

Abstract
Bacteria utilize a large multi-protein chemosensory array to sense attractants and repellents in their environment. The array is a hexagonal lattice formed from three core proteins: a transmembrane receptor, the His kinase CheA, and the adaptor protein CheW. The resulting, highly networked array architecture yields several advantages including strong positive cooperativity in the attractant response and rapid signal transduction through the preformed, integrated signalling circuit. Moreover, when isolated from cells or reconstituted in isolated bacterial membranes, the array possesses extreme kinetic stability termed ultra-stability (Erbse & Falke (2009) Biochemistry 48:6975-87) and is the most long-lived multi-protein enzyme complex described to date. The isolated array retains kinase activity, attractant regulation and its bound core proteins for days or more at 22° C. The present work quantitates this ultra-stability and investigates its origin. The results demonstrate that arrays consist of two major components: (i) a quasi-stable component with a lifetime of 1–2 days that decays due to slow proteolysis of CheA kinase in the lattice, and (ii) a truly ultra-stable component with a lifetime of ~20 days that is substantially more protected from proteolysis. Following proteolysis of the quasi-stable component the apparent positive cooperativity of the array increases, arguing the quasi-stable component is not as cooperative as the ultra-stable component. Introduction of structural defects into the array by coupling a bulky probe to a subset of receptors reveals that modification of only 2% of the receptor population is sufficient to abolish ultra-stability, supporting the hypothesis that the ultra-stable component requires a high level of array spatial order. Overall, the findings are consistent with a model in which the quasi- and ultra-stable components arise from distinct regions of the array, such that the ultra-stable regions possess more extensive, better-ordered, multi-valent interconnectivities between core components, thereby yielding extraordinary stability and cooperativity. Furthermore, the findings indicate that the chemosensory array is a promising platform for the development of ultra-stable biosensors.
Keywords: protein complex, protein array, protein stability, sensory transduction, chemoreceptor, histidine-kinase, CheA
INTRODUCTION
Two-component signalling pathways are the predominant signalling systems in bacteria. At the simplest level, two component pathways typically contain a histidine kinase fused or bound to a transmembrane receptor, and an aspartate kinase that functions as a response regulator (1–3). Bacteria have adapted two component systems to sense chemical gradients in their environment and bias their movements up gradients of attractant and down gradients of repellants (4–6). Sensing of chemical attractants and repellants by the closely related Escherichia coli and Salmonella typhimurium chemosensory pathways is accomplished by transmembrane chemoreceptors that detect simple ligands including sugars and amino acids (7, 8). Receptor monomers assemble into stable homodimers. These homodimers associate into trimers-of-dimers, and ultimately yield extended hexagonal arrays containing thousands of receptors with a trimer-of-dimers at each vertex (9–13). The hexagonal arrays require two additional core proteins for stable assembly and signal transduction: the homodimeric histidine kinase CheA, and the adaptor protein CheW, which both dock to the cytoplasmic tips of the receptors (10,11, 14–20).
The signal output of the array is controlled by the ligand occupancy of the receptors and by the covalent modification state of the receptor adaptation sites (8, 21–23). In the absence of attractant, the apo receptors stimulate CheA kinase, triggering autophosphorylation of its active site histidine (1–6). Subsequently this phosphate is transferred to the active site aspartate of the response regulator CheY. The resulting phospho-CheY diffuses to the rotary swimming motor where it binds and modulates the rotational bias and swimming state, thereby controlling cell migration. Attractant binding to the chemoreceptors triggers a transmembrane signal that switches off CheA kinase autophosphorylation, thereby reducing the cytoplasmic phospho-CheY level and modulating the swimming state. Covalent modification of the adaptation site glutamates, either by methyl esterification or amidation, opposes the attractant signal and stimulates CheA kinase.
The assembled array is a simple, biological integrated circuit or sensory chip with high positive cooperativity and rapid signal transmission (21, 23–27). Previous localization and structural studies have proposed that extensive interconnectivity exists between the three core proteins in the array, particularly at the cytoplasmic tips of the receptors where interlocked rings of CheA-CheW are coupled to the receptor lattice (21, 23, 25, 28–35). The resulting working model of array structure, illustrated in Figure 1, has a very precise three dimensional architecture with all of the core components exhibiting a high degree of spatial order in relationship to one another. The combination of precise order and extensive interconnectivity afford yet another unique advantage: ultra-stability. Such ultra-stability is hypothesized to result from the locking together of the interconnected array core proteins, much like the stability conferred to a jig-saw puzzle once all of its pieces are snapped together (25).
Figure 1.
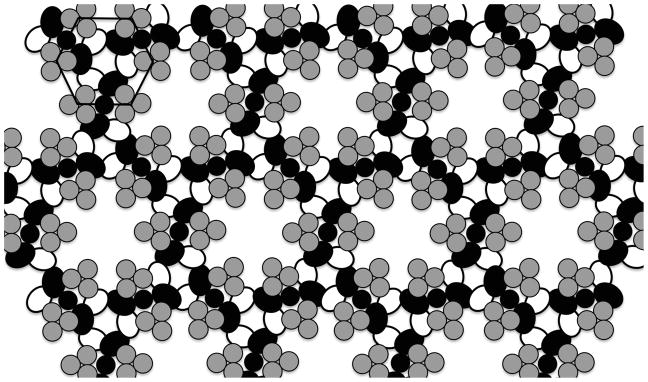
Working model of the core bacterial chemosensory array (20, 31, 32). Receptor trimer-of-dimers (gray) are arrayed in a hexagonal pattern with one trimer at each vertex. Homodimers of the multi-domain CheA His kinase (black) pack between the long edges of adjacent trimers and also contact CheW (white oval). CheW and the structurally homologous P5 domain of CheA (black oval) interact with one another in a head to tail fashion forming an interconnected ring. This basic hexagonal unit is propagated throughout the array, forming a network of interconnected rings.
The present study begins by characterizing the remarkably slow decay kinetics of arrays in vitro by measuring the loss of kinase activity as well as changes in the stoichiometries of the array core proteins. The results indicate that arrays are composed of two stable components with lifetimes of days and weeks, respectively, where the more labile component is lost due to proteolysis. Moreover, there is a strong correlation between array order, cooperativity and stability.
MATERIALS AND METHODS
Materials
All chemicals were highly pure reagent-grade as obtained from Sigma Aldrich with the following exceptions: [γ-32P]ATP was from Perkin-Elmer, DTT was from Research Products International, Ni-NTA agarose resin was from Qiagen, Bicinchoninic Acid (BCA) Assay reagents were from Bio-rad.
Protein Expression and Purification
Cysless CheA and CheW from S. typhimurium were expressed with 6×His tags on their N-termini from the plasmids pET6H-CheA and pET6H-CheW respectively (20). CheY from E. coli was expressed with an N-terminal YFP and C-terminal 6×His from the plasmid pVSYFP-CheY-6H (36). All soluble proteins were isolated as previously described with standard Ni-NTA agarose affinity chromatography (20, 36). Protein concentrations for CheA and CheW were estimated using the BCA assay according to the manufacturers instructions. The YFP-CheY concentration was estimated from its YFP absorbance at 514 nm and the YFP extinction coefficient at this wavelength, 18,000 M−1cm−1.
E. coli serine receptor (Tsr) was overexpressed in the gutted RP3098 strain of E. coli— which lacks all chemotaxis proteins including receptors and adaptation enzymes—using the plasmid pJC3 (37). Inside out, inner membrane vesicles containing Tsr were isolated as previously described (37). The total protein concentration in the membranes was determined with the BCA assay and the fraction of receptors in the total protein content was determined by quantifying SDS-PAGE gels with ImageJ. Receptor concentration was estimated from the receptor fraction of the total protein concentration.
Reconstitution of Signaling Arrays In Vitro
Signaling arrays were reconstituted by combining 6.7 μM Tsr, 7.5 μM CheA, 10 μM CheW and fresh PMSF (1 mM) in activity buffer (160 mM NaCl, 5 mM MgCl2, 50 mM Tris, 0.5 mM EDTA, pH 7.4) for 1 hr at 22°C. Samples were centrifuged at 180,000 × g for 6 min and pellets were washed twice by resuspending in a 10-fold excess of activity buffer and re-pelleting. After the final wash, pellets were resuspended in the original volume of activity buffer.
Array Aging and Exogenous Protease Conditions
Reconstituted signaling arrays were stored at 22°C in an insulated water bath and protected from light. All eppendorf tubes and pipet tips were autoclaved prior to use and aseptic technique was used during time points to minimize the chance of contamination. Identical results were obtained when 0.01 % azide was included, indicating microbial contamination was not a factor.
Exogenous protease experiments were carried out by addition of 3 nM trypsin immediately after array reconstitution. Trypsin stocks were prepared fresh for each experiment from lyophilized powder and dissolved in 1 mM HCl at pH 3 (38).
Fractional Labeling of Receptors
Receptors (6.7 μM) resuspended in activity buffer with fresh PMSF (1 mM) were mixed with 5-iodoacetamido-fluorescein (50 μM) and immediately vortexed. Aliquots of receptors were removed every 10 seconds and quenched by addition of 80 mM β-mercaptoethanol. The staggered timecourse provided labeling efficiencies in the 2–10% labeling range. Labeling efficiency was determined from the ratio of the specific fluorescence intensities of receptor bands on SDS PAGE for a) the labeled protein and b) the same protein labeled 100% following denaturation by SDS. Specific fluorescence intensities were generated by normalizing the integrated fluorescence intensity to the integrated absorbance of the same band following Coomassie staining. Labeled receptors were reconstituted with CheA and CheW using the same procedure outlined for standard Cysless components.
Determination of Attractant Regulated Kinase Activity
The relative kinase activities of arrays were measured as previously described with a few modifications (39). Five microliters of washed and resuspended arrays were mixed with five microliters of YFP-CheY yielding final concentrations of 3.3 μM receptor and 40 μM YFP-CheY, the latter sufficient to ensure that CheA autophosphorylation was the rate determining step and not phosphotransfer from CheA~P to CheY. Sensitivity to attractant was measured by addition of 1 mM serine. Kinase reactions were carried out by addition of 1 mM [γ-32P]ATP (4000–8000 cpm/pmol) followed by incubation for 10 sec. Reactions were quenched in 30 μL of 2 × Laemmli sample buffer containing 50 mM EDTA. Samples were snap frozen in liquid nitrogen and stored until timecourses were completed. At the end of the timecourse samples were thawed and resolved on denaturing SDS PAGE gels. Once resolved, gels were extensively dried and then the γ-32P -labeled CheYband was imaged using phosphorimaging.
Determination of Changes in Total Protein
Changes in the total protein content were followed by removing 25 μL aliquots of reconstituted arrays, pelleting the arrays by centrifugation at 180,000 × g for 6 min, and removing the supernatant. Pellets were then snap frozen in liquid nitrogen and stored at −80°C until completion of the timecourse. Once complete, pellets were resuspended in 25 μL of 2 × Laemmli sample buffer, boiled, resolved on 10% SDS PAGE gels, and stained with Coomassie Brilliant Blue. Gels were imaged using a digital camera (Alpha Innotech) and relative changes in CheA and receptor bands with time were determined by densitometry with ImageJ software. Relative changes in CheW were followed by western blotting using polyclonal antibodies against CheW (a gift from Dr. G. Hazelbauer). The proteolysis products of CheA were similarly monitored by western blotting using a polyclonal antibody against CheA (a gift from Dr. J. Stock). Briefly, dilute samples were resolved on 15% or 10% SDS PAGE gels and transferred to PVDF membranes. Membranes were blocked overnight in tris-buffered saline tween-20 (50 mM Tris, 160 mM NaCl, 0.1% Tween-20, TBST) with 3% dry milk powder before blotting with anti-CheW or anti-CheA primary and anti-rabbit, alkaline phosphatase conjugated secondary. Extensive washing with TBST was performed after incubations. Washed membranes were incubated with ECF substrate and dried extensively before scanning with a Typhoon 9500 scanner. Blots were quantified by densitometry using ImageJ.
Measuring Changes in Cooperativity
Each type of array was reconstituted and its Hill coefficient was immediately measured by mixing arrays with CheY and increasing quantities of serine from 0.005 μM to 1000 μM with a total of 40 data points. Samples were incubated with serine for 30 minutes to ensure full equilibration with the membrane-associated arrays. After the incubation, samples were immediately tested for kinase activity. Measurement of the Hill coefficient was repeated after the arrays were aged for 72 hours following the same procedure.
Determining the Symmetry of CheA Proteolysis
To determine whether proteolysis typically clips CheA subunits in the same or different CheA dimers, arrays were formed with CheA E311C, a mutation that rapidly forms disulfide crosslinks across the P3-P3′ dimerization interface between the two CheA subunits in the same dimer. Arrays containing CheA E311C were formed and aged in the presence of 3 mM TCEP to prevent damage to thiols and also prevent the formation of crosslinks during the timecourse. At regular intervals, 10 μL aliquots of arrays were removed and mixed with 90 μL of activity buffer to dilute the TCEP. A final concentration of 6 mM CuCl2 buffered with 3 mM EDTA was added to catalyze the disufide crosslinking reaction. Reactions were allowed to proceed for 10 minutes before arrays were pelleted at 180,000 × g, snap frozen in liquid nitrogen, and stored at −80°C until the conclusion of the timecourse. Once complete samples were resuspended in 2 × Laemmli sample buffer, diluted, and resolved on 8% SDS PAGE gels by Western blotting. Proteins were transferred to PVDF membranes and blotted with polyclonal anti-CheA (a gift from Dr. J. Stock).
Data Averaging and Error Analysis
All data points shown are averages of three replicates unless otherwise specified. Fit parameters are reported as an average of at least three replicates except where noted in the relevant legend. Error bars and ranges indicate the standard deviation unless otherwise specified.
RESULTS
Experimental Strategy
The goals of this study are to investigate the timescale and molecular mechanism of the striking ultra-stability exhibited by bacterial chemosensory arrays. The study focuses on arrays formed in vitro from the three core proteins: receptor, CheA kinase, and CheW adaptor protein. These in vitro arrays have been previously shown to exhibit decay properties indistinguishable from ex vivo arrays formed in live cells, then isolated in bacterial membranes (25).
The study begins by quantitating the timescale of array stability. CheA kinase is highly activated in the intact, apo-array lacking bound attractant. As the array ages, its kinase activity slowly decreases and the timecourse of this decay can be measured. At the same time, the core protein stoichiometries are monitored to detect changes in array composition. By directly comparing the decay timecourses of kinase activity and protein composition, any correlations between activity and protein loss will be revealed.
Subsequently, the study investigates the relationships between array order, stability, and cooperativity. The current working model proposes that both ultra-stability and inter-receptor cooperativity arise from extensive interconnections between highly ordered array components. In this model, regions of lower array spatial order are hypothesized to exhibit lower stability, less steric protection from proteolysis, lower connectivity and cooperativity. By contrast, regions of higher array order are hypothesized to possess greater stability and more protection from proteolysis, as well as higher connectivity and cooperativity. If this picture is valid, then proteolysis should degrade the less-ordered regions of the array first, leaving the well-ordered, more highly cooperative regions intact. Finally, to directly examine the link between array order and ultra-stability, varying levels of engineered local defects are introduced into the array to systematically decrease its overall order, and the effects on ultra-stability are examined.
Decay of Kinase Activity as the Array Ages
Reconstituted arrays were assembled in vitro from serine receptor in E. coli inner-membranes, CheA and CheW, then their decay timecourses were quantitated by kinase activity measurements. Figure 2 displays a representative 21-day timecourse. Approximately 50% of the initial kinase activity decays during the first 1–2 days, but the remaining 50% of the kinase activity is truly ultra-stable and decays very slowly over the remaining weeks. By the end of the three week timecourse much, but not all, of the ultra-stable activity has decayed. Impressively, throughout the three weeks the kinase activity remains easily measurable and is fully regulated by the attractant serine. Nonlinear least squares, best-fit analysis reveals the timecourse is well-described by a two-component model consisting of two exponential decay terms:
| (Eq. 1) |
where Af and As are the amplitudes, and τf and τs are the time constants, of the fast-decaying and slow-decaying components termed hereafter quasi-stable and ultra-stable, respectively. Comparison of different best-fit models indicates these two-component fits are adequate, since additional exponential terms are not statistically justified. Averaging the best fit parameters from four independent timecourses reveals that 50 ± 3% of the array exhibits a mean kinase activity lifetime of 29 ± 9 hours while 50 ± 6% of the array is ultra-stable with a mean kinase lifetime of 460 ± 50 hours (Table 1).
Figure 2.

Kinase activity as array ages. Serine receptor-containing E. coli membranes were reconstituted in vitro with Cysless CheA kinase and CheW coupling protein to yield triplicate arrays, then were incubated at 22°C for 504 hr (21 days). At the indicated timepoints a sample was removed from each triplicate and mixed with CheY and ATP to measure the array kinase activity (closed circles). An identical sample was mixed with attractant serine to measure attractant regulation (open circles). The data were best fit either with the double exponential decay of Eq. 1 (curve, no serine) or with a straight line (serine). Error bars indicate the standard deviation of each triplicate mean.
Table 1.
Summary of two component fit parameters for 21-day array decay timecourse
A series of control experiments were carried out to ascertain whether the decay kinetics, including the ratio of the quasi- and ultra-stable components, were modulated by specific factors. Attractant serine or protein stabilizer glycerol present during array formation or during array aging has little or no effect on the decay kinetics (Fig. S1, S2). Similarly, varying the array formation time from 15 to 90 min, or varying the array formation temperature from 15° to 30° C, has little or no effect on decay kinetics (Fig. S3, S4). However, decay kinetics are sensitive to the temperature during array aging, such that the kinase activity decays faster as the aging temperature is raised from 15° to 30° (Fig. S5). The latter observation suggests that the array decay involves a thermally activated process. Finally, consistent with previous observations (25), the ultra-stable component is observed both for arrays formed in vitro and for arrays assembled in live cells prior to ex vivo isolation (Fig. S6). In both preparations the array possesses quasi- and ultra-stable components, each representing 50% (within error) of the total array. Interestingly, the quasi-stable component exhibits a slightly longer lifetime in the ex vivo arrays, but this lifetime remains within ~2-fold of that observed for corresponding component of in vitro arrays. In short, the decay lifetimes of both the quasi- and ultra-stable array components are quite invariant to a variety of perturbations.
Determining the Mechanism of Kinase Activity Loss During Array Aging
The decay of kinase activity following array assembly could arise from kinase dissociation, since the free kinase is over 100-fold less active (40). Alternatively, the kinase could remain in the array but be inactivated by alteration of the kinase molecule such as proteolysis or unfolding. To distinguish these possibilities, the protein composition of the array was monitored in parallel with kinase activity. Throughout the timecourse, identical aliquots of arrays were a) subjected to kinase assays and b) centrifuged at high speed to isolate array pellets, which were subjected to SDS-PAGE analysis of core protein levels.
Figure 3A summarizes the relative levels of serine receptor, CheW adapter protein and CheA kinase during a representative 21 day timecourse. The total levels of receptor and CheW both decay slowly following a single exponential decay, with approximately 60% and 32% of the starting full length protein retained at 21 days, respectively. Over the same period, the level of full length CheA kinase drops to 20% of its starting level, and the timecourse of full-length CheA loss is highly correlated with the loss of kinase activity. Both the loss of full-length CheA and the decay of its kinase activity are best fit by the same, within error, double-exponential decay process (Eq. 1, Table 1). To directly test the correlation between loss of CheA and loss of kinase activity, Figure 3B plots the relative level of full-length, array-bound CheA against relative kinase activity, as determined for the large number of time points in the decay timecourses. Least-squares best fit analysis confirms the correlation is highly linear (slope of 0.98 ± 0.06, R = 0.99), indicating there is a direct, one-to-one relationship between kinase activity loss and the decreasing level of full-length CheA kinase in the array with time.
Figure 3.

Comparing the kinetics of kinase activity loss and protein loss. A) Triplicate, membrane-bound arrays reconstituted from serine receptor, CheA kinase and CheW coupling protein were incubated at 22°C for 504 hrs (21 days). At the indicated timepoints a sample was removed from each triplicate and mixed with CheY and ATP to determine the kinase activity of the array (circles). Another sample was pelleted to separate and quantitate array bound CheA (closed squares) and CheA in the supernate (open squares). Pelleted samples were also utilized to quantitate array bound, full-length CheW (diamonds) and full-length Tsr (triangles). The data were best fit with the double exponential decay of Eq. 1 yielding the indicated best fit curves (kinase activity and pellets), or were best fit with a straight line (supernatant). B) Plot investigating the correlation between the average kinase activity and level of full length CheA measured at a given timepoint for each triplicate in (A), as well as 3 other independent experiments like (A). Error bars indicate the standard deviation of the triplicate mean for each parameter.
The depletion of full-length CheA in arrays was further examined to ascertain whether the loss arose from proteolysis or dissociation from the array. Examination of the supernatant from array pellets indicates that no detectable quantity of full-length CheA appears in the supernatant over the duration of the timecourse (Fig. 3A). Instead, western blot analysis of array pellets with polyclonal anti-CheA antibody reveals that two CheA proteolysis fragments build up in the pellets on the same timescale as the loss of full length CheA, as illustrated in Figure 4A, B. These CheA fragments—corresponding to the P2+P3+P4+P5 fragment missing the P1 domain, and the P3+P4+P5 fragment missing the P1+P2 domains—have been previously observed and are soluble. Thus the presence of the P2+P3+P4+P5 and the P3+P4+P5 fragments in the pellets indicates they remain stably incorporated in the arrays. The missing P1 and P1+P2 fragments are highly susceptible to removal from the other domains by proteolysis of the long linkers separating the P1-P2 (27 residues) and P2-P3 (37 residues) domains (41).
Figure 4.

Timecourse of full-length CheA decay and the appearance of CheA proteolytic fragments. A) Triplicate, membrane-bound arrays reconstituted from serine receptor, CheA kinase and CheW coupling protein were incubated at 22°C for 504 hrs (21 days). Atthe indicated timepoints a sample was removed from each triplicate and pelleted to separate array-bound and supernatant CheA. Pelleted samples were resolved on SDS gels and western blotted with polyclonal anti-CheA antibody. The timecourses of full-length CheA decay were similar, generally within error, when monitored by western blotting (filled circles) and Coomassie staining (open circles). In addition to the full-length CheA band, two smaller bands were observed building up in the array western blots. These bands are observed at the molecular weights expected for CheA lacking the P1 domain (triangles, P2-P3-P4-P5) and for CheA lacking the P1-P2 domains (squares, P3-P4-P5). The sum of the western blot densities for full-length CheA, P2-P3-P4-P5, and P3-P4-P5 were also determined (diamonds). All data are relative to the initial level of full-length CheA. B) Sample western blot showing the full-length CheA band and the two degradation products initially and after 48 hours.
When the western blot densities of the remaining full-length CheA and its two fragments are added, the total density nearly equals the initial density of full-length CheA for the first 5 days of array aging (Fig. 4A). The small loss of total density over this period is consistent with the release of the small P1 or P1+P2 domains, while the remaining large fragments remain bound to the array where they are protected from further proteolysis. Such retention is reasonable given the known ability of CheA and CheW to remain for days in the array without dissociation (25). After five days, the total quantity of array-bound CheA, including full-length and the two fragments, begins to slowly decay, reaching 41% of its initial level by 21 days. During this second phase of decay the previously detected, array-bound P2+P3+P4+P5 and P3+P4+P5 fragments slowly disappear, presumably due to further proteolysis, and eventually are lost almost completely. It is not clear whether the presumed final cleavages occur in the array or after dissociation from the array.
Addition of Exogenous Protease to Test for Steric Protection by the Array
During the first five days of array aging, the rapid proteolysis of only half of the CheA sub units is hypothesized to arise from the existence of distinct quasi- and ultra-stable spatial regions within the array. This hypothesis predicts that CheA dimers in the quasi-stable regions will be fully clipped on both subunits during the first phase of proteolysis, yielding the observed 50% loss of full length CheA after five days, while the CheA dimers in the ultra-stable regions will be protected and remain full length during this five day period.
To test these predictions, a small quantity of trypsin (3 nM) was added immediately after array formation and the array kinase activity was tracked over a five day timecourse. Addition of the exogenous trypsin initially causes a more rapid loss of kinase activity, significantly faster than the loss observed due to endogenous proteolysis as illustrated in Figure 5. Strikingly, however, after this initial rapid loss of kinase activity, the loss slows and by the end of the five day timecourse the trypsin and endogenous proteolysis reactions yield nearly the same level of remaining kinase activity. For both reactions, about half of the kinase activity remains after five days, indicating that half of the CheA subunits are protected from proteolysis by trypsin or by endogenous proteases (each CheA subunit in the dimer is catalytically active) (42). The five day timecourse for the endogenous proteolysis reaction is well-fit by a variation of Equation 1 with the two exponential decay constants fixed to the predetermined averages of Table 1:
| (Eq. 2) |
Figure 5.

Kinetic comparison of endogenous and exogenous proteolysis. Triplicate, membrane-bound arrays reconstituted from serine receptor, CheA kinase and CheW coupling protein were incubated at 22°C for 120 hrs (5 days) under normal conditions (filled circles) or with 3 nM trypsin (filled squares). Atthe indicated timepoints a sample was removed from each triplicate and its kinase activity was determined in the absence (filled circles, squares) or presence (open circles, squares) of attractant serine. Error bars indicate the standard deviation of each triplicate mean. The (−) trypsin data (curve) were best fit by Eq. 2, a variation of the double exponential decay of Eq. 1 where the lifetime (τf and τs) was fixed (29 and 460 hours) to the averages obtained from Figure 1., Eq. 1. The (+) trypsin data (curve) were best fit by Eq. 3, a variation of Eq. 2 that contained a third exponential term that was not fixed to any predetermined value.
As before, the quasi- and ultra-stable components each account for about half of the total array (Table 2). For the trypsin proteolysis reaction a third exponential term is required for an adequate fit:
| (Eq. 3) |
Table 2.
Summary of multi-component fit parameters for arrays undergoing endogenous or exogenous proteolysis.
The observed three terms reveal (i) a very fast, 0.7 hour component (τvf) presumably degraded by trypsin and representing 32 ± 6% of the array, (ii) a typical quasi-stable component presumably degraded by the endogenous protease representing 20 ± 6% of the array, and (iii) the usual ultra-stable component representing 48 ± 6% of the array (Table 2). Throughout the entire timecourse the array kinase activity retains sensitivity to attractant (Fig. 5).
In principle, the observed loss of half the full length CheA sub units could arise either from (i) proteolysis of both sub units in half the CheA dimers, or (ii) proteolysis of only one subunit in the entire dimer population. Mechanism (i), consistent with distinct quasi- and ultra-stable spatial regions, predicts that at the end of the five day timecourse, both subunits of nearly all the CheA dimers are proteolyzed in the quasi-stable regions, while in the ultra-stable regions nearly all the CheA dimers remain intact. Mechanism (ii), an asymmetric cleavage model, predicts that after the initial five days, nearly all the CheA dimers in the entire array possess one proteolyzed subunit and one intact subunit. The timecourse of kinase activity decay does not resolve these mechanisms, since both would yield loss of half the total kinase activity after the initial five days (a CheA dimer missing both P1 substrate domains is completely inactive, while a dimer missing just one P1 domain retains 50% kinase activity)(42). To resolve the two mechanisms, a CheA homodimer possessing a symmetric pair of engineered Cys residues (E311C, E311C′) known to form an intra-dimer disulfide bond (43) was incorporated into arrays. Subsequently, the array decay timecourse was analyzed as usual, except that each sample removed at a given timepoint was oxidized to form the intra-dimer disulfide crosslink within the CheA homodimers prior to analysis on non-reducing SDS PAGE. The results in Figure 6A,B indicate that the major crosslinked product is the full-length CheA dimer in which both subunits are intact, and that about half of this full length dimer is lost during the five day timecourse. These findings rule out the asymmetric cleavage model, since in this model nearly all CheA dimers would be clipped on one sub unit by the end of the five day timecourse, leading to complete loss of the full length dimer, which is not observed. Instead, the symmetric cleavage of the CheA homodimer supports the existence of two distinct spatial regions within arrays, such that CheA dimers in ultra-stable regions are nearly fully protected from proteolysis during the initial five days of decay, while the CheA dimers in quasi-stable regions are clipped on both subunits, accounting for the loss of half the full length dimers.
Figure 6.

Decay timecourse of full length CheA dimers detected by disulfide crosslinking. Triplicate, membrane-bound arrays reconstituted from serine receptor, E311C CheA kinase and CheW coupling protein were incubated at 22°C for 120 hrs (5 days). A) At the indicated timepoints a sample was removed from each triplicate and incubated with 6 mM CuCl2 for 10 minutes to covalently link the subunits of the E311C CheA homodimer. Samples were pelleted, resolved on SDS gels, and western blotted with polyclonal anti-CheA yielding the decay timecourse of the full length CheA dimer (squares). For comparison, the decay timecourse of Cysless kinase activity is also plotted (circles). Error bars indicate the standard deviation of each triplicate mean. B) Western blot showing the oxidative crosslinking of E311C CheA in reconstituted arrays to yield covalent homodimer.
Relationship Between Stability and Cooperativity in the Array
The differences observed in the stability and proteolytic protection of two classes of CheA dimers may arise from differences in the degrees of order and networking in different regions of the array. In this model, the extra stability of ultra-stable regions arises from their superior order and connectivity, which confers protection from proteolysis. In contrast, quasi-stable regions are less ordered and possess a lower level of connectivity between proteins, yielding decreased proteolytic resistance. To test this model, the positive cooperativity of arrays with respect to ligand-induced kinase regulation was measured (i) immediately after array formation and (ii) after days of aging to eliminate the quasi-stable component. In both cases, the array kinase activity was titrated with attractant serine to determine the apparent Hill coefficient, a measure of average inter-receptor cooperativity over the heterogeneous array (21). Titrations were completed about 1 hour or 72 hours after the formation for the new and aged arrays, respectively.
For three independent experiments illustrated in Figure 7, the apparent Hill coefficient and K1/2 of serine-induced array kinase inhibition were Happ = 1.4 ± 0.2 and K1/2app = 25 ± 7 μM for new arrays, and Happ = 2.5 ± 0.5 and K1/2 app = 95 ± 9 μM for arrays aged three days, respectively. Given the above findings, both the new and aged arrays are mixtures of quasi- and ultra-stable regions, with the aged arrays possessing a higher ratio of ultra-stable regions. Thus the measured apparent Happ and K1/2 app values are complex averages over the properties of heterogeneous arrays. Since it is not clear whether the weighting factors in the bulk averaging are simply the fractions of the two types of array, the microscopic H and K1/2 values cannot be determined at present. However, these microscopic parameters likely satisfy H < 1.4, K1/2 < 25 μM for the quasi-stable regions, and H > 2.5, K1/2 > 95 μM for the ultra-stable regions. It follows that at least in the ultra-stable regions, the Hill coefficient is significantly over 1, indicating the presence of significant positive cooperativity, consistent with previous observations (21, 22) and with theoretical predictions that attractant binding modulates multiple receptor-kinase units within the array (30). Notably, both H and K1/2 are larger in the ultra-stable regions. Similarly, when receptor adaptation sites are modified the H and K1/2 values increase in parallel (21, 22). Such parallel increases of H and K1/2 can be simply explained by a thermodynamic coupling model in which ligand binding to a more cooperative array must expend more energy to drive conformational changes in larger numbers of receptors, thereby reducing the binding affinity.
Figure 7.

Relationship between stability and cooperativity in arrays. Triplicate, membrane-bound arrays were reconstituted from serine receptor, CheA kinase and CheW coupling protein. On the initial day (New, circles) and third day (Aged, squares) following array formation, triplicate aliquots were mixed with the indicated final concentrations of serine and the array kinase activity was quantitated. Error bars indicate the standard deviation of each triplicate mean.
Examining the Effects of Engineered Defects on Array Stability
To directly examine the relationship between array stability and order, different densities of engineered defects were introduced into the array and the effects on ultra-stability were quantitated. A single cysteine mutant (V398C, selected based on the properties of the homologous side chain V396C in Tar (19,44)) was introduced into the serine receptor and co-expressed in a roughly 1:1 ratio with wild-type receptor – which lacks Cys residues – to minimize formation of inter-receptor disulfide bonds. The engineered Cys side chain of the V398C mutant is non-perturbing, yielding native kinase activity and other array parameters. However, this side chain is located on the receptor surface that binds CheA and CheW, thus labeling the Cys with a bulky fluorescein probe blocks array assembly and kinase activation. As a result, fluorescein labeling of V398C provides a convenient way to introduce a local defect into the array. To test the effects of different levels of defects on array stability, receptor-containing membranes were labeled with different densities of fluorescein ranging from 2 % to 50 %. The resulting modified receptor samples were reconstituted with CheA and CheW into membrane-bound arrays as usual, and the effects of the defects on array stability were quantitated by decay timecourses of kinase activity.
Figure 8A shows kinase activity decays for arrays possessing 0 %, 2.4 %, 6.0 %, and 50 % levels of fluorescein-modified receptors, here termed defects. The 0 % defect arrays yield a kinase decay timecourse indistinguishable from that of the native array (Fig. 8A), confirming that the V398C mutation itself is not significantly perturbing. However, labeling as little as 2.4 % of the receptor population causes kinase activity to decay significantly more rapidly and largely eliminates the ultra-stable component. Fitting the data with Equation 4 confirms these qualitative observations:
| (Eq. 4) |
where Af is the amplitude and τf is the time constant for the fast decaying population and c is the amplitude of the ultrastable population. The pure Cysless receptor and unlabeled V398C mixed arrays yield indistinguishable levels of quasi- and ultrastable components, each within error of half the total (Table 3). Labeling 2.4 % of the V398C cysteines causes 91 ± 7 % of the lattice to exhibit the quasi-stable lifetime while the ultra-stable component is nearly negligible (9 ± 7 %). Additional labeling decreases the kinase activity lifetime of the nearly pure quasi-stable array from ~40 hr (0 % or 2.4 % label) to ~20 hr (6.0 % label) to just ~5 hr (50 % label). The loss of virtually all the ultra-stable component triggered by just a small density of engineered defects (2.4 %) strongly supports a model in which a highly ordered array is essential for ultra-stability.
Figure 8.
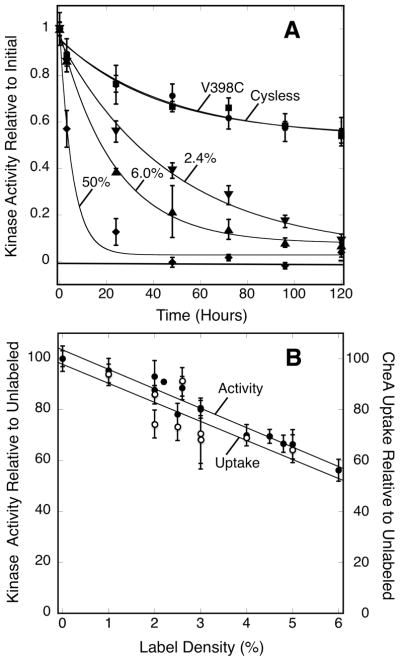
Examining the relationship between defects, stability and assembly. A) Membrane-bound receptor populations containing the single Cys mutant V398C were labeled with the indicated levels of 5-iodoacetamido-fluorescein to introduce local defects, then reconstituted arrays were formed with CheA and CheW. Triplicate samples were incubated at 22°C for 120 hrs (5 days), and at the indicated timepoints a sample was removed from each triplicate and its kinase activity was determined. Error bars indicate the standard deviation of each triplicate mean. Normal attractant regulation was confirmed on the initial, second, and fifth day of the experiment by addition of 1 mM serine (open symbols). B) The initial timepoint samples generated for different defect densities in (A) were also analyzed for their CheA incorporation (open circles) and kinase activities (closed circles) just after array reconstitution. Error bars indicate the standard deviation of each triplicate mean. Data were best fit by Eq. 5 consisting of a single exponential decay term to model the fast quasi-stable population and a constant term to approximate the amplitude of the ultra-stable population.
Table 3.
Summary of two-component fit parameters for arrays containing engineered defects.
| f(x) = Ae−x/τ +c | |||
|---|---|---|---|
| Receptor | Label Density (%) | τ (A) | c |
| Tsr-WT | 0 | 46 ± 9 (0.47 ± 0.06) | 0.53 ± 0.06 |
| Tsr-V398C/WT | 0 | 40 ± 10 (0.46 ± 0.06) | 0.54 ± 0.05 |
| Tsr-V398C/WT | 2.4 ± 0.3 | 47 ± 8 (0.91 ± 0.07) | 0.09 ± 0.07 |
| Tsr-V398C/WT | 6.0 ± 0.2 | 23 ± 3 (0.92 ± 0.02) | 0.08 ± 0.09 |
| Tsr-V398C/WT | 50 ± 2 | 5 ± 2 (0.96 ± 0.06) | 0.04 ± 0.03 |
The indicated values are averages ± standard deviations. The receptor population in isolated bacterial membranes was labeled at a specific, engineered, non-perturbing Cys residue (V398C) with the indicated final density of 5-iodoacetamido-fluorescein. The resulting chemical modification prevents CheA and/or CheW binding to the labeled receptor. Finally, the effect of the indicated labeling density on the decay kinetics of CheA kinase activity (Eq. 4) was quantitated in a five-day timecourse, yielding the lifetime (τ) and amplitude (A) of the quasi-stable component, as well as the amplitude (c) of the ultra-stable component.
Given the observation that array ultra-stability is highly sensitive to defects, the effect of increasing levels of defects on CheA incorporation into the reconstituted array, and on the resulting initial array kinase activity, was also examined. A roughly linear decrease in kinase activity and CheA incorporation was observed as labeling increased from 0 % up to 6.0 %, as illustrated in Figure 8B, but the slope of the linear fit is not unitary. Instead, the slope indicates that a 1 % level of defect causes an 8 ± 1 % decrease in kinase activity and CheA incorporation. These results indicate that array assembly is highly cooperative, such that even a low level of receptor defects can significantly reduce the incorporation of CheA into the array.
DISCUSSION
The results presented here indicate that isolated chemosensory arrays in bacterial membranes are composed of two components nearly identical in size: a quasi-stable population with a lifetime of 29 ± 9 hours and an ultra-stable population with a lifetime of 460 ± 50 hours (at 22° C). The decay of the quasi-stable population is due to proteolysis of array-bound CheA by an endogenous protease that releases the N-terminal P1 or P1+P2 domains while the CheA core domains remain bound to the array. Eventually, within five days, both N-terminal domains are clipped from most CheA dimers in the quasi-stable population. In contrast, the proteolysis of the CheA in ultra-stable population is much slower, indicating these CheA molecules are largely protected from proteolysis, such that little CheA cleavage occurs in this population during the first five days of aging. The ultra-stable population exhibits significantly greater positive cooperativity (H > 2.5) than the quasi-stable population (H < 1.4). Ultra-stability is lost when a low density of receptor defects is chemically introduced into the array structure: a 2 % (or perhaps even lower) level of receptor defects is sufficient to eliminate ultra-stability. Similarly, in the absence of engineered defects, the very slow decay of the ultra-stable population is proposed to arise from a slow build-up of rare defects during aging.
In the simplest molecular model explaining the existence of both quasi- and ultra-stable populations, these two components are proposed to represent different spatial regions of the same array. In this picture the ultra-stable regions possess better order, fewer structural flaws, more extensive networking greater proteolytic resistance, and higher receptor-receptor cooperativity than the quasi-stable regions. One possibility is that the quasi- and ultra-stable regions are the edge and the interior, respectively, of a large expanse of well-ordered array composed of interconnected rings of receptor, CheA and CheW (Fig. 1). In this model the edge of the array lacks the full complement of connectivities found in the interior, leading to the observed differences between the quasi- and ultra-stable components. Alternatively, the quasi-and ultra-stable regions could be arranged in a mosaic pattern, in which defined regions of poorly ordered array are interspersed with regions of well-ordered array. Theoretical modeling studies have suggested that receptors form strongly coupled neighborhoods where all of the receptors in the neighborhood signal together (33, 45). Both the edge and mosaic models propose such strongly coupled neighborhoods co-exist with weakly coupled regions. Cryo-EM imaging of native arrays in wild type cells yields at least some images in which the lateral density of receptors in an array is not uniform but rather divided between less ordered and more ordered regions consistent with a mosaic arrangement (see Fig. 7 in (46)), but further work is needed to resolve the edge and mosaic models. Notably, cryo-EM studies of average array properties indicate that the array geometry and lateral receptor density are unaffected by changes in receptor signaling state, consistent with the idea that on-off switching does not alter the global architectures of the quasi- and ultra-stable regions (47, 48). Such findings are also consistent with the known small spatial amplitude of receptor transmembrane signaling (7, 8).
The protection of CheA in the ultra-stable regions against cleavage by endogenous or exogenous proteases has strong implications for the dynamics and/or accessibility of the N-terminal CheA domains and their inter-domain linkers in the array. The P1 substrate domain is separated from the P2 CheY binding domain by a 27 residue linker, and P2 is separated from the P3-P4-P5 CheA core domains by a 37 residue linker (3, 43). If these two long linkers were extended random coils, their lengths could total up to 220 Å, allowing full access of the P1 substrate domain to the P4 catalytic domain of both subunits. For the CheA dimer in solution these linkers do exhibit detectable dynamics and are hotspots for cleavage by exogeneous proteases (41, 43, 49), but constraints exist that prevent the P1 domain of one sub unit from colliding with the P4 domain of the other sub unit in the same dimer (43). In the ultra-stable regions of the array the two linkers are protected from endogenous and exogenous proteases for at least five days following array formation in both the absence and presence of attractant. The molecular nature of this protection is unknown, but presumably would involve the packing of the linkers in folded regions of the array, or steric exclusion of proteases.
The isolated chemosensory arrays characterized herein display many features that have clear functional importance in native cellular arrays. The present evidence indicates the highly ordered, defect-free regions of these isolated arrays possess high levels of positive cooperativity and receptor networking—properties known to play central roles in the ultra-sensitivity and signaling speed of the native, cellular pathway. The simplest model proposes these highly ordered regions also possess ultra-stability as an inherent property, both in isolated arrays and live cell arrays. Supporting this model, ultra-stability is observed both in arrays reconstituted in vitro on isolated bacterial membranes (herein and previously), and in ex vivo arrays formed within cells then isolated by disrupting the cells and purifying the bacterial membranes. In cells, however, it has been observed that free, cytoplasmic core proteins (CheA, CheW) fused to fluorescent reporter proteins exchange with array proteins on a timescale of tens of minutes (50), in sharp contrast to the complete lack of detectable core protein exchange observed for isolated arrays in vitro (25). The more rapid exchange in cells is hypothesized to arise from an unidentified cellular chaperone or degradation machinery that limits the ultra-stability of the array in growing cells (25), thereby preventing excessive array growth that could fill or perturb the cell membrane. Intracellular array turnover could also ensure that damaged components are replaced, preventing loss of array sensitivity and stability with age. When this turnover machinery is removed by isolating membranes from cells, the inherent ultra-stability of the array architecture becomes apparent.
Ultra-stability may have evolved as an accidental side-effect of the interconnectivity essential for array cooperativity and ultra-sensitivity. Yet, ultra-stability could itself serve an important function when a starving cell is searching for a food source: one can hypothesize that the starving cell would shut down protein synthesis and turn off the turnover machinery that normally disassembles the array in cells. Under such conditions the array would exhibit its inherent ultra-stability observed in vitro, thereby enabling the cell to continue its search as long as it can generate enough ATP to run the chemosensory pathway and enough proton motive force to power the swimming motors. Under these conditions, the proteolytic resistance of the array would become useful as the cell degrades its other components for energy production. Thus, the array ultra-stability that would be toxic in growing cells would become an important survival tool in starving cells. Notably, such ultra-stability provides an ideal platform for the development of long-lived biosensors.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr. A. Erbse, Dr. K. Piasta, Dr. K. Swain, A. Natale, and A. Berlinberg for helpful discussions and careful reading of the manuscript. P. Slivka would like to acknowledge the Interdepartmental Molecular Biophysics Program at the University of Colorado for training and funding.
Support provided by NIH R01 GM-040731 (to JJF) and NIH T32 GM065103 (to PFS)
ABBREVIATIONS
- ATP
adenosine triphosphate
- PMSF
phenylmethylsulfonylfluoride
- YFP
yellow fluorescent protein
- DTT
Dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- PMSF
Phenylmethanesulfonylfluoride
- HCl
hydrochloric cid
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- 5-IAF
5-(iodoacetamido)fluorescein
- Tris
2-Amino-2-hydroxymethyl-propane-1,3-diol
- TBS
tris buffered saline
- TBST
tris buffered saline and tween-20
- TCEP
tris(2-carboxyethyl)phosphine
- Ni-NTA
nickel-nitrilotriacetic acid, nickel-charged resin
- BCA
bicinchoninic acid
- PVDF
polyvinyldifluoride)
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supplementary Figures S1–S6 investigate the effects of various perturbations on array stability, including: attractant, glycerol, formation time, formation temperature, aging temperature, and intracellular vs. in vitro assembly, as discussed in Results. All figures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bourret RB, Silversmith RE. Two-component signal transduction. Curr Opin Microbiol. 2010;13:113–115. doi: 10.1016/j.mib.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao R, Stock AM. Biological insights from structures of two-component proteins. Annu Rev Microbiol. 2009;63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wuichet K, Alexander RP, Zhulin IB. Comparative genomic and protein sequence analyses of a complex system controlling bacterial chemotaxis. Methods Enzymol. 2007;422:1–31. doi: 10.1016/S0076-6879(06)22001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazelbauer GL. Microbiology: Adaptation by target remodelling. Nature. 2012;484:173–175. doi: 10.1038/484173a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sourjik V, Wingreen NS. Responding to chemical gradients: bacterial chemotaxis. Curr Opin Cell Biol. 2012;24:262–268. doi: 10.1016/j.ceb.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hazelbauer GL, Falke JJ, Parkinson JS. Bacterial chemoreceptors: high-performance signaling in networked arrays. Trends Biochem Sci. 2008;33:9–19. doi: 10.1016/j.tibs.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chervitz SA, Falke JJ. Molecular mechanism of transmembrane signaling by the aspartate receptor: a model. Proc Natl Acad Sci USA. 1996;93:2545–2550. doi: 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falke JJ, Hazelbauer GL. Transmembrane signaling in bacterial chemoreceptors. Trends Biochem Sci. 2001;26:257–265. doi: 10.1016/s0968-0004(00)01770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Studdert CA, Parkinson JS. Crosslinking snapshots of bacterial chemoreceptor squads. Proc Natl Acad Sci USA. 2004;101:2117–2122. doi: 10.1073/pnas.0308622100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briegel A, Ortega DR, Tocheva EI, Wuichet K, Li Z, Chen S, Muller A, Iancu CV, Murphy GE, Dobro MJ, Zhulin IB, Jensen GJ. Universal architecture of bacterial chemoreceptor arrays. Proc Natl Acad Sci USA. 2009;106:17181–17186. doi: 10.1073/pnas.0905181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khursigara CM, Wu X, Subramaniam S. Chemoreceptors in Caulobacter crescentus: trimers of receptor dimers in a partially ordered hexagonally packed array. J Bacteriol. 2008;190:6805–6810. doi: 10.1128/JB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massazza DA, Parkinson JS, Studdert CA. Cross-Linking Evidence for Motional Constraints within Chemoreceptor Trimers of Dimers. Biochemistry. 2011;50:820–827. doi: 10.1021/bi101483r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 14.Maddock JR, Shapiro L. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science. 1993;259:1717–1723. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- 15.Sourjik V, Berg HC. Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol Microbiol. 2000;37:740–751. doi: 10.1046/j.1365-2958.2000.02044.x. [DOI] [PubMed] [Google Scholar]
- 16.Vu A, Wang XQ, Zhou HJ, Dahlquist FW. The Receptor-CheW Binding Interface in Bacterial Chemotaxis. Journal of Molecular Biology. 2012;415:759–767. doi: 10.1016/j.jmb.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Vu A, Lee K, Dahlquist FW. CheA-Receptor Interaction Sites in Bacterial Chemotaxis. J Mol Biol. 2012;422:282–290. doi: 10.1016/j.jmb.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Hazelbauer GL. Core unit of chemotaxis signaling complexes. Proc Natl AcadSci USA. 2011;108:9390–9395. doi: 10.1073/pnas.1104824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehan RS, White NC, Falke JJ. Mapping out regions on the surface of the aspartate receptor that are essential for kinase activation. Biochemistry. 2003;42:2952–2959. doi: 10.1021/bi027127g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller AS, Kohout SC, Gilman KA, Falke JJ. CheA Kinase of bacterial chemotaxis: chemical mapping of four essential docking sites. Biochemistry. 2006;45:8699–8711. doi: 10.1021/bi060580y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bornhorst JA, Falke JJ. Quantitative analysis of aspartate receptor signaling complex reveals that the homogeneous two-state model is inadequate: development of a heterogeneous two-state model. J Mol Biol. 2003;326:1597–1614. doi: 10.1016/s0022-2836(03)00026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li G, Weis RM. Covalent modification regulates ligand binding to receptor complexes in the chemosensory system of Escherichia coli. Cell. 2000;100:357–365. doi: 10.1016/s0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]
- 23.Sourjik V, Berg HC. Receptor sensitivity in bacterial chemotaxis. Proc Natl AcadSciUSA. 2002;99:123–127. doi: 10.1073/pnas.011589998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segall JE, Block SM, Berg HC. Temporal comparisons in bacterial chemotaxis. Proc Natl Acad Sci USA. 1986;83:8987–8991. doi: 10.1073/pnas.83.23.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erbse AH, Falke JJ. The core signaling proteins of bacterial chemotaxis assemble to form an ultrastable complex. Biochemistry. 2009;48:6975–6987. doi: 10.1021/bi900641c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter SL, Wadhams GH, Armitage JP. Signal processing in complex chemotaxis pathways. Nat Rev Microbiol. 2011;9:153–165. doi: 10.1038/nrmicro2505. [DOI] [PubMed] [Google Scholar]
- 27.Tindall MJ, Gaffney EA, Maini PK, Armitage JP. Theoretical insights into bacterial chemotaxis. Wiley Interdiscip Rev Syst Biol Med. 2012;4:247–259. doi: 10.1002/wsbm.1168. [DOI] [PubMed] [Google Scholar]
- 28.Amin DN, Hazelbauer GL. Chemoreceptors in signalling complexes: shifted conformation and asymmetric coupling. Mol Microbiol. 78:1313–1323. doi: 10.1111/j.1365-2958.2010.07408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 30.Hansen CH, Sourjik V, Wingreen NS. A dynamic-signaling-team model for chemotaxis receptors in Escherichia coli. Proc Natl Acad Sci USA. 107:17170–17175. doi: 10.1073/pnas.1005017107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Briegel A, Li X, Bilwes AM, Hughes KT, Jensen GJ, Crane BR. Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins. Proc Natl Acad Sci USA. 2012;109:3766–3771. doi: 10.1073/pnas.1115719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Hu B, Morado DR, Jani S, Manson MD, Margolin W. Molecular architecture of chemoreceptor arrays revealed by cryoelectron tomography of Escherichia coli minicells. Proc Natl Acad Sci USA. 2012;109:E1481–1488. doi: 10.1073/pnas.1200781109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bray D, Levin MD, Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393:85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- 34.Lai RZ, Manson JM, Bormans AF, Draheim RR, Nguyen NT, Manson MD. Cooperative signaling among bacterial chemoreceptors. Biochemistry. 2005;44:14298–14307. doi: 10.1021/bi050567y. [DOI] [PubMed] [Google Scholar]
- 35.Sourjik V. Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 2004;12:569–576. doi: 10.1016/j.tim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Miller AS, Falke JJ. Side chains at the membrane-water interface modulate the signaling state of a transmembrane receptor. Biochemistry. 2004;43:1763–1770. doi: 10.1021/bi0360206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gegner JA, Graham DR, Roth AF, Dahlquist FW. Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell. 1992;70:975–982. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- 38.Walsh KA, Gertrude E, Perlmann LL. Methods in Enzymology. Academic Press; 1970. [4] Trypsinogens and trypsins of various species; pp. 41–63. [Google Scholar]
- 39.Chervitz SA, Lin CM, Falke JJ. Transmembrane Signaling by the Aspartate Receptor - Engineered Disulfides Reveal Static Regions of the Subunit Interface. Biochemistry. 1995;34:9722–9733. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levit MN, Grebe TW, Stock JB. Organization of the receptor-kinase signaling array that regulates Escherichia coli chemotaxis. J Biol Chem. 2002;277:36748–36754. doi: 10.1074/jbc.M204317200. [DOI] [PubMed] [Google Scholar]
- 41.Morrison TB, Parkinson JS. Liberation of an Interaction Domain from the Phosphotransfer Region of Chea, a Signaling Kinase of Escherichia-Coli. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:5485–5489. doi: 10.1073/pnas.91.12.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swanson RV, Bourret RB, Simon MI. Intermolecular complementation of the kinase activity of CheA. Mol Microbiol. 1993;8:435–441. doi: 10.1111/j.1365-2958.1993.tb01588.x. [DOI] [PubMed] [Google Scholar]
- 43.Gloor SL, Falke JJ. Thermal domain motions of CheA kinase in solution: Disulfide trapping reveals the motional constraints leading to trans-autophosphorylation. Biochemistry. 2009;48:3631–3644. doi: 10.1021/bi900033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bass RB, Coleman MD, Falke JJ. Signaling domain of the aspartate receptor is a helical hairpin with a localized kinase docking surface: cysteine and disulfide scanning studies. Biochemistry. 1999;38:9317–9327. doi: 10.1021/bi9908179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hansen CH, Sourjik V, Wingreen NS. A dynamic-signaling-team model for chemotaxis receptors in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17170–17175. doi: 10.1073/pnas.1005017107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khursigara CM, Lan GH, Neumann S, Wu XW, Ravindran S, Borgnia MJ, Sourjik V, Milne J, Tu Y, Subramaniam S. Lateral density of receptor arrays in the membrane plane influences sensitivity of the E. coli chemotaxis response. Embo Journal. 2011;30:1719–1729. doi: 10.1038/emboj.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briegel A, Beeby M, Thanbichler M, Jensen GJ. Activated chemoreceptor arrays remain intact and hexagonally packed. Molecular Microbiology. 2011;82:748–757. doi: 10.1111/j.1365-2958.2011.07854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sferdean FC, Weis RM, Thompson LK. Ligand affinity and kinase activity are independent of bacterial chemotaxis receptor concentration: insight into signaling mechanisms. Biochemistry. 2012;51:6920–6931. doi: 10.1021/bi3007466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamel DJ, Zhou H, Starich MR, Byrd RA, Dahlquist FW. Chemical-shift-perturbation mapping of the phosphotransfer and catalytic domain interaction in the histidine autokinase CheA from Thermotoga maritima. Biochemistry. 2006;45:9509–9517. doi: 10.1021/bi060798k. [DOI] [PubMed] [Google Scholar]
- 50.Schulmeister S, Ruttorf M, Thiem S, Kentner D, Lebiedz D, Sourjik V. Protein exchange dynamics at chemoreceptor clusters in Escherichia coli. Proc Natl Acad SciUSA105. 2008:6403–6408. doi: 10.1073/pnas.0710611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.