

Abstract
Improved processes for the preparation of biphenyl-based phosphine ligands t-BuBrettPhos, RockPhos, and BrettPhos are presented. The new methods, featuring the use of Grignard reagents and catalytic amounts of copper, are superior to the previous methods, which require the use of t-butyllithium and stoichiometric amounts of copper. Specifically, the use of less dangerous reagents provides a safer process, while the use of catalytic amounts of copper allows for the isolation of pure products in high yield. These improvements are particularly significant for the large scale preparation of these ligands.
Keywords: P ligands, Synthetic methods, Cross-coupling, Copper
Introduction
Catalytic methods that combine phosphine ligands with a source of Pd(0) are important strategies for effecting carbon-carbon and carbon-heteroatom bond formation.[1] Considerable effort has been focused on the development of new phosphine ligands,[2] and these ligands often play a key role in development of new methodology.[3] Among these ligands, the biaryl-based phosphines developed in our laboratory have emerged as a particularly effective class of ligands[4], and catalysts based on them have been widely employed both in academic settings as well as industrially in the synthesis of active pharmaceutical ingredients.[5] We have developed several such ligands bearing bulky dialkyl phosphine moieties, t-BuBrettPhos (1), RockPhos (2), and BrettPhos (3), (Figure 1), that are uniquely effective for certain classes of challenging carbon-heteroatom coupling reactions. For example, the combination of 1 with a palladium source provides a catalyst system capable of effecting a Pd-catalyzed fluorination of aryl triflates,[6] among other important transformations.[7] A similar Pd system based on 2 allows for the intermolecular arylation of secondary and primary alcohols.[8] BrettPhos, 3, has been used for the Pd-catalyzed amination of aryl electrophiles,[9] as well as the Pd-catalyzed trifluoromethylation of aryl chlorides.[10] Given the utility of these ligands for industrially-relevant processes, there is a significant demand for them in medium to large quantities. While 1 - 3 are commercially available, the current procedures for their preparation, due to issues of safety and product purity, do not lead themselves to efficient large-scale preparations. Herein we describe and demonstrate a common strategy for the preparation of all three ligands in high yields which is more amenable to their larger scale production.
Figure 1.
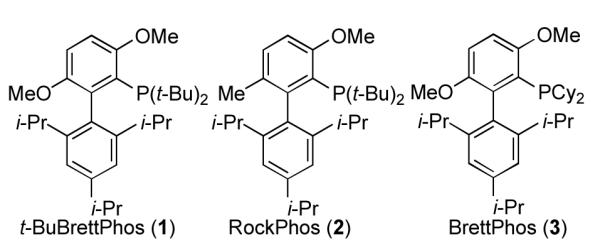
Biaryl-based phosphine ligands.
In our previous report, t-BuBrettPhos, 1, was prepared in 53% yield when iodoarene precursor 4 was treated with 2.0 equivalents of t-BuLi and the resulting aryllithium subsequently was allowed to react with 1.1 equivalents of t-Bu2PCl in the presence of 1.0 equivalent of CuCl at 70°C for 60 h (Scheme 1; R = OMe).[11] Thus, the preparation of 1 from 50 grams of 4 would require approximately 120 mL of t-BuLi (1.7 M in pentane). Similarly, the synthesis of RockPhos, 2, required 2.0 equivalents of t-BuLi and 1.0 equivalent CuCl in combination with biaryl iodide precursor 5 and 1.5 equivalents of t-Bu2PCl in toluene at 140°C for 20 h to provide the ligand in 72% yield (Scheme 1; R = Me).[8] Not only does the use of pyrophoric reagents in high quantities require specific safety equipment and extremely careful handling, but these methods provide the ligands in moderate yields and require extensive reaction times sometimes at high temperatures. Further, the need for stoichiometric amounts of CuCl often results in residual copper-containing species in the product, the presence of which has been shown to negatively affect the palladium-catalyzed reactions which depend on these ligands. We therefore set out to develop methods to address and circumvent these problems and thus allow for facile large-scale production.
Scheme 1.

First-generation preparations of t-BuBrettPhos (1) and RockPhos (2).
Results/discussion
We began by focusing on improving the preparation of t-BuBrettPhos, 1. To avoid the use of t-BuLi and a stoichiometric amount of CuCl, we examined an alternate route based on the formation of a biaryl Grignard reagent and its subsequent reaction with t-Bu2PCl (Scheme 2).
Scheme 2.
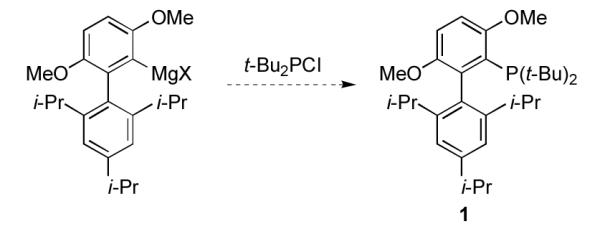
Preparation of 1 via formation of a Grignard reagent.
We chose 6 as our starting material, since the nucleophilicity of Grignard reagents generated from aryl bromides are generally higher than those generated from aryl iodides (Scheme 3).[12] Precursor 6 was prepared via our reported protocol as shown in Scheme 3.[13] Benzyne 8, generated by the treatment of 1,4-dimethoxy-2-fluorobenzene 7 with n-BuLi, was trapped with Grignard reagent 9 and the resulting intermediate was subsequently quenched with bromine to give 6 in 63% yield. We next attempted the conversion of 6 to 1 via the Grignard intermediate (10) and without the use of any copper co-catalyst. Unfortunately, all of our attempts to use copper-free conditions failed to produce any of the desired ligand 1.
Scheme 3.
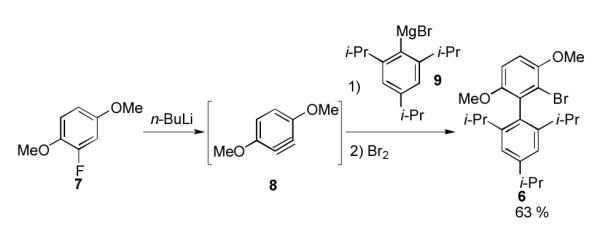
Synthesis of bromide precursor 6.
We next decided to investigate a process that employed a catalytic amount of copper. Such copper-catalyzed reactions have been reported by our group[14] and others.[15] However, prior to this report, there had been no reports of copper-catalyzed couplings of biphenyl derivatives bearing substituents on the top aryl ring. Our attempts to optimize this process are summarized in Table 1. Grignard reagent 10, generated by the treatment of 6 with magnesium in THF at 80°C for 2 hours, was treated with t-Bu2PCl (1.1 equiv.) in the presence of various copper reagents and LiBr.[15b] The reaction of 10 and t-Bu2PCl using CuI (20 mol%) and LiBr (40 mol%) in THF at 100°C for 24 h provided 1 in 13% (Entry 1). Changing the copper source to CuBr (20 mol%), provided 1 in 67% yield (Entry 2). With CuCl (20 mol%) as the copper source, the yield of 1 increased to 74% (Entry 3). In an attempt to improve this yield further, the reaction of 10 with t-Bu2PCl was carried out in toluene by performing a solvent exchange following the formation of 10 in THF. The solution of 10 in toluene was treated with t-Bu2PCl, and the reaction mixture at 140 °C for 12 h in the presence of CuCl (10 mol%) and LiBr (20 mol%). In this way 1 was produced in 93% yield (Entry 4). While we were satisfied with the improvement in yield, the process of switching solvents is undesirable particularly on large scale. To address this issue, we investigated the preparation of 10 in a mixture of toluene and THF. We found that 10 could be formed effectively in a 7.5 : 1 mixture of toluene and THF (1.5 h reaction time), and that subsequent treatment of the resulting reaction mixture with t-Bu2PCl in the presence of CuCl (5 mol%) and LiBr (10 mol%) provided 1 in 95% yield (Entry 5). Finally, we evaluated the coupling using iodide precursor 4 in lieu of bromide precursor 6. Under identical reaction conditions, 1 was obtained in 83% yield from 4 (Entry 6). We thus chose bromide 6 as the optimal precursor for 1.
Table 1.
Optimization of copper-catalyzed reaction.
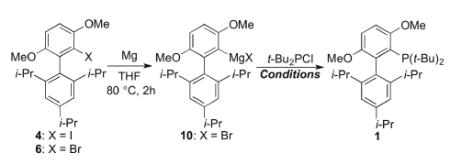
| Entry | X | Cat. [mol%] |
LiBr [mol%] |
Temp. [°C] |
Solvent | Time [h] |
Yield[a] [%] |
|---|---|---|---|---|---|---|---|
| 1 | Br | CuI[20] | 40 | 100 | THF | 24 | 13 |
| 2 | Br | CuBr[20] | 40 | 100 | THF | 24 | 67 |
| 3 | Br | CuCl[20] | 40 | 100 | THF | 24 | 74 |
| 4 | Br | CuCl[10] | 20 | 140 | toluene | 12 | 93 |
| 5[b] | Br | CuCl[5] | 10 | 120 | toluene | 12 | 95 |
| 6[b] | I | CuCl[5] | 10 | 120 | toluene | 12 | 83 |
Yields were calculated by 1H NMR using dibenzyl ether as an internal standard.
Grignard reagents were prepared in a mixture of toluene and THF (7.5:1)
To assess the utility of the improved method on a moderate scale, we carried out the reaction sequence using beginning with 50 grams of biaryl bromide 6. Even on this scale, Grignard reagent 10 was formed smoothly through the treatment of 6 with Mg in a 7.5 : 1 mixture of toluene : THF for 3 hours. The subsequent reaction of 10 with t-Bu2PCl proceeded efficiently to provide 1 in 87% yield (Scheme 4; Route a, R = OMe).
Scheme 4.
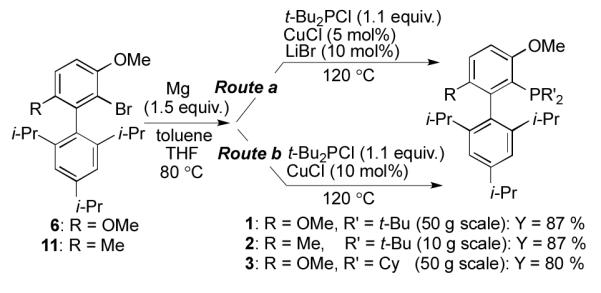
Large-scale preparations of 1, 2, and 3.
To evaluate the generality of the new method, we next attempted the analogous preparation of RockPhos (2) and BrettPhos (3). From 10 grams of RockPhos precursor 11[16], 2 was obtained in 87% yield (Scheme 4; Route a, R = Me). For the preparation of 3, additional optimization of the reaction conditions was required. Specifically, it was found that the use of CuCl (10 mol%) in the absence of LiBr was optimal and 3 was obtained in 80% isolated yield (Scheme 4; Route b, R = OMe).
Conclusion
In summary, we have developed improved methods for the preparation of t-BuBrettPhos, RockPhos and BrettPhos. The methods provide these ligands in high yields and without the use of t-BuLi or a stoichiometric amount of copper halide. Thus, these methods are superior to the previously-reported ones from the viewpoints of safety, yield, and economy. We anticipate that the new methods will lead to a more accessible supply of these ligands in larger quantity, leading to an increase in their use.
Experimental Section
General Reagent Information
All reactions were carried out under an argon atmosphere. THF and toluene were purchased from J.T. Baker in CYCLE-TAINER® solvent-delivery kegs and vigorously purged with argon for 1 h. The solvent was further purified by passing through successive alumina and Q5 reactant-packed columns on a solvent purification system. Commercial materials were used as received unless otherwise noted. 1,4-Dimethoxy-2-fluorobenzene was purchased from Synquest Labs, Inc. 4-Methoxy-1-methyl-2-fluorobenzene was purchased form TCI America. n-BuLi (2.5 M in hexane), Bromine, and 1,2-dibromoethane were purchased from Aldrich Chemical Co. CuCl and CuI were purchased from Acros Organics. CuBr and LiBr were purchased from Strem Chemicals Inc. Magnesium turnings (99.8%; 2.3 mm (0.09 in) wide) was purchased from Alfa Aesar. 2,4,6-Triisopropylbromobenzene was purchased from ABCR GmbH & Co. KG.
General Analytical Information
All compounds were characterized by 1H NMR, 13C NMR and IR spectroscopy, melting point, and elemental analysis. Nuclear Magnetic Resonance spectra were recorded on a Bruker 400 MHz instrument. All 1H NMR are reported in δ units, parts per million (ppm), and were measured relative to the signals for residual chloroform (7.26 ppm) in the deuterated solvent, unless otherwise stated. All 13C NMR are reported in ppm relative to deuterochloroform (77.0 ppm), unless otherwise stated, and all were obtained with 1H decoupling. All 31P NMR spectra are reported in ppm relative to 85% phosphoric acid external standard (0 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, m = multiplet. All IR spectra were taken on a Perkin – Elmer 2000 FTIR. All GC analyses were performed on an Agilent 6890 gas chromatograph with an FID detector using a J & W DB-1 column (10 m, 0.1 mm ID). Elemental analyses were performed by Atlantic Microlabs Inc., Norcross, GA.
Preparation of 2-bromo-2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl (6)
An oven-dried three-neck 500 mL round bottom flask, which was equipped with a magnetic stir bar and charged with magnesium turnings (2.8 g, 115 mmol) was fitted with a reflux condenser, glass stopper and rubber septum. The flask was purged with argon and then THF (100 mL) and 2,4,6-triisopropylbromobenzene (24.3 mL, 95.9 mmol) were added via syringe. The reaction mixture was heated to reflux and 1,2-dibromoethane (40 μL) was added via syringe. The reaction mixture was allowed to stir at reflux for 1.5 h and was then cooled to room temperature. A separate oven-dried 2L round bottom flask, which was equipped with a magnetic stir bar and fitted with a rubber septum, was purged with argon and then THF (500 mL) and 1,4-dimethoxy-2-fluorobenzene (7.49 g, 48.0 mmol) were added to the flask via syringe. The reaction mixture was cooled to −78°C and n-BuLi (2.5 M in hexane, 19.4 mL, 48.5 mmol) was added in a dropwise fashion over a 30 min period. The solution was stirred for 1 h and the Grignard reagent, which was prepared in the first reaction vessel, was added via cannula over a 40 min period. The resulting mixture was allowed to stir at −78°C for 1 h. The cooling bath was then removed and the reaction mixture was allowed to slowly warm to room temperature over a 1.5 h period after it was stirred for an additional 12 h. The mixture was then cooled to 0°C with the aid of an ice/water bath and bromine (5.4 mL, 105 mmol) was slowly added dropwise via syringe over 20 min and the resulting dark purple solution was stirred for 1 h. At this point a 10% Na2SO3 aqueous solution (300 mL) was added to the reaction mixture and the solution was stirred for 10 min. The mixture was transferred to a separatory funnel and then the aqueous layer was removed. The organic layer was washed with 10% Na2SO3 aqueous solution (300 mL) and subsequently washed with brine (300 mL). The individual aqueous layers were then back-extracted with EtOAc (400 mL) and the combined organic layers were dried over MgSO4, filtered, and then solvent was removed with the aid of a rotary evaporator. The resulting solid was triturated with cold hexane and filtered to give the crude product as a pale pink solid. The solid was crystallized from hot EtOAc (first crystallization; 100 mL, second crystallization; 50 mL) at 80°C to give the desired product 6 (12.7 g, 63%); mp = 191-192°C. 1H NMR (400 MHz, CDCl3) δ: 7.05 (s, 2H), 6.89 (d, J = 9.0 Hz, 1H), 6.85 (d, J = 9.0 Hz, 1H), 3.91 (s, 3H), 3.66 (s, 3H), 2.95 (septet, J = 6.8 Hz, 1H), 2.40 (septet, J = 6.9 Hz, 2H), 1.31 (d, J = 6.8 Hz, 6H), 1.14 (d, J = 6.9 Hz, 6H), 1.01 (d, J = 6.9 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3) δ: 152.5, 150.2, 147.9, 145.9, 132.2, 131.9, 120.5, 116.0, 110.1, 109.0, 56.5, 55.5, 34.0, 30.8, 24.3, 23.9, 23.7 ppm. IR (neat, cm−1): 2957, 5865, 1734, 1698, 1651, 1572, 1457, 1429, 1358, 1260, 1089, 1037, 874, 791, 760. Anal. Calcd. for C23H31BrO2: C, 65.87; H, 7.45. Found: C, 65.97; H, 7.61.
Representative procedure for the examples described in Table 1 (Entry 1)
An oven-dried test tube, which was equipped with a magnetic stir bar, fitted with a teflon screw cap, and charged with magnesium tunings (33 mg, 1.35 mmol), was evacuated and backfilled with argon (this process was repeated a total of three times). THF (0.5 mL) was then added via syringe. The solution was heated at 80°C for 5 min, and then 1,2-dibromoethane (2 drops) was added to the solution via syringe. After stirring for 15 min, a solution of 2-bromo-2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl (150 mg, 0.358 mmol) in THF (0.5 mL) was added dropwise to the mixture. The reaction mixture was allowed to stir at 80°C for 2 h and was then cooled to room temperature. A separate oven-dried test tube, which was equipped with a magnetic stir bar and fitted with a teflon screw cap, was purged with argon. CuI (13.6 mg, 0.0714 mmol) and LiBr (12.4 mg, 0.143 mmol) were added to the reaction vessel after which time it was purged with argon a second time. The Grignard reagent, which was prepared in the first reaction vessel, was transferred to the second reaction vessel via cannula, and t-Bu2PCl (75 μL, 0.394 mmol) was then added to the solution. The reaction mixture was heated to 100°C for 24 h, then cooled to room temperature and diluted with EtOAc. The mixture was washed with 28% aqueous NH4OH (this process was repeated a total of three times), washed with brine, dried over MgSO4, and concentrated in vacuo to give crude product. Dibenzyl ether was then added as an internal standard and the reaction mixture was analyzed by 1H NMR. The data indicated that the product was provided in 13% yield.
Large-scale preparation of t-BuBrettPhos (1):[11]
An oven-dried flask, which was equipped with a magnetic stir bar and charged with magnesium turnings (4.34 g, 179 mmol), was fitted with a reflux condenser and rubber septum. The flask was purged with argon and then THF (20 mL) and 1,2-dibromoethane (500 μL) were added via syringe. The solution was heated at 80°C for 30 min. The solution was then cooled to room temperature and toluene (220 mL) and 2-bromo-2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl (50 g, 119 mmol) were added to the solution. The mixture was then heated at 80°C for 3 h. A separate oven-dried Schlenk flask was fitted with a rubber septum and purged with argon, after which CuCl (590 mg, 5.96 mmol) and LiBr (1.03 g, 11.9 mmol) were quickly added to the flask. The septum was replaced and the Grignard reagent, which was prepared in the first reaction vessel, was transferred to the Schlenk flask via cannula after which t–Bu2PCl (24.8 mL, 130 mmol) was added to the solution via syringe. The rubber septum was quickly exchanged for a teflon screw cap and the reaction mixture was heated to 120°C for 20 h. The reaction mixture was then cooled to room temperature and diluted with EtOAc (900 mL). The solution was poured into a mixture of 28% aqueous NH4OH (200 mL), brine (200 mL), and water (200 mL) and the mixture was stirred for 10 min. The mixture was transferred to a separatory funnel and the aqueous layer was removed. The organic layer was washed three times with a mixture of 28% aqueous NH4OH (400 mL) and brine (400 mL) and was then washed one final time with brine (800 mL). The individual aqueous layers were then back-extracted with EtOAc (800 mL). The two organic layers were combined, dried over MgSO4, and concentrated in vacuo to provide the crude product. The crude material was then crystallized from hot methanol (100 mL) and EtOAc (100 mL) to give the desired product as white crystals (50.5 g, 87%); mp = 170-171°C. 1H NMR (400 MHz, CDCl3) δ: 6.98 (s, 2H), 6.88 (d, J = 8.8 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 3.79 (s, 3H), 3.56 (s, 3H), 2.96 (septet, J = 6.8 Hz, 1H), 2.51 (septet, J = 6.6 Hz, 2H), 1.33 (d, J = 6.8 Hz, 6H), 1.23 (d, J = 6.6 Hz, 6H), 1.16 (s, 9H), 1.13 (s, 9H), 0.95 (d, J = 6.4 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3) δ: 155.68, 155.66, 152.38, 152.27, 146.96, 146.46, 140.18, 139.80, 132.79, 132.72, 127.37, 126.91, 119.85, 110.66, 110.65, 108.00, 54.17, 53.77, 33.91, 33.88, 33.62, 31.81, 31.64, 31.02, 30.99, 25.47, 24.08, 23.36 ppm. 31P NMR (161 MHz, CDCl3) δ: 34.56 ppm. IR (neat, cm−1): 2956, 2862, 1735, 1698, 1578, 1559, 1458, 1421, 1382, 1360, 1252, 1171, 1086, 1041, 1018, 877, 801, 733. Anal. Calcd. for C31H49O2P: C, 76.82; H, 10.19. Found: C, 77.03; H, 10.10.
Preparation of 2-bromo-2′,4′,6′-triisopropyl-3-methoxy-6-methylbiphenyl (11)
An oven-dried three-neck 500 mL round bottom flask, which was equipped with a magnetic stir bar and charged with magnesium turnings (2.8 g, 115 mmol), was fitted with a reflux condenser, glass stopper and rubber septum. The flask was purged with argon and then THF (190 mL) and 2,4,6-triisopropylbromobenzene (24.1 mL, 96.1 mmol) were added via syringe. The reaction mixture was heated to reflux and 1,2-dibromoethane (60 μL) was added via syringe. The reaction was allowed to stir at reflux for 2 h and was then cooled to room temperature. A separate oven-dried 2L round bottom flask, which was equipped with a magnetic stir bar and fitted with a rubber septum, was purged with argon and then THF (250 mL) and 1-methoxyl-4-methyl-3-fluorobenzene (6.73 g, 48.0 mmol) were added to the flask via syringe. The reaction mixture was cooled to −78 °C and n-BuLi (2.5 M in hexane, 20 mL, 50.0 mmol) was added in a dropwise fashion over a 1 h period. The solution was stirred for 1 h and then the Grignard reagent, which was prepared in the first reaction vessel, was added via cannula over a 30 min period and the mixture was then allowed to stir at −78°C for 1 h. The reaction mixture was then allowed to warm slowly to room temperature over 1.5 h, where it was stirred for an additional 12 h. The mixture was then cooled to 0°C and bromine (5.9 mL, 115 mmol) was added dropwise via syringe slowly over 20 min, and the resulting dark purple solution was allowed to stir for 1 h. At this time a 10% Na2SO3 aqueous solution (300 mL) was added to the reaction mixture and the solution was stirred for 10 min. The mixture was transferred to a separatory funnel and the aqueous layer was removed. The organic layer was washed with a 10% Na2SO3 aqueous solution (300 mL) and was then washed with brine (300 mL). Then combined aqueous layers were then extracted with EtOAc (400 mL). The individual organic layers were dried over MgSO4, filtered, and then the solvent was removed with the aid of a rotary evaporator. The crude material was triturated with cold MeOH and filtered to give the crude product as a white solid. The solid was crystallized from a combination of hot EtOAc (20 mL) and hot MeOH (80 mL) at 80°C to give the desired product (10.6 g, 54%); mp = 146-147°C. 1H NMR (400 MHz, CDCl3) δ: 7.18 (d, J = 8.4 Hz, 1H), 7.08 (s, 2H), 6.83 (d, J = 8.4 Hz, 1H), 3.94 (s, 3H), 2.96 (septet, J = 6.9 Hz, 1H), 2.38 (septet, J = 6.9 Hz, 2H), 1.94 (s, 3H), 1.32 (d, J = 6.9 Hz, 6H), 1.16 (d, J = 6.9 Hz, 6H), 1.08 (d, J = 6.9 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3) δ: 154.1, 148.2, 145.3, 142.5, 134.7, 130.8, 128.8, 120.9, 114.8, 109.9, 56.1, 34.0, 30.6, 24.5, 24.2, 24.0, 20.5 ppm. IR (neat, cm−1): 2956, 2868, 1734, 1653, 1606, 1559, 1464, 1436, 1382, 1361, 1292, 1264, 1210, 1083, 1055, 1019, 878, 801, 760, 738. Anal. Calcd. for C23H31BrO: C, 68.48; H, 7.75. Found: C, 68.28; H, 7.75.
Large-scale preparation of RockPhos (2):[8]
An oven-dried flask, which was equipped with a magnetic stir bar and charged with magnesium turnings (904 mg, 37.2 mmol), was fitted with a reflux condenser and a rubber septum. The flask was purged with argon and THF (4 mL) and 1,2-dibromoethane (110 μL) were added via syringe. The solution was heated at 80°C for 30 min. The solution was then cooled to room temperature and toluene (45 mL) and 2-bromo-2′,4′,6′-triisopropyl-3-methoxy-6-methylbiphenyl (10.0 g, 24.8 mmol) were added to the flask. The solution was heated at 80°C for 3 h. A separate oven-dried Schlenk flask was fitted with a rubber septum and was purged with argon, after which CuCl (123 mg, 1.24 mmol) and LiBr (215 mg, 2.48 mmol) were quickly added to the flask. The septum was replaced and the Grignard reagent, which was prepared in the first reaction vessel, was transferred to the Schlenk flask via cannula and then t-Bu2PCl (5.2 mL, 27.3 mmol) was added via syringe. The rubber septum was quickly exchanged for a teflon screw cap and the reaction mixture was then heated to 120°C for 18 h. The solution was then cooled to room temperature and diluted with EtOAc (200 mL). The solution was poured into a mixture of 28% aqueous NH4OH (90 mL) and water (90 mL) and the mixture was stirred for 10 min. The mixture was transferred to a separatory funnel and the aqueous layer was removed. The organic layer was washed three times with a mixture of 28% aqueous NH4OH (80 mL) and brine (80 mL) and was then washed with brine (170 mL). The individual aqueous layers were back-extracted with EtOAc (total 800 mL). The combined organic layers were then dried over MgSO4 and concentrated in vacuo to give the crude product. The crude material was crystallized from hot methanol to provide the desired product 2 as white crystals (10.2 g, 87%); mp = 129-130°C. 1H NMR (400 MHz, CDCl3) δ: 7.22 (d, J = 8.4 Hz, 1H), 7.01 (s, 2H), 6.81 (d, J = 8.4 Hz, 1H), 3.81 (s, 3H), 2.95 (septet, J = 6.8 Hz, 1H), 2.51 (septet, J = 6.7 Hz, 2H), 1.80 (s, 3H), 1.32 (d, J = 6.8 Hz, 6H), 1.24 (d, J = 6.7 Hz, 6H), 1.18 (s, 9H), 1.15 (s, 9H), 1.00 (d, J = 6.7 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3) δ: 160.05, 160.03, 150.97, 150.59, 147.33, 145.72, 145.71, 136.55, 136.48, 131.91, 131.90, 130.54, 130.46, 125.46, 125.01, 120.74, 108.19, 53.58, 34.13, 33.99, 33.84, 32.01, 31.85, 30.79, 30.77, 25.35, 24.66, 24.64, 24.17, 22.17, 22.15 ppm. 31P NMR (161 MHz, CDCl3) δ: 35.71 ppm. IR (neat, cm−1): 2958, 2866, 1716, 1606, 1561, 1459, 1426, 1382, 1362, 1278, 1262, 1168, 1145, 1054, 1021, 877, 804, 738. Anal. Calcd. for C31H49OP: C, 79.44; H, 10.54. Found: C, 79.53; H, 10.47.
Large-scale preparation of BrettPhos (3):[9c]
An oven-dried flask, which was equipped with a magnetic stir bar and charged with magnesium turnings (4.34 g, 179 mmol), was fitted with a reflux condenser and a rubber septum. The flask was purged with argon and then THF (20 mL) and 1,2-dibromoethane (500 μL) were added via syringe. The solution was heated at 80°C for 30 min. The solution was then cooled to room temperature and toluene (220 mL) and 2-bromo-2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl (50 g, 119 mmol) were added to the flask via syringe. The solution was heated at 80°C for 3 h. A separate oven-dried Schlenk flask, fitted with a rubber septum, was purged with argon and then CuCl (1.18 g, 11.9 mmol) was quickly added to the flask. The septum was replaced and the Grignard reagent, which was prepared in the first reaction vessel, was added to the Schlenk flask via cannula and then Cy2PCl (28.9 mL, 130 mmol) was added to the solution via syringe. The rubber septum was then quickly exchanged for a teflon screw cap and then heated to 120°C and it was stirred for 20 h. The solution was then cooled to room temperature and diluted with EtOAc (900 mL). The reaction mixture was poured into a mixture of 28% aqueous NH4OH (400 mL) and water (400 mL) and the mixture was stirred for 10 min. The mixture was transferred to a separatory funnel and the aqueous layer was removed. The organic layer was washed three times with a mixture of 28% aqueous NH4OH (400 mL) and brine (400 mL) and was then washed with brine (800 mL). The combined aqueous layers were extracted with EtOAc (800 mL). The individual organic layers were then dried over MgSO4, and concentrated in vacuo to provide the crude product. The crude material was crystallized from hot acetone to give the desired product as white crystals (51.3 g, 80%); mp = 193-194°C. 1H NMR (400 MHz, CDCl3) δ: 6.89 (s, 2H), 6.86 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 3.83 (s, 3H), 3.56 (s, 3H), 2.95 (septet, J = 7.0 Hz, 1H), 2.44 (septet, J = 6.8 Hz, 2H), 2.25-2.17 (m, 2H), 1.85-1.81 (m, 2H), 1.73-1.63 (m, 6H), 1.43-0.94 (m, 10H), 1.33 (d, J = 7.0 Hz, 6H), 1.22 (d, J = 6.8 Hz, 6H), 0.95 (d, J = 6.8 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3) δ: 156.29, 156.27, 152.29, 152.18, 146.82, 145.88, 145.86, 139.19, 138.83, 132.64, 132.56, 126.79, 126.51, 120.04, 110.60, 108.51, 55.05, 54.53, 36.63, 36.49, 33.76, 32.94, 32.71, 30.96, 30.85, 30.47, 27.97, 27.88, 27.59, 27.45, 26.49, 26.48, 25.05, 23.95, 23.52 ppm. 31P NMR (161 MHz, CDCl3) δ: 1.61 ppm. IR (neat, cm−1): 2958, 2847, 1581, 1459, 1422, 1380, 1359, 1294, 1173, 1091, 1051, 1021, 878, 802, 730. Anal. Calcd. for C35H53O2P: C, 78.32; H, 9.95. Found: C, 78.23; H, 9.85.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (GM46059) for financial support of this project. N.H. thanks the Astellas Foundation for Research on Metabolic Disorders for a Fellowship and is a Japan Society for the Promotion of Science (JSPS) for Young Scientist Research Fellow for which he is grateful. We thank Dr. Meredeth A. McGowan for help with the preparation of this manuscript.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- [1] a).Miyaura N. Cross-Coupling Reactions. A Practical Guide, Topics in Current Chemistry. SpringerVerlag; Berlin: 2002. [Google Scholar]; b) de Meijere A, Diederich F. Metal Catalyzed Cross-Coupling Reactions. 2nd ed Wiley-VCH; Weinheim: 2004. [Google Scholar]; c) Special issue on cross-coupling. Acc. Chem. Res. 2008;41:1439. doi: 10.1021/ar8001798. [DOI] [PubMed] [Google Scholar]
- [2] a).Shen Q, Ogata T, Hartwig JF. J. Am. Chem. Soc. 2008;130:6586. doi: 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J. Am. Chem. Soc. 2006;128:4101. doi: 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]; c) Nishiyama M, Yamamoto T, Koie Y. Tetrahedron Lett. 1998;39:617. [Google Scholar]; d) Fleckenstein CA, Plenio H. Chem. Soc. Rev. 2010;39:694. doi: 10.1039/b903646f. [DOI] [PubMed] [Google Scholar]; e) Wolfe JP, Wagaw S, Buchwald SL. J. Am. Chem. Soc. 1996;118:7215. [Google Scholar]; f) Wolfe JP, Buchwald SL. J. Org. Chem. 2000;65:1144. doi: 10.1021/jo9916986. [DOI] [PubMed] [Google Scholar]; g) Guari Y, van Es DS, Reek JNH, Kamer PCJ, van Leeuwen P. Tetrahedron Lett. 1999;40:3789. [Google Scholar]; h) Driver MS, Hartwig JF. J. Am. Chem. Soc. 1996;118:7217. [Google Scholar]; i) Sadighi JP, Harris MC, Buchwald SL. Tetrahedron Lett. 1998;39:5327. [Google Scholar]; j) Yin J, Zhao MM, Huffman MA, McNamara JM. Org. Lett. 2002;4:3481. doi: 10.1021/ol0265923. [DOI] [PubMed] [Google Scholar]; k) Zhao M, Yin J, Huffman MA, McNamara JM. Tetrahedron. 2006;62:1110. [Google Scholar]; l) Bonala R, Torres MC, Iden CR, Johnson F. Chem. Res. Toxicol. 2006;19:734. doi: 10.1021/tx0600191. [DOI] [PubMed] [Google Scholar]; m) Schulte JP, II, Tweedie SR. Synlett. 2007;15:2331. [Google Scholar]; n) Monguchi Y, Kitamoto K, Ikawa T, Maegawa T, Sajiki H. Adv. Synth. Catal. 2008;350:2767. [Google Scholar]; o) Shen Z, Hong Y, He X, Mo W, Hu B, Sun N, Hu X. Org. Lett. 2010;12:552. doi: 10.1021/ol902759k. [DOI] [PubMed] [Google Scholar]
- [3] a).Muller WE. The Benzodiazepine Receptor. Cambridge University Press; New York: 1988. [Google Scholar]; b) Belciug M, Ananthanarayanan VS. J. Med. Chem. 1994;37:4392. doi: 10.1021/jm00051a017. [DOI] [PubMed] [Google Scholar]
- [4] a).Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Surry DS, Buchwald SL. Chem. Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] a).Busacca CA, Fandrick DR, Song JJ, Senanayake CH. Adv. Synth. Catal. 2011;353:1825. [Google Scholar]; b) Torborg C, Beller M. Adv. Synth. Catal. 2009;351:3027. [Google Scholar]
- [6].Watson DA, Su M, Teverovskiy G, Zhang Y, Garcia-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a).Dooleweerdt K, Fors BP, Buchwald SL. Org. Lett. 2010;12:2350. doi: 10.1021/ol100720x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Breitler S, Oldenhuis NJ, Fors BP, Buchwald SL. Org. Lett. 2011;13:3262. doi: 10.1021/ol201210t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Maimone TJ, Buchwald SL. J. Am. Chem. Soc. 2010;132:9990. doi: 10.1021/ja1044874. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Fors BP, Buchwald SL. J. Am. Chem. Soc. 2009;131:12898. doi: 10.1021/ja905768k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Pan J, Wang X, Zhang Y, Buchwald SL. Org. Lett. 2011;13:4974. doi: 10.1021/ol202098h. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Shen X, Hyde AM, Buchwald SL. J. Am. Chem. Soc. 2010;132:14076. doi: 10.1021/ja107481a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fors X. Wu. B. P., Buchwald SL. Angew. Chem. Int. Ed. 2011;50:9943. doi: 10.1002/anie.201104361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9] a).Maiti D, Fors BP, Henderson JL, Nakamura Y, Buchwald SL. Chem. Sci. 2011;2:57. doi: 10.1039/C0SC00330A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fors BP, Buchwald SL. J. Am. Chem. Soc. 2010;132:15914. doi: 10.1021/ja108074t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fors BP, Watson DA, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Maiti D, Buchwald SL. J. Am. Chem. Soc. 2009;131:17423. doi: 10.1021/ja9081815. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Henderson JL, Buchwald SL. Org. Lett. 2010;12:4442. doi: 10.1021/ol101929v. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Henderson JL, McDermott SM, Buchwald SL. Org. Lett. 2010;12:4438. doi: 10.1021/ol101928m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fors BP, Dooleweerdt KD, Zeng Q, Buchwald SL. Tetrahedron. 2009;65:6576. doi: 10.1016/j.tet.2009.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Silverman GS, Rakita PE. Handbook of Grignard reagents. Marcel Dekker; New York: 1996. [Google Scholar]
- [13].Barder TE, Buchwald SL. J. Am. Chem. Soc. 2007;129:12003. doi: 10.1021/ja073747z. [DOI] [PubMed] [Google Scholar]
- [14] a).Kaye S, Fox JM, Hicks FA, Buchwald SL. Adv. Synth. Catal. 2001;343:789. [Google Scholar]; b) Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2003;125:6653. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- [15] a).Liu L, Wu H-C, Yu J-Q. Chem. Eur. J. 2011;17:10828. doi: 10.1002/chem.201101467. [DOI] [PubMed] [Google Scholar]; b) Stambuli JP, Stauffer SR, Shaughnessy KH, Hartwig JF. J. Am. Chem. Soc. 2001;123:2677. doi: 10.1021/ja0058435. [DOI] [PubMed] [Google Scholar]
- [16].11 was synthesized by similar procedure to that used for 6. See experimental section.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.