

Abstract
To explore the acceptor regioselectivity of OleD-catalyzed glucosylation, the products of OleD-catalyzed reactions with the six structurally diverse acceptors - flavones (daidzein), isoflavones (flavopiridol), stilbenes (resveratrol), indole alkaloids (10-hydroxycamptothecin), and steroids (2-methoxyestradiol) - were determined. This study highlights the first synthesis of flavopiridol and 2-methoxyestradiol glucosides and confirms the ability of OleD to glucosylate both aromatic and aliphatic nucleophiles. In all cases, molecular dynamics simulations were consistent with the determined product distribution and suggest the potential to develop a virtual screening model to identify additional OleD substrates.
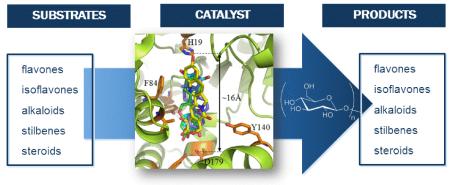
The glucosyltransferase OleD from Streptomyces antibioticus catalyzes the glucosylation of oleandomycin using UDP-D-glucose (UDP-Glc) as the glycosyl donor (Figure 1). This enzyme, first studied by Salas and coworkers, exists as part of a prototype system for macrolide inactivation and secretion in macrolide-producing microorganisms.1 Consistent with its role in detoxification, wild-type OleD (wtOleD) displays relatively broad substrate tolerance with a bias toward small aromatic hydroxy groups and recent OleD directed evolution and engineering efforts dramatically improved upon this catalyst’s proficiency and range of accessible substrates.2,3 The availability of enhanced OleD mutants and simple activated aromatic glycoside donors also enabled efforts to modulate the corresponding reaction equilibrium as a unique strategy for sugar nucleotide synthesis, glycodiversified small molecules and even a high-throughput screen for glycosylation.3c Cumulatively, these studies revealed OleD variants to function as a multifunctional and iterative O-/S-/N-GT capable of glucosylating well over 100 diverse acceptors. However, with a few exceptions2,3b,4 the product characterization for these studies was limited to LC-MS and thus, the regio-/stereospecificity of OleD-catalyzed glycosyltransfer with ‘non-native’ substrates remains poorly understood.5 To address this limitation, herein we describe the characterization of glycosides produced via the OleD-catalyzed glucosylation of a set of six representative structural classes – flavones (daidzein), isoflavones (flavopiridol), stilbenes (resveratrol), indole alkaloids (10-hydroxycamptothecin), and steroids (2-methoxyestradiol). This study revealed OleD to glycosylate both aromatic and aliphatic nucleophiles, the regioselectivity of which was dictated by a range factors, including reaction thermodynamics, enzyme mutation, and the acceptor architecture. A parallel molecular dynamics simulation for each reaction study was consistent with the corresponding product distribution observed and sets the stage to use virtual screening as a means to identify additional OleD substrates.
Figure 1.

The native OleD-catalyzed reaction as a macrolide-producing host resistance mechanism.
RESULTS AND DISCUSSION
Glucosylation of Daidzein
Isoflavones occur naturally in legumes and are consumed regularly in the human diet.6 Isoflavones often exist naturally as O-glycosides, and have attracted considerable pharmaceutical interest. 7 Daidzein is one of the most commonly occurring isoflavones with the corresponding 4′- and 7- O-glucosides as well as the 7,4′-di-O-glucosides of daidzein isolated from numerous sources.8 Daidzein and its corresponding O-glucosides are believed to be the major effective components of a traditional Chinese medicine Kudzu (Pueraria lobata) for the treatment of a wide range of disorders since 600 AD.9 While daidzein has limited solubility in water, the aqueous solubility of corresponding 7-O-glucoside is ~30-fold improved.10 The unique activities of these glycosides have inspired several targeted syntheses of daidzein 4′- and 7-O-glucosides.11 However, a convergent method to produce both mono- and di-glucosides in parallel has not been reported.
The pilot reaction for this study utilized UDP-Glc as the donor and OleD ASP3 as the catalyst under standard conditions (0.5 mM UDP-Glc, 0.1 mM aglycon, 16h). Based upon LC-MS, three products were observed (two monoglucosides and one diglucoside) with the diglucoside emerging as the major product over time (Figure 2b). To maximize the production of all three products for subsequent characterization, a 25 h reaction was selected for the preparative scale reaction. For this, daidzein (6.4 mg, 28.0 μmol) was dissolved in 1.25 mL DMSO and transferred to 25 mL assay buffer solution (50 mM Tris HCl, 5 mM MgCl2, pH 8.0). The reaction was initiated via the addition of UDP-Glc (38 mg, 62.3 μmol) and 25 mg of OleD ASP. After 25 h of gentle agitation at room temperature (rt), the reaction was frozen and lyophilized to dryness. HPLC purification of the crude reaction mixture provided daidzein 7,4′-di-O-β-D-diglucoside 6 (5 mg, 8.6 μmol, 31%), daidzein 4′-O-β-D-glucoside 5 (1 mg, 2.4 μmol, 9%) and daidzein 7-O-β-D-glucoside 4 (2 mg, 4.8 μmol, 17%). The 1H and 13C NMR and HR-MS data of the isolated glucosides were consistent with previously reported characterization data.11
Figure 2.

(a) Products deriving from OleD ASP-catalyzed glucosylation of daidzein. (b) Product distribution over time (the standard deviation of three trials was ± 3%).
Glucosylation of Resveratrol
Resveratrol, a naturally occurring phytoalexin found in various plants, grape skin, peanuts, cranberries, and red wine,12 reportedly exhibits multiple effects including life extension, 13 neuroprotection, 14 anti-inflammatory activity, 15 cardioprotection, 16 anti-diabetic activity, 17 viral inhibition,18 and cancer chemoprevention.19 Recent work also revealed resveratrol to inhibit Wnt/β-catenin signaling which inspired the synthesis of stilbene analogues that inhibit a unique target (methionine adenosyltransferase) and display dramatic anticancer activity.20 Despite these beneficial effects, the low bioavailability of resveratrol limits therapeutic application.21 In humans and rats, less than 5% of an oral dose was observed as free resveratrol with the most abundant metabolites comprised of resveratrol 3-O-glucuronide and resveratrol 3-O-sulfate.22 The β-D-glucosides of resveratrol, 8,239,24 and 1025 (Figure 3) are also naturally occurring products which possess anti-platelet,26 antioxidant,27 and prolyl endopeptidase inhibitory activities, 28 and these activities stimulated the pursuit of various resveratrol glycosylation strategies.29 Among these, the four glycosides 8, 9, 10 and 11 were synthesized in parallel using multi-step trifluoroacetimidate methodology.29d
Figure 3.

(a) Products deriving from OleD ASP-catalyzed glucosylation of resveratrol. (b) Product distribution over time (the standard deviation of three trials was ± 2.5%).
The pilot reaction utilized UDP-Glc as the donor and OleD ASP as catalyst under standard conditions (2.5 mM UDP-Glc, 1 mM aglycon). The reaction was nearly complete within 3 h leading to two diglucosides and two monoglucosides based upon LC-MS. A continuation of the reaction for longer periods of time (48 h) led to the production of two diglucosides 10 and 11 as the only products (data not shown). Based upon this pilot reaction, 3 h was selected as the optimal reaction time for a preparative scale reaction. For the preparative scale reaction, resveratrol (5.7 mg, 25.0 μmol) was dissolved in 1.25 mL DMSO and transferred to 25 mL assay buffer solution (50 mM Tris HCl, 5 mM MgCl2, pH 8.0). The reaction was initiated via addition of UDP-Glc (38 mg, 62.3 μmol) and 30 mg of OleD ASP. After 3 h of gentle agitation at rt, the reaction was frozen and lyophilized to dryness. HPLC purification of the crude reaction gave four products - resveratrol 4′-O-β-D-glucoside (8, 1.8 mg, 4.6 μmol, 18%), resveratrol 3-O-β-D-glucoside (9, 2.0 mg, 5.1 μmol, 20%), resveratrol 3,4′-di-O-β-D-glucoside (10, 2.2 mg, 4.0 μmol, 16%), and resveratrol 3,5-di-O-β-D-glucoside (11, 1.2 mg, 2.2 μmol, 9%). The 1H and 13C NMR and HR-MS data were consistent with previously reported data.29d
Glucosylation of Flavopiridol
Flavopiridol (also known as Alvocidib, HMR-1275, NSC 649890) is a semisynthetic analogue of the alkaloid rohitukine, a compound derived from the indigenous Indian plant Dysoxylum binectarife. 30 Flavopiridol is a cyclin-dependent kinase inhibitor that targets the positive transcription elongation factor P-TEFb, preventing activation of RNA polymerase II. Flavopiridol is cytotoxic to a range of cancer cell lines and initiates cell cycle arrest and p53-independent apoptosis through down-regulation of Mcl-1 and X-linked inactivator of apoptosis (XIAP).31 Preclinical studies demonstrated the capacity of flavopiridol to induce programmed cell death, promote differentiation, inhibit angiogenic processes, and modulate transcriptional events. 32 These unique characteristics inspired extensive clinical investigation of flavopiridol33 Flavopiridol is eliminated via excretion in the form of both the parent drug and the C-5- or C-7-glucuronide.34
Using UDP-Glc as the donor and OleD ASP as catalyst (1.25 mM UDP-Glc, 0.25 mM aglycon, 16 h), the formation of a single monoglucoside (10% conversion) was observed by HPLC and LC-MS analysis (Figure 4). In an effort to boost production of this desired product, reactions catalyzed by a panel of OleD mutants were examined (Figure 4b) which surprisingly revealed wtOleD to enable the best conversion (35%). Thus, flavopiridol (11.1 mg, 25.0 μmol) was dissolved in 1.25 mL DMSO and transferred to 50 mL assay buffer solution (50 mM Tris HCl, 5 mM MgCl2, pH 8.0). The reaction was initiated via addition of UDP-Glc (76 mg, 0.125 mmol) and 15 mg of wtOleD. After 24 h of gentle agitation at rt, the reaction was frozen and lyophilized to dryness. The residue was dissolved in MeOH and subjected to HPLC purification to give flavopiridol monoglucoside product (4.9 mg, 8.7 μmol) in 35% yield.
Figure 4.

(a) OleD-catalyzed glucosylation of flavopiridol. (b) Flavopiridol glucosylation mediated by different OleD variants.
HRESIMS analysis of purified glucoside yielded an [M + H]+ ion at m/z 564.1644, confirming a monoglucoside of flavopiridol with a formula of C27H30ClNO10. 1D and 2D NMR data support the 3′-O–β-D-glucosidic structure presented in Figure 4a. The key evidence for C-3′ glucosylation derives from the HMBC correlation between the anomeric proton and the C-3′ carbon with the large coupling constant (8.0 Hz) of the anomeric proton (δH 4.28, doublet) as a key signature for the β-anomer (Figure S1). That OleD catalysis led to the glucosylation of the C-3′ aliphatic hydroxy was surprising given the typical bias of OleD for aromatic nucleophiles and the previously reported accessibility of flavopiridol C-5- and C-7-OH for glucuronidation.34
Glucosylation of 10-Hydroxycamptothecin
The indole alkaloid 10-hydroxycamptothecin from the Chinese tree Camptotheca acuminata inhibits the activity of DNA topoisomerase I and has a broad spectrum of anticancer activity in vitro and in vivo. 35 The unique mode of action of 10-hydroxycamptothecin has inspired many structure activity relationship studies, which ultimately led to the discovery of two water-soluble drugs used for the treatment of ovarian and lung cancer (topotecan),36 colon cancer (irinotecan).37 Although these drugs are notably effective, their use suffers from dose-limiting toxicities prompting continuing efforts to improve upon the properties of this drug class via structural modification, including glyconjugation.38
Using both wtOleD and OleD ASP, the pilot glucosylation of 10-hydroxycamptothecin under standard conditions (2.5 mM UDP-Glc, 1 mM aglycon) revealed the OleD ASP-catalyzed production of a single monoglucoside product (Figure 5, 50% conversion). In contrast, OleD ASP glucosylation of topotecan (Figure 5a) was low (<3%, data not shown), suggesting steric infringement imposed by substitution at C-9. Thus, OleD ASP was selected to catalyze the preparative scale reaction. For subsequent product characterization, the preparative scale reaction was conducted with 10-hydroxycamptothecin (6 mg, 16.25 μmol), UDP-Glc (50 mg, 81.9 μmol) and OleD ASP (16 mg) in the assay buffer solution (50 mM Tris HCl, 5 mM MgCl2, pH 8.0, 50 mL total volume). After 20 h of gentle agitation at rt, the reaction was frozen and lyophilized to dryness. The residue was dissolved in MeOH and subjected to HPLC purification to give monoglucoside 15 (1.1 mg, 2.1 μmol) in 13% yield.
Figure 5.

(a) OleD ASP-catalyzed glucosylation of 10-hydroxycamptothecin. (b) Glucosylation of 10-hydroxycamptothecin mediated by different OleD variants.
HRESIMS analysis of purified glucoside yielded an [M + Na]+ ion at m/z 549.1484, confirming a monoglucoside of 10-hydroxycamptothecin with a formula of C26H26N2O10. 1D and 2D NMR data support the 10-O-β–D-glucoside structure presented in Figure 5a. The key evidence for C-10 glucosylation derives from the HMBC correlation between the anomeric proton and the C-10 carbon (Figure S1). The anomeric β-configuration is supported by the large coupling constant of anomeric proton (δH 5.12, d, J = 6.0 Hz) (Figure S1). The 1H NMR spectra was consistent with previously reported data.38e,f
Glucosylation of 2-Methoxyestradiol
The naturally occurring estrogen metabolite 2-methoxyestradiol exists at low levels in human blood serum.39 This metabolite is notable as it displays antiproliferative, apoptotic and anti-angiogenesis activities mediated via pathways independent of estrogen receptors.40 Mechanistically, 2-methoxyestradiol was found to invoke microtubule stabilization via the colchicine binding site.41 The in vitro GI50 of 2-methoxyestradiol against the NCI 60 cancer cell ranges from 0.08-5.0 μM and this molecule has led to promising outcomes in phase I and II clinical trials as a new cancer chemotherapy.40a, 42 In addition, 2-methoxyestradiol inhibits vascular smooth muscle cell growth in arteries and induces the expression of endothelial nitric oxide synthase and production of nitric oxide.43 However, the low aqueous solubility (5 nM in H2O) and rapid clearance of 2-methoxyestradiol as the C-3 or C-17-glucuronide 44 has compelled continuing efforts to develop analogues with improved properties.41,45
Using UDP-Glc as the donor, 2-methoxyestradiol was assessed as a substrate for a panel of OleD variants under standard pilot conditions (0.5 mM UDP-Glc, 0.1 mM aglycon, 4 h). Four products, two mono-glucosides and two di-glucosides, were observed by HPLC and LC-MS analysis (Figure 6a) wherein product distribution was dependent on the catalyst employed. To maximize the yield of each product for subsequent characterization, OleD ASP was selected for the preparative scale reaction.
Figure 6.

(a) OleD-catalyzed glucosylation of 2-methoxyestradiol. (b) The glucosylation of 2-methoxyestradiol catalyzed by a panel of OleD variants (the standard deviation of 3 trials was ± 1.5%).
The preparative scale reaction contained 2-methoxyestradiol (7.6 mg, 25.0 μmol), UDP-Glc (76 mg, 0.125 mmol) in 50 mM Tris HCl, 5 mM MgCl2, pH 8.0 (50 mL of total volume) and was initiated with the addition of 20 mg ASP OleD. After gentle agitation at rt for 16 h, the reaction was frozen and lyophilized to dryness. The residue was dissolved in MeOH and subjected to HPLC purification to give 17 (4 mg, 8.6 μmol, 34.4%), 18 (0.1 mg, 0.2 μmol, 0.8%), 19 (1 mg, 1.6 μmol, 6.4%), and 20 (1 mg, 1.6 μmol, 6.4%). The identification of compounds 17-20 was confirmed by 1D, 2D NMR and HRESIMS analysis. The key evidence for C-3, C-17 or C-2′ glucosylation derives from the HMBC correlation between the anomeric proton and the carbon which was glycosylated (Figure S1). The anomeric β-configuration is supported by the large coupling constant (7.0-8.0 Hz) of the anomeric proton (Figure S1). Importantly, this study highlights the first synthesis of 2-methoxyestradiol glucosides. Also particularly intriguing from this study is the apparent C-3 glucosylation (17) en route to disaccharide 19 versus a switch to C-17 glucosylation (18) en route the same product with the AIP variant.
Molecular Dynamics Simulations
The binding modes of all 18 structures by OleD have been studied by molecular docking and include all five native aglycons (3, 7, 12, 14 and 16), all monoglucoside products/intermediates (4, 5, 8, 9, 13, 15, 17 and 18) and all diglucosides (6, 10, 11, 19, 20) (Figure 7). This cumulative analysis revealed all compounds studied, with the exception of flavopiridol (12), to bind in a manner recently described for steroidal glycosides.4b Specifically, ligand binding within this model is mediated via distal hydrogen bonding interactions with key residues (Y114, Y162 and D179) deep within a polar pocket located ~16 Å from the proximal catalytic H19. This analysis indicates an average length of substrates examined to be ~12.5 Å and, in cases where diglucosides were formed, modeling revealed the distal pocket to readily accommodate the larger glucosides with only minor shifts in the H19-aglycon nucleophile distance/alignment. Also consistent with the product distribution observed, docking simulations with 3, 7, and 16 each revealed two modes of binding (head first versus tail first) with similar binding affinity. While flavopiridol was found to occupy the same substrate cavity, the model revealed unique proximal interactions – specifically, side chain hydrogen bonding with Y116 and van der Waals contacts with V81 and F84 – to accommodate the substrate’s smaller size. In addition, this docking model revealed the C-3′ aliphatic hydroxy of flavopiridol as the only aglycon nucleophile in near proximity of the critical H19. Importantly, this collective analysis highlights common binding constraints of a range of non-native substrates for this unique glycosyltransferase and as such, serves as a basis for the application of virtual screening in an attempt to rapidly identify new potential substrates for enzyme-catalyzed glycoconjugation.46
Figure 7.

Binding modes based upon molecular dynamics simulations. A common extended binding mode (panel a) was observed for all compounds highlighted within this study with the exception flavopiridol which adapted a truncated binding mode (panel b).
In summary, this work is an important key follow-up to studies which revealed the uniquely permissive nature of a range of engineered/evolved OleD variants. Specifically, the full characterization of product distributions deriving from OleD-catalyzed reactions with the diverse set of non-native substrates highlighted within this study provides important insights into the regioselectivity of each target reaction and a critical basis for the development of future predictive models for the substrate specificity scope of this fascinating catalyst.
EXPERIMENTAL SECTION
General Experimental Procedures
All chemicals and reagents were purchased from Sigma-Aldrich, unless otherwise stated. NMR spectra, including 1H, 13C, gCOSY, TOCSY, gHSQC, gHMBC were recorded on Varian UNITYINOVA 400 or 500 MHz spectrometer. Mass spectra were acquired on a Bruker MaXis high resolution quadrupole time of flight mass spectrometer.
Protein Expression and Purification
A single colony of E. coli BL21(DE3)pLysS (Stratagene) transformed with the pET28a-based OleD expression vector (wt, ASP, AIP, TDP16, 3-1-H12)3 was used to inoculate 3 mL LB medium supplemented with 50 μg/mL kanamycin and cultured overnight at 37 °C with shaking (250 rpm). The starter culture was then transferred to 1 L LB medium supplemented with 50 μg/mL kanamycin and grown at 37 °C with shaking (250 rpm) until the OD600 reached ~0.7. Isopropyl β-D-thiogalactoside (IPTG) was subsequently added to a final concentration of 0.4 mM and the culture was incubated at 28 °C for approximately 18 h with shaking at 250 rpm. Cell pellets were collected by centrifugation at 10,000 g and 4 °C for 20 min and the supernatant discarded. Cell pellets were resuspended in 10 mL chilled lysis buffer (20 mM phosphate buffer, 0.5 M NaCl, 10 mM imidazole, pH 7.4) and were lysed by sonication (8 pulses of 40 seconds each) in an ice bath. The cell debris was removed by centrifugation at 10,000 g and 4 °C for 20 min. The cleared supernatant was immediately applied to 3 mL of nickel nitrilotriacetic acid (Ni-NTA) resin (QIAgen) pre-equilibrated with wash buffer (20 mM phosphate buffer, pH 7.4, 0.3 M NaCl, 10 mM imidazole). Protein was allowed to bind for 30 min at 4 °C with gentle agitation and the resin was subsequently washed with 4 × 50 mL wash buffer. Finally, the enzyme was eluted from the resin via incubation with 2 mL wash buffer containing 250 mM imidazole for 15 min at 4 °C with gentle agitation. The purified protein was applied to a PD-10 desalting column (Amersham Biosciences) equilibrated with 50 mM Tris-HCl (pH 8.0) and eluted as described by the manufacturer. Protein aliquots were immediately flash frozen in liquid nitrogen and stored at −80 °C. Protein purity was confirmed by SDS-PAGE to be >95% and protein concentration for all studies was determined using the Bradford Protein Assay Kit from Bio-Rad.
General Pilot Scale Reaction
Reactions were conducted in a final volume of 100 μL under standard conditions (50 mM Tris HCl, 5 mM MgCl2, pH 8.0, 0.5 μg/μL purified enzyme, 2.5-5 equiv. UDP-Glc, 1 equvi. aglycon, 25 °C) Two separate control reactions that withheld either enzyme or UDP-Glc were performed in parallel. Reactions were allowed to proceed at 25 °C for specific time period, quenched with 100 μL of cold MeOH, centrifuged at 10,000 g for 10 min and the supernatant was removed for analysis. The clarified reaction mixtures were analyzed by analytical reverse-phase HPLC [Phenomenex 250 mm × 4.6 mm Gemini 5μ C18 column; 1 ml/min; gradient of solvents A (0.1% TFA) and B (100% CH3CN): (a) 0–20 min, 10–75% B; (b) 20–21 min, 75–100% B; (c) 21–26 min, 100% B; (d) 26–29 min, 100–10% B; and (e) 29–35 min, 10% B; A254 detection]. HPLC peak areas were integrated using the Star Chromatography Workstation software (Varian) and the total percent conversion calculated as a percent of the total peak area of substrate and products.
General Procedure for Preparative Scale OleD Catalyzed Glycosylation Reaction
Aglycon was dissolved in 5% DMSO and transferred to pH 8 buffer solution (50 mM Tris HCl, 5 mM MgCl2). UDP-Glc was added followed by OleD catalyst. After the designated time of agitation at rt, the reaction was subsequently frozen and lyophilized, the debris was resuspended in 2 mL of ice cold MeOH, and filtered. Product O-glucosides were isolated by collecting fractions from semi-preparative reverse-phase HPLC [Phenomenex 250 mm × 10 mm Gemini 5μ C18 column; 4.5 mL/min; solvent A (0.1% trifluoroacetic acid) and B (100% CH3CN) using the gradient: 0–20 min, 10–75% B; 20–21 min, 75–100% B; 21–26 min, 100% B; 26–29 min, 100–10% B; and 29–35 min, 10% B; A254 detection]. The product-containing fractions were subsequently collected and lyophilized to provide the corresponding O-glucosides. The compound was then characterized using 1D and 2D NMR, including 1H, 13C, gCOSY, TOCSY, gHSQC, gHMBC.
Flavopiridol 3′-O-β-D-glucoside (13)
white powder, 1H NMR (CD3OD, 500 MHz): 7.81 (dd, J = 7.5, 2.0 Hz, 1H), 7.63 (dd, J = 7.0, 2.0 Hz, 1H), 7.60-7.50 (m, 2H), 6.48 (s, 1H), 6.37 (s, 1H), 4.33 (m, 1H), 4.28 (d, J = 8.0 Hz, 1H), 3.71 (m, 1H), 3.67-3.57 (m, 3H), 3.54 (dd, J = 11.0, 3.0 Hz, 1H), 3.30-3.24 (m, 2H), 3.19 (dd, J = 11.0, 6.0 Hz, 1H), 3.06 (ddd, J = 9.5, 6.0, 3.0 Hz, 1H), 3.00 (dd, J = 9.5, 8.5 Hz, 1H), 2.99 (dd, J = 9.0, 8.0 Hz, 1H), 2.89 (s, 3H), 2.06 (m, 2H); 13C NMR (CD3OD, 125 MHz): 184.0, 164.5, 163.0, 162.7, 162.2, 133.6, 133.4, 133.1, 132.4, 131.8, 129.0, 111.8, 106.8, 105.9, 101.7, 100.0, 77.9, 77.6, 75.3, 74.2, 72.3, 63.5, 58.8, 56.7, 44.2, 37.5, 24.5; HRESIMS m/z 564.16438 [M+H]+ (cacld forC27H31ClNO10, 564.1631).
2-Methoxyestradiol 3-O-β-D-glucoside (17)
white powder, 1H NMR (CD3OD, 500 MHz): 6.91 (s, 1H), 6.88 (s, 1H), 4.84 (d, J = 7.5 Hz, 1H), 3.89 (m, 1H), 3.84 (s, 3H), 3.71 (m, 1H), 3.68 (m, 1H), 3.48 (m, 2H), 3.40 (m, 2H), 2.78 (m, 2H), 2.35 (dd, J = 14.0, 3.0 Hz, 1H), 2.20 (m, 1H), 2.06 (m, 1H), 2.00 (m, 1H), 1.90 (dd, J = 12.0, 2.5 Hz, 1H), 1.72 (m, 1H), 1.60-1.17 (m, 7H), 0.80 (s, 3H); 13C NMR (CD3OD, 100 MHz): 148.8, 146.1, 136.2, 130.9, 118.8, 111.6, 103.3, 82.6, 78.4, 78.0, 75.1, 71.6, 62.7, 57.2, 51.4, 45.9, 44.5, 40.5, 38.2, 30.9, 30.3, 28.7, 27.8, 24.2, 11.8; HRESIMS m/z 487.2304 [M+Na]+ (cacld for C25H36O8 Na, 487.2302).
2-Methoxyestradiol 17-O-β-D-glucoside (18)
white powder, 1H NMR (CD3OD, 500 MHz): 8.51 (s, 1H), 6.77 (s, 1H), 6.45 (s, 1H), 4.34 (d, J = 8.0 Hz, 1H), 3.83 (dd, J = 12.0, 2.0 Hz, 1H), 3.80 (d, J = 8.0 Hz, 1H), 3.77 (s, 3H), 3.65 (dd, J = 12.0, 6.0 Hz, 1H), 3.40-3.15 (m, 4H), 2.68 (m, 2H), 2.63 (s, 1H), 2.17-2.10 (m, 3H), 1.66 (m, 2H), 1.54-1.15 (m, 7H), 0.86 (s, 3H); 13C NMR (CD3OD, 125 MHz): 147.1, 145.4, 132.8, 130.4, 116.6, 110.5, 104.9, 90.0, 78.3, 78.0, 75.6, 71.9, 63.0, 56.7, 51.3, 45.4, 44.4, 40.4, 39.0, 30.2, 30.1, 28.7, 28.0, 24.2, 12.3; HRESIMS m/z 487.2304 [M+Na]+(cacld for C25H36O8 Na, 487.2302).
2-Methoxyestradiol 3,17-di-O-β-D-glucoside (19)
white powder, 1H NMR (CD3OD, 500 MHz): 6.86 (s, 1H), 6.83 (s, 1H), 4.80 (d, J = 7.0 Hz, 1H), 4.34 (d, J = 7.5 Hz, 1H), 3.83 (m, 1H), 3.79 (s, 3H), 3.66 (d, J = 5.0 Hz, 1H), 3.64 (d, J = 5.0 Hz, 1H), 3.73-3.63 (m, 3H), 3.54 (dd, J = 12.0, 4.0 Hz, 1H), 3.45 (m, 2H), 3.38 (m, 2H), 3.30 (m, 1H), 3.26 (m, 1H), 3.19 (m, 1H), 2.79 (m, 2H), 2.35 (dd, J = 14.0, 3.0 Hz, 1H), 2.21 (m, 1H), 2.06 (m, 1H), 2.00 (m, 1H), 1.90 (dd, J = 12.0, 2.0 Hz, 1H), 1.72 (m, 1H), 1.60-1.17 (m, 7H), 0.81 (s, 3H); 13C NMR (CD3OD, 125 MHz): 148.7, 145.3, 136.1, 130.8, 118.7, 111.5, 104.9, 103.2, 89.9, 78.36, 78.32, 78.04, 78.00, 75.6, 75.1, 71.8, 71.6, 62.9, 62.7, 57.1, 51.3, 45.9, 44.7, 40.5, 40.2, 30.3, 30.1, 28.6, 27.9, 24.1, 12.3; HRESIMS m/z 649.2838 [M+Na]+ (cacld for C31H46O13Na, 649.2831).
2-Methoxyestradiol 3-(O-β-D-glucopyranosyl)-(1″→ 2′)-(O-β-D-glucoside) (20)
white powder, 1H NMR (CD3 OD, 500 MHz): 6.91 (s, 1H), 6.85 (s, 1H), 5.13 (d, J = 7.5 Hz, 1H), 4.78 (d, J = 8.0 Hz, 1H), 3.90 (m, 1H), 3.82 (s, 3H), 3.78 (dd, J = 9.0, 8.0 Hz, 1H), 3.73-3.63 (m, 3H), 3.54 (dd, J = 12.0, 4.0 Hz, 1H), 3.45 (m, 2H), 3.38 (m, 2H), 3.30 (m, 1H), 3.26 (m, 1H), 3.19 (m, 1H), 2.79 (m, 2H), 2.35(dd, J = 14.0, 3.0 Hz, 1H), 2.21 (m, 1H), 2.06 (m, 1H), 2.00 (m, 1H), 1.90 (dd, J = 12.0, 2.0 Hz, 1H), 1.72 (m, 1H), 1.60-1.17 (m, 7H), 0.81 (s, 3H); 13C NMR (CD3OD, 125 MHz): 148.0, 145.8, 135.6, 130.7, 117.0, 111.4, 104.2, 100.9, 82.6, 82.5, 78.0, 77.93, 77.88, 77.78, 75.7, 71.2, 71.0, 62.7, 62.0, 57.1, 51.4, 45.9, 44.5, 40.5, 38.3, 30.9, 30.4, 28.7, 28.0, 24.2, 11.9; HRESIMS m/z 649.2835 [M+Na]+ (cacld for C31H46O13Na, 649.2831).
Molecular Dynamics Simulations
Multiple conformations of each ligand were generated using OMEGA (Open Eye Scientific Software). The conformations of each ligand were fitted in the binding cavity of OleD (PDBID 2IYF)1f using Sabre and Fred software packages. 47 The docking strategy exhaustively explored all possible positions of each ligand in the binding site and generally focused upon two parameters - shape and optimization. During the shape fitting, the ligand was placed into a grid box encompassing all active-site atoms (including hydrogen atoms) using smooth Gaussian potential. 48 Two optimization filters were subsequently processed - rigid-body optimization and optimization of the ligand pose in the dihedral angle space. The pose ensemble was then filtered to first reject poses that did not have sufficient shape complementarity with the active site of the protein followed by rejection of those lacking at least one heavy atom hydrogen bond with the His19 imidazole.
In separate docking runs, the binding poses of the ligand structure were refined by MD simulations followed by MM-GBSA calculations using the Sander module from Amber11 package49 as previously described.46 Briefly, the OleD-ligand binding complex was neutralized by adding appropriate counter ions and was solvated in a rectangular box of TIP3P H2O molecules with a minimum solute-wall distance of 10 Å.50 The partial atomic charges used for the ligand were the electrostatic potential (ESP)-fitted atomic charges, calculated at ab initio HF/6-31G* level using the Gaussian03 program.51 The solvated systems were energy-minimized and carefully equilibrated. These systems were gradually heated from T = 10 K to T = 298.15 K in 100 ps before running the MD simulation. The MD simulations were performed with a periodic boundary condition in the NPT ensemble at T = 298.15 K with Berendsen temperature coupling and constant pressure (P=1 atm) with isotropic molecule-based scaling.52 A time step of 2.0 fs was used, with a cutoff of 12 Å for the nonbonded interactions, and the SHAKE algorithm was employed to keep all bonds involving hydrogen atoms rigid.53 Long-range interactions were handled using the particle mesh Ewald (PME) algorithm.54 As we did in our previous work,4b only the ligand and residue side chains in the binding pocket were permitted to move during the energy minimization and MD simulations. We used the constraint to prevent any changes in the OleD structure due to the presence of residues in the loops on the top of the protein active site. A residue-based cutoff of 12 Å was utilized for non-covalent interactions. MD simulations were then carried out for 4.0 ns. During the simulations, the coordinates of the system were collected every 10 ps.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH R37 AI52218 (JST) and the National Center for Advancing Translational Sciences (UL1TR000117). We thank the University of Wisconsin College of Pharmacy Analytical Instrumentation Center for analytical support and Professor G. Williams (North Carolina State University), Dr. S. Singh and Dr. R. W. Gantt (Dupont) for materials and helpful discussion.
Footnotes
ASSOCIATED CONTENT
Supporting Information. 1H and 13C spectroscopic data, molecular docking data and individual docking illustrations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): The authors report competing interests. J.S.T. is a co-founder of Centrose (Madison, WI).
REFERENCES
- (1).(a) Hernández C, Olano C, Méndez C, Salas JA. Gene. 1993;134:139–140. doi: 10.1016/0378-1119(93)90189-a. [DOI] [PubMed] [Google Scholar]; (b) Olano C, Rodriguez AM, Mendez C, Salas JA. Mol. Microbiol. 1995;16:333–343. doi: 10.1111/j.1365-2958.1995.tb02305.x. [DOI] [PubMed] [Google Scholar]; (c) Quirós LM, Aguirrezabalaga I, Olano C, Méndez C, Salas JA. Mol. Microbiol. 1998;6:1177–1185. doi: 10.1046/j.1365-2958.1998.00880.x. [DOI] [PubMed] [Google Scholar]; (d) Quirós LM, Carbajo RJ, Braña AF, Salas JA. J. Biol. Chem. 2000;275:11713–11720. doi: 10.1074/jbc.275.16.11713. [DOI] [PubMed] [Google Scholar]; (e) Zhao L, Beyer NJ, Borisova SA, Liu HW. Biochemistry. 2003;42:14794–14804. doi: 10.1021/bi035501m. [DOI] [PubMed] [Google Scholar]; (f) Bola DN, Roberts S, Proctor MR, Turkenburg JP, Dodson EJ, Martinez-Fleites C, Yang M, Davis BG, Davies GJ, Gilbert H. Proc. Natl. Acad. Sci. USA. 2007;104:5336–5341. doi: 10.1073/pnas.0607897104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Yang M, Proctor MR, Bolam DN, Errey JC, Field RA, Gilbert HJ, Davis BG. J. Am. Chem. Soc. 2005;127:9336–9337. doi: 10.1021/ja051482n. [DOI] [PubMed] [Google Scholar]; (b) Gantt RW, Goff RD, Williams GJ, Thorson JS. Angew. Chem. Int. Ed. 2008;47:8889–8892. doi: 10.1002/anie.200803508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Williams GJ, Zhang C, Thorson JS. Nat. Chem. Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]; (b) Williams GJ, Goff RD, Zhang C, Thorson JS. Chem Biol. 2008;15:393–401. doi: 10.1016/j.chembiol.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gantt RW, Peltier-Pain P, Cournoyer WJ, Thorson JS. Nat. Chem. Biol. 2011;7:685–691. doi: 10.1038/nchembio.638. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gantt RW, Peltier-Pain P, Thorson JS. Nat. Prod. Rep. 2011;28:1811–1853. doi: 10.1039/c1np00045d. [DOI] [PubMed] [Google Scholar]
- (4).(a) Zhou M, Thorson JS. Org. Lett. 2011;13:2786–2788. doi: 10.1021/ol200977u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou M, Hou Y, Hamza A, Zhan C-G, Bugni TS, Thorson JS. Org. Lett. 2012;14:5424–5427. doi: 10.1021/ol3024924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Choi SH, Ryu M, Yoon YJ, Kim DM, Lee EY. Biotechnol. Lett. 2012;34:499–505. doi: 10.1007/s10529-011-0789-z. [DOI] [PubMed] [Google Scholar]
- (6).Hendrich S, Lee K-W, Xu X, Wang H-J, Murphy PA. J. Nutr. 1994;124:1789S–1792S. doi: 10.1093/jn/124.suppl_9.1789S. [DOI] [PubMed] [Google Scholar]
- (7).(a) Galati G, O’Brien PJ. Free Radical Biol. Med. 2004;37:287–303. doi: 10.1016/j.freeradbiomed.2004.04.034. [DOI] [PubMed] [Google Scholar]; b) Barnes S, Prasain J. Curr.Opin. Plant. Biol. 2005;8:324–328. doi: 10.1016/j.pbi.2005.03.010. [DOI] [PubMed] [Google Scholar]
- (8).(a) Shibuya Y, Tahara S, Kimura Y, Miyzutani J. Z. Naturforsch. 1991;C46:513–518. [Google Scholar]; (b) Hwang M-H, Kwon Y-S, Kim C-M. Nat. Med. 1998;52:527–528. [Google Scholar]
- (9).Keung WM, Vallee BL. Phytochemistry. 1998;47:499–506. doi: 10.1016/s0031-9422(97)00723-1. [DOI] [PubMed] [Google Scholar]
- (10).Shimoda K, Hamada H, Hamada H. Int. J. Mol. Sci. 2011;12:5616–5625. doi: 10.3390/ijms12095616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Lewis P, Kaltia S, a K. J. Chem. Soc., Perkin Trans. 1998;1:2481–2484. [Google Scholar]; (b) Lewis PT, Wähälä K. Tetrahedron Lett. 1998;39:9559–9562. [Google Scholar]; (c) Weis M, Lim E-K, Bruce N, Bowles D. Angew. Chem. 2006;118:3614–3618. doi: 10.1002/anie.200504505. [DOI] [PubMed] [Google Scholar]
- (12).Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- (13).Howitz1 KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang L-L, Scherer B, Sinclair DA. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- (14).Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE. Neurochem. Int. 2009;54:111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Elmali N, Baysal O, Harma A, Esenkaya I, Mizrak B. Inflammation. 2007;30:1–6. doi: 10.1007/s10753-006-9012-0. [DOI] [PubMed] [Google Scholar]
- (16).(a) Kopp P. Eur. J. Endocrinol. 1998;138:619–620. doi: 10.1530/eje.0.1380619. [DOI] [PubMed] [Google Scholar]; (b) Ferrières J. Heart. 2004;90:107–111. doi: 10.1136/heart.90.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Su HC, Hung LM, Chen JK. Am. J. Physiol. Endocrinol. Metab. 2006;290:E1339–E1346. doi: 10.1152/ajpendo.00487.2005. [DOI] [PubMed] [Google Scholar]
- (18).Docherty JJ, Sweet TJ, Bailey E, Faith SA, Booth T. Antiviral Res. 2006;72:171–177. doi: 10.1016/j.antiviral.2006.07.004. [DOI] [PubMed] [Google Scholar]
- (19).(a) Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]; (b) Cal C, Garban H, Jazirehi A, Yeh C, Mizutani Y, Bonavida B. Curr. Med. Chem. Anticancer Agents. 2003;3:77–93. doi: 10.2174/1568011033353443. [DOI] [PubMed] [Google Scholar]; (c) Huang X, Zhu H-L. Med. Chem. 2011;1 doi:10.4172/2161-0444.1000104. [Google Scholar]
- (20).Zhang W, Sviripa V, Kril LM, Chen X, Yu T, Shi J, Rychahou P, Evers BM, Watt DS, Liu C. J. Med. Chem. 2011;54:1288–1297. doi: 10.1021/jm101248v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Goldberg DA, Yan J, Soleas GJ. Clin. Biochem. 2003;36:79–87. doi: 10.1016/s0009-9120(02)00397-1. [DOI] [PubMed] [Google Scholar]; (b) Walle T, Hsieh F, DeLegge MH, Oatis JE, Jr, Walle UK. Drug Metab. Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- (22).(a) Yu C, Shin YG, Chow A, Li Y, Kosmeder JW, Lee YS, Hirschelman WH, Pezzuto JM, Mehta RG, van Breemen RB. Pharm Res. 2002;19:1907–1914. doi: 10.1023/a:1021414129280. [DOI] [PubMed] [Google Scholar]; (b) Goldberg DM, Yan J, Soleas GJ. Clin.Biochem. 2003;36:79–87. doi: 10.1016/s0009-9120(02)00397-1. [DOI] [PubMed] [Google Scholar]
- (23).Orsini F, Pelizzoni F, Verotta L, Aburjai T. J. Nat. Prod. 1997;60:1082–1087. doi: 10.1021/np970069t. [DOI] [PubMed] [Google Scholar]
- (24).Nonaka G, Minani M, Nishioka I. Chem. Pharm. Bull. 1977;25:2300–2305. doi: 10.1248/cpb.25.2300. [DOI] [PubMed] [Google Scholar]
- (25).(a) Hano Y, Goi K, Nomura T, Ueda S. Cell Mol. LifeSci. 1997;53:237–241. [Google Scholar]; (b) Zhou CX, Kong LD, Ye WC, Cheng CHK, Tan RX. Planta Med. 2001;67:158–161. doi: 10.1055/s-2001-11500. [DOI] [PubMed] [Google Scholar]
- (26).Aburjai TA. Phytochemistry. 2000;55:407–410. doi: 10.1016/s0031-9422(00)00341-1. [DOI] [PubMed] [Google Scholar]
- (27).Teguo PW, Fauconneau B, Deffieux G, Huguet F, Vercauteren J, Merillon J-M. J. Nat. Prod. 1998;61:655–657. doi: 10.1021/np9704819. [DOI] [PubMed] [Google Scholar]
- (28).Fan W, Tezuka Y, Kadota S. Chem. Pharm. Bull. 2000;48:1055–1061. doi: 10.1248/cpb.48.1055. [DOI] [PubMed] [Google Scholar]
- (29).(a) Orsini F, Pelizzoni F, Bellini B, Miglierini G. Carbohydr. Res. 1997;301:95–109. doi: 10.1016/s0008-6215(97)00087-6. [DOI] [PubMed] [Google Scholar]; (b) Brandolini V, Maietti A, Tedeschi P, Durini E, Vertuani S, Manfredini S. J. Agric. Food Chem. 2002;50:7407–7411. doi: 10.1021/jf0256384. [DOI] [PubMed] [Google Scholar]; (c) Learmonth DA. Synth. Commun. 2004;34:1565–1575. [Google Scholar]; (d) Zhang Z, Yu B, Schmidt RR. Synthesis. 2006;8:1301–1306. [Google Scholar]
- (30).Wirger A, Perabo FGE, Burgemeister LH, Schmidt DH, Doehn CD, Jocham D. Anticancer Res. 2005;25:4341–4348. [PubMed] [Google Scholar]
- (31).(a) Ni W, Ji J, Dai Z, Papp A, Johnson AJ, Ahn S, Farley KL, Lin TS, Dalton JT, Li X, Jarjoura D, Byrd JC, Sadee W, Grever MR, Phelps MA. PLoS One. 2010;5:e13792. doi: 10.1371/journal.pone.0013792. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jain SK, Bharate SB, Vishwakarma RA. Mini-Reviews in Med. Chem. 2012;12:632–649. doi: 10.2174/138955712800626683. [DOI] [PubMed] [Google Scholar]
- (32).(a) Sedlacek H, Czech J, Naik R, Kaur G, Worland PJ, Losiewiecz MD, Parker B, Carlson BA, Smith A, Senderowicz A, Sausville EA. Int. J. Oncol. 1996;9:1143–1168. doi: 10.3892/ijo.9.6.1143. [DOI] [PubMed] [Google Scholar]; (b) Shapiro GI. Clin. Cancer Res. 2004;10:4270S–4275S. doi: 10.1158/1078-0432.CCR-040020. [DOI] [PubMed] [Google Scholar]; (c) Newcomb EW. Anticancer Drugs. 2004;15:411–419. doi: 10.1097/01.cad.0000127332.06439.47. [DOI] [PubMed] [Google Scholar]
- (33).(a) Sedlacek HH, Homann D, Czech J, Kolar C, Seeman G, Gussow D, Bosslet K. Chimia. 1991;45:311–316. [Google Scholar]; (b) Czech J, Homann D, Naik R, Sedlacek HH. Int. J. Oncol. 1995;6:31–36. [PubMed] [Google Scholar]; (c) http://clinicaltrials.gov/ct2/results?term=flavopiridol.
- (34).(a) Jager W, Zembsch B, Wolschann P, Pittenauer E, Senderowicz AM, Sausville EA, Sedlacek HH, Graf J, Thalhammer T. Life Sci. 1998;62:1861–1873. doi: 10.1016/s0024-3205(98)00152-0. [DOI] [PubMed] [Google Scholar]; (b) Jager W, Gehringa E, Hagenauera B, Austb S, Senderowiczc A, Thalhammer T. Life Sci. 2003;73:2841–2854. doi: 10.1016/s0024-3205(03)00699-4. [DOI] [PubMed] [Google Scholar]
- (35).(a) Ling YH, Andersson BS, Nelson JA. Cancer Biochem. Biophys. 1990;11:23–30. [PubMed] [Google Scholar]; (b) Zhang R, Cai Q, Lindsey JR, Li Y, Chambless B, Naguib FNM. Int. J. Oncol. 1997;10:1147–1156. doi: 10.3892/ijo.10.6.1147. [DOI] [PubMed] [Google Scholar]; (c) Zhou JJ, Liu J, Xu B. Acta Pharmacol Sinica. 2001;22:827–830. [PubMed] [Google Scholar]; (d) Liang YJ, Fu LW, Ding Y, Xiong HY, Chen LM, Yang XP, Pan QC. Aizheng. 2003;22:368–371. [PubMed] [Google Scholar]; (e) Ping YH, Lee HC, Lee JY, Wu PH, Ho LK, Chi CW, Lu MF, Wang JJ. Oncol. Rep. 2006;15:1273–1279. [PubMed] [Google Scholar]
- (36).Kingsbury WD, Boehm JC, Jakas DR, Holden KG, Hecht SM, Gallagher G, Caranfa MJ, McCabe FL, Faucette LF, Johnson RK, Hertzberg RP. J. Med. Chem. 1991;34:98–107. doi: 10.1021/jm00105a017. [DOI] [PubMed] [Google Scholar]
- (37).Sawada S, Okajima S, Aiyama R, Nokata K-I, Furuta T, Yokokura T, Sugino E, Yamaguchi K, Miyasaka T. Chem. Pharm. Bull. 1991;39:1446–1450. doi: 10.1248/cpb.39.1446. [DOI] [PubMed] [Google Scholar]
- (38).(a) Hecht JR. Oncology (Huntingt) 1998;12:72–78. [PubMed] [Google Scholar]; (b) Lerchen HG, Baumgarten J, von dem Bruch K, Lehmann TE, Sperzel M, Kempka G, Fiebig HH. J. Med. Chem. 2001;44:4186–95. doi: 10.1021/jm010893l. [DOI] [PubMed] [Google Scholar]; (c) Ulukan H, Swaan PW. Drugs. 2002;62:2039–2057. doi: 10.2165/00003495-200262140-00004. [DOI] [PubMed] [Google Scholar]; (d) Thomas CJ, Rahier NJ, Hecht SM. Bioorg. Med. Chem. 2004;12:1585–604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]; (e) Li Q, Zhang Y, Yao L, Fu Y, Zu Y, Chen X, Zheng C. Acta Pharmaceutica Sinica. 2005;40:1116–1121. [PubMed] [Google Scholar]; (f) Xu C, Cui J, Yang L, Huang M. Chin. Pharmaceut. J. 2006;37:299–300. [Google Scholar]; (g) Leu YL, Chen CS, Wu YJ, Chern JW. J. Med. Chem. 2008;51:1740–1746. doi: 10.1021/jm701151c. [DOI] [PubMed] [Google Scholar]; (h) Jiao Y, Liu H, Geng M, Duan W. Bioorg. Med. Chem. Lett. 2011;21:2071–2074. doi: 10.1016/j.bmcl.2011.02.005. [DOI] [PubMed] [Google Scholar]; (i) Leu YL, Chen CS, Wu YJ, Chern JW. J. Med. Chem. 2008;51:1740–1746. doi: 10.1021/jm701151c. [DOI] [PubMed] [Google Scholar]
- (39).(a) Lakhani NJ, Sarkar MA, Venitz J, Figg WD. Pharmacotherapy. 2003;23:165–172. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]; (b) Xu X, Roman JM, Issaq HJ, Keefer LK, Veenstra TD, Ziegler RG. Anal. Chem. 2007;79:7813–7821. doi: 10.1021/ac070494j. [DOI] [PubMed] [Google Scholar]; (c) Dubey RK, Jackson EK. Trends Endocrinol. Metab. 2009;20:374–379. doi: 10.1016/j.tem.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).(a) Pribluda VS, Gubish ER, LaVallee TM, Treston A, Swartz GM, Green SJ. Cancer Metastasis Rev. 2000;19:173–179. doi: 10.1023/a:1026543018478. [DOI] [PubMed] [Google Scholar]; (b) LaVallee TM, Zhan XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS. Cancer Res. 2002;62:3691–3697. [PubMed] [Google Scholar]
- (41).(a) Verenich S, Gerk PM. Mol. Pharmaceutics. 2010;7:2030–2039. doi: 10.1021/mp100190f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mueck AO, Seeger H. Steroids. 2010;75:625–631. doi: 10.1016/j.steroids.2010.02.016. [DOI] [PubMed] [Google Scholar]
- (42).(a) Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, Fife K, Armstrong V, Treston A, Sidor C, Wilding G. Clin. Cancer Res. 2005;11:6625–6633. doi: 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]; (b) Dahut WL, Lakhani NJ, Gulley JL, Arlen PM, Kohn EC, Kotz H, McNally D, Parr A, Nguyen D, Yang SX, Steinberg SM, Venitz J, Sparreboom A, Figg WD. Cancer Biol. Ther. 2006;5:22–27. doi: 10.4161/cbt.5.1.2349. [DOI] [PubMed] [Google Scholar]; (c) James J, Murry DJ, Treston AM, Storniolo AM, Sledge GW, Sidor C, Miller KD. Invest. New Drugs. 2006;25:41–48. doi: 10.1007/s10637-006-9008-5. [DOI] [PubMed] [Google Scholar]; (d) Rajkumar SV, Richardson PG, Lacy MQ, Dispenzieri A, Greipp PR, Witzig TE, Schlossman R, Sidor CF, Anderson KC, Gertz MA. Clin. Cancer Res. 2007;13:6162–6167. doi: 10.1158/1078-0432.CCR-07-0807. [DOI] [PubMed] [Google Scholar]
- (43).(a) Lakhani NJ, Sarkar MA, Venitz J, Figg WD. Pharmacotherapy. 2003;23:165–172. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]; (b) Kurokawa A, Azuma K, Mita T, Toyofuko Y, Fujitani Y, Hirose T, Iwabuchi K, Ogawa H, Takeda S, Kawamori R, Watada H. Endocr. J. 2007;54:1027–1031. doi: 10.1507/endocrj.k07e-034. [DOI] [PubMed] [Google Scholar]; (c) Dubey RK, Jackson EK. Trends Endocrinol. Metab. 2009;20:374–379. doi: 10.1016/j.tem.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).(a) Lakhani NJ, Sparreboom A, Xu X, Veenstra TD, Venitz J, Dahut WL, Figg WD. J. Pharm. Sci. 2007;96:1821–1831. doi: 10.1002/jps.20837. [DOI] [PubMed] [Google Scholar]; (b) Shi X, Lee I, Chen X, Shen M, Xiao S, Zhu M, Baker RJ, Jr, Wang SH. Soft Matter. 2010;6:2539–2545. doi: 10.1039/b925274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).(a) Basu NK, Kubota S, Meselhy MR, Ciotti M, Chowdhury B, Hartori M, Owens IS. J. Biol. Chem. 2004;279:28320–28329. doi: 10.1074/jbc.M401396200. [DOI] [PubMed] [Google Scholar]; (b) Lépine J, Bernard O, Plante M, Têtu B, Pelletier G, Labrie F, Bélanger A, Guillemette C. J. Clin. Endocrin. Metab. 2004;89:5222–5232. doi: 10.1210/jc.2004-0331. [DOI] [PubMed] [Google Scholar]; (c) Seeger H, Mueck AO. J. Reproduktionsmed. Endokrinol. 2010;7:62–66. [Google Scholar]; (d) Peyrat J-F, Brion J-D, Alami M. Curr. Med. Chem. 2012;19:4142–4156. doi: 10.2174/092986712802430072. [DOI] [PubMed] [Google Scholar]
- (46).(a) Hamza A, Zhao X, Tong M, Tai H-H, Zhan C-G. Bioorg. Med. Chem. 2011;19:6077–6086. doi: 10.1016/j.bmc.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang W, AbdulHameed MDM, Hamza A, Zhan C-G. Bioorg. Med. Chem. Lett. 2012;22:1629–1632. doi: 10.1016/j.bmcl.2011.12.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).(a) Fred v. OpenEye Scientific Software, Inc.; Santa Fe, NM, USA: 2007. www.eyesopen.com. [Google Scholar]; (b) Hamza A, Wei NN, Zhan CG. J. Chem. Inf. Model. 2012;52:963–974. doi: 10.1021/ci200617d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Biopolymers. 2003;68:76–90. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- (49).Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 11. University of California; San Francisco, CA: 2010. [Google Scholar]
- (50).Jorgensen WL. J. Am. Chem. Soc. 1981;103:335–340. [Google Scholar]
- (51).(a) Singh UC, Kollman PA. J. Comput. Chem. 1984;5:129–145. [Google Scholar]; (b) Besler BH, Merz KM, Kollman PA. J. Comput. Chem. 1990;11:431–439. [Google Scholar]; (c) Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Stratmann RE, Burant JC, Dapprich S, Millan JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malich DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzales C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andreas JL, Head-Gordon M, Reploge ES, Pople JA. Gaussian 03, revision B-03. Gaussian, Inc.; Pittsburgh, PA: 2003. [Google Scholar]
- (52).Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- (53).Ryckaert JP, Ciccotti G, Berendsen HJC. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- (54).Darden T, York D, Pedersen L. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.