules that interact with purified ECM molecules could be affinity isolated from tissue or cells. Details of the MS-based proteomic pipeline are provided in Box 1.
Proteomic analysis of ECM from tissue
Striving towards complete characterization of the ECM, many studies have analysed ECM from healthy or diseased whole tissues (Figure 1). Tissues that can be readily dissected away from other tissues yield samples of reduced complexity, which simplifies analysis by MS-based methods. Examples of such tissues include cartilage and bone, and a number of studies have analysed the composition of these ECM-rich tissues (Belluoccio et al. 2006; Garcia et al. 2006; Lammi et al. 2006; Vincourt et al. 2006; Pecora et al. 2007; Schreiweis et al. 2007; Wu et al. 2007; Wilson et al. 2008; Alves et al. 2011; Wilson et al. 2012). As a consequence of being dominated by highly crosslinked ECM components, tissues such as cartilage are difficult to solubilize. A combination of physical tissue disruption (pulverization), sequential chemical extraction [sodium chloride and guanidinium chloride (GuHCl)] and enzymatic deglycosylation (chondroitinase ABC) enabled the identification of 703 proteins from cartilage (Wilson et al. 2012). The study used spectral counting, a label-free method of MS quantification that correlates the number of observed mass spectra to the relative abundance of the protein from which the spectra were derived (Box 1), to quantify changes in cartilage ECM components during postnatal mouse development. A beta-binomial test, which incorporates both within-sample (technical) and between-sample (biological) variations (Pham et al. 2010), coupled to false discovery rate correction for multiple testing was used to assess differences between protein abundances at distinct developmental stages, resulting in the identification of 146 proteins as differentially expressed. Thirty-four of the differentially expressed proteins were categorized as ECM or ECM related according to Gene Ontology annotations (Gene Ontology Consortium 2012), with 22 proteins upregulated and 12 proteins downregulated during development, indicating fundamental roles for ECM synthesis and remodelling in cartilage maturation (Wilson et al. 2012). Using a similar biochemical methodology, cartilage proteins were isolated from various tissues by pulverization and GuHCl extraction (Önnerfjord et al. 2012). The isolated proteins were digested using trypsin, and the resulting peptides were chemically labelled with iTRAQ tags that can be quantified by MS to determine relative protein abundance (Box 1). Analysis of eight cartilaginous tissues resulted in the identification and quantification of 340 proteins, whereof 92 were manually selected as relevant to the ECM based on UniProt Knowledgebase annotation (UniProt Consortium 2012) and quantification in at least half of the tissues tested (which excluded several bona fide ECM components that may have been tissue specific). Comparisons of the filtered protein profiles using t–tests revealed both similarities and differences in tissue composition that may reflect distinct tissue mechanical properties (Önnerfjord et al. 2012).
Proteomic analysis of the ECM of the eye is simplified by the ability to separate and isolate the retinal basement membrane from the vitreous body. Mass spectrometry–based proteomic analysis of embryonic chick eye identified 27 retinal basement membrane ECM proteins and 48 vitreous body ECM proteins according to manual and Gene Ontology annotation (Balasubramani et al. 2010). Apart from these ECM proteins, the study reported a considerable number of non-ECM proteins in the retinal basement membrane (228) and vitreous body (252), which may stringently interact with the basement membrane or vitreous or, more likely, were contaminants that persisted during sample preparation, indicating a major challenge in biochemical fractionation of tissue. The ECM proteomes of other basement membrane–rich tissues, such as the mammary gland, have also been analysed by MS-based approaches. Mammary glands of adult rats were pulverized and extracted using a high-salt buffer, and the remaining ECM components were solubilized in urea (Hansen et al. 2009). To aid the preparation of ECM samples for analysis by MS, an ultrasonication-assisted tryptic digestion method was used, which, although causing a partial loss of trypsin activity, improved the sequence coverage for collagen I. As with similar methodologies, there were a large number of cellular proteins that contaminated the extracted ECM; in total, 46 of the 248 proteins identified in the rat mammary gland ECM preparations were manually annotated as recognized ECM components (Hansen et al. 2009). Incorporation of label-free MS quantification into this method, using a combination of spectral counting and spectral intensity measurements (Box 1), enabled the relative quantification of changes in rat mammary gland ECM composition during involution and in response to the non-steroidal anti-inflammatory drug ibuprofen (O'Brien et al. 2012). Mammary gland involution after pregnancy involves considerable ECM remodelling and has been shown to enhance tumour progression (Lyons et al. 2011). The study identified 884 proteins, of which 59 were annotated as ECM components (excluding abundant plasma components, such as serum albumin and fibrinogens). A t-test, coupled to a false discovery rate–corrected F-test to determine whether samples differed from one another, was used to assess the statistical significance of protein abundance changes. Interestingly, several of the extracellular proteins reported as upregulated in involuting glands are associated with tumour progression, such as collagen VII, laminin β1 and tenascin-C, whereas proteins with roles in growth factor availability (e.g. fibrillin-1) and collagen organization (e.g. tenascin-X) were downregulated in involuting glands, although the functional significance of these changes is not clear. Furthermore, treatment with ibuprofen, which has been shown to inhibit pregnancy-associated breast tumour progression (Lyons et al. 2011), decreased levels of laminins and tenascin-C, as quantified by MS, suggesting protective changes in mammary gland ECM composition induced by ibuprofen (O'Brien et al. 2012).
Tissues that are poorer in ECM require different extraction approaches to reduce or eliminate the contribution of cellular components that otherwise overwhelm data acquired by MS. Differential detergent extraction, a commonly used method for sequential partitioning of cytosolic, nuclear, membrane and cytoskeletal fractions, results in a remaining insoluble fraction that is enriched for ECM molecules. Using this method to analyse murine lung and colon tissues, Naba et al. (2012) identified over 100 ECM proteins in each tissue, with tissue-specific proteins representing 10–30% of the total. To identify the origin of components of the tumour microenvironment, ECM extracted from human tumours was implanted in mice, and a semi-quantitative label-free MS approach based on ratios of ion intensities for species-specific peptides was employed. This analysis showed that both the tumour- and stroma-derived ECM differed in composition depending on the metastatic potential of the implanted tumour, suggesting potential crosstalk between tumour and stromal cells, although the statistical and functional significance of these findings were not determined (Naba et al. 2012). In a different study of tumour-associated ECM changes, the transition from fibrosis and steatohepatitis to hepatocellular carcinoma (HCC) in the liver was characterized using MS-based proteomics (Lai et al. 2011). In this work, ECM-enriched fractions from livers of two mouse models relevant to human HCC were isolated after solubilization with detergents and chaotropic agents. Extensive sample fractionation using two-dimensional high-performance liquid chromatography and one-dimensional gel electrophoresis in combination with label-free MS quantification by peptide counting (Box 1) revealed numerous HCC-associated changes in the composition and relative abundance of both collagenous and non-collagenous ECM components, although the statistical significance of these changes was not reported. The majority of changes in ECM components were similar in both mouse models, suggesting a common molecular mechanism, which may involve platelet-derived growth factor, a major player in angiogenesis and fibrosis. In contrast, a subset of laminins and the laminin receptors integrins α3β1 and α6β1 were specifically upregulated in one HCC subtype, implicating these cell–ECM interactions in distinct stages of hepatocarcinogenesis (Lai et al. 2011).
To modulate the flow of blood through the body, the ECM of the vascular system must be precisely deposited and remodelled to maintain robust, appropriate mechanical properties. To study the composition of heart valves, which control blood outflow from the heart, murine pulmonary and aortic valves were microdissected and decellularized using detergents and sonication (Angel et al. 2011). Further protein extraction was not performed so as not to solubilize, and hence oversample by MS, the abundant structural collagens. Sequence-based subcellular localization prediction of proteins identified by MS enabled the annotation of over 200 ECM proteins in each valve proteome (of over 2000 total proteins in each valve proteome) (Angel et al. 2011). With the aim of improving the characterization of all aortic ECM and ECM-associated proteins, Didangelos et al. (2010) used a three-stage strategy to isolate extracellular components from human aortas obtained during cardiac surgery. Loosely associated extracellular molecules were extracted using sodium chloride and collected, then bulk cellular material was removed using the ionic detergent sodium dodecyl sulphate (below the critical micelle concentration to avoid disrupting ECM molecules), and then the remaining insoluble ECM-enriched fraction was extracted using GuHCl and collected. Interestingly, independent work reported a loss of ECM components in a phosphate-buffered saline pre-extraction wash step, which was omitted from the final workflow (Önnerfjord et al. 2012), but the report by Didangelos et al. (2010) demonstrates that collection and analysis of this material can augment a tissue-derived ECM proteomic data set. Enzymatic deglycosylation of the ECM-enriched samples permitted the identification of 103 extracellular proteins, classified using Gene Ontology annotation, one-third of which had not been previously reported in proteomics studies of vascular tissues. Almost all of the new proteins were identified in the sodium chloride and GuHCl extracts, which also captured ECM protein degradation products, demonstrating the value of methodical extraction to the characterization of the ECM and enabling screening for ECM proteolysis in clinical samples (Didangelos et al. 2010). This tissue subfractionation technique was used to describe ECM changes that occur in human abdominal aortic aneurysms and enabled the identification of a similar number of extracellular proteins (125) (Didangelos et al. 2011). Abdominal aortic aneurysms are characterized by extensive remodelling of the aortic ECM, and label-free MS quantification by spectral counting revealed statistically significant differences between diseased and healthy tissue. The proteomic analysis identified scarce secreted proteins, including the matrix metalloproteinase (MMP) macrophage metalloelastase, in aneurysmal tissue, in which proteolytic products of ECM components were also observed, implicating substrates of macrophage metalloelastase as markers of pathological ECM degeneration (Didangelos et al. 2011). Selective cleavage of ECM molecules by MMPs regulates ECM architecture and integrity, and MMPs play a key role in tumour invasion and metastasis, because tumour cells must degrade ECM to migrate across basement membrane, and in injury and infection (Page-McCaw et al. 2007; Sorokin 2010). The high-throughput discovery of targets of extracellular proteases such as MMPs, termed ‘degradomics’ (Morrison et al. 2009), has been used to examine changes in ventricular ECM after myocardial infarction (Lindsey et al. 2012). In a more recent study of cardiac ECM, subfractionation of cardiovascular tissue from a porcine model of myocardial ischaemia and reperfusion injury identified 139 cardiac ECM and ECM-associated proteins (Barallobre-Barreiro et al. 2012). Quantification by spectral counting and statistical analysis enabled a signature of ECM changes during early- and late-stage cardiac ECM remodelling to be defined. Moreover, previously unreported cardiac ECM components were detected, including the TGF-β regulators asporin, cartilage intermediate layer protein 1 and dermatopontin, which were validated in ischaemic human left ventricular tissue (Barallobre-Barreiro et al. 2012).
Proteomic analysis of ECM from cell culture
To overcome the complexities of extracting and analysing ECM from tissue, a number of MS-based proteomic studies have used ECM derived from cell culture models (Figure 1). The main advantages of this approach are ease of use and the ability to assign ECM production to specific cell types. Moreover, cell-derived ECM provides a three-dimensional matrix system that has features relevant to tissue physiology and cell behaviour in vivo (Cukierman et al. 2001). The underlying methodology for most proteomic studies of cell-derived ECM is the rapid removal of the bulk cellular material, either with hypotonic buffers (Pflieger et al. 2006; Yang et al. 2011), ammonium hydroxide (Todorović et al. 2010) or detergents (Burns et al. 2011), which retains an ECM-enriched fraction that can be collected for analysis by MS. Using an adapted method that combines ammonium hydroxide and detergent extraction and DNase treatment to remove nuclei (Beacham et al. 2007), we have analysed cell-derived ECM from a cell culture model of liver fibrosis using MS (Rashid et al. 2012). Fibrotic cell–specific ECM components were identified by a label-free quantitative comparison to ECM derived from a non-fibrotic control. Forty-eight extracellular proteins were detected in the fibrotic ECM (of 258 total proteins), of which 16 were statistically enriched over the non-fibrotic control. In addition to differences in the abundance of ECM components, interaction network analysis revealed differences in the connectivity of inferred protein networks between the ECM niches, which suggest the presence of specific cell microenvironments related to pathological abnormality. The analysis of fibrotic cell–derived ECM identified the vast majority of previously characterized fibrotic liver ECM components and discovered a number of putative novel components, two of which, CYR61 and Wnt-5a, were validated by immunohistochemical staining of human and mouse fibrotic tissues (Rashid et al. 2012). In other recent proteomic studies, we have analysed cell-derived ECMs from glomerular endothelial cells and podocytes and embryonic stem cell feeder layers, which identified hundreds of ECM proteins with cell type–specific distribution patterns (unpublished data). In addition to our work, proteomic analyses of cell-derived ECM or commercially available ECM preparations commonly used in stem cell studies have identified specific ECM components capable of supporting stem cell pluripotency (Abraham et al. 2010; Hughes et al. 2011). These studies could aid the optimization of cell culture conditions that either maintain stem cell self-renewal or drive differentiation down specific cell lineages, which has the potential to facilitate the development of stem cell therapies.
Murine neocartilage cell culture models have important applications in tissue engineering, such as autologous cartilage repair in arthritis. To investigate the neocartilage ECM, sequential extraction was used to fractionate the extracellular molecules based on differential solubility (Wilson et al. 2010). Neocartilage cultured for 3 weeks was compared to 3-day postnatal (juvenile) cartilage tissue by spectral counting and statistical analysis, which revealed a change from largely readily soluble proteins in juvenile cartilage (extracted using sodium chloride) to largely poorly soluble proteins in neocartilage (extracted using GuHCl). The authors concluded that neocartilage formation involves the production and integration of multiple ECM components into increasingly insoluble networks, which has implications for the development of transplantation and other biomedical applications (Wilson et al. 2010). To compare cartilage maturation in vitro to development in vivo, the cell culture model data were reanalysed, which resulted in the identification of 27 differentially expressed ECM or ECM-related proteins in the juvenile or neocartilage data sets, whereas proteomic analysis of cartilage development in vivo identified 34 differentially expressed ECM or ECM-related proteins (Wilson et al. 2012). This meta-analysis revealed distinct, statistically significant subsets of proteins involved in the development of cartilage in vitro and in vivo and demonstrated an orthogonal approach for the selection of significant candidates for validation and further investigation.
Proteomic analysis of purified ECM molecules
To investigate specific subsets of ECM molecules or molecular interactions, purified or recombinantly expressed ECM proteins have been used in a number of proteomic studies (Figure 1). Sputum is an accessible biological fluid, and MS-based analysis of human induced sputum has identified 191 proteins (Nicholas et al. 2006). Sputum is rich in the major mucosal structural components, mucins, which are heavily glycosylated, high–molecular weight proteins. The analysis of mucins is challenged by their composition and size, which can lead to underrepresentation in compositional studies. To address this issue, Kesimer et al. (2009) solubilized mucosal secretions in GuHCl and used caesium chloride density-gradient centrifugation to separate mucins from other proteins based on their buoyant density. Using this approach, MS identified a total of 136 proteins in human induced sputum, from which proteins from potential extraneous sources, such as saliva and blood, were excluded. Analysis of the mucin-rich fraction enabled the detection of both epithelial mucins (MUC1, MUC4 and MUC16) and gel-forming mucins (MUC5B and MUC5AC). Furthermore, the authors used this system to identify 134 proteins in a tracheobronchial air–liquid interface culture model, including the same complement of mucins, and 87% of proteins shared between both data sets were associated with innate immunity, thus validating the use of the model as a tool to investigate innate defence of the airways (Kesimer et al. 2009).
Mass spectrometry–based proteomics was used to analyse the composition of fibrillin-rich microfibrils extracted from ciliary zonules, aorta and skin under non-denaturing conditions with bacterial collagenase type 1A or under denaturing conditions using GuHCl (Cain et al. 2006). In addition to identifying new ciliary microfibrillar components, this study confirmed that fibrillin-1 was the only fibrillin isoform detected in all of the tissue microfibril preparations. To overcome technical limitations of this study and to gain insights into the molecular interactions of elastic tissue ECM components, Cain et al. (2009) recombinantly expressed the elastic fibre proteins fibrillin-1, microfibril-associated glycoprotein–1, fibulin-5 and lysyl oxidase and used them as bait in affinity purification experiments. Extracellular matrix proteins from cell culture that specifically copurified with each bait were identified by MS. This approach validated known molecular interactions, permitted the detection of novel elastic fibre components and interactions and resulted in the construction of an elastic fibre interaction network (Cain et al. 2009).
Proteins that associate with collagens have been isolated and analysed using MS-based proteomics. The vitreous body of the eye comprises a hydrated, acellular ECM containing collagen II and other collagens and non-collagenous components. Enrichment of collagens from the vitreous body using sodium acetate and hyaluronan lyase followed by release of associated proteins using GuHCl led to the identification by MS of a novel small leucine-rich repeat protein family member termed opticin (Reardon et al. 2000). More recently, opticin was shown to be anti-angiogenic and to inhibit preretinal neovascularization by modulating cell–ECM adhesion (Le Goff et al. 2012a,b). Within cartilage, collagen fibrils interact with other ECM components to maintain and regulate tissue development, structural integrity and fibrillogenesis, and the absence or mutation of collagen type XI within cartilage leads to disease (Table 2). To investigate molecular interactions with collagen XI, recombinantly expressed N-terminal domain of the collagen XI α1 chain was used as bait in an affinity purification experiment (Brown et al. 2011). From cartilage-extracted protein, 15 extracellular proteins (including other collagens, non-collagenous glycoproteins and proteoglycans) and 16 cellular proteins (including several enzymes known to regulate collagen biosynthesis) associated directly or indirectly with the recombinant collagen XI N-terminal domain. Importantly, a number of the identified proteins have previously been implicated in disorders relating to the development of bone or cartilage, suggesting that the approach identified functionally relevant interactions (Brown et al. 2011).
Computational analysis of ECM
Mass spectrometry–based proteomic approaches generate large sets of data that present considerable challenges for data analysis, visualization and interpretation. These are further complicated by protein sequence complexity imparted by alternatively spliced variants and post-translational modifications, which are common in extracellular proteins. Such modifications alter the mass-to-charge ratio of the peptide ions that the mass spectrometer detects, and so potential mass shifts must be accounted for when searching MS data against protein sequence databases to identify proteins successfully (Box 1). Hydroxylation of proline and lysine residues, for example, is prevalent in collagens (Table 1) and must be considered for comprehensive analysis of ECM MS data in silico (unpublished data). The increased computational search space resulting from specifying multiple potential peptide modifications, however, increases the potential for false discoveries, although some search engines are able to perform unrestricted identification of post-translational modifications (Tanner et al. 2005; Shilov et al. 2007; Han et al. 2011). Furthermore, the recent discovery by MS of a sulphilimine bond in collagen IV (Vanacore et al. 2009), a bond not previously found in biomolecules, emphasizes that current knowledge of the repertoire of ECM post-translational modifications is likely incomplete.
Despite advances in the biochemical isolation of extracellular molecules, ECM preparations derived from cells or tissues virtually never contain pure ECM. Consequently, the interrogation of ECM proteomic data sets to identify genuine extracellular molecules is an important analytical step. The Gene Ontology database (Gene Ontology Consortium 2012) provides structured, controlled vocabularies that are used to describe reported functions and locations of gene products. The database of annotation terms can be queried to capture knowledge of proteins reported in a data set, such as those that may be extracellular, and tools such as those implemented in DAVID Bioinformatics Resources (Huang da et al. 2009) enable the determination of statistical enrichment of specific annotation terms within a data set. The evidence used by the Gene Ontology to annotate a protein with a term can range from experimental characterization in vivo to text mining inference in silico and, although the evidence source is not a measure of annotation quality, care must be taken in the interpretation of results based on disparate sources of evidence. Indeed, some Gene Ontology terms are underrepresented or misassigned, including those of ECM components (Naba et al. 2012). This motivated the bioinformatic definition and prediction of ECM components based on diagnostic protein domains, in addition to manual curation (Naba et al. 2012); the incorporation of expert knowledge and biological validation remains an important aspect of any such bioinformatic workflow. The ECM catalogue defined in silico by Naba et al. (termed the ‘matrisome’) included all previously known ECM components and several potential novel extracellular proteins. Furthermore, unbiased proteomic analysis lends itself to the discovery of unanticipated cellular roles for proteins (Byron et al. 2012a). It is, therefore, critical that analytical approaches are open to the possibility of identifying new functions and subcellular locations of proteins to procure unexpected insights into regulation of the ECM.
To enable interrogation of data reporting interactions between ECM components, which are often poorly represented in general protein interaction databases, the MatrixDB database was created (Chautard et al. 2009). MatrixDB incorporates experimental data from the literature and interactions reported in several databases involving extracellular proteins and polysaccharides. The resulting interaction network enables the computational analysis of the complex structural and functional properties of the ECM, and provides a step towards a global view of extracellular molecular interactions (Figure 2), although spatial and temporal context remain important considerations in the interpretation of such networks. Updates to MatrixDB have incorporated additional data sets and model organisms (Chautard et al. 2011), and the database provides a valuable resource for the assimilation of proteomics and other data. MatrixDB and other databases can be supplemented with further experimental data to refine our understanding of ECM networks, such as the network of interactions involving the C-terminal collagen fragment endostatin (Faye et al. 2009), the glycosaminoglycan heparan sulphate (Ori et al. 2011) or low-affinity extracellular interactions (Bushell et al. 2008; Martin et al. 2010). In addition to experimental approaches to limit the impact of non-specific proteins in a data set, such as rigorous sample preparation, determination of specific protein enrichment over appropriate control samples and multiple replicate analyses (Box 1), high-confidence interaction networks built from data validated by multiple approaches or sources can be used to minimize false positive interactions, and interaction likelihoods can be estimated using various scoring methods (Nesvizhskii 2012). Indeed, common contaminants can be highlighted or filtered out of data sets based on the frequency of their detection in a library of other experiments (Boulon et al. 2010). Systematic curation of proteomic databases is important to consolidate knowledge and enable progress in the field, and such strategies can enhance our understanding of the organization of extracellular systems (Cromar et al. 2012).
Figure 2.
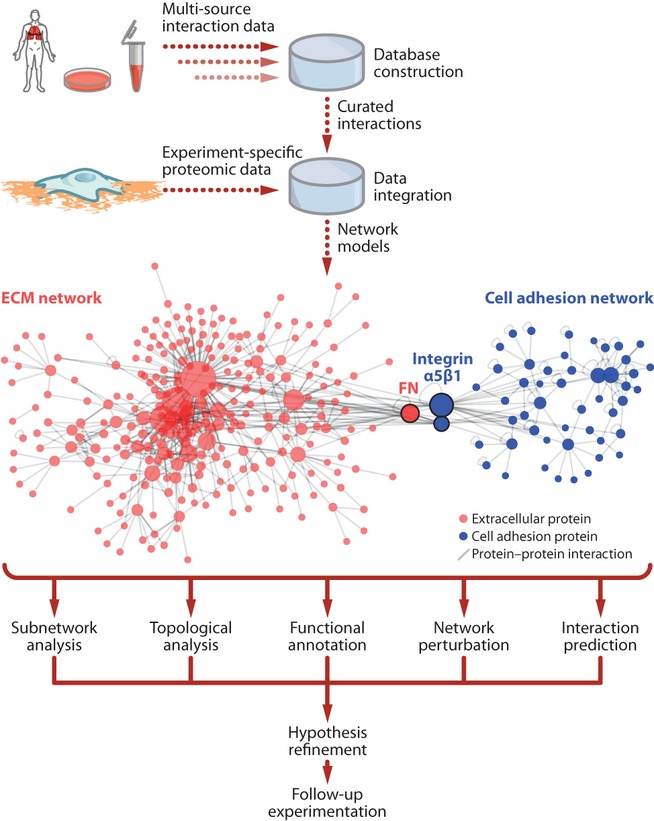
Integration and modelling of extracellular matrix (ECM) proteomics data. The schematic depicts the integration of new experimental (context-specific) data with databases of curated interactions from multiple sources to enable the further analysis, modelling and interpretation of those data. For example, analysis of interaction networks, based on previously reported protein–protein interactions, can provide insights into the functional roles of the identified components and the general organizing principles of the molecular networks under study. The displayed hypothetical interaction network merges protein–protein interactions extracted from the MatrixDB database (release 2010-08-26) (Chautard et al. 2009) (ECM network, red) with a model of a fibronectin-induced adhesion complex (Humphries et al. 2009) (cell adhesion network, blue). Circles represent detected proteins, coloured according to the data set, and grey lines represent reported interactions between the detected proteins. Fibronectin (FN) and the fibronectin receptor integrin α5β1 are arranged at the cell–ECM network interface and are highlighted with black borders. Resulting interaction network models can be analysed using various computational methods to enable biological interpretation. For example, focussed subnetworks can be extracted to interrogate potential protein complexes. Proteins in subnetworks with many common interaction partners are more likely to function together. Annotation of network models with enriched protein functions or signalling pathways can reveal unpredicted roles for components of the networks. Protein–protein interactions can be predicted on the basis of protein domains or network structural parameters. Analysis of network topology can also afford insights into signalling hubs, which are key signalling control points, and subnetwork robustness, which can be tested by inhibiting or depleting a candidate protein in silico, in vitro or in vivo and assessing the resultant network perturbation. In the displayed hypothetical interaction network, the networks were visualized using the force-directed layout implemented in cytoscape (version 2.8.2) (Shannon et al. 2003), which clusters together proteins with common interaction partners, and circle diameter was sized proportionally to the number of interaction partners. The visualization reveals clusters of many proteins with few interaction partners (small circles) around relatively few proteins with many interaction partners (large circles), which suggests that these highly connected proteins may be important for cell–ECM network structure or function.
Perspectives and future directions
Methodological challenges to the comprehensive proteomic analysis of ECM still remain, notably the detection of a substantial proportion of contaminating cellular proteins from ECM isolated from both tissue and cell culture, as described above. Indeed, a simple survey of the applicable studies cited herein reveals that the majority report approximately one quarter (typically 12–30%) of total protein identifications as ECM components. However, analysis of numbers of identified proteins is often not the most appropriate approach to determine the enrichment of ECM within these preparations. Naba et al. (2012) noted that more than 75% of the total precursor ion intensity (the sum of peptide ion peak areas for all identified peptides) from their MS data corresponded to ECM proteins. O'Brien et al. (2012) showed that ECM proteins accounted for the highest number of spectral counts in their data sets, despite detecting a greater number of non-ECM proteins. Together, these data indicate that the ECM preparations were quantitatively enriched for ECM proteins but qualitatively dominated by contaminating intracellular proteins. In this context, what is current best practice for the isolation and detection of ECM proteins using MS-based proteomics? Methods that improve the purity and completeness of ECM preparations, the specificity and sensitivity of detection of ECM proteins, enable more comprehensive proteomic data sets. The additional collection of loosely bound components using sodium chloride washes can substantially supplement ECM protein identifications from insoluble fractions of tissue or cells (Didangelos et al. 2010; Barallobre-Barreiro et al. 2012). Extensive separation of proteins and peptides prior to MS analysis can improve data from complex samples (Box 1) (Lai et al. 2011). Development and application of ECM-specific databases to curate lists of identified proteins can improve annotation and analysis of ECM proteins and the tissues from which they were derived (Figure 2) (Chautard et al. 2009, 2011; Naba et al. 2012). Although challenges to analytical robustness still exist, current available ECM isolation strategies permit the effective identification and quantification of ECM proteins and, furthermore, show that more defined cell culture models can replicate ECM composition in vivo.
Both in tissue and in culture, cells are intimately integrated with their microenvironment, and transmembrane integrin receptors link the ECM to the cytoskeleton of the cell (Byron 2011). This link acts as a physical tether, which serves to compartmentalize signalling events; it enables transmission of signals bidirectionally across the plasma membrane, which provides an informational link between the extracellular milieu and the cell; and it transduces force. Most integrin receptors recognize a wide variety of extracellular ligands, and many ECM molecules bind to multiple integrins (Humphries et al. 2006). Thus, there is a combinatorial complexity that enables and determines specific cell behavioural responses. To interpret this complexity, it is important to understand the nature of ECM receptors and their associated adhesion complexes. We and others developed proteomic strategies to analyse the integrin adhesion environment by MS, which revealed unanticipated molecular complexity and insights into mechanisms of cell adhesion to the ECM (Humphries et al. 2009; Byron et al. 2011; Kuo et al. 2011; Schiller et al. 2011; Byron et al. 2012b). In addition, interaction network analyses in silico have led to an appreciation of the subnetwork properties of sites of cell adhesion (Zaidel-Bar et al. 2007; Paris & Bazzoni 2008). Ultimately, a combination of complementary approaches and integration of extracellular and adhesion signalling data sets will lead to a more context-specific understanding of ECM regulation and dynamics (Figure 2), which is critical for a systems-level appreciation of the ECM and its interactions with cells (Adra et al. 2010).
Many questions remain open with regard to the genetic, molecular, physiological and pathological mechanisms of ECM regulation. To address these questions and to enable systems-level analyses of the ECM, quantitative data are key. Although MS is not inherently quantitative (owing to the variability of the physicochemical properties of peptide ions and undersampling by mass spectrometers), various approaches have been developed to perform relative or absolute quantification using MS (Box 1). Several of these approaches have been applied to the quantitative analysis of extracellular molecules, although appropriate statistical corrections for multiple testing are not consistently implemented. To gain insight into ECM regulation and dysregulation, quantitative data recorded at multiple developmental or disease stages can enable detailed comparisons of ECM dynamics (Lai et al. 2011; Wilson et al. 2012), but an understanding of the perturbations to and dynamic rewiring of extracellular protein networks in disease is lacking. The systematic, global, quantitative analysis of extracellular molecular networks has the potential to reveal emergent properties of molecularly complex ECMs that arise from interactions between ECM components and between cells and the ECM. Furthermore, the integration of proteomic data with complementary genomic, transcriptomic and metabolomic data could serve to build more robust computational models that can describe and predict the control mechanisms of extracellular systems. As such, the transition from molecular cataloguing to computational modelling modalities represents a major underexplored avenue in the field.
In summary, working towards a comprehensive understanding of ECM regulation in health and disease, recent research efforts have embraced global analyses of extracellular systems using proteomic strategies. It often remains practically difficult or ethically unacceptable to collect large quantities of primary material for proteomic studies, which continues to drive the increased efficiency and sensitivity of proteomics protocols and instrumentation. Recent methodological advances have improved our understanding of the catalogues of components that are important for control and modification of the extracellular milieu. Looking ahead, proteomic analysis of ECM provides the opportunity to measure complementary sets of markers of disease on a large scale and thus the prospect of refined disease biomarkers. This offers the potential of a more complete prognostic view of pathological processes, which may open the way to predictive and personalized therapies. As the volume and complexity of proteomics data continue to increase, researchers must rise to the challenge of integrating, communicating and sharing results so as to maximize the insight that can be gained from these large-scale experiments.
Acknowledgments
We apologize to authors whose work could not be discussed here because of space limitations. We thank C. Baldock, P.N. Bishop, K.E. Kadler, D. Knight, D.J. Thornton and G.A. Wallis for discussions and suggestions. A.B. is the recipient of a British Society for Matrix Biology Young Investigator Award.
Conflicts of interest
The authors declare no conflicts of interest.
Funding source
This work was supported by grants from the Wellcome Trust.
References
- Abraham S, Riggs MJ, Nelson K, Lee V, Rao RR. Characterization of human fibroblast-derived extracellular matrix components for human pluripotent stem cell propagation. Acta Biomater. 2010;6:4622–4633. doi: 10.1016/j.actbio.2010.07.029. [DOI] [PubMed] [Google Scholar]
- Adra S, Sun T, MacNeil S, Holcombe M, Smallwood R. Development of a three dimensional multiscale computational model of the human epidermis. PLoS ONE. 2010;5:e8511. doi: 10.1371/journal.pone.0008511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R, Mann M. Mass spectrometry–based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Alves RD, Demmers JA, Bezstarosti K, et al. Unraveling the human bone microenvironment beyond the classical extracellular matrix proteins: a human bone protein library. J. Proteome Res. 2011;10:4725–4733. doi: 10.1021/pr200522n. [DOI] [PubMed] [Google Scholar]
- Angel PM, Nusinow D, Brown CB, et al. Networked-based characterization of extracellular matrix proteins from adult mouse pulmonary and aortic valves. J. Proteome Res. 2011;10:812–823. doi: 10.1021/pr1009806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszódi A, Legate KR, Nakchbandi I, Fässler R. What mouse mutants teach us about extracellular matrix function. Annu. Rev. Cell Dev. Biol. 2006;22:591–621. doi: 10.1146/annurev.cellbio.22.010305.104258. [DOI] [PubMed] [Google Scholar]
- Balasubramani M, Schreiber EM, Candiello J, Balasubramani GK, Kurtz J, Halfter W. Molecular interactions in the retinal basement membrane system: a proteomic approach. Matrix Biol. 2010;29:471–483. doi: 10.1016/j.matbio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, Didangelos A, Schoendube FA, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125:789–802. doi: 10.1161/CIRCULATIONAHA.111.056952. [DOI] [PubMed] [Google Scholar]
- Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer. 2012;12:540–552. doi: 10.1038/nrc3319. [DOI] [PubMed] [Google Scholar]
- Bateman JF, Boot-Handford RP, Lamandé SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- Beacham DA, Amatangelo MD, Cukierman E. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr. Protoc. Cell Biol. 2007;10:10. doi: 10.1002/0471143030.cb1009s33. [DOI] [PubMed] [Google Scholar]
- Belluoccio D, Wilson R, Thornton DJ, Wallis TP, Gorman JJ, Bateman JF. Proteomic analysis of mouse growth plate cartilage. Proteomics. 2006;6:6549–6553. doi: 10.1002/pmic.200600191. [DOI] [PubMed] [Google Scholar]
- Boulon S, Ahmad Y, Trinkle-Mulcahy L, et al. Establishment of a protein frequency library and its application in the reliable identification of specific protein interaction partners. Mol. Cell. Proteomics. 2010;9:861–879. doi: 10.1074/mcp.M900517-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., 3rd Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RJ, Mallory C, McDougal OM, Oxford JT. Proteomic analysis of Col11a1-associated protein complexes. Proteomics. 2011;11:4660–4676. doi: 10.1002/pmic.201100058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JS, Kristiansen M, Kristensen LP, et al. Decellularized matrix from tumorigenic human mesenchymal stem cells promotes neovascularization with galectin-1 dependent endothelial interaction. PLoS ONE. 2011;6:e21888. doi: 10.1371/journal.pone.0021888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell KM, Söllner C, Schuster-Boeckler B, Bateman A, Wright GJ. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res. 2008;18:622–630. doi: 10.1101/gr.7187808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron A. Analyzing the anatomy of integrin adhesions. Sci. Signal. 2011;4:jc3. doi: 10.1126/scisignal.2001896. [DOI] [PubMed] [Google Scholar]
- Byron A, Morgan MR, Humphries MJ. Adhesion signalling complexes. Curr. Biol. 2010;20:R1063–R1067. doi: 10.1016/j.cub.2010.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron A, Humphries JD, Bass MD, Knight D, Humphries MJ. Proteomic analysis of integrin adhesion complexes. Sci. Signal. 2011;4:pt2. doi: 10.1126/scisignal.2001827. [DOI] [PubMed] [Google Scholar]
- Byron A, Humphries JD, Humphries MJ. Alternative cellular roles for proteins identified using proteomics. J. Proteomics. 2012a;75:4184–4185. doi: 10.1016/j.jprot.2012.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron A, Humphries JD, Craig SE, Knight D, Humphries MJ. Proteomic analysis of α4β1 integrin adhesion complexes reveals α-subunit-dependent protein recruitment. Proteomics. 2012b;12:2107–2114. doi: 10.1002/pmic.201100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SA, Morgan A, Sherratt MJ, Ball SG, Shuttleworth CA, Kielty CM. Proteomic analysis of fibrillin-rich microfibrils. Proteomics. 2006;6:111–122. doi: 10.1002/pmic.200401340. [DOI] [PubMed] [Google Scholar]
- Cain SA, McGovern A, Small E, et al. Defining elastic fiber interactions by molecular fishing: an affinity purification and mass spectrometry approach. Mol. Cell. Proteomics. 2009;8:2715–2732. doi: 10.1074/mcp.M900008-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chautard E, Ballut L, Thierry-Mieg N, Ricard-Blum S. MatrixDB, a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics. 2009;25:690–691. doi: 10.1093/bioinformatics/btp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chautard E, Fatoux-Ardore M, Ballut L, Thierry-Mieg N, Ricard-Blum S. MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res. 2011;39:D235–D240. doi: 10.1093/nar/gkq830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromar GL, Xiong X, Chautard E, Ricard-Blum S, Parkinson J. Toward a systems level view of the ECM and related proteins: a framework for the systematic definition and analysis of biological systems. Proteins. 2012;80:1522–1544. doi: 10.1002/prot.24036. [DOI] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Gygi SP. Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signal. 2012;5:rs2. doi: 10.1126/scisignal.2002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell. Proteomics. 2010;9:2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Yin X, Mandal K, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol. Cell. Proteomics. 2011;10:M111.008128. doi: 10.1074/mcp.M111.008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle AD, Wang FW, Matsumoto K, Yamada KM. One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol. 2009;184:481–490. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011;12:308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Faye C, Chautard E, Olsen BR, Ricard-Blum S. The first draft of the endostatin interaction network. J. Biol. Chem. 2009;284:22041–22047. doi: 10.1074/jbc.M109.002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J. Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresquet M, Jackson GC, Loughlin J, Briggs MD. Novel mutations in exon 2 of MATN3 affect residues within the alpha-helices of the A-domain and can result in the intracellular retention of mutant matrilin-3. Hum. Mutat. 2008;29:330. doi: 10.1002/humu.9518. [DOI] [PubMed] [Google Scholar]
- Gan CS, Chong PK, Pham TK, Wright PC. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ) J. Proteome Res. 2007;6:821–827. doi: 10.1021/pr060474i. [DOI] [PubMed] [Google Scholar]
- Garcia BA, Platt MD, Born TL, Shabanowitz J, Marcus NA, Hunt DF. Protein profile of osteoarthritic human articular cartilage using tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006;20:2999–3006. doi: 10.1002/rcm.2692. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium. The Gene Ontology: enhancements for 2011. Nucleic Acids Res. 2012;40:D559–D564. doi: 10.1093/nar/gkr1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PM, Havenstrite KL, Magnusson KE, et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin NM, Yu J, Long F, et al. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010;28:83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Aslanian A, Yates JR., 3rd Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008;12:483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, He L, Xin L, Shan B, Ma B. PeaksPTM: mass spectrometry-based identification of peptides with unspecified modifications. J. Proteome Res. 2011;10:2930–2936. doi: 10.1021/pr200153k. [DOI] [PubMed] [Google Scholar]
- Hansen KC, Kiemele L, Maller O, et al. An in-solution ultrasonication-assisted digestion method for improved extracellular matrix proteome coverage. Mol. Cell. Proteomics. 2009;8:1648–1657. doi: 10.1074/mcp.M900039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartog AW, Franken R, Zwinderman AH, Groenink M, Mulder BJ. Current and future pharmacological treatment strategies with regard to aortic disease in Marfan syndrome. Expert Opin. Pharmacother. 2012;13:647–662. doi: 10.1517/14656566.2012.665446. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hughes CS, Radan L, Betts D, Postovit LM, Lajoie GA. Proteomic analysis of extracellular matrices used in stem cell culture. Proteomics. 2011;11:3983–3991. doi: 10.1002/pmic.201100030. [DOI] [PubMed] [Google Scholar]
- Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J. Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries JD, Byron A, Bass MD, et al. Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci. Signal. 2009;2:ra51. doi: 10.1126/scisignal.2000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Evolution: the evolution of metazoan extracellular matrix. J. Cell Biol. 2012;196:671–679. doi: 10.1083/jcb.201109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, Oda Y, Tabata T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- Karp NA, Spencer M, Lindsay H, O'Dell K, Lilley KS. Impact of replicate types on proteomic expression analysis. J. Proteome Res. 2005;4:1867–1871. doi: 10.1021/pr050084g. [DOI] [PubMed] [Google Scholar]
- Kesimer M, Kirkham S, Pickles RJ, et al. Tracheobronchial air-liquid interface cell culture: a model for innate mucosal defense of the upper airways? Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296:L92–L100. doi: 10.1152/ajplung.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey R, Williamson MR, Chaudhry S, et al. Fibrillin-1 microfibril deposition is dependent on fibronectin assembly. J. Cell Sci. 2008;121:2696–2704. doi: 10.1242/jcs.029819. [DOI] [PubMed] [Google Scholar]
- Kuo JC, Han X, Hsiao CT, Yates JR, 3rd, Waterman CM. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KK, Shang S, Lohia N, et al. Extracellular matrix dynamics in hepatocarcinogenesis: a comparative proteomics study of PDGFC transgenic and Pten null mouse models. PLoS Genet. 2011;7:e1002147. doi: 10.1371/journal.pgen.1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamandé SR, Chessler SD, Golub SB, et al. Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl-terminal propeptide which impair subunit assembly. J. Biol. Chem. 1995;270:8642–8649. doi: 10.1074/jbc.270.15.8642. [DOI] [PubMed] [Google Scholar]
- Lammi MJ, Häyrinen J, Mahonen A. Proteomic analysis of cartilage- and bone-associated samples. Electrophoresis. 2006;27:2687–2701. doi: 10.1002/elps.200600004. [DOI] [PubMed] [Google Scholar]
- Le Goff MM, Lu H, Ugarte M, et al. The vitreous glycoprotein opticin inhibits preretinal neovascularization. Invest. Ophthalmol. Vis. Sci. 2012a;53:228–234. doi: 10.1167/iovs.11-8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff MM, Sutton MJ, Slevin M, Latif A, Humphries MJ, Bishop PN. Opticin exerts its anti-angiogenic activity by regulating extracellular matrix adhesiveness. J. Biol. Chem. 2012b;287:28027–28036. doi: 10.1074/jbc.M111.331157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ML, Weintraub ST, Lange RA. Using extracellular matrix proteomics to understand left ventricular remodeling. Circ. Cardiovasc. Genet. 2012;5:o1–o7. doi: 10.1161/CIRCGENETICS.110.957803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons TR, O'Brien J, Borges VF, et al. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat. Med. 2011;17:1109–1115. doi: 10.1038/nm.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nat. Biotechnol. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- Martin S, Söllner C, Charoensawan V, et al. Construction of a large extracellular protein interaction network and its resolution by spatiotemporal expression profiling. Mol. Cell. Proteomics. 2010;9:2654–2665. doi: 10.1074/mcp.M110.004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CJ, Butler GS, Rodríguez D, Overall CM. Matrix metalloproteinase proteomics: substrates, targets, and therapy. Curr. Opin. Cell Biol. 2009;21:645–653. doi: 10.1016/j.ceb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics. 2012;12:1639–1655. doi: 10.1002/pmic.201100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Nicholas B, Skipp P, Mould R, et al. Shotgun proteomic analysis of human-induced sputum. Proteomics. 2006;6:4390–4401. doi: 10.1002/pmic.200600011. [DOI] [PubMed] [Google Scholar]
- Nundlall S, Rajpar MH, Bell PA, et al. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones. 2010;15:835–849. doi: 10.1007/s12192-010-0193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JH, Vanderlinden LA, Schedin PJ, Hansen KC. Rat mammary extracellular matrix composition and response to ibuprofen treatment during postpartum involution by differential GeLC-MS/MS analysis. J. Proteome Res. 2012;11:4894–4905. doi: 10.1021/pr3003744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Önnerfjord P, Khabut A, Reinholt FP, Svensson O, Heinegård D. Quantitative proteomic analysis of eight cartilaginous tissues reveals characteristic differences as well as similarities between subgroups. J. Biol. Chem. 2012;287:18913–18924. doi: 10.1074/jbc.M111.298968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Wilkinson MC, Fernig DG. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem. 2011;286:19892–19904. doi: 10.1074/jbc.M111.228114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC. The evolution of extracellular matrix. Mol. Biol. Cell. 2010;21:4300–4305. doi: 10.1091/mbc.E10-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris L, Bazzoni G. The protein interaction network of the epithelial junctional complex: a system-level analysis. Mol. Biol. Cell. 2008;19:5409–5421. doi: 10.1091/mbc.E08-05-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecora F, Forlino A, Gualeni B, et al. A quantitative and qualitative method for direct 2-DE analysis of murine cartilage. Proteomics. 2007;7:4003–4007. doi: 10.1002/pmic.200700276. [DOI] [PubMed] [Google Scholar]
- Pelham RJ, Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl Acad. Sci. USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflieger D, Chabane S, Gaillard O, et al. Comparative proteomic analysis of extracellular matrix proteins secreted by two types of skin fibroblasts. Proteomics. 2006;6:5868–5879. doi: 10.1002/pmic.200402040. [DOI] [PubMed] [Google Scholar]
- Pham TV, Piersma SR, Warmoes M, Jimenez CR. On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics. Bioinformatics. 2010;26:363–369. doi: 10.1093/bioinformatics/btp677. [DOI] [PubMed] [Google Scholar]
- Piróg-Garcia KA, Meadows RS, Knowles L, et al. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum. Mol. Genet. 2007;16:2072–2088. doi: 10.1093/hmg/ddm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpar MH, McDermott B, Kung L, et al. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet. 2009;5:e1000691. doi: 10.1371/journal.pgen.1000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid ST, Humphries JD, Byron A, et al. Proteomic analysis of extracellular matrix from the hepatic stellate cell line LX-2 identifies CYR61 and Wnt-5a as novel constituents of fibrotic liver. J. Proteome Res. 2012;11:4052–4064. doi: 10.1021/pr3000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon AJ, Le Goff M, Briggs MD, et al. Identification in vitreous and molecular cloning of opticin, a novel member of the family of leucine-rich repeat proteins of the extracellular matrix. J. Biol. Chem. 2000;275:2123–2129. doi: 10.1074/jbc.275.3.2123. [DOI] [PubMed] [Google Scholar]
- Righetti PG, Castagna A, Antonioli P, Boschetti E. Prefractionation techniques in proteome analysis: the mining tools of the third millennium. Electrophoresis. 2005;26:297–319. doi: 10.1002/elps.200406189. [DOI] [PubMed] [Google Scholar]
- Rinner O, Mueller LN, Hubálek M, Müller M, Gstaiger M, Aebersold R. An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat. Biotechnol. 2007;25:345–352. doi: 10.1038/nbt1289. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Schiller HB, Friedel CC, Boulegue C, Fässler R. Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep. 2011;12:259–266. doi: 10.1038/embor.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiweis MA, Butler JP, Kulkarni NH, et al. A proteomic analysis of adult rat bone reveals the presence of cartilage/chondrocyte markers. J. Cell. Biochem. 2007;101:466–476. doi: 10.1002/jcb.21196. [DOI] [PubMed] [Google Scholar]
- Shang S, Plymoth A, Ge S, et al. Identification of osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology. 2012;55:483–490. doi: 10.1002/hep.24703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilov IV, Seymour SL, Patel AA, et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics. 2007;6:1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- Silva JC, Denny R, Dorschel CA, et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 2005;77:2187–2200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010;26:397–419. doi: 10.1146/annurev-cellbio-100109-104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010;10:712–723. doi: 10.1038/nri2852. [DOI] [PubMed] [Google Scholar]
- Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner S, Shu H, Frank A, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- Todorović V, Desai BV, Eigenheer RA, et al. Detection of differentially expressed basal cell proteins by mass spectrometry. Mol. Cell. Proteomics. 2010;9:351–361. doi: 10.1074/mcp.M900358-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Boulon S, Lam YW, et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 2008;183:223–239. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, Christiano AM, McLean WH, McGrath JA. Novel molecular therapies for heritable skin disorders. J. Invest. Dermatol. 2012;132:820–828. doi: 10.1038/jid.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanacore R, Ham AJ, Voehler M, et al. A sulfilimine bond identified in collagen IV. Science. 2009;325:1230–1234. doi: 10.1126/science.1176811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincourt JB, Lionneton F, Kratassiouk G, et al. Establishment of a reliable method for direct proteome characterization of human articular cartilage. Mol. Cell. Proteomics. 2006;5:1984–1995. doi: 10.1074/mcp.T600007-MCP200. [DOI] [PubMed] [Google Scholar]
- Wilson R. The extracellular matrix: an underexplored but important proteome. Expert Rev. Proteomics. 2010;7:803–806. doi: 10.1586/epr.10.93. [DOI] [PubMed] [Google Scholar]
- Wilson R, Belluoccio D, Little CB, Fosang AJ, Bateman JF. Proteomic characterization of mouse cartilage degradation in vitro. Arthritis Rheum. 2008;58:3120–3131. doi: 10.1002/art.23789. [DOI] [PubMed] [Google Scholar]
- Wilson R, Diseberg AF, Gordon L, et al. Comprehensive profiling of cartilage extracellular matrix formation and maturation using sequential extraction and label-free quantitative proteomics. Mol. Cell. Proteomics. 2010;9:1296–1313. doi: 10.1074/mcp.M000014-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Norris EL, Brachvogel B, et al. Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Mol. Cell. Proteomics. 2012;11:M111.014159. doi: 10.1074/mcp.M111.014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Liu W, Bemis A, et al. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 2007;56:3675–3684. doi: 10.1002/art.22876. [DOI] [PubMed] [Google Scholar]
- Yang KE, Kwon J, Rhim JH, et al. Differential expression of extracellular matrix proteins in senescent and young human fibroblasts: a comparative proteomics and microarray study. Mol. Cells. 2011;32:99–106. doi: 10.1007/s10059-011-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]