

Abstract
Despite substantial progress in screening, early diagnosis, and the development of noninvasive technology, gastrointestinal (GI) cancer remains a major cause of cancer-associated mortality. Chemoprevention is thought to be a realistic approach for reducing the global burden of GI cancer, and efforts have been made to search for chemopreventive agents that suppress acid reflux, GI inflammation and the eradication of Helicobacter pylori. Thus, proton pump inhibitors, statins, monoclonal antibodies targeting tumor necrosis factor-alpha, and nonsteroidal anti-inflammatory agents have been investigated for their potential to prevent GI cancer. Besides the development of these synthetic agents, a wide variety of the natural products present in a plant-based diet, which are commonly called phytoceuticals, have also sparked hope for the chemoprevention of GI cancer. To perform successful searches of chemopreventive agents for GI cancer, it is of the utmost importance to understand the factors contributing to GI carcinogenesis. Emerging evidence has highlighted the role of chronic inflammation in inducing genomic instability and telomere shortening and affecting polyamine metabolism and DNA repair, which may help in the search for new chemopreventive agents for GI cancer.
Keywords: Chemoprevention, Gastrointestinal neoplasms, Phytoceuticals, Molecular target
INTRODUCTION
1. The general principle for chemoprevention of gastrointestinal (GI) cancers
1) General concept of chemoprevention
Since the incidence of new cancer cases as well as the rate of cancer mortality are increasing worldwide,1 the prevention of cancer is ranked as a prime importance to reduce the global burden of cancer. The perception of preventing cancer was first outset by Wattenberg2 in 1960s, after which a wide array of population-based as well as preclinical studies has been conducted to evaluate the cancer prevention potential of diverse classes of natural as well as synthetic compounds. In 1976, Sporn3 first coined the term "chemoprevention" that refers to the use of nontoxic chemical substances of either natural or synthetic origin to delay, retard or reverse the process of carcinogenesis. Over the last few decades, numerous preclinical and clinical studies demonstrated the success of the chemoprevention strategy in curbing the cancer incidence and mortality. Chemoprevention is the strategy to intervene multistage carcinogenesis process, which comprises of apparently three distinct phases: initiation, promotion, and progression. Tumor initiation is a rapid and irreversible process that involves damage of cellular DNA by various known and unknown carcinogens. Many carcinogens, either endogenous or exogenous, are inactive per se and are activated through biotransformation inside the body. Biotransformation is a process of eliminating relatively nonpolar carcinogenic substances by converting them into water soluble entities, hence called detoxification. Extensive metabolic activation of carcinogens and compromised detoxification leads to the accumulation of highly reactive carcinogens, which cause covalent modification of genomic DNA, thereby activating various oncogenes and inactivating tumor suppressor genes, which leads to the initiation of cell transformation. Therefore, chemopreventive agents that can either inhibit carcinogen activation or promote detoxification are generally termed as anti-initiating agents or blocking agents. Tumor promotion, a reversible process, is the clonal expansion of initiated or transformed cells to grow as a population of preneoplastic cells forming the benign tumor. Abnormal biochemical reactions encompassing inappropriate amplification and/or inactivation of cell signaling pathways underlie tumor promotion stage that often spans over 10 years. Thus, the normalization of aberrant cell signaling pathways by chemopreventive agents can reverse or halt the journey of premalignant cells to become malignant. Tumor progression involves malignant conversion of preneoplastic cells with characteristic features of increased angiogenesis, invasion and metastasis.4,5 In summary, cancer can be prevented by intervening any of these three stages of carcinogenesis and chemopreventive agents that can interfere with tumor promotion or progression are known as suppressing agents. The aforementioned stage-specific prevention of cancer is a simplistic view of chemoprevention strategy. Conclusively, accumulating evidence of the success of chemopreventive agents in reducing the risk of various cancers suggests that chemoprevention is the first line of defense against carcinogenesis.6
2) The basis for chemoprevention of GI cancers
GI cancers include cancers of the esophagus, stomach, intestine, colon, rectum, pancreas, and liver. Among these, the esophageal squamous cell carcinoma accounts for approximately one-sixth of all cancer-related mortality worldwide.7 Esophageal adenocarcinoma (EAC) has received considerable attention because of the dramatically increased incidence in the past 2 decades and the poor prognoses with a 5-year survival rate of 10% to 20%.8,9 In spite of relative decline in the incidence and mortality, gastric cancer is still the fourth most common cancer worldwide and ranks the third most common cause of cancer-related deaths.10 Likewise, hepatocellular carcinoma (HCC) remains as the fifth common cancers and a major cause of cancer-related deaths.11 Pancreatic cancer, the most lethal form of GI cancer, is the fourth leading cause of cancer mortality with an overall median 5-year survival rate of only 5%.12 Colorectal cancers are also increasing worldwide with an estimate of 1,200,000 new cases in the year 2011 and half of them are going to die from the disease.13,14 Based on the looming scenario of cancer mortality, chemoprevention appears to be the forefront in fighting against GI cancers. Majority of the GI cancers have etiologic link with dietary and life style factors, of which point is critical basis for prevention. For instance, the association between gastric cancer and high salt diet intake or Helicobacter pylori infection, the positive corelation between HCC and aflatoxin-B1 contaminated food consumption or chronic hepatitis virus infection or excessive alcohol consumption, and the causal relationship between colon cancer and the intake of burnt meat are notable examples. Thus, GI cancers can be prevented by changing dietary habit and life style. Whereas the maintenance of proper hygiene, abstinence from alcohol and smoking, increasing physical activity and avoidance of high salt and brunt food intake are commonly advocated, the pharmacological intervention for chemoprevention of GI cancers have been sought for over last several decades. Since oxidative stress and chronic inflammation, in general, play a key role in carcinogenesis, antioxidants, and anti-inflammatory agents have been shown to prevent various GI cancers. Nonsteroidal anti-inflammatory drugs (NSAIDs) have been extensively investigated for the chemoprevention of colorectal cancers.15,16
MOLECULAR BASIS IMPLICATED IN THE PREVENTION OF GI CANCERS
1. Inflammation and oxidative stress
Despite having unique etiology, all forms of GI cancers share the common mechanisms of oxidative stress-induced damage of genomic DNA, modification of cellular proteins and lipids, altered cell signaling and persistent local tissue inflammation. Whereas oxidative stress incites local tissue inflammation, persistent inflammation leads to the generation of reactive oxygen species (ROS). Excessive ROS as well as reactive nitrogen species (RNS) perturbs cellular homeostasis by inducing genetic and epigenetic changes and amplifying and/or inactivating cell signaling network, thereby inducing premalignant transformation of cells. ROS and RNS generate other reactive species, such as malondialdehyde and 4-hydroxynonenal (4-HNE), which can cause DNA damage by forming DNA adducts,17 thereby initiating the tumor formation. For example, 4-HNE forms 1,N6-ethenodeoxyadenosine (εdA) and 3,N4-ethenodeoxycytidine (εdC) DNA adducts in inflamed human pancreas and colon, respectively.18 DNA damage caused by oxidative stress is a major contributor to colorectal cancer development in ulcerative colitis patients.19 Peroxynitrite, a powerful oxidant, causes DNA damage through the formation of 8-nitroguanine (8-NG).20,21 In addition, elevated expression of nitrative and oxidative DNA lesion products, 8-NG and 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxodG), and iNOS was detected in inflammation sites of HCC, gastric cancer and cholangiocarcinomas.22 The incidence of EAC in patients with Barrett's esophagus with dysplasia is increased by 30- to 125-fold.23 Likewise, inflammatory bowel diseases (IBD) (ulcerative colitis and Crohn's disease) are associated with about 10-fold increase in the risk of colorectal cancer24 and the use of anti-inflammatory therapy reduces this risk.15 The fact that inflammation precedes tumor development has recently been reported in a mouse model of pancreatic cancer.25 According to this study, authors developed a method to tag pancreatic cancer cells in order to track the movement of these cells. It was found that tagged pancreatic cells acquired mesenchymal phenotype and appeared in circulation much before the development of pancreatic tumor, and these cells were seeded into liver. This phenomenon was aggravated in the presence of pancreatitis and was most abundant at the inflammatory foci.25 Mouse pancreatic ductal adenocarcinomas arising from pancreatic acinar cells, which are resistant to transformation by oncogene activation or tumor suppressor gene inactivation, have been reported to form pancreatic intraepithelial neoplasia when exposed to limited bouts of nonacute pancreatitis and harbor K-ras oncogene.26
2. Genomic instability
Inflammation in the GI epithelial cells induces genomic instability as a mechanism of turning into tumor development. Barrett's esophagus and EACs showed 64% and 43% changes in chromosome 11 and 12, respectively. Nondysplastic multilayered epithelium and Barrett's esophagus retaining normal levels p53 and adenomatous polyposis coli showed aneusomy of these chromosomes, suggesting that inflammation caused genomic instability prior to the inactivation of tumor suppressor genes.27 The loss of heterozygosity at chromosome three and microsatellite instability at chromosome 12 were observed in 50% and 62%, respectively, in chronically inflamed, nondysplastic epithelium of ulcerative colitis patients, corroborating the fact that inflammation induces microsatellite instability as an initial event for dysplasia and consequent development of colonic adenocarcinoma.28 Likewise, chronic pancreatitis was found to exhibit increased incidence of microsatellite instability29 and chromosomal instability.30 In the early stage of hepatocarcinogenesis, infection with hepatitis C leads to hepatic inflammation that is characterized by aneuploidy in hepatocyte DNA.31
3. Telomere shortening
Several studies have reported that certain forms of GI cancers are associated with telomere shortening. For instance, the relative telomeric repeat content was substantially reduced in inflamed liver tissues as well as in HCC.32 The risk of gastric cancer was increased by 2-fold in H. pylori-infected patients, who had shortened telomere.33 Likewise, telomere shortening was noted in colon biopsies from ulcerative colitis patients with low grade dysplasia.34
4. Impaired DNA repair system
Mice deficient in alkyladenine DNA glycosylase (AAG), a base excision repair enzyme, exhibited more intense gastric lesions as upon infection with H. pylori.35 The AAG-mediated DNA repair protected mice against dextran sodium sulphate-induced colon carcinogenesis.35 In a murine colitis model, homozygous deletion of mismatch repair gene MSH2 caused 60% incidence of high-grade dysplasia or adenocarcinoma as compared to 29% of the animals harboring wild type MSH2. Moreover, tumors in MSH2-/- mice exhibited high index of microsatellite instability than that in wild type mice, suggesting that deficient mismatch repair plays a key role in inflammation-associated colon carcinogenesis.36 A similar phenomenon was observed in mice deficient of another mismatch repair gene hMlh1. The incidence of colon tumors in male and female Mlh1-null mice challenged with DSS was 63% and 44%, respectively. However, Mlh1 heterozygous and wild type mice were free from tumors. Thus, a strong association exists between Mlh1 deficiency and colon carcinogenesis.37 Edwards et al.38 reported that colon tumors in mice lacking the heterotrimeric G protein alpha subunit Giα2 showed increased microsatellite instability arising from epigenetic silencing of Mlh1. Homozygous deletion of 8-oxoguanine glycosylase (Ogg1), a base excision repair enzyme, increased the susceptibility to DSS-induced colitis and the formation of colorectal adenocarcinomas as compared to Ogg1 wild type animals.39 The Ogg1 activity was inhibited by NO in human cholangiocarcinoma cells.40 In contrast to the protective role of DNA repair enzymes in GI carcinogenesis, Hofseth et al.41 demonstrated that AAG and apurinic/apyrimidinic endonuclease (APE1) are elevated in colonic epithelium of ulcerative colitis patients as compared to normal epithelium. The elevated levels of AAG and APE1 were associated with increased microsatellite instability in inflamed colon tissue. One possible mechanism may be the role of APE1 in enhancing inflammatory signaling by functioning as a redox chaperone to cause thiol reduction of proinflammatory transcription factors nuclear factor-kappaB and AP-1, thereby increasing DNA binding of these transcription factors.42
5. Polyamine metabolism
The imbalance in cellular polyamine pool is also associated with GI carcinogenesis. Significant reduction in colonic adenomas in patients receiving a combination of diferuloylmethane, polyamine biosynthesis inhibitor, and sulindac suggest that increased polyamine synthesis contributes to colon carcinogenesis.43 Gobert et al.44 reported that H. pylori infection, the major cause of gastric cancer, induces the expression and activity of enzymes utilized for polyamine biosynthesis. Moreover, increased cellular polyamine pool has been noted in tumor cells.45 In contrast, spermine has been reported to inhibit lipopolysaccharide-induced expression of iNOS, formation of nitrotyrosine, and the release of inflammatory mediators in mice.46 Moreover, increased catabolic depletion of polyamine pool led to acute pancreatitis,47 which was prevented by treatment with a polyamine analogue.48 Whereas the induction of polyamine catabolic enzymes, such as spermidine/spermine-N1-acetyltransferase, N1-acetylpolyamine oxidase, and spermine oxidase may prevent carcinogenesis by depleting cellular polyamine pool, H2O2 generated as a by-product of polyamine catabolism may cause DNA damage leading to cell transformation.49,50 Thus, more rigorous investigation is warranted to delineate the role of polyamine metabolism in GI carcinogenesis.
6. Activation-induced cytidine deaminase (AID)
Recently the role of AID, an enzyme that naturally functions as a DNA and RNA sequence editor, in GI carcinogenesis has been reported. The expression of AID is elevated in chronic inflammatory conditions, such as ulcerative colitis51 and H. pylori-induced gastritis.52 It has been reported that increased levels of AID causes mutation of many tumor-related genes53 in hepatocytes of hepatitis C virus infected patients54 and in gastric epithelial cells in patients with H. pylori infection.52 The expression of AID is increased in response to several cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-4, and IL-13 in colonic epithelial cells.51 Shimizu et al.55 and Marusawa et al.56 strongly concluded that AID might play an integral role in inflammation-associated GI carcinogenesis and is therefore a potential target molecule for the prevention and treatment of cancers since the activity of AID as a genome mutator provides a new avenue for studies aimed at understanding mutagenesis mechanisms during carcinogenesis.
REALITY OF CANCER PREVENTION ACCORDING TO AGENTS
1. Prevention of GI cancers with proton pump inhibitors (PPIs) beyond acid suppression
Another class of agents studied for the chemoprevention of GI cancers is PPI, which act beyond authentic acid suppression. Epidemiological studies suggest that PPIs can reduce the risk of neoplastic progression in patients with Barrett's esophagus.57,58 Miyashita et al.59 reported that subcutaneous administration of rabeprazole protected against the development esophageal cancers in a rat surgical reflux model. Contrary to these observations, a case control study from the UK demonstrated an increased risk of EACs in patients receiving PPI therapy for GI reflux disease symptoms or Barrett's esophagus.60 Although the controversy hailed over the tumorigenic potential of PPIs, Obszynska et al.61 have conducted a large clinical trial and reported that long term use of PPIs has no clinical evidence of extending esophagus length and is clinically safe. The major concern with PPIs is the hypergastrinemia that might play role in inducing colorectal tumorigenesis after the use of PPIs. However, our recent study demonstrated that intraperitoneal administration of omeprazole reduced the incidence and the multiplicity of colitis-associated colon tumors in C57BL/6 mice.62 Our study revealed that omeprazole can block the trophic effect of gastrin on colonic epithelial cells.63 Moreover, the ability of PPIs in inducing apoptosis in gastric cancer cells64 or the enhanced eradication of H. pylori with a combination of lansoprazole, amoxicillin, and rebamipide65 suggest the potential of PPIs in the chemoprevention of GI cancers.
2. Prevention of GI cancers with NSAIDs
A notable example is the emergence of aspirin or NSAIDs in the prevention and therapy of colorectal cancer.66 According to a recent randomized placebo-controlled chemoprevention trial, regular administration of aspirin reduced the polyp formation and disease progression in young patients with high risk of familial adenomatous polyposis (FAP).67 Regular intake of a low dose aspirin has also been shown to reduce the risk of sporadic colorectal cancers in patients with IBDs.68 A randomized controlled trial comprising 17,285 participants reported that daily aspirin can prevent distant metastasis of certain cancers.69 In a prospective human study, Vaughan et al.70 demonstrated that NSAIDs can reduce the risk of neoplastic progression in Barrett's esophagus, a metaplastic disease that serves as the precursor for EACs. The clinic- or hospital-based studies demonstrated that aspirin, but not nonaspirin NSAIDs, reduced the risk of developing pancreatic cancer.71-73 The history of developing aspirin or NSAIDs is rather long (Fig. 1A), starting from drastic trepanation to the discovery of the efficacy of willow tree bark on the relief of pain as well as inflammation by Hipocrates. These experiences were completed by Dr. John Vane, who got Nobel Prize through the identification of prostaglandin and cyclooxygenase (COX) as core mechanisms of aspirin and NSAIDs. These great achievements were proven to be chemopreventive way of NSAIDs in diverse GI cancers including colon cancer and gastric cancer (Fig. 1B). Diverse mechanisms of NSAIDs on GI cancer prevention had been identified (Fig. 1C), including COX dependent mechanisms and COX-independent mechanisms (Fig. 1).
Fig. 1.

Chemopreventive actions of nonsteroidal anti-inflammatory drugs (NSAIDs). (A) The history of the development of NSAIDs as chemopreventive agents has shown that they can inhibit tumor development in some cancers, although various side effects, such as gastrointestinal (GI) bleeding and renal disorders, have been shown to occur. Selective cyclooxygenase (COX)-2 inhibitors (coxibs) were originally developed as anti-inflammatory drugs to avoid the side effects of NSAIDs. (B, C) Similar to NSAIDs, the coxibs also proved to have an inhibitory effect on tumorigenesis in many experimental studies using cell lines and animal models. Because a randomized study for polyp chemoprevention with celecoxib in familial adenomatous polyposis (FAP) patients demonstrated a significant reduction in the number of colorectal polyps, the clinical use of celecoxib was approved for FAP patients. (C) The role of COX-2 in carcinogenesis of the GI tract and the chemopreventative mechanisms of NSAIDs, including coxibs, are shown.
PPI, proton pump inhibitor; NF-κB, nuclear factor kappa B; STAT3, signal transducer and activator of transcription-3; APC, adenomatous polyposis coli; PGE, prostaglandin E2; EP, prostaglandin E receptor; VEGF, vascular endothelial growth factor.
3. Prevention of GI cancers with natural products
Although several monoclonal antibodies targeting specific gene products have been developed as chemotherapeutic agents, only few studies have demonstrated their chemoprevention potential. We have recently reported that infliximab, a monoclonal antibody against TNF-α, can prevent the development of colitis-associated colorectal cancers in rodents.63 Besides these synthetic products, a large body of evidence suggests that regular consumption of fruits and vegetables can prevent different forms of GI cancers (Table 1).74-132 Because it is estimated that two thirds to three quarters of gastric cancer worldwide are associated with H. pylori infection,133 it seems that the eradication of H. pylori and the control of gastric inflammation induced by the pathogen are essential in the prevention of gastric cancer. Previous our studies have shown that Korea red ginseng could rescue H. pylori-induced cytotoxicity,76 suppress H. pylori-induced 5-lipoxygenase pathway,77 and attenuate gastric inflammation through augmented eradication rates of H. pylori in the clinical study.78 These results suggest that Korean red ginseng administration after the eradication of H. pylori would be helpful to prevent H. pylori-associated inflammation and carcinogenesis. Moreover, Korean red ginseng powder also has shown to the inhibitory effects on aberrant crypt foci formation and the preneoplastic lesions in the rat colon treated with azoxymethane.79 In addition to Korean red ginseng, a wide variety of antioxidant and anti-inflammatory substances present in our regular plant-based diet are effective in preventing GI cancers. Examples include, but not limited to, epigallocatechin gallate (EGCG) from green tea,134,135 anthocyanins from raspberry,136 curcumin from turmeric,137,138 resveratrol from grapes,44,80-83 etc. Tea and tea constituents, especially EGCG from green tea has well known to the cancer preventive activities in various models for GI carcinogenesis.134,135 Chemopreventive activities of EGCG were associated with the inhibition of tumorigenic signaling including β-catenin, c-Myc, pAkt, cyclin D1, etc.84,85 However, a few studies which covered different areas in Japan did not observe a relationship between green tea consumption and stomach cancer.139,140 Several clinical trials with curcumin have been reported in GI cancers,141,142 among which curcumin could decrease lymphocytic glutathione-S-transferase86 and prostaglandin E2 production87 in patiens with colorectal cancer and reduce polyp number and size in patients with FAP.88 Resveratrol has potent anti-inflammatory and anticarcinogenic effect through mainly scavenge of free radicals, by which its unique aromatic structure.143 Several studies demonstrates that resveratrol suppresses proliferation of colon cancer cells82,89 and gastric cancer cells.80 Liang et al.90 suggested the cell cycle arrest at G2/M phase through inhibition of Cdk7 kinase activity as a mechanism of apoptosis in cancer cells treated with resveratrol.
Table 1.
Promising Phytoceuticals for Gastrointestinal Cancer Types
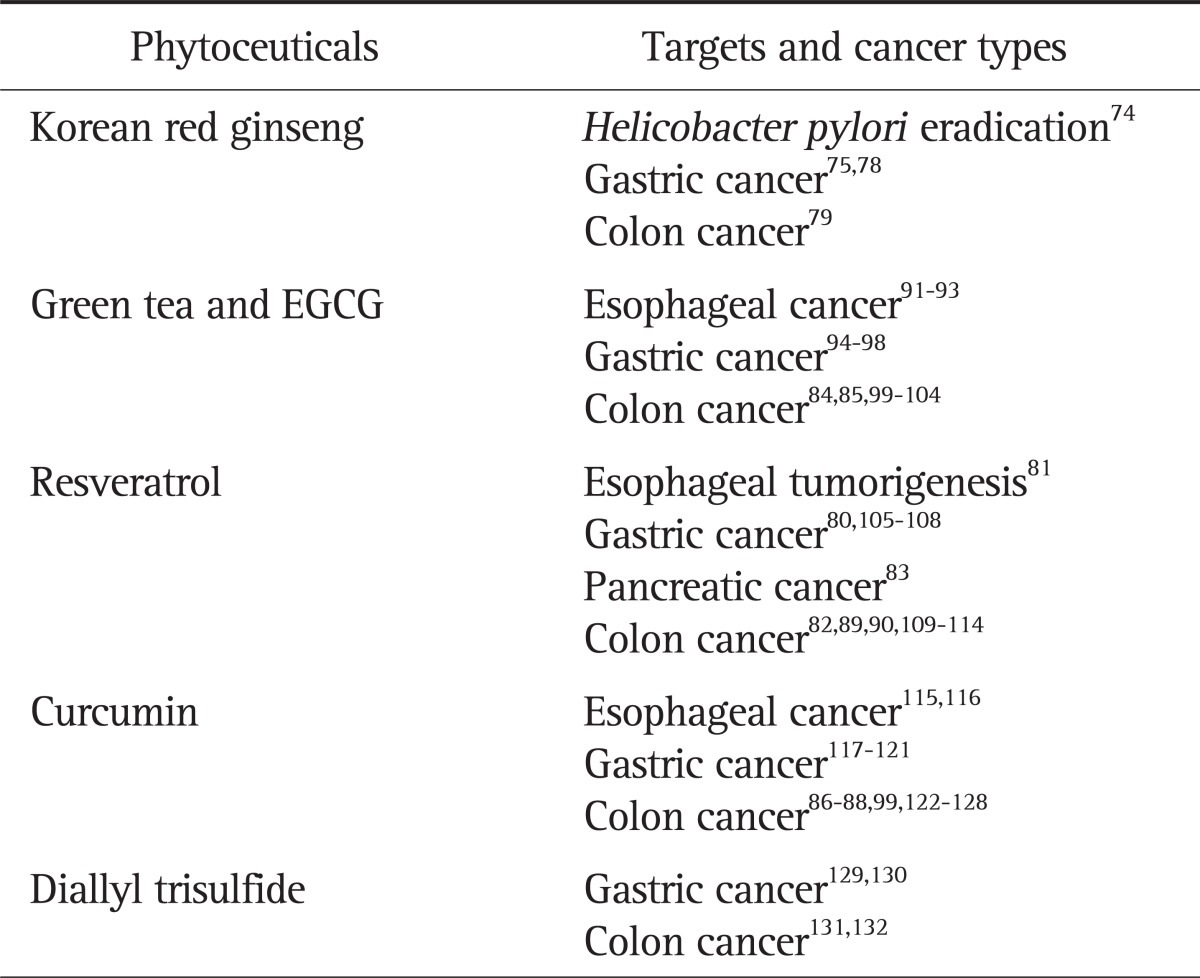
EGCG, epigallocatechin gallate.
4. Prevention of GI cancers with other promising agents
Focus has also been given to statins for the chemoprevention of GI carcinogenesis. A population-based case control study suggests the ability of statins in reducing the risk of gastric cancer.144 In contrast, lovastatin did not show any beneficial effects in patients with advanced gastric cancer.145 Moreover, administration of pravastatin failed to inhibit H. pylori-induced gastric cancer in rodents.146 Likewise, statins were found to have no protective effects against colorectal cancers.147 However, simvastatin was found to inhibit the proliferation and growth of HCC cells in vitro.148,149 Additional studies are warranted to establish the potential of statins in the chemoprevention of GI cancer. In addition, prebiotics, selectively fermented food ingredients that induce specific changes in the composition or activity in the GI microbiota, has been acknowledged as beneficial to the host well being and health, showed a protective effect and may be the useful for colon cancer prevention and treatment.150 Similarly, probiotics can carry broad spectrum of GI cancer prevention, particularly H. pylori-associated gastric cancer and IBD-associated colon cancer.151 Recently we have published cancer preventive effect of exogenous 8-hydroxydeoxyguanosine against colitis-associated cancer, where we have shown that strong anti-inflammatory and antioxidative agents administered at early phase of inflammation-associated carcinogenesis could suppressed inflammation-based mutagenic actions strongly, supporting the thesis that top-down strategy might be better in a wide spectrum of cancer prevention rather than bottom-up strategy.152 As seen in Fig. 2, accumulating evidences are available showing that n-polyunsaturated fatty acids (n-PUFAs) were very essential in either ameliorating carcinogenesis-associated inflammation or spurting rejuvenation to revert precancerous lesion.153-155 In order to clearly document the cancer preventive efficacy of n-3 PUFA, Kang et al.156 generated fat-1 transgenic mice, which was made through over-expressing n-3 desaturase (Fig. 2B), and proved the chemopreventive actions of n-PUFA in diverse clinical diseases from inflammation based GI diseases to diverse GI cancers (Fig. 2A). Several mechanisms by which n-3 PUFA can exert the chemopreventive effects have recently been identified; inhibition of COX activity,154 production of novel anti-inflammatory lipid mediators; resolvins, protectins, maresins, etc,157 direct fatty acid signaling via G protein-coupled receptors,158 alteration of membrane dynamics and cell surface receptor function,159 decreased cellular oxidative stress,160 antioxidative function,161 and restorative and rejuvenating action (Fig. 2B).162 Currently our group is continuing to prove the efficacy of n-3 PUFA to halt the progression of precancerous to cancer lesions in gastroenterology (Fig. 2C).
Fig. 2.

Chemopreventative actions of n-3 polyunsaturated fatty acids (PUFAs). (A) Diverse clinical effects of n-3 PUFA beyond the prevention of atherosclerosis or the use of healthy food supplements have been demonstrated. (B) Antitumoric actions of n-PUFAs. Thus far, the following effects of n-3 PUFA have been documented: inhibition of cyclooxygenase (COX) activity; production of novel anti-inflammatory lipid mediators, such as resolvins, protectins, maresins, etc.; direct fatty acid signaling via G protein-coupled receptors (GPCR); alterations of membrane dynamics and cell surface receptor functions; decreased cellular oxidative stress; anti-oxidative function; and restorative and rejuvenating actions. (C) Future prospects for the use of n-3 PUFAs in the prevention of various gastrointestinal (GI) diseases, such as gastric ulcers, ulcerative colitis, gastric cancer and colon cancer. Additional detailed clinical trials should be performed to rank n-3 PUFAs as potent chemopreventive agents.
CONCLUSIONS
In spite of advancement and development in screening and early diagnosis as well as minimally invasive treatment technology, GI cancers still remain a leading cause of death worldwide, a challenge for the clinicians to search more effective preventive strategies. The increasing burden of cancer treatment as evidenced with the failure of cancer act to conquest has led to a significant paradigm shift in more intense efforts from treatment to prevention. As potential candidates postulated as having a chemopreventive effect on GI cancers, aspirin, and NSAID had been most widely studied and has gained universal acknowledgement. However, complications such as peptic ulceration, upper GI bleeding, and perforation as well as increased cardiovascular threatening posed serious threats to the routine administration of NSAIDs for the purpose of cancer prevention, still awaiting serious improvement in formulation for chronic use. Finally though a balance between the risks and benefits must be struck accompanied with extensive clinical trials to weight advantage, the safety, cost effectiveness and optimal dose will be solved in a near future. Another hope comes from cancer metabolism, for instance, Warburg effect can shed promising chance for cancer prevention. As shown in the main text, we have high levels of evidence that PPI as well as acid pump antagonist might be evolved to cover the villainous characteristics of cancer cells into a virtuous feature of noncancer cells. Finally, natural products or phytoceuticals cover the long journey to the prevention of cancers through chronic safe administration.
ACKNOWLEDGEMENTS
This work was supported by a specialized research fund of institute for new drug development, Keimyung University and the grants from Korean Heath Industry Development Institute (KHIDI) and also by National Center of Efficacy Evaluation for the Development of Health Products Targeting Digestive Disorders (NCEED).
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Mann JR, Backlund MG, DuBois RN. Mechanisms of disease: inflammatory mediators and cancer prevention. Nat Clin Pract Oncol. 2005;2:202–210. doi: 10.1038/ncponc0140. [DOI] [PubMed] [Google Scholar]
- 2.Wattenberg LW. Chemoprophylaxis of carcinogenesis: a review. Cancer Res. 1966;26:1520–1526. [PubMed] [Google Scholar]
- 3.Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res. 1976;36(7 Pt 2):2699–2702. [PubMed] [Google Scholar]
- 4.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 5.Moolgavkar SH. The multistage theory of carcinogenesis and the age distribution of cancer in man. J Natl Cancer Inst. 1978;61:49–52. doi: 10.1093/jnci/61.1.49. [DOI] [PubMed] [Google Scholar]
- 6.Gravitz L. Chemoprevention: first line of defence. Nature. 2011;471:S5–S7. doi: 10.1038/471S5a. [DOI] [PubMed] [Google Scholar]
- 7.Stoner GD, Wang LS, Chen T. Chemoprevention of esophageal squamous cell carcinoma. Toxicol Appl Pharmacol. 2007;224:337–349. doi: 10.1016/j.taap.2007.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pera M, Manterola C, Vidal O, Grande L. Epidemiology of esophageal adenocarcinoma. J Surg Oncol. 2005;92:151–159. doi: 10.1002/jso.20357. [DOI] [PubMed] [Google Scholar]
- 9.Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst. 2008;100:1184–1187. doi: 10.1093/jnci/djn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 11.Jemal A, Ward E, Hao Y, Thun M. Trends in the leading causes of death in the United States, 1970-2002. JAMA. 2005;294:1255–1259. doi: 10.1001/jama.294.10.1255. [DOI] [PubMed] [Google Scholar]
- 12.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 13.Moshkowitz M, Shapira S, Arber N. Chemoprevention for advanced CR neoplasia. Best Pract Res Clin Gastroenterol. 2011;25:623–630. doi: 10.1016/j.bpg.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Albeniz X, Chan AT. Aspirin for the prevention of colorectal cancer. Best Pract Res Clin Gastroenterol. 2011;25:461–472. doi: 10.1016/j.bpg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eaden J, Abrams K, Ekbom A, Jackson E, Mayberry J. Colorectal cancer prevention in ulcerative colitis: a case-control study. Aliment Pharmacol Ther. 2000;14:145–153. doi: 10.1046/j.1365-2036.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 16.DuBois RN, Giardiello FM, Smalley WE. Nonsteroidal anti-inflammatory drugs, eicosanoids, and colorectal cancer prevention. Gastroenterol Clin North Am. 1996;25:773–791. doi: 10.1016/s0889-8553(05)70274-0. [DOI] [PubMed] [Google Scholar]
- 17.Bartsch T, Levy MJ, Knight YE, Goadsby PJ. Inhibition of nociceptive dural input in the trigeminal nucleus caudalis by somatostatin receptor blockade in the posterior hypothalamus. Pain. 2005;117:30–39. doi: 10.1016/j.pain.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 18.Nair J, Gansauge F, Beger H, Dolara P, Winde G, Bartsch H. Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn's disease, ulcerative colitis, and chronic pancreatitis. Antioxid Redox Signal. 2006;8:1003–1010. doi: 10.1089/ars.2006.8.1003. [DOI] [PubMed] [Google Scholar]
- 19.Seril DN, Liao J, Yang GY, Yang CS. Oxidative stress and ulcerative colitis-associated carcinogenesis: studies in humans and animal models. Carcinogenesis. 2003;24:353–362. doi: 10.1093/carcin/24.3.353. [DOI] [PubMed] [Google Scholar]
- 20.Ohshima H, Sawa T, Akaike T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid Redox Signal. 2006;8:1033–1045. doi: 10.1089/ars.2006.8.1033. [DOI] [PubMed] [Google Scholar]
- 21.Yermilov V, Rubio J, Becchi M, Friesen MD, Pignatelli B, Ohshima H. Formation of 8-nitroguanine by the reaction of guanine with peroxynitrite in vitro. Carcinogenesis. 1995;16:2045–2050. doi: 10.1093/carcin/16.9.2045. [DOI] [PubMed] [Google Scholar]
- 22.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365–372. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 23.Wild CP, Hardie LJ. Reflux, Barrett's oesophagus and adenocarcinoma: burning questions. Nat Rev Cancer. 2003;3:676–684. doi: 10.1038/nrc1166. [DOI] [PubMed] [Google Scholar]
- 24.Prior P, Gyde SN, Macartney JC, Thompson H, Waterhouse JA, Allan RN. Cancer morbidity in ulcerative colitis. Gut. 1982;23:490–497. doi: 10.1136/gut.23.6.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y, Fruehauf J, Xiang S, Li CJ. Genomic instability in precancerous lesions before inactivation of tumor suppressors p53 and APC in patients. Cell Cycle. 2006;5:1443–1447. doi: 10.4161/cc.5.13.2897. [DOI] [PubMed] [Google Scholar]
- 28.Park WS, Pham T, Wang C, et al. Loss of heterozygosity and microsatellite instability in non-neoplastic mucosa from patients with chronic ulcerative colitis. Int J Mol Med. 1998;2:221–224. [PubMed] [Google Scholar]
- 29.Brentnall TA, Chen R, Lee JG, et al. Microsatellite instability and K-ras mutations associated with pancreatic adenocarcinoma and pancreatitis. Cancer Res. 1995;55:4264–4267. [PubMed] [Google Scholar]
- 30.Moskovitz AH, Linford NJ, Brentnall TA, et al. Chromosomal instability in pancreatic ductal cells from patients with chronic pancreatitis and pancreatic adenocarcinoma. Genes Chromosomes Cancer. 2003;37:201–206. doi: 10.1002/gcc.10189. [DOI] [PubMed] [Google Scholar]
- 31.Werling K, Szepesi A, Szentirmay Z, Schaff Z, Tulassay Z, Szalay F. Effect of hepatitis C virus on hepatocyte proliferation and DNA ploidy in patients with chronic hepatitis C. Z Gastroenterol. 2000;38:553–558. doi: 10.1055/s-2000-7448. [DOI] [PubMed] [Google Scholar]
- 32.Isokawa O, Suda T, Aoyagi Y, et al. Reduction of telomeric repeats as a possible predictor for development of hepatocellular carcinoma: convenient evaluation by slot-blot analysis. Hepatology. 1999;30:408–412. doi: 10.1002/hep.510300211. [DOI] [PubMed] [Google Scholar]
- 33.Hou L, Savage SA, Blaser MJ, et al. Telomere length in peripheral leukocyte DNA and gastric cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18:3103–3109. doi: 10.1158/1055-9965.EPI-09-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Risques RA, Lai LA, Himmetoglu C, et al. Ulcerative colitis-associated colorectal cancer arises in a field of short telomeres, senescence, and inflammation. Cancer Res. 2011;71:1669–1679. doi: 10.1158/0008-5472.CAN-10-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohonen-Corish MR, Daniel JJ, te Riele H, Buffinton GD, Dahlstrom JE. Susceptibility of Msh2-deficient mice to inflammation-associated colorectal tumors. Cancer Res. 2002;62:2092–2097. [PubMed] [Google Scholar]
- 37.Taniguchi K, Kakinuma S, Tokairin Y, et al. Mild inflammation accelerates colon carcinogenesis in Mlh1-deficient mice. Oncology. 2006;71:124–130. doi: 10.1159/000100522. [DOI] [PubMed] [Google Scholar]
- 38.Edwards RA, Witherspoon M, Wang K, et al. Epigenetic repression of DNA mismatch repair by inflammation and hypoxia in inflammatory bowel disease-associated colorectal cancer. Cancer Res. 2009;69:6423–6429. doi: 10.1158/0008-5472.CAN-09-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao J, Seril DN, Lu GG, et al. Increased susceptibility of chronic ulcerative colitis-induced carcinoma development in DNA repair enzyme Ogg1 deficient mice. Mol Carcinog. 2008;47:638–646. doi: 10.1002/mc.20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal M, LaRusso NF, Gores GJ. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G626–G634. doi: 10.1152/ajpgi.2001.281.3.G626. [DOI] [PubMed] [Google Scholar]
- 41.Hofseth LJ, Khan MA, Ambrose M, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112:1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ando K, Hirao S, Kabe Y, et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyskens FL, Jr, McLaren CE, Pelot D, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila) 2008;1:32–38. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gobert AP, Cheng Y, Wang JY, et al. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol. 2002;168:4692–4700. doi: 10.4049/jimmunol.168.9.4692. [DOI] [PubMed] [Google Scholar]
- 45.Koza RA, Megosh LC, Palmieri M, O'Brien TG. Constitutively elevated levels of ornithine and polyamines in mouse epidermal papillomas. Carcinogenesis. 1991;12:1619–1625. doi: 10.1093/carcin/12.9.1619. [DOI] [PubMed] [Google Scholar]
- 46.ter Steege JC, Forget PP, Buurman WA. Oral spermine administration inhibits nitric oxide-mediated intestinal damage and levels of systemic inflammatory mediators in a mouse endotoxin model. Shock. 1999;11:115–119. doi: 10.1097/00024382-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Alhonen L, Parkkinen JJ, Keinanen T, Sinervirta R, Herzig KH, Jänne J. Activation of polyamine catabolism in transgenic rats induces acute pancreatitis. Proc Natl Acad Sci U S A. 2000;97:8290–8295. doi: 10.1073/pnas.140122097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rasanen TL, Alhonen L, Sinervirta R, et al. A polyamine analogue prevents acute pancreatitis and restores early liver regeneration in transgenic rats with activated polyamine catabolism. J Biol Chem. 2002;277:39867–39872. doi: 10.1074/jbc.M205967200. [DOI] [PubMed] [Google Scholar]
- 49.Babbar N, Murray-Stewart T, Casero RA., Jr Inflammation and polyamine catabolism: the good, the bad and the ugly. Biochem Soc Trans. 2007;35(Pt 2):300–304. doi: 10.1042/BST0350300. [DOI] [PubMed] [Google Scholar]
- 50.Xu H, Chaturvedi R, Cheng Y, et al. Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res. 2004;64:8521–8525. doi: 10.1158/0008-5472.CAN-04-3511. [DOI] [PubMed] [Google Scholar]
- 51.Endo Y, Marusawa H, Chiba T. Involvement of activation-induced cytidine deaminase in the development of colitis-associated colorectal cancers. J Gastroenterol. 2011;46(Suppl 1):6–10. doi: 10.1007/s00535-010-0326-1. [DOI] [PubMed] [Google Scholar]
- 52.Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 53.Takaishi S, Wang TC. Providing AID to p53 mutagenesis. Nat Med. 2007;13:404–406. doi: 10.1038/nm0407-404. [DOI] [PubMed] [Google Scholar]
- 54.Kou T, Marusawa H, Kinoshita K, et al. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer. 2007;120:469–476. doi: 10.1002/ijc.22292. [DOI] [PubMed] [Google Scholar]
- 55.Shimizu T, Marusawa H, Endo Y, Chiba T. Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012;103:1201–1206. doi: 10.1111/j.1349-7006.2012.02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marusawa H, Takai A, Chiba T. Role of activation-induced cytidine deaminase in inflammation-associated cancer development. Adv Immunol. 2011;111:109–141. doi: 10.1016/B978-0-12-385991-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 57.Hillman LC, Chiragakis L, Shadbolt B, Kaye GL, Clarke AC. Proton-pump inhibitor therapy and the development of dysplasia in patients with Barrett's oesophagus. Med J Aust. 2004;180:387–391. doi: 10.5694/j.1326-5377.2004.tb05991.x. [DOI] [PubMed] [Google Scholar]
- 58.Hillman LC, Chiragakis L, Shadbolt B, Kaye GL, Clarke AC. Effect of proton pump inhibitors on markers of risk for high-grade dysplasia and oesophageal cancer in Barrett's oesophagus. Aliment Pharmacol Ther. 2008;27:321–326. doi: 10.1111/j.1365-2036.2007.03579.x. [DOI] [PubMed] [Google Scholar]
- 59.Miyashita T, Shah FA, Marti GP, et al. Rabeprazole impedes the development of reflux-induced esophageal cancer in a surgical rat model. Dig Dis Sci. 2011;56:1309–1314. doi: 10.1007/s10620-010-1465-1. [DOI] [PubMed] [Google Scholar]
- 60.García Rodríguez LA, Lagergren J, Lindblad M. Gastric acid suppression and risk of oesophageal and gastric adenocarcinoma: a nested case control study in the UK. Gut. 2006;55:1538–1544. doi: 10.1136/gut.2005.086579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Obszynska JA, Atherfold PA, Nanji M, et al. Long-term proton pump induced hypergastrinaemia does induce lineage-specific restitution but not clonal expansion in benign Barrett's oesophagus in vivo. Gut. 2010;59:156–163. doi: 10.1136/gut.2009.186775. [DOI] [PubMed] [Google Scholar]
- 62.Kim YJ, Lee JS, Hong KS, Chung JW, Kim JH, Hahm KB. Novel application of proton pump inhibitor for the prevention of colitis-induced colorectal carcinogenesis beyond acid suppression. Cancer Prev Res (Phila) 2010;3:963–974. doi: 10.1158/1940-6207.CAPR-10-0033. [DOI] [PubMed] [Google Scholar]
- 63.Kim YJ, Hong KS, Chung JW, Kim JH, Hahm KB. Prevention of colitis-associated carcinogenesis with infliximab. Cancer Prev Res (Phila) 2010;3:1314–1333. doi: 10.1158/1940-6207.CAPR-09-0272. [DOI] [PubMed] [Google Scholar]
- 64.Yeo M, Kim DK, Kim YB, et al. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin Cancer Res. 2004;10:8687–8696. doi: 10.1158/1078-0432.CCR-04-1065. [DOI] [PubMed] [Google Scholar]
- 65.Hahm KB, Lee KJ, Kim YS, et al. Augmented eradication rates of Helicobacter pylori by new combination therapy with lansoprazole, amoxicillin, and rebamipide. Dig Dis Sci. 1998;43:235–240. doi: 10.1023/a:1018825532059. [DOI] [PubMed] [Google Scholar]
- 66.Thun MJ, Jacobs EJ, Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol. 2012;9:259–267. doi: 10.1038/nrclinonc.2011.199. [DOI] [PubMed] [Google Scholar]
- 67.Burn J, Bishop DT, Chapman PD, et al. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res (Phila) 2011;4:655–665. doi: 10.1158/1940-6207.CAPR-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang J, Sharif O, Pai C, Silverman AL. Mesalamine protects against colorectal cancer in inflammatory bowel disease. Dig Dis Sci. 2010;55:1696–1703. doi: 10.1007/s10620-009-0942-x. [DOI] [PubMed] [Google Scholar]
- 69.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 70.Vaughan TL, Dong LM, Blount PL, et al. Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett's oesophagus: a prospective study. Lancet Oncol. 2005;6:945–952. doi: 10.1016/S1470-2045(05)70431-9. [DOI] [PubMed] [Google Scholar]
- 71.Tan XL, Reid Lombardo KM, Bamlet WR, et al. Aspirin, nonsteroidal anti-inflammatory drugs, acetaminophen, and pancreatic cancer risk: a clinic-based case-control study. Cancer Prev Res (Phila) 2011;4:1835–1841. doi: 10.1158/1940-6207.CAPR-11-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bonifazi M, Gallus S, Bosetti C, et al. Aspirin use and pancreatic cancer risk. Eur J Cancer Prev. 2010;19:352–354. doi: 10.1097/CEJ.0b013e32833b48a4. [DOI] [PubMed] [Google Scholar]
- 73.Menezes RJ, Huber KR, Mahoney MC, Moysich KB. Regular use of aspirin and pancreatic cancer risk. BMC Public Health. 2002;2:18. doi: 10.1186/1471-2458-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee SJ, Park JY, Choi KS, et al. Efficacy of Korean red ginseng supplementation on erdication rate and gastric volatile sulfur compound levles after Helicobacter pylori eradication therapy. J Ginseng Res. 2010;34:122–131. [Google Scholar]
- 75.Choi KS, Song H, Kim EH, et al. Inhibition of hydrogen sulfide-induced angiogenesis and inflammation in vascular endothelial cells: potential mechanisms of gastric cancer prevention by Korean red ginseng. J Ginseng Res. 2012;36:135–145. doi: 10.5142/jgr.2012.36.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park S, Yeo M, Jin JH, et al. Rescue of Helicobacter pylori-induced cytotoxicity by red ginseng. Dig Dis Sci. 2005;50:1218–1227. doi: 10.1007/s10620-005-2763-x. [DOI] [PubMed] [Google Scholar]
- 77.Park S, Yeo M, Jin JH, et al. Inhibitory activities and attenuated expressions of 5-LOX with red ginseng in Helicobacter pylori-infected gastric epithelial cells. Dig Dis Sci. 2007;52:973–982. doi: 10.1007/s10620-006-9440-6. [DOI] [PubMed] [Google Scholar]
- 78.Kim DK, Lee JA, Kim YB, Lee KM, Hahm KB. A randomized controlled trial assessing Korean red ginseng treatment of Helicobacter pylori-associated chronic gastritis. Korean J Med. 2007;72:20–28. [Google Scholar]
- 79.Wargovich MJ. Colon cancer chemoprevention with ginseng and other botanicals. J Korean Med Sci. 2001;16(Suppl):S81–S86. doi: 10.3346/jkms.2001.16.S.S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holian O, Wahid S, Atten MJ, Attar BM. Inhibition of gastric cancer cell proliferation by resveratrol: role of nitric oxide. Am J Physiol Gastrointest Liver Physiol. 2002;282:G809–G816. doi: 10.1152/ajpgi.00193.2001. [DOI] [PubMed] [Google Scholar]
- 81.Li ZG, Hong T, Shimada Y, et al. Suppression of N-nitrosomethylbenzylamine (NMBA)-induced esophageal tumorigenesis in F344 rats by resveratrol. Carcinogenesis. 2002;23:1531–1536. doi: 10.1093/carcin/23.9.1531. [DOI] [PubMed] [Google Scholar]
- 82.Cui X, Jin Y, Hofseth AB, et al. Resveratrol suppresses colitis and colon cancer associated with colitis. Cancer Prev Res (Phila) 2010;3:549–559. doi: 10.1158/1940-6207.CAPR-09-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roy SK, Chen Q, Fu J, Shankar S, Srivastava RK. Resveratrol inhibits growth of orthotopic pancreatic tumors through activation of FOXO transcription factors. PLoS One. 2011;6:e25166. doi: 10.1371/journal.pone.0025166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ju J, Hong J, Zhou JN, et al. Inhibition of intestinal tumorigenesis in Apcmin/+ mice by (-)-epigallocatechin-3-gallate, the major catechin in green tea. Cancer Res. 2005;65:10623–10631. doi: 10.1158/0008-5472.CAN-05-1949. [DOI] [PubMed] [Google Scholar]
- 85.Xiao H, Hao X, Simi B, et al. Green tea polyphenols inhibit colorectal aberrant crypt foci (ACF) formation and prevent oncogenic changes in dysplastic ACF in azoxymethane-treated F344 rats. Carcinogenesis. 2008;29:113–119. doi: 10.1093/carcin/bgm204. [DOI] [PubMed] [Google Scholar]
- 86.Sharma RA, McLelland HR, Hill KA, et al. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin Cancer Res. 2001;7:1894–1900. [PubMed] [Google Scholar]
- 87.Sharma RA, Euden SA, Platton SL, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10:6847–6854. doi: 10.1158/1078-0432.CCR-04-0744. [DOI] [PubMed] [Google Scholar]
- 88.Cruz-Correa M, Shoskes DA, Sanchez P, et al. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2006;4:1035–1038. doi: 10.1016/j.cgh.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 89.Delmas D, Rébé C, Lacour S, et al. Resveratrol-induced apoptosis is associated with Fas redistribution in the rafts and the formation of a death-inducing signaling complex in colon cancer cells. J Biol Chem. 2003;278:41482–41490. doi: 10.1074/jbc.M304896200. [DOI] [PubMed] [Google Scholar]
- 90.Liang YC, Tsai SH, Chen L, Lin-Shiau SY, Lin JK. Resveratrol-induced G2 arrest through the inhibition of CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem Pharmacol. 2003;65:1053–1060. doi: 10.1016/s0006-2952(03)00011-x. [DOI] [PubMed] [Google Scholar]
- 91.Ye F, Zhang GH, Guan BX, Xu XC. Suppression of esophageal cancer cell growth using curcumin, (-)-epigallocatechin-3-gallate and lovastatin. World J Gastroenterol. 2012;18:126–135. doi: 10.3748/wjg.v18.i2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hou Z, Sang S, You H, et al. Mechanism of action of (-)-epigallocatechin-3-gallate: auto-oxidation-dependent inactivation of epidermal growth factor receptor and direct effects on growth inhibition in human esophageal cancer KYSE 150 cells. Cancer Res. 2005;65:8049–8056. doi: 10.1158/0008-5472.CAN-05-0480. [DOI] [PubMed] [Google Scholar]
- 93.Li ZG, Shimada Y, Sato F, et al. Inhibitory effects of epigallocatechin-3-gallate on N-nitrosomethylbenzylamine-induced esophageal tumorigenesis in F344 rats. Int J Oncol. 2002;21:1275–1283. [PubMed] [Google Scholar]
- 94.Inoue M, Sasazuki S, Wakai K, et al. Green tea consumption and gastric cancer in Japanese: a pooled analysis of six cohort studies. Gut. 2009;58:1323–1332. doi: 10.1136/gut.2008.166710. [DOI] [PubMed] [Google Scholar]
- 95.Gutierrez-Orozco F, Stephens BR, Neilson AP, Green R, Ferruzzi MG, Bomser JA. Green and black tea inhibit cytokine-induced IL-8 production and secretion in AGS gastric cancer cells via inhibition of NF-kappaB activity. Planta Med. 2010;76:1659–1665. doi: 10.1055/s-0030-1249975. [DOI] [PubMed] [Google Scholar]
- 96.Wu H, Xin Y, Xiao Y, Zhao J. Low-dose docetaxel combined with (-)-epigallocatechin-3-gallate inhibits angiogenesis and tumor growth in nude mice with gastric cancer xenografts. Cancer Biother Radiopharm. 2012;27:204–209. doi: 10.1089/cbr.2011.1103. [DOI] [PubMed] [Google Scholar]
- 97.Zhu BH, Chen HY, Zhan WH, et al. (-)-Epigallocatechin-3-gallate inhibits VEGF expression induced by IL-6 via Stat3 in gastric cancer. World J Gastroenterol. 2011;17:2315–2325. doi: 10.3748/wjg.v17.i18.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tanaka T, Ishii T, Mizuno D, et al. (-)-Epigallocatechin-3-gallate suppresses growth of AZ521 human gastric cancer cells by targeting the DEAD-box RNA helicase p68. Free Radic Biol Med. 2011;50:1324–1335. doi: 10.1016/j.freeradbiomed.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 99.Xu G, Ren G, Xu X, et al. Combination of curcumin and green tea catechins prevents dimethylhydrazine-induced colon carcinogenesis. Food Chem Toxicol. 2010;48:390–395. doi: 10.1016/j.fct.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 100.Lambert JD, Sang S, Hong J, Yang CS. Anticancer and anti-inflammatory effects of cysteine metabolites of the green tea polyphenol, (-)-epigallocatechin-3-gallate. J Agric Food Chem. 2010;58:10016–10019. doi: 10.1021/jf102311t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stingl JC, Ettrich T, Muche R, et al. Protocol for minimizing the risk of metachronous adenomas of the colorectum with green tea extract (MIRACLE): a randomised controlled trial of green tea extract versus placebo for nutriprevention of metachronous colon adenomas in the elderly population. BMC Cancer. 2011;11:360. doi: 10.1186/1471-2407-11-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Larsen CA, Dashwood RH. Suppression of Met activation in human colon cancer cells treated with (-)-epigallocatechin-3-gallate: minor role of hydrogen peroxide. Biochem Biophys Res Commun. 2009;389:527–530. doi: 10.1016/j.bbrc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Adachi S, Shimizu M, Shirakami Y, et al. (-)-Epigallocatechin gallate downregulates EGF receptor via phosphorylation at Ser1046/1047 by p38 MAPK in colon cancer cells. Carcinogenesis. 2009;30:1544–1552. doi: 10.1093/carcin/bgp166. [DOI] [PubMed] [Google Scholar]
- 104.Shimizu M, Shirakami Y, Sakai H, et al. (-)-Epigallocatechin gallate suppresses azoxymethane-induced colonic premalignant lesions in male C57BL/KsJ-db/db mice. Cancer Prev Res (Phila) 2008;1:298–304. doi: 10.1158/1940-6207.CAPR-08-0045. [DOI] [PubMed] [Google Scholar]
- 105.Wang Z, Li W, Meng X, Jia B. Resveratrol induces gastric cancer cell apoptosis via reactive oxygen species, but independent of sirtuin1. Clin Exp Pharmacol Physiol. 2012;39:227–232. doi: 10.1111/j.1440-1681.2011.05660.x. [DOI] [PubMed] [Google Scholar]
- 106.Riles WL, Erickson J, Nayyar S, Atten MJ, Attar BM, Holian O. Resveratrol engages selective apoptotic signals in gastric adenocarcinoma cells. World J Gastroenterol. 2006;12:5628–5634. doi: 10.3748/wjg.v12.i35.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Atten MJ, Attar BM, Milson T, Holian O. Resveratrol-induced inactivation of human gastric adenocarcinoma cells through a protein kinase C-mediated mechanism. Biochem Pharmacol. 2001;62:1423–1432. doi: 10.1016/s0006-2952(01)00788-2. [DOI] [PubMed] [Google Scholar]
- 108.Zhou HB, Chen JJ, Wang WX, Cai JT, Du Q. Anticancer activity of resveratrol on implanted human primary gastric carcinoma cells in nude mice. World J Gastroenterol. 2005;11:280–284. doi: 10.3748/wjg.v11.i2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Juan ME, Alfaras I, Planas JM. Colorectal cancer chemoprevention by trans-resveratrol. Pharmacol Res. 2012;65:584–591. doi: 10.1016/j.phrs.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 110.Panaro MA, Carofiglio V, Acquafredda A, Cavallo P, Cianciulli A. Anti-inflammatory effects of resveratrol occur via inhibition of lipopolysaccharide-induced NF-kappaB activation in Caco-2 and SW480 human colon cancer cells. Br J Nutr. 2012;108:1623–1632. doi: 10.1017/S0007114511007227. [DOI] [PubMed] [Google Scholar]
- 111.Miki H, Uehara N, Kimura A, et al. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int J Oncol. 2012;40:1020–1028. doi: 10.3892/ijo.2012.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vanamala J, Radhakrishnan S, Reddivari L, Bhat VB, Ptitsyn A. Resveratrol suppresses human colon cancer cell proliferation and induces apoptosis via targeting the pentose phosphate and the talin-FAK signaling pathways-A proteomic approach. Proteome Sci. 2011;9:49. doi: 10.1186/1477-5956-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Radhakrishnan S, Reddivari L, Sclafani R, Das UN, Vanamala J. Resveratrol potentiates grape seed extract induced human colon cancer cell apoptosis. Front Biosci (Elite Ed) 2011;3:1509–1523. doi: 10.2741/e352. [DOI] [PubMed] [Google Scholar]
- 114.Patel KR, Brown VA, Jones DJ, et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70:7392–7399. doi: 10.1158/0008-5472.CAN-10-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tian F, Zhang C, Tian W, Jiang Y, Zhang X. Comparison of the effect of p65 siRNA and curcumin in promoting apoptosis in esophageal squamous cell carcinoma cells and in nude mice. Oncol Rep. 2012;28:232–240. doi: 10.3892/or.2012.1777. [DOI] [PubMed] [Google Scholar]
- 116.Subramaniam D, Ponnurangam S, Ramamoorthy P, et al. Curcumin induces cell death in esophageal cancer cells through modulating Notch signaling. PLoS One. 2012;7:e30590. doi: 10.1371/journal.pone.0030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sintara K, Thong-Ngam D, Patumraj S, Klaikeaw N. Curcumin attenuates gastric cancer induced by N-methyl-N-nitrosourea and saturated sodium chloride in rats. J Biomed Biotechnol. 2012;2012:915380. doi: 10.1155/2012/915380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu LL, Wu JG, Dai N, Yu HG, Si JM. Curcumin reverses chemoresistance of human gastric cancer cells by downregulating the NF-kappaB transcription factor. Oncol Rep. 2011;26:1197–1203. doi: 10.3892/or.2011.1410. [DOI] [PubMed] [Google Scholar]
- 119.Zaidi SF, Yamamoto T, Refaat A, et al. Modulation of activation-induced cytidine deaminase by curcumin in Helicobacter pylori-infected gastric epithelial cells. Helicobacter. 2009;14:588–595. doi: 10.1111/j.1523-5378.2009.00724.x. [DOI] [PubMed] [Google Scholar]
- 120.Cai XZ, Wang J, Li XD, et al. Curcumin suppresses proliferation and invasion in human gastric cancer cells by downregulation of PAK1 activity and cyclin D1 expression. Cancer Biol Ther. 2009;8:1360–1368. doi: 10.4161/cbt.8.14.8720. [DOI] [PubMed] [Google Scholar]
- 121.Koo JY, Kim HJ, Jung KO, Park KY. Curcumin inhibits the growth of AGS human gastric carcinoma cells in vitro and shows synergism with 5-fluorouracil. J Med Food. 2004;7:117–121. doi: 10.1089/1096620041224229. [DOI] [PubMed] [Google Scholar]
- 122.Johnson JJ, Mukhtar H. Curcumin for chemoprevention of colon cancer. Cancer Lett. 2007;255:170–181. doi: 10.1016/j.canlet.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 123.Subramaniam D, May R, Sureban SM, et al. Diphenyl difluoroketone: a curcumin derivative with potent in vivo anticancer activity. Cancer Res. 2008;68:1962–1969. doi: 10.1158/0008-5472.CAN-07-6011. [DOI] [PubMed] [Google Scholar]
- 124.Kanwar SS, Yu Y, Nautiyal J, et al. Difluorinated-curcumin (CDF): a novel curcumin analog is a potent inhibitor of colon cancer stem-like cells. Pharm Res. 2011;28:827–838. doi: 10.1007/s11095-010-0336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nautiyal J, Banerjee S, Kanwar SS, et al. Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. Int J Cancer. 2011;128:951–961. doi: 10.1002/ijc.25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Patel BB, Gupta D, Elliott AA, Sengupta V, Yu Y, Majumdar AP. Curcumin targets FOLFOX-surviving colon cancer cells via inhibition of EGFRs and IGF-1R. Anticancer Res. 2010;30:319–325. [PMC free article] [PubMed] [Google Scholar]
- 127.Mudduluru G, George-William JN, Muppala S, et al. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci Rep. 2011;31:185–197. doi: 10.1042/BSR20100065. [DOI] [PubMed] [Google Scholar]
- 128.Villegas I, Sánchez-Fidalgo S, de la Lastra CA. Chemopreventive effect of dietary curcumin on inflammation-induced colorectal carcinogenesis in mice. Mol Nutr Food Res. 2011;55:259–267. doi: 10.1002/mnfr.201000225. [DOI] [PubMed] [Google Scholar]
- 129.Li N, Guo R, Li W, et al. A proteomic investigation into a human gastric cancer cell line BGC823 treated with diallyl trisulfide. Carcinogenesis. 2006;27:1222–1231. doi: 10.1093/carcin/bgi306. [DOI] [PubMed] [Google Scholar]
- 130.Yang YH, Zhao M, Li WM, Lu YY, Chen YY, Kang B. Expression of programmed cell death 5 gene involves in regulation of apoptosis in gastric tumor cells. Apoptosis. 2006;11:993–1001. doi: 10.1007/s10495-006-6714-6. [DOI] [PubMed] [Google Scholar]
- 131.Wu PP, Liu KC, Huang WW, et al. Diallyl trisulfide (DATS) inhibits mouse colon tumor in mouse CT-26 cells allograft model in vivo. Phytomedicine. 2011;18:672–676. doi: 10.1016/j.phymed.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 132.Hosono T, Fukao T, Ogihara J, et al. Diallyl trisulfide suppresses the proliferation and induces apoptosis of human colon cancer cells through oxidative modification of beta-tubulin. J Biol Chem. 2005;280:41487–41493. doi: 10.1074/jbc.M507127200. [DOI] [PubMed] [Google Scholar]
- 133.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 134.Pan MH, Chiou YS, Wang YJ, Ho CT, Lin JK. Multistage carcinogenesis process as molecular targets in cancer chemoprevention by epicatechin-3-gallate. Food Funct. 2011;2:101–110. doi: 10.1039/c0fo00174k. [DOI] [PubMed] [Google Scholar]
- 135.Kumar N, Shibata D, Helm J, Coppola D, Malafa M. Green tea polyphenols in the prevention of colon cancer. Front Biosci. 2007;12:2309–2315. doi: 10.2741/2233. [DOI] [PubMed] [Google Scholar]
- 136.Zikri NN, Riedl KM, Wang LS, Lechner J, Schwartz SJ, Stoner GD. Black raspberry components inhibit proliferation, induce apoptosis, and modulate gene expression in rat esophageal epithelial cells. Nutr Cancer. 2009;61:816–826. doi: 10.1080/01635580903285148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Surh YJ, Chun KS. Cancer chemopreventive effects of curcumin. Adv Exp Med Biol. 2007;595:149–172. doi: 10.1007/978-0-387-46401-5_5. [DOI] [PubMed] [Google Scholar]
- 138.Goel A, Kunnumakkara AB, Aggarwal BB. Curcumin as "Curecumin": from kitchen to clinic. Biochem Pharmacol. 2008;75:787–809. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 139.Hoshiyama Y, Kawaguchi T, Miura Y, et al. Green tea and stomach cancer: a short review of prospective studies. J Epidemiol. 2005;15(Suppl 2):S109–S112. doi: 10.2188/jea.15.S109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsubono Y, Nishino Y, Komatsu S, et al. Green tea and the risk of gastric cancer in Japan. N Engl J Med. 2001;344:632–636. doi: 10.1056/NEJM200103013440903. [DOI] [PubMed] [Google Scholar]
- 141.Sung B, Prasad S, Yadav VR, Aggarwal BB. Cancer cell signaling pathways targeted by spice-derived nutraceuticals. Nutr Cancer. 2012;64:173–197. doi: 10.1080/01635581.2012.630551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 143.Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24(5A):2783–2840. [PubMed] [Google Scholar]
- 144.Chiu HF, Ho SC, Chang CC, Wu TN, Yang CY. Statins are associated with a reduced risk of gastric cancer: a population-based case-control study. Am J Gastroenterol. 2011;106:2098–2103. doi: 10.1038/ajg.2011.277. [DOI] [PubMed] [Google Scholar]
- 145.Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma. Invest New Drugs. 2001;19:81–83. doi: 10.1023/a:1006481423298. [DOI] [PubMed] [Google Scholar]
- 146.Toyoda T, Tsukamoto T, Takasu S, et al. Pitavastatin fails to lower serum lipid levels or inhibit gastric carcinogenesis in Helicobacter pylori-infected rodent models. Cancer Prev Res (Phila) 2009;2:751–758. doi: 10.1158/1940-6207.CAPR-09-0082. [DOI] [PubMed] [Google Scholar]
- 147.Cheng MH, Chiu HF, Ho SC, Tsai SS, Wu TN, Yang CY. Statin use and the risk of colorectal cancer: a population-based case-control study. World J Gastroenterol. 2011;17:5197–5202. doi: 10.3748/wjg.v17.i47.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Relja B, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int J Mol Med. 2010;26:735–741. doi: 10.3892/ijmm_00000520. [DOI] [PubMed] [Google Scholar]
- 149.Relja B, Meder F, Wang M, et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol. 2011;38:879–885. doi: 10.3892/ijo.2010.892. [DOI] [PubMed] [Google Scholar]
- 150.Bertkova I, Hijova E, Chmelarova A, et al. The effect of probiotic microorganisms and bioactive compounds on chemically induced carcinogenesis in rats. Neoplasma. 2010;57:422–428. doi: 10.4149/neo_2010_05_422. [DOI] [PubMed] [Google Scholar]
- 151.Goossens D, Jonkers D, Stobberingh E, van den Bogaard A, Russel M, Stockbrügger R. Probiotics in gastroenterology: indications and future perspectives. Scand J Gastroenterol Suppl. 2003:15–23. doi: 10.1080/00855920310002645. [DOI] [PubMed] [Google Scholar]
- 152.Ock CY, Kim EH, Hong H, et al. Prevention of colitis-associated colorectal cancer with 8-hydroxydeoxyguanosine. Cancer Prev Res (Phila) 2011;4:1507–1521. doi: 10.1158/1940-6207.CAPR-11-0161. [DOI] [PubMed] [Google Scholar]
- 153.Lim K, Han C, Xu L, Isse K, Demetris AJ, Wu T. Cyclooxygenase-2-derived prostaglandin E2 activates beta-catenin in human cholangiocarcinoma cells: evidence for inhibition of these signaling pathways by omega 3 polyunsaturated fatty acids. Cancer Res. 2008;68:553–560. doi: 10.1158/0008-5472.CAN-07-2295. [DOI] [PubMed] [Google Scholar]
- 154.Lim K, Han C, Dai Y, Shen M, Wu T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol Cancer Ther. 2009;8:3046–3055. doi: 10.1158/1535-7163.MCT-09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Song KS, Jing K, Kim JS, et al. Omega-3-polyunsaturated fatty acids suppress pancreatic cancer cell growth in vitro and in vivo via downregulation of Wnt/Beta-catenin signaling. Pancreatology. 2011;11:574–584. doi: 10.1159/000334468. [DOI] [PubMed] [Google Scholar]
- 156.Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427:504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- 157.Weylandt KH, Krause LF, Gomolka B, et al. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-alpha. Carcinogenesis. 2011;32:897–903. doi: 10.1093/carcin/bgr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hawcroft G, Volpato M, Marston G, et al. The omega-3 polyunsaturated fatty acid eicosapentaenoic acid inhibits mouse MC-26 colorectal cancer cell liver metastasis via inhibition of PGE(2)-dependent cell motility. Br J Pharmacol. 2012;166:1724–1737. doi: 10.1111/j.1476-5381.2012.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Barbosa DS, Cecchini R, El Kadri MZ, Rodríguez MA, Burini RC, Dichi I. Decreased oxidative stress in patients with ulcerative colitis supplemented with fish oil omega-3 fatty acids. Nutrition. 2003;19:837–842. doi: 10.1016/s0899-9007(03)00162-x. [DOI] [PubMed] [Google Scholar]
- 161.King VR, Huang WL, Dyall SC, Curran OE, Priestley JV, Michael-Titus AT. Omega-3 fatty acids improve recovery, whereas omega-6 fatty acids worsen outcome, after spinal cord injury in the adult rat. J Neurosci. 2006;26:4672–4680. doi: 10.1523/JNEUROSCI.5539-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cockbain AJ, Toogood GJ, Hull MA. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut. 2012;61:135–149. doi: 10.1136/gut.2010.233718. [DOI] [PubMed] [Google Scholar]