

Abstract
Structure-based virtual screening (SBVS) methods often rely on docking score. The docking score is an over-simplification of the actual ligand-target binding. Its capability to model and predict the actual binding reality is limited. Recently, interaction fingerprinting (IFP) has come and offered us an alternative way to model reality. IFP provides us an alternate way to examine protein-ligand interactions. The docking score indicates the approximate affinity and IFP shows the interaction specificity. IFP is a method to convert three dimensional (3D) protein-ligand interactions into one dimensional (1D) bitstrings. The bitstrings are subsequently employed to compare the protein-ligand interaction predicted by the docking tool against the reference ligand. These comparisons produce scores that can be used to enhance the quality of SBVS campaigns. However, some IFP tools are either proprietary or using a proprietary library, which limits the access to the tools and the development of customized IFP algorithm. Therefore, we have developed PyPLIF, a Python-based open source tool to analyze IFP. In this article, we describe PyPLIF and its application to enhance the quality of SBVS in order to identify antagonists for estrogen α receptor (ERα).
Availability
PyPLIF is freely available at http://code.google.com/p/pyplif
Keywords: Virtual screening, interaction fingerprinting, docking software, Python, open source
Background
Interaction fingerprinting (IFP) is a relatively new method in virtual screening (VS) and proven to be able to increase VS quality. This method is matching the protein-ligand interaction from the output of molecular docking against the reference (usually from experimental study). In fact, the current world record for prospective fragment-based VS study was aided by IFP [1]. Unfortunately the IFP software is usually proprietary, or using a proprietary library. Therefore, we have attempted to develop a Python-based IFP software which depends on OpenBabel [2], an open source chemical library to give a completely free IFP tool that anyone can use and freely modify/develop according to their need.
Methodology
Basically PyPLIF accomplishes IFP by converting the molecular interaction of ligand-protein into bit array according to the residue of choice and the interaction type [3]. For every residue there are seven bits which represent seven type of interactions: (i) Apolar (van der Waals), (ii) aromatic face to face, (iii) aromatic edge to face, (iv) hydrogen bond (protein as hydrogen bond donor), (v) hydrogen bond (protein as hydrogen bond acceptor), (vi) electrostatic interaction (protein positively charged), and (vii) electrostatic interaction (protein negatively charged) (Figure 1a). Subsequently, the bit arrays from the docking pose are compared against the reference and checked for the similarity using Tanimoto coefficient (Tc) (Figure 1B), which give the result between 0.000 – 1.000 where 0.000 means no similarity, and 1.000 means the docking pose interaction fingerprints (within the selected residues) are identical with the reference.
Figure 1.
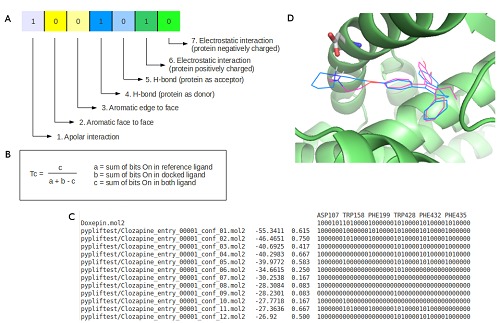
PyPLIF results: (A) 7 bits that represent 7 different interactions for each residue, 1 (one) means the interaction is exist (on) while 0 (zero) means the interaction is not exist (off); (B) Tanimoto coefficient (Tc) which is used to measure interaction similarity; (C) An example of PyPLIF result; and (D) Best ligand pose screened with PyPLIF and additional ASP351 filter, the ligand (ZINC03815477 conformation #9) gives not only high overlap but also hydrogen bond with ASP351. The 3D figure was generated using PyMOL 1.2r1 (http://www.pymol.org).
Input
Aside from the docking output from PLANTS [4], PyPLIF requires three files: Configuration file (config.txt), protein binding site file, and ligand reference. The configuration file consists of five lines each with a keyword-value pair, where the keywords are protein_reference, ligand_reference, protein_ligand_folder, residue_of_choice, and output_file (available in supplementary material).
Output
After a run has completed, PyPLIF generates an output file in .csv format (Figure 1C), which is best opened using a text editor. This file contains many lines, the first line shows the list of residue of choice, the subsequent line shows the ligand reference and its bitstring, while the rest of the lines are the ligand output from PLANTS. Each line of the ligand output from PLANTS consists of 4 columns: The first one is the name of the ligand, the second one is the docking score, the third is the Tc, and the last column presents the bitstrings. A simple shell script can be employed to PyPLIF to increase the quality of SBVS.
Results & Discussion
PyPLIF version 0.1.1 has been tested by running it in Ubuntu with three different versions of Open Babel libraries: (i) 2.2.3, (ii) 2.3.0, and (iii) 2.3.1. These Open Babel library versions were selected as they are available in the recent Ubuntu versions as the default version [5]. For the input we used the docking results of retrospective validation of SBVS protocols to identify estrogen α receptor (ERα) antagonists, which were kindly provided by Anita, et al. [6]. Despite the code and data differences among three Open Babel versions, the output has shown that the bit arrays and the Tc's are identical. This means that PyPLIF is stable and robust enough, at least for the dataset used in the retrospective validation of SBVS protocols to identify estrogen α receptor (ERα) antagonists [6].
In order to see the applicability of PyPLIF to enhance the SBVS quality, the enrichment factor at 1% false positives (EF1%) values were examined by sorting the ligands based on their Tc's. In case of multiple ligands with the same Tc's values appear, those ligands were sorted by the docking score. This method gives EF1% value of 17.94, whereas the previous study showed EF1% value of 21.2 [6]. In this attempt, PyPLIF could not enhance the SBVS quality. Then, to demonstrate another way of using PyPLIF we tried another approach employing the knowledge of molecular determinants of ligand binding to ERα. This approach is similar to the one used by de Graaf et al. [1]. Since residue ASP351 has been particularly important for ligand binding to ERα [7, 8], we added a hydrogen bond filter of the residue ASP351 using a simple shell script (available in supplementary material) which surprisingly increased EF1% value to 53.84. Thus, it is clear that post-dock analysis using PyPLIF could significantly increase VS campaign quality.
Caveat & Future Development
Since this tool is still very new, the feature is quite limited. First, this tool works only for the output from PLANTS. Currently, the tool is developed to support for Autodock Vina [9]. Second, this tool is still based on command-line interface that needs additional skill to run and analyze the output of PyPLIF. We would like to integrate a graphical user interface (GUI) to assist any medicinal chemists to easily run PyPLIF and analyze the results.
Supplementary material
Acknowledgments
The authors thank Digikom Multimedia Pratama (http://digikom.co.id/) for providing instruments used in the research and Anita, et al. for providing the docking results of the retrospective validation of SBVS protocols to identify estrogen α receptor (ERα) antagonists [6] that we used here as the input files.
Footnotes
Citation:Radifar et al, Bioinformation 9(6): 325-328 (2013)
References
- 1.de Graaf C, et al. J Med Chem. 2011;54:8195. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Boyle NM, et al. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcou G, Rognan D. J Chem Inf Model. 2007;47:195. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 4.Korb O, et al. J Chem Inf Model. 2009;49:84. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 5. http://packages.ubuntu.com/search?keywords=openbabel.
- 6.Anita Y, et al. Bioinformation. 2012;8:901. doi: 10.6026/97320630008901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayan G, et al. Mol Pharmacol. 2006;70:579. doi: 10.1124/mol.105.021931. [DOI] [PubMed] [Google Scholar]
- 8.Maximov, et al. J Med Chem. 2010;53:3273. doi: 10.1021/jm901907u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.