

Abstract
BACKGROUND
A significant number of medulloblastomas (MBs) originate from abnormal activation of the sonic hedgehog/patched (SHH/PTC) signaling pathway. Although p53 deficiency enhances tumor formation in mice, inactivation of the p53 gene is seen in a minority of MBs. Wild-type p53-induced phosphatase 1 (WIP1) downregulates p53 expression and has been shown to be overexpressed in MBs.
OBJECTIVE
We tested the hypothesis that overexpression of WIP1 enhances tumor formation in an SHH-dependent model of MB.
METHODS
We used the RCAS/Ntv-a system to study the effect of WIP1 in vitro and in vivo. We transfected A375-TVA cells with RCAS-WIP1 and then exposed these cells to cisplatin to determine the effect on p53 expression. We modeled ectopic WIP1 expression independently and in combination with SHH in the cerebella of newborn mice to assess the effect on tumor formation. Mice were observed for 12 weeks or until neurological symptoms developed. The brains were examined for tumor formation.
RESULTS
A375-TVA cells infected with RCAS-WIP1 demonstrated reduced p53 expression after exposure to cisplatin compared with controls. We detected tumors in 12 of 35 mice (34%) injected with RCAS-WIP1 and RCAS-SHH. Tumors were detected in 3 of 40 mice (8%) injected with RCAS-SHH alone. The difference in tumor formation rates was significant (×2 test, P = < .01). Tumors did not form in mice injected with RCAS-WIP1 alone.
CONCLUSION
We show that ectopic expression of WIP1 cooperates with SHH to enhance formation of MB, although it is insufficient to induce tumors independently. Our results verify the role of WIP1 in MB formation and provide a crucial link to the inactivation of p53 in MBs.
Keywords: Brain tumor, Medulloblastoma, Mouse model, p53, Sonic hedgehog, WIP1
Medulloblastoma (MB) is the most common malignant brain tumor in children. Although 5-year survival rates currently approach 75%, the deleterious consequences of treatment leave many children with significant long-term side effects.1,2 Novel therapeutic strategies are needed to improve treatment outcome, and their development requires an improved understanding of the molecular basis for MB tumorigenesis.
A subset of MBs, accounting for 25% to 30% of tumors, results from activation of the sonic hedgehog/patched (SHH/PTC) signaling pathway.3–5 Patients with Gorlin syndrome who have inherited a defective copy of the Ptc gene, are predisposed to MB.6 MBs will develop in Ptc± mice that recapitulate histological features of the human disease.7 Retroviral transfer and expression of SHH into the developing cerebellum induces MB.8,9 SHH/PTC signaling is crucial to the normal development of the cerebellum as mitogenic stimulation by SHH causes granule neuron precursors, the presumed cell of origin for SHH-associated MB, to undergo rapid expansion in the external granule layer.10 During this period, granule neuron precursors may be more vulnerable to transforming genetic events, initiating MB formation.
Although increased SHH/PTC pathway activity is sufficient to form MBs in animal models, tumor penetrance is limited. Tumors form in only 14% to 25% of Ptc± mice. Retroviral transfer of SHH results in MB formation in 9% to 15% of mice, suggesting that although increased SHH/PTC signaling is sufficient to generate MBs, defects in other mechanisms are also required for tumor growth. Signaling molecules implicated in MBs have been investigated in animal models to determine their impact on tumorigenesis. For example, insulin-like growth factor-2 expression has been observed in human MBs and retroviral transfer of insulin-like growth factor 2 enhances the tumor formation rate in mice in an SHH-dependent model of MBs, indicating that combined activation of the SHH/PTC and insulin-like growth factor/phosphoinositide 3-kinase signaling pathways contribute to the development of MB.11,12
One of the most common cytogenetic lesions, found in approximately one third of cases of MB, is isochromosome 17q, which consists of 17p deletion with duplication of 17q.13,14 The presence of isochromosome 17q is strongly associated with high-risk MB.4,5,15 As a consequence of 17p deletion, the subsequent loss of the tumor suppressor gene p53, found on 17p13.1, was suspected to contribute to MB tumorigenesis. Several lines of evidence suggest that the p53 pathway is perturbed in MB. Frank et al16 described the activation of the p53–p14ARF pathway in the large-cell/anaplastic variant of MB. The spontaneous tumor formation rate in Ptc± mice increases to 100% when the mice are bred into a p53−/− background.17 Tumor formation in other murine models of MB, including those dependent on knockout of Ku80 and DNA ligase 4, is also significantly enhanced by p53 deficiency.18,19 Individuals with Li-Fraumeni syndrome who carry germline p53 mutations are also at increased risk of the development of MB.20,21 However, only 7% to 16% of sporadic MBs display p53 mutations, suggesting that p53 function may be abrogated by alternate mechanisms as observed in other cancers with wild-type p53.22–25
A negative regulator of p53 function, wild-type p53-induced phosphatase 1 (WIP1 or PPM1D) is overexpressed in various tumor types including neuroblastoma, breast cancer, and ovarian cancer.26–29 WIP1 expression is induced by p53 as a response to DNA damage. Accumulation of WIP1 results in dephosphorylation of p53 and inhibition of p53 kinases and facilitates MDM2-dependent degradation of p53.26,30 In both classic and desmoplastic variants of MB, comparative genomic hybridization studies have shown amplification of PPM1D.31,32 Recent work has shown that WIP1 mRNA and protein levels from human MB samples are significantly higher than that of normal brain.33 Castellino et al33 described WIP1 copy gain and amplification in a series of 33 newly diagnosed primary MB specimens. WIP1 copy numbers in these specimens ranged from 2 to 11. The WIP1 chromosomal location of 17q22/23 is significant because, as described previously, aberrations of chromosome 17 are commonly observed in MB, and WIP1 mRNA levels are nearly 20-fold higher in tumors with a gain of 17q or isochromosome 17q compared with those without these cytogenetic lesions.13,34–38
To determine what effect WIP1 overexpression has on SHH-induced MB, we modeled ectopic WIP1 expression in mice using the RCAS/Ntv-a transgenic mouse system. In this system, a gene is cloned into a modified avian retrovirus (RCAS) that is replication defective in mammalian cells. The vector is injected in the brains of transgenic mice (Ntv-a) that express TVA (the avian leukosis virus subtype A receptor, the receptor for RCAS) under control of the Nestin gene promoter. Nestin-expressing cells include neuronal precursors such as the granule neuron precursor. The gene is randomly incorporated into the host cell's genome and is expressed by the constitutive retroviral promoter long terminal repeat. This method of somatic cell gene transfer can assess the functional consequence of gene expression in vivo. Further, multiple RCAS vectors can be co-injected, permitting the study of cooperative effects of different genes on tumorigenesis. This well-established mouse model has been used to understand the contributions of various signaling pathways to SHH-dependent MB formation. We hypothesized that WIP1 would promote tumor formation in this system. Here, we show that although independent overexpression of WIP1 is insufficient to induce tumors, ectopic expression of WIP1 enhances tumor formation in a SHH-dependent model of MB. These results suggest that WIP1 signaling contributes to tumor formation in the SHH-dependent subtype of MB and validate it as a potential therapeutic target.
MATERIALS AND METHODS
Mice
The Ntv-a transgenic mouse line has been described previously.39 The breeding strategy used results in mice composed of a combination of C57BL/6, BALB/C, FVB/N, and CD1 strains. Expression of the TVA receptor (the receptor for ALV-A) is driven by the Nestin gene promoter on multipotent neural stem cells.
Vector Constructs
RCAS-SHH contains full-length chicken SHH with an in-frame carboxy hemagglutinin epitope (HA tag). RCAS-Y (provided by Dr Yi Li, Baylor College of Medicine, Houston, Texas) was converted into a Gateway destination vector using the Gateway Vector Conversion System (Invitrogen, Carlsbad, California) following the manufacturer's protocol to create the RCAS-Y destination vector. RCAS-WIP1, encoding full-length human WIP1, was constructed by a Gateway LR Clonase II recombination reaction between the ultimate ORF clone IOH 25777 and RCAS-Y destination vector. RCAS-GFP, containing full-length GFP (green fluorescent protein), was used as a control (provided by Dr Yi Li).
Tissue Culture
The DF-1 cell line, an immortalized chicken fibroblast line, was obtained from the ATCC and grown at 37°C, 5% CO2 in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum. To produce live virus, DF-1 cells were transfected using Fugene6 (Roche, Nutley, New Jersey) with plasmid versions of the RCAS vectors and allowed to replicate as viral vectors in culture. The A375-TVA cell line, a human melanoma cell line stably expressing the TVA receptor, was obtained from Dr Cindy Miranti (Van Andel Research Institute, Grand Rapids, Michigan). A375 is a human melanoma cell line that has been engineered to express the TVA receptor by transfection with a pcDNA3.1/hygro construct (Invitrogen) containing the TVA gene and selection in medium containing 300-μg/m: Hygromycin B (Invitrogen). The cell line is maintained in Dulbecco modified Eagle medium: F12 supplemented with 5% fetal bovine serum, 2 mM l-glutamine, 0.1 mM NEAA, 1 mM sodium pyruvate, 30 U/mL Pen-Strep (Invitrogen), and 300 μg/mL Hygromycin B to maintain the TVA receptor expression, and grown at 37°C, 5% CO2.
Immunofluorescence
DF-1 cells transfected with RCAS-WIP1 were grown to 80% to 90% confluency on cover slips. Cells were washed with phosphate-buffered saline and fixed with 4% paraformaldehyde in phosphate-buffered saline followed by cold methanol. Immunocytochemistry was performed using standard procedures. A rabbit polyclonal antibody against human WIP1 (H-300; 1:200; Santa Cruz Biotechnology, Santa Cruz, California) and goat anti-rabbit Alexa Fluor 594 fluorescent conjugate (1:500; Molecular Probes, Carlsbad, California) were used for detection. Cell nuclei were labeled and the cover slips mounted using Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (Molecular Probes). Staining was observed with a Zeiss Axioskop 40 microscope.
Western Blot Analysis
Cell lysates from DF-1 cells transfected with RCAS-WIP1 were prepared after several rounds of viral replication. Protein samples (10 μg) were resolved on 10% sodium dodecyl sulfate–polyacrylamide electrophoresis gels, transferred to polyvinylidene fluoride membranes, and probed with rabbit anti-human PPM1D antibody BL3066 (Bethyl Laboratories, Inc, Montgomery, Texas). Detection was done using horseradish peroxidase–conjugated secondary antibody (goat anti-rabbit IgG) and ECL Plus detection reagents (GE Healthcare, Piscataway, New Jersey).
WIP1 Functionality Assay
A375-TVA cells were plated in 100-mm tissue culture dishes and grown overnight to 20% to 30% confluency. For exposure to RCAS-WIP1, medium was removed from the cells and replaced with 4.5 mL of filtered RCAS-WIP1 viral supernatant obtained from DF-1 transfected cells and 8 μg/mL Polybrene (Sigma Aldrich, Milwaukee, Wisconsin). A375 cells not exposed to viral supernatant had their media replaced with Dulbecco modified Eagle medium/fetal bovine serum and 8 μg/mL of Polybrene. After 3.5 hours, 10 mL of complete growth media containing Hygromycin B was added to each plate and the cells grown for 48 hours. At this time, the media containing the RCAS-WIP1 virus was removed and the cells exposed to 30 μM of cisplatin in 10 mL of complete growth media for 24 to 48 hours. Cell lysates were then harvested and protein samples (20 μg) resolved on 10% Tris-HCl SDS-PAGE gels (Bio-Rad, Hercules, California) followed by detection of WIP1 as previously described. The experiment was performed in triplicate to verify reproducibility.
In Vivo Infection of Transgenic Mice
DF-1 virus producer cells were harvested by trypsin digestion, collected by centrifugation, and resuspended in phosphate-buffered saline. One microliter (105 cells) was injected by a gas-tight Hamilton syringe bilaterally into the cerebellum within 48 hours of birth because the population of Nestin-positive cells is highest during the immediate postnatal period. The mice were killed 12 weeks after injection or earlier if signs of hydrocephaly and/or debilitation were present. The brains were removed and fixed in 10% phosphate-buffered formalin (Fisher Scientific, Pittsburgh, Pennsylvania) for at least 24 hours. After fixation, each brain was sectioned and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin.
RESULTS
To test whether ectopic overexpression of WIP1 could suppress p53 expression in cells susceptible to RCAS vector infection, we created an RCAS vector to express human WIP1. Expression of WIP1 by RCAS in DF-1 producer cells was confirmed by immunofluorescence and Western blot analysis (Figure 1A, B). We wanted to know whether the WIP1 protein expressed by RCAS retained its ability to inhibit p53 expression. To test this, we analyzed the functional effect of WIP1 in vitro by assaying p53 expression in A375-TVA cells after exposure to cisplatin. A375-TVA is a human melanoma cell line that has been engineered to express the TVA receptor and has an excellent efficiency of infection by RCAS vectors with more than 90% of cells demonstrating positive infection by RCAS-GFP.40
FIGURE 1.
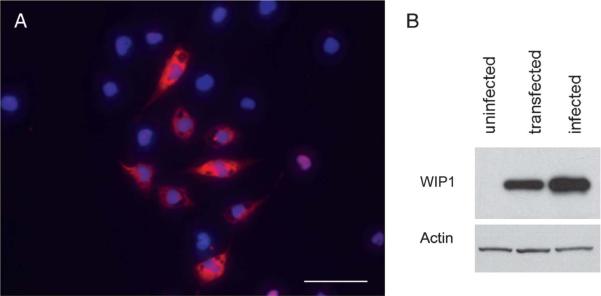
Verification of wild-type p53-induced phosphatase 1 (WIP1) expression by RCAS vector in DF-1 cells. A, immunofluorescence photomicrograph (×400) of DF-1 cells after infection with RCAS-WIP1. WIP1 (scale bar = 50 μm). B, Western blot demonstrating expression of WIP1 in DF-1 cells after infection with RCAS-WIP1. Uninfected cells demonstrate no expression of WIP1. Transfected cells demonstrate WIP1 expression. Infected cells demonstrate increased WIP1 expression.
We exposed A375-TVA cells to supernatant from DF-1 cells infected with RCAS-WIP1 or RCAS-GFP. After 48 hours of exposure to the supernatant, the A375-TVA cells were plated with cisplatin to induce expression of p53 (and subsequently WIP1). We harvested cell lysates at 24 and 48 hours after exposure to cisplatin and assayed them for WIP1 and p53 expression (Figure 2). Control A375-TVA cells (either uninfected or infected with RCAS-GFP) demonstrated increased p53 expression after exposure to cisplatin compared to unexposed cells. WIP1 was also induced in A375-TVA cells (both uninfected and infected with RCAS-GFP) after exposure to cisplatin at 24 and 48 hours. This was more than the level of expression observed in control A375-TVA cells (either uninfected or infected with RCAS-GFP). Cells infected with RCAS-WIP1 demonstrated significantly increased WIP1 expression, which was enhanced further after exposure to cisplatin at both 24 and 48 hours. A375-TVA cells infected with RCAS-WIP1 demonstrated suppressed p53 expression at 24 and 48 hours. The level of p53 expression in cells infected with RCAS-WIP1 and exposed to cisplatin was comparatively less than uninfected cells exposed to cisplatin. These results (confirmed in triplicate) verified that the RCAS-WIP1 construct was capable of infecting TVA+ cells and demonstrated an inhibitory effect of WIP1 on p53 expression in vitro.
FIGURE 2.
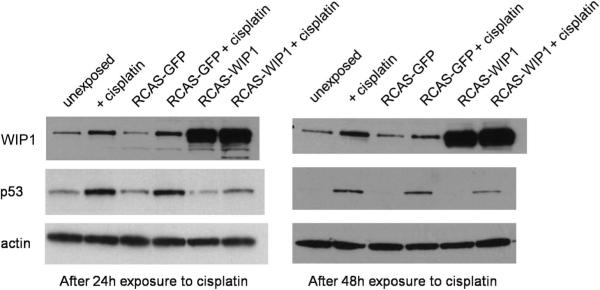
RCAS wild-type p53-induced phosphatase 1 (WIP1) suppresses p53 expression after exposure to cisplatin in A375-TVA cells. Western blot of WIP1, p53 and actin (control) in A375-TVA cells. Cells unexposed to cisplatin demonstrate minimal WIP1 and p53 expression at 24 and 48 hours. Expression of WIP1 and p53 increases after exposure to cisplatin at 24 and 48 hours. A375-TVA cells infected with RCAS-green fluorescent protein (GFP) demonstrate a similar increase in both WIP1 and p53 expression after exposure to cisplatin at both 24 and 48 hours. A375-TVA cells infected with RCAS-WIP1 demonstrate decreased p53 expression at both 24 and 48 hours after exposure to cisplatin, verifying the suppressive effect of WIP1 on p53.
To determine the effect of WIP1 overexpression on tumor formation in a SHH-dependent model of MB, we co-injected RCAS-WIP1 and RCAS-SHH into Ntv-a mice. Independent expression of SHH delivered by RCAS is sufficient to induce MB with histological features consistent with classic MB. We injected equivalent amounts of RCAS-WIP1 and RCAS-SHH virus–producing cells in the cerebellar hemispheres of newborn Ntv-a mice. The mice were followed for 90 days or killed sooner if they displayed signs of neurological dysfunction, and their brains were analyzed for tumor formation. We identified tumors consistent with MBs in the cerebella of 12 of 35 mice (34%) (Figure 3A, B) injected with both RCAS-SHH and RCAS-WIP1. This was significantly higher than the tumor formation rate in a contemporaneous cohort of mice injected with RCAS-SHH alone in which tumors formed in 3 of 40 mice (8%) (χ2 test, P < .01) (Table). The tumors generated by the combination of RCAS-WIP1 + and RCAS-SHH were histologically indistinguishable from tumors formed by those injected with RCAS-SHH alone and displayed characteristic features of MBs including monomorphous sheets of small, undifferentiated cells (Figure 3C). We characterized these tumors further by staining them for the neuronal tumor marker synaptophysin, which was present throughout the tumor (Figure 3D). To verify WIP1 expression in tumors generated by the combination of RCAS-WIP1 and RCAS-SHH, we stained tumors with an antibody for WIP1 and identified positive staining in the nucleus as well as throughout the cytoplasm of tumor cells (Figure 3E). We did not detect any tumors in the 30 mice injected with RCAS-WIP1 alone. The median symptom-free survival was 90 days (range, 27–90 days) in the cohort of mice injected with RCAS-SHH alone, and 90 days (range, 16–90 days) in the cohort of mice injected with RCAS-SHH and RCAS-WIP1. The difference was not statistically significant (log rank test, P = .55).
FIGURE 3.

RCAS wild-type p53-induced phosphatase 1 (WIP1) cooperates with RCAS sonic hedgehog (SHH) to induce MB in the cerebellum of Ntv-a mice. A, gross specimen of brain of Ntv-a mouse 3 months after co-injection of RCAS-WIP1 and RCAS-SHH demonstrating tumor (Tu) in the cerebellar hemisphere. B, whole-mount hematoxylin and eosin of Ntv-a mouse cerebellum (Cb) after injection of RCAS-WIP1 and RCAS-SHH demonstrating tumor formation with mass effect on the fourth ventricle (4V) and brainstem (Pons). C, photomicrograph (×400) of tumor induced by RCAS-WIP1 and RCAS-SHH demonstrating histological features consistent with MB including sheets of monomorphous small cells (Scale bar = 100 μm). D, photomicrograph (×400) of tumor induced by RCAS-WIP1 and RCAS-SHH demonstrating positive staining for synaptophysin, a marker of neuronal tumors (scale bar = 100 μm). E, photomicrograph (×400) of tumor induced by RCAS-WIP1 and RCAS-SHH. Arrow indicates positive staining for WIP1 in the nucleus of a tumor cell. (Scale bar = 100 μm).
TABLE.
Incidence of Tumor Formation in Ntv-a Mice Injected With RCAS-SHH, RCAS-SHH + RCAS-WIP1, and RCAS-WIP1a
| Vector | Tumor Incidence |
|---|---|
| RCAS-SHH | 3/40 (8%) |
| RCAS-SHH + RCAS-WIP1 | 12/35 (34%) |
| RCAS-WIP1 | 0/30 (0%) |
The difference in tumor formation between mice injected with RCAS-SHH and RCAS-SHH + RCAS-WIP1 is significant (χ2 test, P = .0038). SHH, sonic hedgehog; WIP1, wild-type p53-induced phosphatase 1.
DISCUSSION
MB is classified as a grade IV embryonal brain tumor by the World Health Organization based on histological features.41 Molecular characteristics can be used to further categorize MBs into 4 or 5 different variants.3–5 Of these variants, a significant number are driven by SHH/PTC pathway activation, but other molecular mechanisms also contribute to tumor formation and growth. Transcription factors such as N-myc and cMyc, growth factors such as insulin-like growth factor 2 and hepatocyte growth factor, and antiapoptotic genes such as Bcl2 have been shown to enhance SHH-dependent MB formation.8,42–44 Recent evidence showed that the type 2C protein phosphatase WIP1 is overex-pressed in many human cancers, including MB. The location of WIP1 in chromosome 17 also raises the possibility of its involvement in tumor formation and progression as cytogenetic aberrations of this chromosome are very common in MB. However, alterations of chromosome 17 are typically observed in subgroups of MB that are not dependent on activation of the SHH signaling pathway, and other genes on chromosome 17q, notably LASP1, have also been associated with poor prognosis in MB.15,32,37,38,45,46 WIP1 modulates multiple signaling pathways, but exerts its major effect on p53 by disabling p53-dependent apoptosis.47 Mice that are WIP1 deficient are resistant to mammary gland tumors, and WIP1 inhibitors that block WIP1 phosphatase activity decrease tumor cell proliferation in xenograft mammary tumors and Erbb2-driven mammary gland tumorigenesis models.48,49 By expressing WIP1 in a murine model that faithfully recapitulates the genotypic and phenotypic features of a SHH-dependent subtype of MB, we were able to study the in vivo effects of WIP1 overexpression on tumor formation. Here, we show that WIP1 cooperates with SHH to enhance MB formation, although it appears insufficient to induce MB independently.
WIP1 directly inhibits p53 through its dephosphorylation and subsequent degradation.50 WIP1 is induced after ionizing or ultraviolet radiation and its expression correlates with a commensurate increase in expression of p53, leading to the suggestion that it is responsible for returning the cell to a homeostatic state after repair of cellular damage.30,51 In this context, WIP1 appears to suppress the response by p53 to genotoxic stress. Our results are consistent with the suppressive effect of WIP1 on p53 inasmuch as cells expressing RCAS-WIP1 demonstrated a significant decrease in expression of p53 after exposure to cisplatin compared with controls. The potential enhancing effect of WIP1 over-expression on the formation of SHH-dependent MBs is made more relevant by the observation that SHH/PTC pathway activation promotes genomic instability.52
The characterization of WIP1 as an oncogene is based on in vitro experiments in which overexpression of WIP1 coupled with RAS overexpression results in transformation of primary mouse embryo fibroblasts (MEFs).28 Further, coexpression of WIP1 with other oncogenes, including H-rasV12, MYC, and or NEU1, results in transformation of MEFs (as analyzed by anchorage-independent growth and ability to form foci in soft agar).26 Multiple studies have shown that WIP1 deficiency decreases tumorgenicity. Chemical inhibition of WIP1 with a compound directed at its phosphatase activity attenuates tumor cell growth in vitro primarily by suppressing cellular proliferation.48 In vivo studies of this compound showed decreased cellular proliferation in mammary gland tumors compared with control tumors. WIP1-deficient mice are resistant to spontaneous tumor formation throughout their life span.53 MEFs derived from WIP1 null mice demonstrate a substantially decreased proliferative capacity in culture.49 The same report showed that WIP1-null MEFs transformed by the oncogenes E1A and Hras1 form tumors after subcutaneous injection in nude mice at a significantly lower rate compared with wild-type MEFs transformed with these same oncogenes. Further, WIP1−/− mice crossed with mice prone to mammary gland tumors were profoundly resistant to tumor formation, and those few tumors that did form appeared to have a lower proliferation potential. In contrast to previous studies demonstrating tumor resistance conferred by WIP1 deficiency, we determined the ability of WIP1 overexpression to induce MB in vivo by expressing it with SHH and independently. The ectopic expression of SHH in Ntv-a mice using the RCAS/Ntv-a system is sufficient to induce MB, albeit at a low rate. In this study, we observed MB in just 3 of 40 mice (8%) injected with RCAS-SHH. The enhancing effect of WIP1 was demonstrated in a cohort of mice co-injected with RCAS-SHH and RCAS-WIP1. In this group, tumors formed in 12 of 35 mice (34%). This enhancing effect is similar to our results combining other signaling molecules such as insulin-like growth factor 2 or Akt, which, when co-injected with RCAS-SHH, increased the tumor formation rate.12
Although these results suggest that WIP1 cooperates with SHH to enhance MB formation in our model system, we examined its expression in the context of the known MB subtypes (ie, WNT, SHH, group C, and group D) by determining the levels of WIP1 in a defined MB dataset45 used for genomic profiling. This analysis shows that although WIP1 expression does occur in the SHH subtype of MB (approximately 15% of cases), its expression is significantly higher (compared with normal fetal cerebellum) in group C and group D MBs (of note, the expression of WIP1 in the WNT subtype of MBs was similar to that of normal fetal cerebellum) (Figure 4). Group C and D MBs have a less favorable outcome compared with the other groups, and a possible explanation may be that the genomic instability engendered by WIP1 expression leads to a more aggressive tumor. Future studies that model the genetic aberrations associated with group C and D MBs may validate WIP1's role in promoting tumorigenesis.
FIGURE 4.
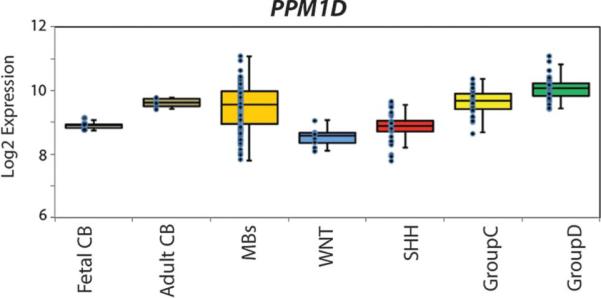
Expression profiling of wild-type p53-induced phosphatase 1 (WIP1) reveals a correlation with group C and D medulloblastomas (MBs). WIP1 expression is approximately 1.5 times higher in all MBs compared with normal fetal cerebellum (CB). Increased WIP1 expression is observed in approximately 15% of sonic hedgehog (SHH) MBs, although not significantly higher than fetal CB (P > .05). WIP1 expression is significantly higher in group C and D MBs (P < .001 for both).
The inability of WIP1 to form tumors after its independent expression in Ntv-a mice is consistent with the results of previous studies investigating the oncogenic nature of WIP1. Although WIP1 cooperates with RAS to transform primary mouse fibroblasts, independent overexpression of WIP1 does not transform NIH-3T3 cells.28 Additionally, infection of either wild-type or p53−/− MEFs by WIP1-expressing virus alone does not cause colony formation or growth in soft agar.26 Although WIP1 suppresses p53 expression, transgenic murine models of MB demonstrate enhancement of tumor formation after being bred into a p53−/− background, but these models are not dependent on p53 deficiency for tumor initiation. We showed previously that in the RCAS/Ntv-a-based, SHH-dependent model of MB, other genes (including Bcl-2, Igf2, c-Myc, and Nmyc) are capable of enhancing tumor formation. However, tumor formation was not observed after ectopic expression of these genes independently. Thus, activation of the SHH signaling pathway likely represents a tumor-initiating event, and we were not surprised that WIP1 was unable to induce MBs in this model.
Here, we have provided evidence that the WIP1 gene contributes to MB growth. Its enhancement of tumor formation in a SHH-dependent model of MB corroborates previous observations that include its increased expression in human MB tumor samples. Although our results show that overexpression of WIP1 independently is insufficient to cause MB (consistent with previous studies), its association with p53 suppression suggests that it may be an important contributor to MB tumorigenesis.
Acknowledgments
This work was supported by an Institutional Research Grant from MD Anderson Cancer Center (G.R.), a Career Development Award Number P50CA127001 (GR), a Mentored Clinical Scientist Development Award Number K08NS070928 (GR) from the National Cancer Institute and Award Number CA108622 (DWF) from the National Institutes of Health.
ABBREVIATIONS
- MB
medulloblastoma
- MEF
mouse embryo fibroblast
Footnotes
Disclosures The authors have no personal financial or institutional interest in any of the drugs, materials, or devices described in this article.
REFERENCES
- 1.Gilbertson RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5(4):209–218. doi: 10.1016/S1470-2045(04)01424-X. [DOI] [PubMed] [Google Scholar]
- 2.Paulino AC. Current multimodality management of medulloblastoma. Curr Probl Cancer. 2002;26(6):317–356. doi: 10.1067/mcn.2002.130267. [DOI] [PubMed] [Google Scholar]
- 3.Northcott PA, Fernandez LA, Hagan JP, et al. The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res. 2009;69(8):3249–3255. doi: 10.1158/0008-5472.CAN-08-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 6.Oliver TG, Read TA, Kessler JD, et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132(10):2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 7.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 8.Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003;5(3):198–204. doi: 10.1016/S1476-5586(03)80052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiner HL, Bakst R, Hurlbert MS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 2002;62(22):6385–6389. [PubMed] [Google Scholar]
- 10.Fogarty MP, Kessler JD, Wechsler-Reya RJ. Morphing into cancer: the role of developmental signaling pathways in brain tumor formation. J Neurobiol. 2005;64(4):458–475. doi: 10.1002/neu.20166. [DOI] [PubMed] [Google Scholar]
- 11.Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415(6870):436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 12.Rao G, Pedone CA, Valle LD, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23(36):6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 13.Biegel JA. Cytogenetics and molecular genetics of childhood brain tumors. Neurooncol. 1999;1(2):139–151. doi: 10.1215/15228517-1-2-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellison D. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol. 2002;28(4):257–282. doi: 10.1046/j.1365-2990.2002.00419.x. [DOI] [PubMed] [Google Scholar]
- 15.Pfister S, Remke M, Benner A, et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol. 2009;27(10):1627–1636. doi: 10.1200/JCO.2008.17.9432. [DOI] [PubMed] [Google Scholar]
- 16.Frank AJ, Hernan R, Hollander A, et al. The TP53-ARF tumor suppressor pathway is frequently disrupted in large/cell anaplastic medulloblastoma. Brain Res Mol Brain Res. 2004;121(1–2):137–140. doi: 10.1016/j.molbrainres.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 17.Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61(2):513–516. [PubMed] [Google Scholar]
- 18.Holcomb VB, Vogel H, Marple T, Kornegay RW, Hasty P. Ku80 and p53 suppress medulloblastoma that arise independent of Rag-1-induced DSBs. Oncogene. 2006;25(54):7159–7165. doi: 10.1038/sj.onc.1209704. [DOI] [PubMed] [Google Scholar]
- 19.Lee Y, McKinnon PJ. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002;62(22):6395–6399. [PubMed] [Google Scholar]
- 20.Benard J, Douc-Rasy S, Ahomadegbe JC. TP53 family members and human cancers. Hum Mutat. 2003;21(3):182–191. doi: 10.1002/humu.10172. [DOI] [PubMed] [Google Scholar]
- 21.Ohgaki H, Eibl RH, Wiestler OD, Yasargil MG, Newcomb EW, Kleihues P. p53 mutations in nonastrocytic human brain tumors. Cancer Res. 1991;51(22):6202–6205. [PubMed] [Google Scholar]
- 22.Adesina AM, Nalbantoglu J, Cavenee WK. p53 gene mutation and mdm2 gene amplification are uncommon in medulloblastoma. Cancer Res. 1994;54(21):5649–5651. [PubMed] [Google Scholar]
- 23.Saylors RL, III, Sidransky D, Friedman HS, et al. Infrequent p53 gene mutations in medulloblastomas. Cancer Res. 1991;51(17):4721–4723. [PubMed] [Google Scholar]
- 24.Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28(8):1345–1350. doi: 10.1200/JCO.2009.23.5952. [DOI] [PubMed] [Google Scholar]
- 25.Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulavin DV, Demidov ON, Saito S, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31(2):210–215. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 27.Hirasawa A, Saito-Ohara F, Inoue J, et al. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin Cancer Res. 2003;9(6):1995–2004. [PubMed] [Google Scholar]
- 28.Li J, Yang Y, Peng Y, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002;31(2):133–134. doi: 10.1038/ng888. [DOI] [PubMed] [Google Scholar]
- 29.Saito-Ohara F, Imoto I, Inoue J, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63(8):1876–1883. [PubMed] [Google Scholar]
- 30.Lu X, Nguyen TA, Zhang X, Donehower LA. The Wip1 phosphatase and Mdm2: cracking the “Wip” on p53 stability. Cell Cycle. 2008;7(2):164–168. doi: 10.4161/cc.7.2.5299. [DOI] [PubMed] [Google Scholar]
- 31.Ehrbrecht A, Muller U, Wolter M, et al. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol. 2006;208(4):554–563. doi: 10.1002/path.1925. [DOI] [PubMed] [Google Scholar]
- 32.Mendrzyk F, Radlwimmer B, Joos S, et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol. 2005;23(34):8853–8862. doi: 10.1200/JCO.2005.02.8589. [DOI] [PubMed] [Google Scholar]
- 33.Castellino RC, De Bortoli M, Lu X, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86(3):245–256. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avet-Loiseau H, Venuat AM, Terrier-Lacombe MJ, Lellouch-Tubiana A, Zerah M, Vassal G. Comparative genomic hybridization detects many recurrent imbalances in central nervous system primitive neuroectodermal tumours in children. Br J Cancer. 1999;79(11–12):1843–1847. doi: 10.1038/sj.bjc.6690293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reardon DA, Michalkiewicz E, Boyett JM, et al. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 1997;57(18):4042–4047. [PubMed] [Google Scholar]
- 36.Russo C, Pellarin M, Tingby O, et al. Comparative genomic hybridization in patients with supratentorial and infratentorial primitive neuroectodermal tumors. Cancer. 1999;86(2):331–339. doi: 10.1002/(sici)1097-0142(19990715)86:2<331::aid-cncr18>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 37.Northcott PA, Nakahara Y, Wu X, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41(4):465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheurlen WG, Schwabe GC, Joos S, Mollenhauer J, Sorensen N, Kuhl J. Molecular analysis of childhood primitive neuroectodermal tumors defines markers associated with poor outcome. J Clin Oncol. 1998;16(7):2478–2485. doi: 10.1200/JCO.1998.16.7.2478. [DOI] [PubMed] [Google Scholar]
- 39.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12(23):3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bromberg-White JL, Webb CP, Patacsil VS, Miranti CK, Williams BO, Holmen SL. Delivery of short hairpin RNA sequences by using a replication-competent avian retroviral vector. J Virol. 2004;78(9):4914–4916. doi: 10.1128/JVI.78.9.4914-4916.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binning MJ, Niazi T, Pedone CA, et al. Hepatocyte growth factor and sonic Hedgehog expression in cerebellar neural progenitor cells costimulate medulloblastoma initiation and growth. Cancer Res. 2008;68(19):7838–7845. doi: 10.1158/0008-5472.CAN-08-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Browd SR, Kenney AM, Gottfried ON, et al. N-myc can substitute for insulin-like growth factor signaling in a mouse model of sonic hedgehog-induced medulloblastoma. Cancer Res. 2006;66(5):2666–2672. doi: 10.1158/0008-5472.CAN-05-2198. [DOI] [PubMed] [Google Scholar]
- 44.McCall TD, Pedone CA, Fults DW. Apoptosis suppression by somatic cell transfer of Bcl-2 promotes Sonic hedgehog-dependent medulloblastoma formation in mice. Cancer Res. 2007;67(11):5179–5185. doi: 10.1158/0008-5472.CAN-06-4177. [DOI] [PubMed] [Google Scholar]
- 45.Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2010;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Traenka C, Remke M, Korshunov A, et al. Role of LIM and SH3 protein 1 (LASP1) in the metastatic dissemination of medulloblastoma. Cancer Res. 2010;70(20):8003–8014. doi: 10.1158/0008-5472.CAN-10-0592. [DOI] [PubMed] [Google Scholar]
- 47.Baxter EW, Milner J. p53 Regulates LIF expression in human medulloblastoma cells. J Neurooncol. 2010;97(3):373–382. doi: 10.1007/s11060-009-0043-x. [DOI] [PubMed] [Google Scholar]
- 48.Belova GI, Demidov ON, Fornace AJ, Jr, Bulavin DV. Chemical inhibition of Wip1 phosphatase contributes to suppression of tumorigenesis. Cancer Biol Ther. 2005;4(10):1154–1158. doi: 10.4161/cbt.4.10.2204. [DOI] [PubMed] [Google Scholar]
- 49.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36(4):343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 50.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19(10):1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiscella M, Zhang H, Fan S, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94(12):6048–6053. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leonard JM, Ye H, Wetmore C, Karnitz LM. Sonic Hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J Cell Biol. 2008;183(3):385–391. doi: 10.1083/jcb.200804042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nannenga B, Lu X, Dumble M, et al. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog. 2006;45(8):594–604. doi: 10.1002/mc.20195. [DOI] [PubMed] [Google Scholar]