

Abstract
The remarkable effects of added salts on the properties of aqueous micelles derived from the amphiphile PTS are described. Most notably Heck reactions run in the presence of NaCl lead to couplings on aryl bromides in water at room temperature. Olefin cross- and ring-closing metathesis reactions run in the presence of small amounts of pH-lowering KHSO4 are also accelerated, another phenomenon that does not apply to typical processes in organic media. These salt effects allow, in general, for synthetically valuable C-C bond-forming processes to be conducted under environmentally benign conditions. Recycling of the surfactant is also demonstrated.
INTRODUCTION
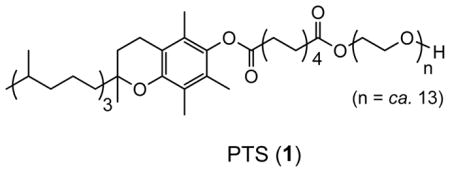
Micellar catalysis is a well-known phenomenon in organic synthesis.1 Typically, an amphiphilic species of the anionic, cationic, or nonionic variety spontaneously aggregates in water at low concentrations forming “normal” (as opposed to “inverted”) micellar arrays. Such particles can be organized into spheres, rods or worms, vesicles, or combinations thereof, with their polar portions outwardly interacting with the surrounding water, while their nonpolar subsection(s) make up the interior. These lipophilic cores function as hosts for organic compounds; in essence, as solvent. Given the huge influence of solvent effects in organic chemistry, variations in the nature of amphiphiles could well determine the level of success realized in various synthetic transformations. We have identified a first generation “designer” surfactant in PTS (Polyoxyethanyl α-Tocopheryl Sebacate; 1)2 as a generally useful nanoparticle-forming reagent that enables several Pd- and Ru-catalyzed “name” reactions,3 as well as asymmetric CuH-catalyzed 1,4-reductions,4 to be carried out in purely aqueous media at room temperature. In this contribution we describe further advances in PTS nanotechnology; advances applied to especially useful cross-coupling chemistry resulting from either control of nanoparticle size via adjustments in ionic strength,5 or through the expediency of changes in pH, of the surrounding water.
RESULTS AND DISCUSSION
Salt Effects on Reactions in Aqueous PTS
Heck Reactions.6
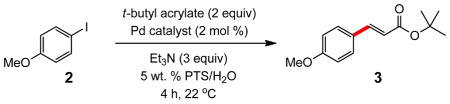
The impetus for studying reactions in solutions of PTS/H2O of varying ionicity followed from early trials using seawater as the reaction medium, in place of standard HPLC-grade water. Unexpectedly, significant rate accelerations were observed in Heck couplings between an acrylate and either an aryl iodide or bromide.7 As shown in Table 1, iodides 2 and t-butyl acrylate, using (PtBu3)2Pd as catalyst, reacted in 5 wt. % PTS/H2O (0.5 M global concentration of aryl halide) at room temperature in the presence of Et3N to afford 3 in 96% yield after four hours (entry 2).8 Of the many catalysts that were screened (Figure 1), only (PtBu3)2Pd was effective. Catalysts originating at the Pd(II) oxidation state (entries 1, 4–6) gave low levels of conversion, or no reaction at all (entries 7–10) even in the presence of excess zinc metal (entry 8). Surprisingly, switching from the Pd(0) species 5 to the analogous Pd(0) catalyst 11, where only one of three t-butyl groups on each phosphorus is replaced by a p-dimethylamino residue, completely inhibits the coupling. Reducing the amount of PTS from 5 to 2 wt. % with the same catalyst led to moderate product formation in the same time frame (entry 3). In seawater, the yield reached 98% within one hour (entry 11).
Table 1.
Catalyst Screening for Heck Reactions
| ||
|---|---|---|
| entry | catalyst | conversion (%)a |
| 1 | 4 | <5 |
| 2 | 5 | 96b |
| 3c | 5 | 68b |
| 4 | 6 | 50 |
| 5 | 7 | 25 |
| 6 | 8 | <5 |
| 7 | 9 | N.R. |
| 8d | 9 | N.R. |
| 9 | 10 | N.R. |
| 10 | 11 | N.R. |
| 11e | 5 | 98b |
Based on GC.
Yield of chromatographically pure materials.
Using 2 wt. % PTS/H2O.
Zn dust (20 mol %) used.
Reaction run in seawater for 1 h.
Figure 1.
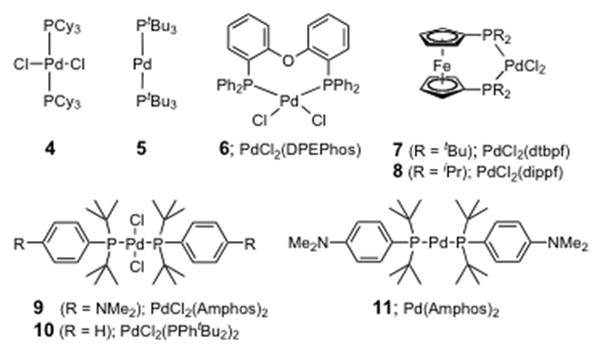
Palladium Catalysts Screened for Heck Reactions.
The effect of adding NaCl (3 M) to HPLC grade water, rather than using seawater, was even more pronounced with iodide 12, where the cinnamate was obtained in 96% yield after three hours at room temperature (Scheme 1). In the absence of salt, this reaction takes eight hours at 50 °C to realize the same yield at the same concentration. A similar effect was also observed for naphthyl iodide 13, where the reaction is done within three hours when NaCl is present in solution; otherwise, 24 hours are required to reach completion.
Scheme 1.

Effect of NaCl on Heck Reactions in PTS/water
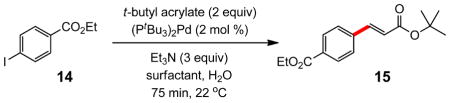
Comparisons between PTS/water and PTS/seawater with other surfactants was investigated in reactions with both aryl iodides and bromides. The former educt type was studied using iodobenzoate 14 along with t-butyl acrylate. As illustrated in Table 2, BASF’s solutol9 as the nanoparticle-forming amphiphile in water, as with PTS in pure water or seawater, led to high-yielding reactions (entries 1–3). Other surfactants including cremophor10 (entry 4) and TPGS11 (entry 5), however, were not as effective. The corresponding reaction “on water”12 (entry 6) gave the lowest level of conversion, which is consistent with observations made in related micellar chemistry.13
Table 2.
Comparison of Amphiphiles for Heck Couplings in Water
| ||
|---|---|---|
| entry | surfactanta | yield (%)b |
| 1 | PTS | 99 |
| 2 | PTS in seawater | 98 |
| 3 | Solutol | 97 |
| 4 | Cremophor | 57 |
| 5 | TPGS | 60 |
| 6 | “on water” | 48 |
5 wt. % used in all cases.
Chromatographically pure materials.
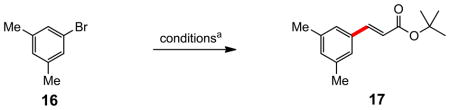
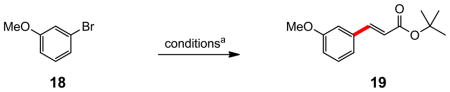
More challenging aryl bromides 16 and 18 were expectedly sluggish at ambient temperature (Table 3), with neither PTS (entries 1 and 8) nor solutol (entries 2 and 9) affording synthetically useful amounts of cinnamates after fourteen hours. However, heating the reaction in PTS/H2O to 40 °C drove the coupling to completion (entry 3). On the other hand, when the aqueous phase contained PTS and NaCl (3 M), essentially complete conversion was observed for both substrates, and high yields of the desired products 17 and 19 were obtained at ambient temperature (entries 4 and 10). Dropping the salt concentration from 3 M in NaCl to either 1 M or 2 M led to far slower reactions (48% and 68% yields, respectively, after the same 14 hours). The salt effect on the corresponding reaction in solutol was also minimal (entry 5), as expected (vide infra). When run “on water,”12 again, limited conversion was noted (entry 6). An identical Heck reaction on 16 run in toluene afforded enoate 17 in only 40% yield, along with a significant amount of the product of reduction of the aryl bromide, in addition to small amounts of unreacted starting halide remaining (entry 7).14
Table 3.
Comparison of Amphiphiles for Heck Couplings in Water
| ||
|---|---|---|
| entry | surfactantb | yield (%)c |
| 1 | PTS | 43 |
| 2 | solutol | 32 |
| 3 | PTS at 40 °C | 94 |
| 4 | 3 M NaCl, PTS/water | 95 |
| 5 | 3M NaCl, solutol/water | 46 |
| 6 | “on water” | 30 |
| 7 | toluene | 40 |
Conditions: tert-butyl acrylate (2 equiv), cat. 5 (2 mol %), Et3N (3 equiv) surfactant/H2O, 22 °C, 14 h.
5 wt. % used in all cases.
Chromatographically pure materials.
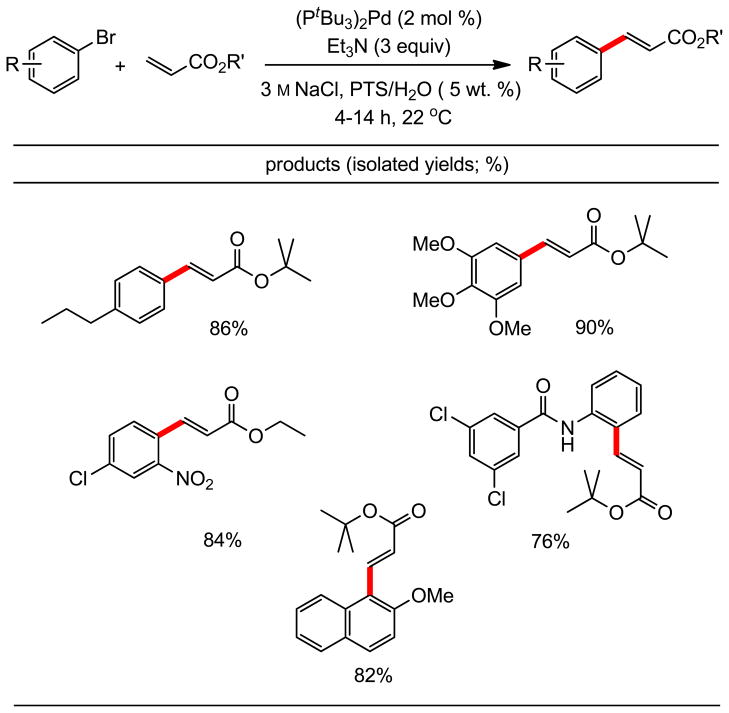
Several examples of related cinnamates prepared using NaCl-containing PTS/HPLC water are illustrated in Table 4. The nature of the ester in the acrylate partners appears to be of no consequence. Both electron-rich and -poor aryl bromides react equally well under these micellar conditions in 4–14 hours (0.5 M global concentration) at ambient temperatures. Lower levels of PTS (e.g., 2 wt. %) led to reactions that were equally rapid but not as clean, judging from TLC analyses.
Table 4.
Heck Couplings of Aryl Bromides with Acrylates
|
Olefin Metathesis.15
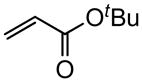
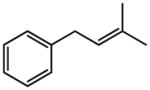
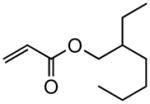
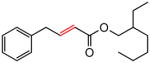
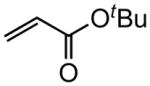
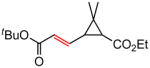
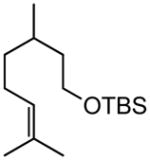
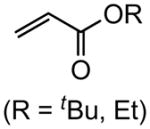
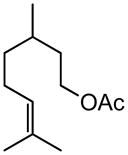
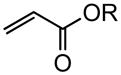
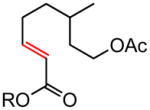
Early in our studies on room temperature olefin ring-closing and cross-metathesis reactions (RCM and CM, respectively) in aqueous nanoparticles of PTS, we described conditions that avoided both chlorinated solvents and heat.16 Type 1 olefins17 studied focused mainly on terminal alkenes, although E- or Z-disubstituted systems readily participated. Not included previously in our work were couplings involving trisubstituted olefins of the isopropylidene variety.17 The impetus to examine Type 1 olefins of this sort derived not only from an interest in extending the limits of metathesis in micelles, but in anticipation that this naturally occurring array might be amenable to analog formation. Table 5 lists several examples of such educts and their derived products from cross-metathesis in water at room temperature using the Grubbs-2 catalyst. Reaction efficiencies are high in all cases, although a tetrazole (entry 5) and ethyl chrysanthemate (entry 7) did not give full conversion under the standard conditions employed (0.5 M, 12 h). While racemic citronellol TBS ether was smoothly converted to the corresponding enoate over twelve hours (84%; entry 8), the identical reaction run in 3 M aqueous NaCl/PTS afforded essentially the same yield (86%) but in half the time. The corresponding acetate derivative also led to good yields in CM reactions with t-butyl and 2-ethylhexyl acrylates (entry 9).
Table 5.
Olefin CM Reactions in 2.5% aqueous PTSa
| entry | substrate | olefinic partner | product | yield (%)b |
|---|---|---|---|---|
| 1 |
|
|
|
92 |
| 2 |
|
|
|
97 |
| 3 |
|
|
|
84 |
| 4 |
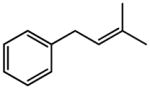
|
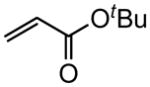
|
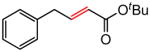
|
88 |
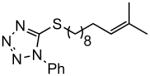
| 5 |
|
|
|
90c (55)d |
| 6 |
|
|
|
88 |
| 7 |
|
|
|
83c (50)d |
| 8 |
|
|

|
84, 86f (tBu) 82(Et) |
| 9 |
|
|
|
80 (tBu) 78(2-ethylhexyl) |
Reaction were conducted at 0.5 M over 12 h at 22 °C using 3 mol % Grubbs-2.
Chromatographically pure materials.
Based on recovered starting material.
Isolated.
Mixture of cis and trans isomers.
Reaction run in 3 M aqueous NaCl/PTS for 6 h.
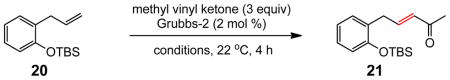
The effect of added NaCl was not significant, in general, for cross metathesis reactions involving Type 1 alkenes other than those containing an isopropylidene unit. Even in the case of especially challenging couplings using Type 2 olefins such as methyl vinyl ketone (MVK),18 reactions in PTS/water with added NaCl were only slightly faster (Table 6, entry 1 vs 2). Trials that replaced water totally (HOAc, entry 7), or in large measure (MeOH, entry 3; EtOH, entry 4; 95% EtOH; entries 6 and 10) gave inferior levels of conversion. On the other hand, rather than adding NaCl, addition of small amounts of KHSO4 (0.02 M) had a much greater impact; e.g., in the case of MVK, the resulting acidic conditions (pH 2)19 drove this reaction to almost full conversion, with either the Grubbs-2 (entry 11) or Neolyst M2 catalyst (entry 12). Replacing water with 95% EtOH led to only trace amounts of product enone (entry 13). Figure 2 illustrates graphically the effect of added KHSO4 relative to both aqueous PTS and CH2Cl2 alone.
Table 6.
Salt Effect on Olefin CM Reactions in 2.5% aq. PTS
| ||
|---|---|---|
| entry | conditions | conversion (%)a |
| 1 | PTS/water | 64 |
| 2 | 3 M NaCl, PTS/water | 75 |
| 3 | PTS/water-methanolb | 59 |
| 4 | PTS/water-ethanolc | 54 |
| 5 | 2.5% solutol/water | 33 |
| 6 | 95% ethanol | 22 |
| 7 | AcOH | 20 |
| 8 | CH2Cl2 | 30 |
| 9 | CH2Cl2+PTSA(10mol%) | <5 |
| 10 | PTS/95% ethanol | 28 |
| 11 | 0.02 M KHSO4, PTS/water | 95 |
| 12 | 0.02 M KHSO4, PTS/waterd | 94 |
| 13 | 0.02 M KHSO4, PTS/95% ethanol | <5 |
Based on 1H NMR.
Water:methanol = 19:1.
Water:ethanol = 19:1.
Neolyst M2 used instead of Grubbs-2.
Figure 2.
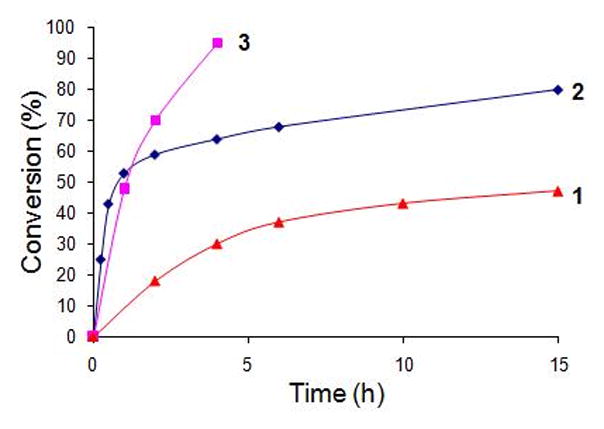
Conversion versus time profile for CM reaction of olefin 20 and MVK with Grubbs-2 catalyst in CH2Cl2(1), PTS/water (2), and 0.02 M KHSO4, PTS/water (3), as measured by 1H NMR spectroscopy.
Under these slightly acidic conditions, acetal 22 remained intact (Scheme 2). Not only MVK but also ethyl vinyl ketone and phenethyl vinyl ketone coupled with olefin 20 to give products 23 and 24 in 88% and 81% yields, respectively, under these standard conditions (Scheme 3). Curiously, no product was observed from the control reaction performed “on water” in the presence of KHSO4 (0.02 M) with phenethyl vinyl ketone.
Scheme 2.
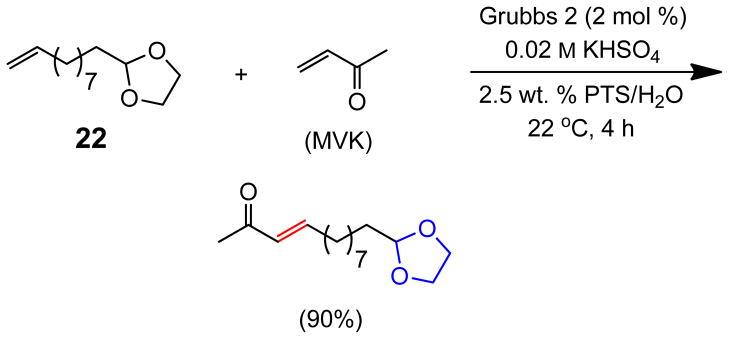
Olefin CM Reaction in the Presence of an Acetal
Scheme 3.
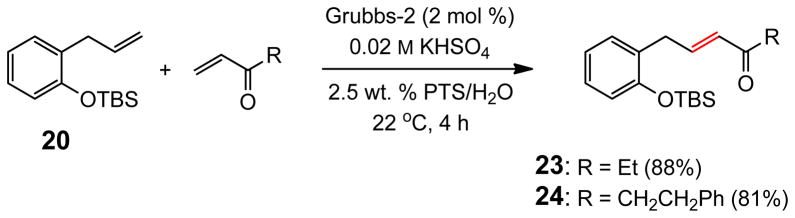
Olefin CM Reactions with Vinyl Ketones
Although 20 reacted with MVK in a highly efficient manner (Table 6, entry 11, and Table 7, entry 1), coupling with acrylonitrile could only be achieved to the extent of 30%. Nonetheless, under these room temperature conditions, reaction in the typically used solvent20 CH2Cl2 gave product in only 4% yield after the same four hours (Table 7, entry 2). Acrolein is another difficult case in CH2Cl2 at ambient temperature (9%),18a,21 and while aqueous PTS led to an improved 43% yield after 8 h, the presence of KHSO4 raised the yield to 70% under otherwise identical conditions (entry 3). Likewise, the corresponding CM with a vinylphosphonate22 gave the best yield (65%) under these pH-adjusted conditions.
Table 7.
More difficult Olefin CM Reactions

| |||||
|---|---|---|---|---|---|
| entry | R | time (h) | yield (%)a,b
|
||
| PTS | PTS/KHSO4c | CH2Cl2 | |||
| 1 | COMe | 4 | 74d | 91 | 30 |
| 2 | CN | 4 | 20 | 30 | 4 |
| 3 | CHO | 8 | 43 | 70 | 9 |
| 4 | P(O)(OEt)2 | 4 | 51 | 65 | 39 |
Chromatographically pure materials.
2.5 wt. % PTS used.
0.02 M KHSO4 used.
Reaction run for 12 h.
Ring-closing metathesis reactions, either in 3 M aqueous NaCl/PTS solutions, or in the presence of KHSO4, were only marginally improved relative to that achieved in PTS/H2O alone (Table 8). The best result (92% yield; entry 3) was obtained through the expediency of added KHSO4. The corresponding experiment in the absence of PTS (i.e., “on water”; entry 4), as in all previous comparisons herein, was not competitive.
Table 8.
RCM Under Aqueous Conditions

| ||
|---|---|---|
| entry | conditions | yield (%)a |
| 1 | PTS/water | 85 |
| 2 | 3 M NaCl, PTS/water | 90 |
| 3 | 0.02 M KHSO4, PTS/water | 92 |
| 4 | 0.02 M KHSO4/water | 42 |
Chromatographically pure materials.
Catalysts other than those in the Grubbs-1, -2, and Grubbs-Hoveyda-1 series, including those shown in Figure 3, have all been tested for their compatibility with micellar conditions, and ultimately, their effectiveness in olefin metathesis reactions. Among the Umicore series of Neolyst catalysts,23 an RCM example (Table 9) suggested that the internally chelated species Neolyst M51 mediates a very efficient cyclization, while catalyst Neolyst M2 is apparently preferred for cross-metathesis (Table 10). Likewise, among the two ruthenium carbene Zhan catalysts24 RC-303 and RC-304 tested in a challenging cross-metathesis example with MVK, RC-303 was found to work well, even in the absence of KHSO4, while being roughly comparable to both the Grubbs-2 and Neolyst M2 analogs at lower pH (Table 11). Not surprisingly, with no phosphine ligand present in RC-303 and GH-2, the rate of their catalysis is unaffected by the presence of acid in the aqueous medium.19a
Figure 3.

Structures of Ru-based catalysts used for olefin metathesis.
Table 9.
Olefin RCM Reactions Using Neolyst Catalysts
| ||
|---|---|---|
| entry | catalyst | conversion (%)a |
| 1 | Neolyst M41 | N.R. |
| 2 | Neolyst M42 | N.R. |
| 3 | Neolyst M3 | 84 |
| 4 | Neolyst M51 | 100(99)b |
| 5 | Neolyst M2 | 82 |
Based on 1H NMR.
Yield of chromatographically pure materials.
Table 10.
Olefin CM Reactions Using Neolyst Catalysts
| ||
|---|---|---|
| entry | catalyst | conversion (%)a |
| 1 | Neolyst M41 | N.R. |
| 2 | Neolyst M42 | N.R. |
| 3 | Neolyst M3 | 27(40)b |
| 4 | Neolyst M51 | 80 |
| 5 | Neolyst M2 | 99(93)c |
Based on 1H NMR.
Homocoupling product.
Yield of chromatographically pure materials.
Table 11.
Effect of pH on Olefin CM Reactions Using Metathesis Catalysts

| |||
|---|---|---|---|
| entry | catalyst | conversion (%)a,b
|
|
| PTS | PTS/KHSO4c | ||
| 1 | RC-304 | 0 | 0 |
| 2 | RC-303 | 88 | 90 |
| 3 | G-2 | 64 | 95 |
| 4 | Neolyst M2 | 64 | 94 |
| 5 | GH-2 | 61 | 62 |
Based on 1H NMR.
2.5 wt. % PTS used.
0.02 M KHSO4 used.
Another option for improving olefin cross metathesis under micellar conditions is inclusion of a copper salt in the medium. Surprisingly, although CuCl is routinely used to assist with formation of Grubbs-Hoveyda-1 or Grubbs-Hoveyda-2 ruthenium carbene complexes,25 use of copper to accelerate metathesis reactions is rare.26 Among several Cu(I) and Cu(II) species investigated, CuI (3 mol %) has been found to enhance the extent of conversion to product alkenes at room temperature.27 Thus, as illustrated in Table 12, three cases were studied: entry 1 is particularly noteworthy, as numerous attempts (by varying the nature and percentage of the surfactant, reaction temperature, and the amount of catalyst) to drive this coupling beyond the 55% reported previously had not been successful.16a The examples in entries 2 and 3 reflect problems normally encountered due to Type 1 olefin homocoupling, especially involving styrenes.17,28 Standard conditions for each of these examples compare well with those typically employed (refluxing CH2Cl2 over ca. 12 hours). Levels of carbene catalyst needed in organic media are also rarely less than 5 mol %,29 whereas with nanomicelles in water, 2 mol % is sufficient. Attempts to apply the benefits of both added KHSO4 and CuI did not lead to a cumulative effect.
Table 12.
Effect of CuI on Olefin CM Reactions

Chromatographically pure materials.
3 mol % CuI.
The potential for applications of this technology to synthesis is demonstrated by the cross-coupling reaction between olefin 25 and ethyl vinyl ketone (Scheme 4). This combination serves as a close model system to that used by Rychnovsky and co-workers in their synthesis of (+)-epicalyxin F, which relied on olefin metathesis (without complication due to the chalcone fragment) as a crucial means of attaching the required carbon framework for subsequent intramolecular conjugate addition.30 Under conditions that employed an organic solvent (PhH), heat (60 °C), time (24 hours), and catalyst (20 mol %), enone 27 was obtained in modest (47%) yield (60% brsm). The corresponding reaction between model enone 25 and ethyl vinyl ketone could be effected in water at room temperature in eight hours using 4 mol % of the Grubbs-2 Ru carbene to give 26 in 75% yield.
Scheme 4.

Comparison of conditions with a close literature example
Salt Effects
The positive impact on reaction rates observed due to the salts present in seawater on Heck reactions suggested that the nature of the PTS micellar nanoreactors was being perturbed. A DLS experiment on PTS in seawater revealed that the average particle size had increased from ca. 25 to 75 nm in diameter. A study on particle size vs. concentration of NaCl in HPLC grade water revealed a steady increase in the average diameter with increasing ionic strength; at 3 M NaCl nanoparticles averaging 110 nm are present (Figure 4). Other salts led to highly variable changes in particle size, although given the influence of factors such as viscosity on DLS measurements,31 no quantitative inferences as to relative particle size should be made. What does follow literature precedent is the qualitative sense of particle growth due to the “salting out” effect for NaCl, as well as several other salts (e.g., Na2SO4, NaBr, KCl, LiCl, etc.), a well known phenomenon for nonionic surfactants.5 On the other hand, weakly hydrated salts such as iodides and thiocyanates lead to a “salting in” effect, where water is directed in and around the micellar array, thereby reducing particle size. Salts such as NaCl, however, effectively compete for, and thus withdraw, water (of hydration) from the PEGylated portions of PTS micelles thereby increasing particle size. These effects of halides are further manifested by additional Heck reactions on 16 run in the presence of 3 M NaBr and 3 M NaI (Scheme 5). Thus, while the corresponding couplings in 3 M NaCl led to product cinnamate 17 in 95% yield, those run in smaller micelles gave considerably slower reactions. Nonionic surfactants other than PTS responded to an increase in ionic strength due to the presence of NaCl in a similar albeit far less pronounced fashion. Particle size increases for solutol, TPGS, and Triton X-100, all initially in the 10–13 nm range in pure water, do not exceed, on average, 42 nm in diameter. Brij 30, which forms 110 nm micelles in aqueous solution at room temperature, forms turbid mixtures of particles of roughly twice their size in the presence of 3 M NaCl. None of these alternative media, with or without added salts, led to consistently competitive results in cross couplings relative to those in PTS.
Figure 4.
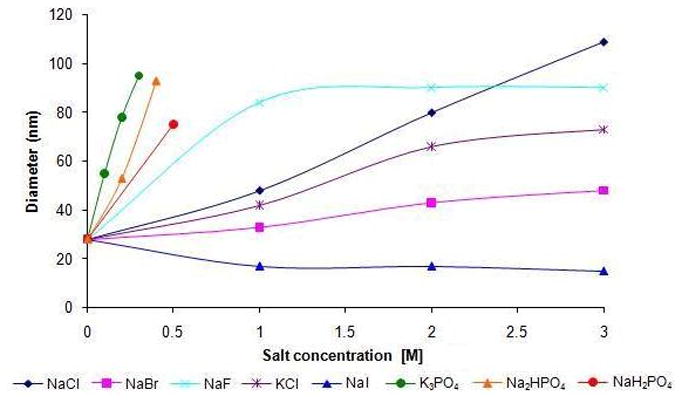
Effect of different salt concentrations on the diameter of PTS micelles.
Scheme 5.
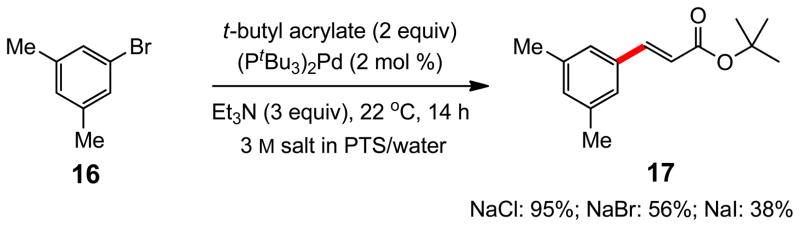
Impact of halides on a Heck coupling in PTS/water at rt
Data on PTS in water acquired from DLS measurements provide insight as to the average particle size in solution, but no information as to particle shape(s) or individual micelle sizes. An alternative technique, cryo-TEM,32 serves well in this capacity, giving a good indication of the nature of the micelles present. For PTS alone, cryo-TEM showed that both spherical micelles of ca. 10 nm are plentiful, along with a mix of worm and rod-shaped particles of considerably longer lengths (Figure 5A). Measurements on PTS in 3 M NaCl, in stark contrast, clearly revealed a fully interconnected, extended network (Figure 5B). In addition, smaller, irregularly shaped closed loops can also be discerned (white arrow). Thus, the enhanced cross-coupling chemistry (vide supra) resulting from an increase in ionic strength of the aqueous medium may be attributable to a dramatic change in the nature of the nanoreactors in which these Pd- and Ru-catalyzed couplings take place. While an explanation as to why such an array might correlate with better catalysis, as opposed to that seen with mainly spherical micelles formed by most other commonly used surfactants, remains to be clarified, there are lessons that can be learned from studies on both ionic and Gemini micelles.33 Rate enhancements are greater in the latter (e.g., for selected hydrolysis reactions), an observation that has been attributed to greater binding constants (Kb) for both substrates and catalysts.34
Figure 5.
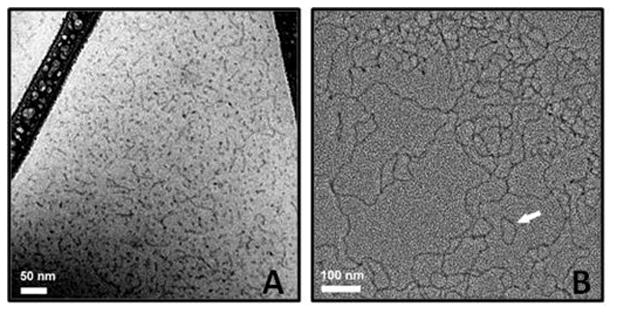
Cryo-TEM image of (A) aqueous PTS (50 nm scale), and (B) aqueous PTS in presence of 3 M NaCl (100 nm scale).
Recycling
Although PTS is used catalytically, the option to recycle this surfactant was examined using three olefin metathesis reactions: RCM; Equation 1; CM; Equations 2 and 3 (Scheme 6). Reaction handling involved the addition of EtOAc once the reaction reached completion, stirring, and in-flask removal of solvent leaving the aqueous layer behind. Re-introduction of fresh educt(s) and catalyst allowed for subsequent metathesis under otherwise identical conditions. In the case of an azadiene, RCM took place smoothly as previously noted (Eq. 1). Using an acrylate as partner with olefin 20, the extent of conversion in 3 M aqueous NaCl remained high and essentially unchanged after eight recycles (Eq. 2). Likewise, with MVK, in this case using the pH-lowering effect of KHSO4, ten recycles did not alter the observed level of conversion to the anticipated enone (Eq. 3).
Scheme 6.

Recycling of PTS
CONCLUSIONS
As viewed within the context of transition metal-mediated C-C bond forming reactions, the observations provided herein foreshadow the potential that spontaneously formed nanomicelles in water hold as alternative media to organic solvents. The dynamic nature of these particles, along with their surrounding aqueous environment through which educts and catalysts must traverse, offer rare opportunities to leverage the influence of both aqueous and non-aqueous solutions on cross-coupling reactions. Additives such as simple salts can significantly realign surfactant monomers, thereby perturbing particle size. These subtle changes can be used to great synthetic advantage in synthesis, impacting reaction rates, in particular with respect to Heck couplings. Changes in pH have also been shown to increase rates of olefin metathesis reactions in water as the only medium, especially when notoriously difficult Type 2 olefins are involved. Several cases demonstrating surfactant recyclability, where workup procedures involve no increase in aqueous waste streams, also support the inexpensive and environmentally friendly nature of this chemistry.
Experimental Section
Typical Procedure for Heck Couplings
Catalyst (PtBu3)2Pd (5.1 mg, 0.01 mmol) and aryl iodide/bromide (0.50 mmol) were added under argon into a 5.0 mL microwave vial equipped with a large stir bar and Teflon lined septum. An aliquot of 3 M NaCl in PTS/(degassed)H2O (1.0 mL; 5.0% PTS by weight) solution, triethylamine (208 μL, 1.50 mmol), and acrylate/styrene (1.0 mmol) were added by syringe, and the resulting solution was allowed to stir at rt until complete. The homogeneous reaction mixture was then diluted with EtOAc (2 mL), filtered through a bed of silica gel, and the bed further washed (3 × 5 mL) with EtOAc to collect all of the coupled material. The volatiles were removed in vacuo to afford the crude product which was subsequently purified by flash chromatography on silica gel.
(E)-t-Butyl 3-(4-methoxyphenyl)acrylate (3)
Following the general procedure using 4-methoxyiodobenzene (117 mg, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 4 h at rt. Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (112 mg, 96%). 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 16.0 Hz, 1H), 7.47 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.25 (d, J = 16.0 Hz, 1H), 3.84 (s, 3H), 1.54 (s, 9H).35
(E)-2-Ethylhexyl 3-(4-(3,7,11-trimethyldodecyloxy)phenyl)acrylate
Following the general procedure using 1-iodo-4-(3,7,11-trimethyldodecyloxy)benzene (215 mg, 0.50 mmol) and 2-ethylhexyl acrylate (208 μL, 1.0 mmol), the reaction was stirred for 3 h at rt. Column chromatography on silica gel (eluting with 4% EtOAc/hexanes) afforded the product as a colorless oil (233 mg, 96%). 1H NMR (400 MHz, CDCl3):δ 7.64 (d, J = 16.0 Hz, 1H), 7.48 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.32 (d, J = 16.0 Hz, 1H), 4.14-4.08 (m, 2H), 4.06-3.98 (m, 2H), 1.86-1.80 (m, 1H), 1.70-1.03 (m, 25H), 0.96-0.85 (m, 18H).8
(E)-1-(2,4-Dimethylstyryl)-2-methoxynaphthalene
Following the general procedure using 1-iodo-2-methoxynaphthalene (142 mg, 0.5 mmol) and 2,4-dimethylstyrene (147 μL, 1.0 mmol), the reaction was stirred for 3 h at rt. Column chromatography on silica gel (eluting with 5% EtOAc/hexanes) afforded the product as a semi-solid (131 mg, 91%). 1H NMR (400 MHz, CDCl3):δ 8.30 (d, J = 8.7 Hz, 1H), 7.81 (t, J = 7.8 Hz, 2H), 7.68 (d, J = 7.8 Hz, 1H), 7.47 (t, J = 7.2 Hz, 1H), 7.40-7.30 (m, 4H), 7.10 (d, J = 8.2 Hz, 1H), 7.05 (s, 1H), 3.98 (s, 3H), 2.40 (s, 3H), 2.37 (s, 3H).8
(E)-t-Butyl 3-(4-carboethoxylphenyl)acrylate (15)
Following the general procedure using ethyl 4-iodobenzoate (84 μL, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 75 min at rt. Column chromatography on silica gel (eluting with 4% EtOAc/hexanes) afforded the product as a colorless oil (137 mg, 99%). 1H NMR (400 MHz, CDCl3):δ 8.05 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 16.0 Hz, 1H), 7.57 (d, J = 8.4 Hz, 2H), 6.46 (d, J = 16.0 Hz, 1H), 4.39 (q, J = 7.2 Hz, 2H), 1.55 (s, 9H), 1.41 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 166.1, 165.9, 142.3, 138.9, 131.6, 130.1, 127.9, 122.6, 80.9, 61.3, 28.3, 14.4; ESI-MS m/z: 299 (M + Na); HRESIMS calcd for C16H20O4Na [M + Na]+ = 299.1259, found 299.1262.
(E)-t-Butyl 3-(3,5-dimethylphenyl)acrylate (17)
Following the general procedure using 3,5-dimethyl bromobenzene (68 μL, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 14 h at rt. Column chromatography on silica gel (eluting with 2% EtOAc/hexanes) afforded the product as a colorless oil (110 mg, 95%). IR (neat): 3006, 2978, 2922, 2870, 1707, 1637, 1603, 1455, 1391, 1366, 1330, 1288, 1255, 1151, 1039, 982, 844 cm−1; 1H NMR (400 MHz, CDCl3):δ 7.54 (d, J = 16.0 Hz, 1H), 7.14 (s, 2H), 7.01 (s, 1H), 6.35 (d, J = 16.0 Hz, 1H), 2.33 (s, 6H), 1.54 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 166.6, 144.0, 138.5, 134.7, 131.9, 126.0, 119.9, 80.5, 28.4, 21.4; EI-MS m/z (%): 232 (M+, 26), 176 (100), 161 (75); HRMS (EI) calcd for C15H20O2 [M]+ = 232.1463, found 232.1464.
(E)-t-Butyl 3-(3-methoxyphenyl)acrylate (19)
Following the general procedure using 3-methoxy bromobenzene (64 μL, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 14 h at rt. Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (115 mg, 98%). 1H NMR (400 MHz, CDCl3):δ 7.54 (d, J = 16.0 Hz, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 7.01 (s, 1H), 6.90 (dd, J = 8.0, 2.4 Hz, 1H), 6.34 (d, J = 16.0 Hz, 1H), 3.81 (s, 3H), 1.52 (s, 9H).36
(E)-t-Butyl 3-(4-propylphenyl)acrylate
Following the general procedure using 4-propyl bromobenzene (77 μL, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 8 h at rt. Column chromatography on silica gel (eluting with 2% EtOAc/hexanes) afforded the product as a colorless oil (106 mg, 86%). IR (neat): 2968, 2872, 1713, 1633, 1609, 1568, 1512, 1456, 1418, 1391, 1367, 1323, 1256, 1210, 1149, 983 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 16.0 Hz, 1H), 7.43 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 6.35 (d, J = 16.0 Hz, 1H), 2.60 (t, J = 7.6 Hz, 2H), 1.65 (sextet, J = 7.6 Hz, 2H), 1.55 (s, 9H), 0.95 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3):δ 166.7, 145.2, 143.8, 132.5, 129.1, 128.1, 119.4, 80.5, 38.1, 28.4, 24.5, 13.9; EI-MS m/z (%): 246 (M+, 12), 190 (88), 173 (24), 161 (100); HRMS(EI) calcd for C16H22O2 [M]+ = 246.1619, found 246.1616.
(E)-t-Butyl 3-(3,4,5-trimethoxyphenyl)acrylate
Following the general procedure using 5-bromo-1,2,3-trimethoxybenzene (124 mg, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 4 h at rt. Column chromatography on silica gel (eluting with 12% EtOAc/hexanes) afforded the product as a white solid (132 mg, 90%). 1H NMR (400 MHz, CDCl3):δ 7.50 (d, J = 16.0 Hz, 1H), 6.74 (s, 2H), 6.28 (d, J = 16.0 Hz, 1H), 3.89 (s, 9H), 1.54 (s, 9H).37
(E)-Ethyl 3-(4-chloro-2-nitrophenyl)acrylate
Following the general procedure using 1-bromo-4-chloro-2-nitrobenzene (118 mg, 0.50 mmol) and ethyl acrylate (109 μL, 1.00 mmol), the reaction was stirred for 14 h at rt. Column chromatography on silica gel (eluting with 5% EtOAc/hexanes) afforded the product as a pale yellow solid (107 mg, 84%). 1H NMR (400 MHz, CDCl3):δ 8.07-8.03 (m, 2H), 7.65-7.59 (m, 2H), 6.37 (d, J = 15.6 Hz, 1H), 4.30 (q, J = 7.2 Hz, 2H), 1.36 (t, J = 7.2 Hz, 2H).38
(E)-t-Butyl 3-(2-(3,5-dichlorobenzamido)phenyl)acrylate
Following the general procedure using N-(2-bromophenyl)-3,5-dichlorobenzamide (173 mg, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 14 h at rt. Column chromatography on silica gel (eluting with 10% EtOAc/hexanes) afforded the product as white solid (149 mg, 76%). IR (neat): 3243, 3065, 2979, 2932, 1705, 1682, 1660, 1635, 1587, 1529, 1483, 1455, 1368, 1322, 1253, 1152, 1104, 1050, 981, 911, 869, 760, 734 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.18 (br s, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 16.0 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 6.32 (d, J = 16.0 Hz, 1H), 1.51 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 166.0, 164.1, 138.0, 135.4, 133.3, 131.9, 130.7, 130.3, 128.0, 127.4, 126.6, 125.2, 123.4, 94.6, 81.1, 28.4; ESI-MS m/z: 414 (M + Na); HRESIMS calcd for C20H19NO3Cl2Na [M + Na]+ = 414.0640, found 414.0647.
(E)-t-Butyl 3-(2-methoxynaphthalen-1-yl)acrylate
Following the general procedure using 1-bromo-2-methoxynaphthalene (119 mg, 0.50 mmol) and t-butyl acrylate (145 μL, 1.00 mmol), the reaction was stirred for 14 h at rt. Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (116 mg, 82%). IR (neat): 3059, 2977, 2935, 2842, 1704, 1622, 1591, 1568, 1513, 1469, 1391, 1367, 1270, 1146, 1102, 1045, 982, 850 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.30 (d, J = 16.0 Hz, 1H), 8.22 (d, J = 8.8 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.53 (t, J = 8.4 Hz, 1H), 7.38 (t, J = 8.4 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 6.70 (d, J = 16.0 Hz, 1H), 4.00 (s, 3H), 1.61 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 167.4, 156.6, 136.8, 132.9, 131.4, 129.1, 128.7, 127.4, 125.4, 124.0, 123.5, 117.0, 112.9, 80.4, 56.3, 28.5; EI-MS m/z (%): 284 (M+, 28), 228 (67), 211 (23), 183 (100); HRMS(EI) calcd for C18H20O3 [M]+ = 284.1412, found 284.1415.
Typical Procedure for Cross Metathesis
Alkene (0.50 mmol), acrylate (1.00 mmol)/ketone (1.50 mmol) and Grubbs-2 catalyst (0.01–0.02 mmol) were sequentially added into a Teflon-coated-stir-bar-containing Biotage 2–5 mL microwave reactor vial at rt, and then sealed with a septum. An aliquot of 0.02 M of KHSO4 in PTS/H2O or PTS/H2O (1.0 mL; 2.5% PTS by weight; all cross-coupling reactions were conducted at 0.5 M unless stated otherwise) was added, via syringe, and the resulting solution was allowed to stir at rt for 4–12 h. The homogeneous reaction mixture was then diluted with EtOAc (2 mL), filtered through a bed of silica gel, and the bed further washed (3 × 5 mL) with EtOAc to collect all of the cross-coupled material. The volatiles were removed in vacuo to afford the crude product that was subsequently purified by flash chromatography on silica gel.
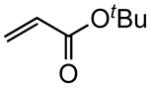
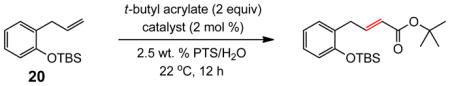
(E)-t-Butyl 12-(t-butyldimethylsilyloxy)dodec-2-enoate (Table 5, entry 1)
The representative procedure above was followed using t-butyldimethyl(11-methyldodec-10-en-1-yloxy)silane (156 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (177 mg, 92%). 1H NMR (400 MHz, CDCl3):δ 6.87 (dt, J = 15.6, 7.2 Hz, 1H), 5.74 (dt, J = 15.6, 1.6 Hz, 1H), 3.60 (t, J = 6.8 Hz, 2H), 2.16 (qd, J = 6.8, 1.6 Hz, 2H), 1.55–1.50 (m, 2H), 1.49 (s, 9H), 1.46–1.40 (m, 2H), 1.33–1.28 (m, 10H), 0.90 (s, 9H), 0.05 (s, 6H).39
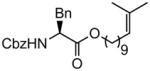
(R,E)-t-Butyl 12-(2-(benzyloxycarbonylamino)-3-phenylpropanoyloxy)dodec-2-enoate (Table 5, entry 2)
The representative procedure above was followed using (R)-11-methyldodec-10-en-1-yl 2-(benzyloxycarbonylamino)-3-phenylpropanoate (240 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (267 mg, 97%). 1H NMR (400 MHz, CDCl3): δ 7.38-7.32 (m, 4H), 7.31-7.24 (m, 4H), 7.11 (dd, J = 7.2, 1.2 Hz, 2H), 6.87 (dt, J = 15.6, 6.8 Hz, 1H), 5.74 (dt, J = 15.6, 1.2 Hz, 1H), 5.24 (d, J = 8.4 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 5.08 (d, J = 12.4 Hz, 1H), 4.66 (dt, J = 8.4, 5.6 Hz, 1H), 4.12 (dd, J = 10.8, 6.8 Hz, 1H), 4.06 (dd, J = 10.8, 6.8 Hz, 1H), 3.14 (dd, J = 13.6, 5.6 Hz, 1H), 3.09 (dd, J = 13.6, 5.6 Hz, 1H), 2.17 (qd, J = 7.2, 1.6 Hz, 2H), 1.60-1.55 (m, 2H), 1.49 (s, 9H), 1.46-1.41 (m, 2H), 1.32-1.28 (m, 10H).16a
(E)-4-Phenylbut-2-en-1-yl acetate (Table 5, entry 3)
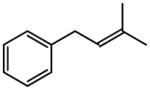
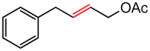
The representative procedure above was followed using (3-methylbut-2-en-1-yl)benzene (73 mg, 0.50 mmol), (Z)-but-2-ene-1,4-diyl diacetate (172 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (80 mg, 84%). 1H NMR (400 MHz, CDCl3):δ 7.33-7.18 (m, 5H), 5.97-5.90 (m, 1H), 5.68-5.61 (m, 1H), 4.55 (d, J = 6.4 Hz, 2H), 3.41 (d, J = 6.4 Hz, 2H), 2.07 (s, 3H).40
(E)-t-Butyl 4-phenyl-2-butenoate (Table 5, entry 4)
The representative procedure above was followed using (3-methylbut-2-en-1-yl)benzene (73 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol), and Grubbs second-generation catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 4% EtOAc/hexanes) afforded the product as a colorless oil (96 mg, 88%). 1H NMR (400 MHz, CDCl3): δ 7.33 (td, J = 8.4, 1.2 Hz, 2H), 7.25 (t, J = 8.4 Hz, 1H), 7.19 (d, J = 8.4 Hz, 2H), 7.00 (dt, J = 15.6, 6.8 Hz, 1H), 5.74 (dt, J = 15.6, 1.6 Hz, 1H), 3.51 (d, J = 6.8 Hz, 2H), 1.48 (s, 9H).41
(E)-t-Butyl 12-((1-phenyl-1H-tetrazol-5-yl)thio)dodec-2-enoate (Table 5, entry 5)
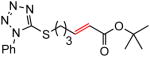
The representative procedure above was followed using 5-((11-methyldodec-10-en-1-yl)thio)-1-phenyl-1H-tetrazole (179 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (118 mg, 55%). IR (neat): 3053, 2977, 2928, 2855, 1713, 1652, 1598, 1500, 1460, 1389, 1367, 1292, 1246, 1156, 1088, 1074, 1053, 1014, 981, 914, 851, 762 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.60-7.52 (m, 5H), 6.85 (dt, J = 15.6, 6.8 Hz, 1H), 5.73 (dt, J = 15.6, 0.8 Hz, 1H), 3.39 (t, J = 7.2 Hz, 2H), 2.15 (qd, J = 6.8, 0.8 Hz, 2H), 1.82 (quint, J = 7.2 Hz, 2H), 1.50-1.43 (m, 4H), 1.48 (s, 9H), 1.32-1.28 (m, 8H); 13C NMR (100 MHz, CDCl3): δ 166.4, 148.4, 133.9, 130.3, 130.0, 124.0, 123.1, 80.2, 33.5, 32.2, 29.5, 29.3, 29.24, 29.18, 28.8, 28.3, 28.2; EI-MS m/z (%): 430 (M+, 8), 357 (19), 329 (14), 118 (76), 57 (100); HRMS(EI) calcd for C23H34O2N4S [M]+ = 430.2402, found 430.2390.
(E)-2-Ethylhexyl 4-phenyl-2-butenoate (Table 5, entry 6)
The representative procedure was followed using (3-methylbut-2-en-1-yl)benzene (73 mg, 0.50 mmol), 2-ethylhexyl acrylate (184 mg, 1.00 mmol), and Grubbs second-generation catalyst (23.4 mg, 0.0275 mmol). Column chromatography on silica gel (eluting with 5% EtOAc/hexanes) afforded the product as a colorless oil (121 mg, 88%). 1H NMR (400 MHz, CDCl3): δ 7.33 (t, J = 7.2 Hz, 2H), 7.25 (tt, J = 7.2, 1.2 Hz, 1H), 7.19 (dd, J = 7.2, 1.2 Hz, 2H), 7.10 (dt, J = 15.6, 6.8 Hz, 1H), 5.84 (dt, J = 15.6, 1.6 Hz, 1H), 4.06 (dd, J = 14.4, 6.0 Hz, 1H), 4.03 (dd, J = 14.4, 6.0 Hz, 1H), 3.54 (dd, J = 6.8, 1.6 Hz, 2H), 1.60 (septet, J = 6.4 Hz, 1H), 1.42-1.26 (m, 8H), 0.90 (t, J = 7.2 Hz, 6H).16a
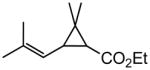
(E)-Ethyl 3-(3-(t-butoxy)-3-oxoprop-1-en-1-yl)-2,2-dimethylcyclopropanecarboxylate (Table 5, entry 7)
The representative procedure above was followed using ethyl chrysamthemate (135 mg, 0.50 mmol, mixture of cis and trans isomers), t-butyl acrylate (128 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 2% EtOAc/hexanes) afforded the mixture of cis and trans isomers as a colorless oil (143 mg, 84%). For cis isomer: IR (neat): 2979, 1727, 1713, 1643, 1455, 1415, 1391, 1368, 1309, 1270, 1189, 1157, 1135, 1086, 985 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.24-7.15 (m, 1H), 5.90 (d, J = 15.2 Hz, 1H), 4.13 (q, J = 7.2 Hz, 2H), 1.87 (d, J = 5.2 Hz, 1H), 1.86 (d, J = 5.2 Hz, 1H), 1.48 (s, 9H), 1.35 (s, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.23 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 170.5, 165.7, 143.5, 124.5, 80.2, 60.5, 35.3, 34.0, 28.9, 28.8, 28.4, 15.2, 14.5; ESI-MS m/z: 307 (M + K), 291 (M + Na); HRESIMS calcd for C15H24O4Na [M + Na]+ = 291.1572, found 291.1559. For trans isomer: IR (neat): 2979, 2933, 1728, 1714, 1645, 1457, 1427, 1392, 1381, 1367, 1313, 1281, 1232, 1197, 1155, 1137, 1114, 982 cm−1; 1H NMR (400 MHz, CDCl3):δ 6.57 (dd, J = 15.2, 10.0 Hz, 1H), 5.91 (d, J = 15.2 Hz, 1H), 4.19-4.07 (m, 2H), 2.14 (dd, J = 10.0, 5.2 Hz, 1H), 1.76 (d, J = 5.2 Hz, 1H), 1.47 (s, 9H), 1.28 (s, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.24 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 171.3, 165.9, 145.3, 124.4, 80.4, 60.8, 35.7, 35.5, 30.4, 28.3, 22.4, 20.5, 14.5; ESI-MS m/z: 291 (M + Na); HRESIMS calcd for C15H24O4Na [M + Na]+ = 291.1572, found 291.1558.
(E)-t-Butyl 8-(tert-butyldimethylsilyloxy)-6-methyloct-2-enoate (Table 5, entry 8, R = tBu)
The representative procedure above was followed using t-butyl(3,7-dimethyloct-6-en-1-yloxy)dimethylsilane42 (135 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 2% EtOAc/hexanes) afforded the product as a colorless oil (143 mg, 84%). IR (neat): 2956, 2929, 2858, 1717, 1654, 1472, 1462, 1390, 1366, 1289, 1255, 1156, 1096, 983, 897, 836, 775 cm−1; 1H NMR (400 MHz, CDCl3): δ 6.86 (dt, J = 15.6 Hz, 1H), 5.74 (dt, J = 15.6, 1.6 Hz, 1H), 3.69-3.59 (m, 2H), 2.26-2.10 (m, 2H), 1.65-1.41 (m, 3H), 1.48 (s, 9H), 1.38-1.23 (m, 2H), 0.89 (s, 9H), 0.892 (d, J = 6.4 Hz, 3H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 166.4, 148.4, 123.0, 80.2, 61.4, 39.9, 35.5, 29.8, 29.2, 28.4, 26.2, 19.6, 18.5, −5.1; MS (CI) m/z (%): 343 (M + H, 100), 287 (89), 269 (83), 229 (65), 109 (29), 59 (76); HRMS (CI) calcd for C19H39O3Si [M + H]+ = 343.2668, found 343.2669.
(E)-Ethyl 8-(t-butyldimethylsilyloxy)-6-methyloct-2-enoate (Table 5, entry 8, R = Et)
The representative procedure above was followed using t-butyl(3,7-dimethyloct-6-en-1-yloxy)dimethylsilane42 (135 mg, 0.50 mmol), ethyl acrylate (0.11 mL, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 50% CH2Cl2/hexanes) afforded the product as a colorless oil (129 mg, 82%). IR (neat): 2957, 2856, 1725, 1656, 1464, 1388, 1367, 1308, 1257, 1198, 1171, 1094, 1048, 984, 939, 897, 836, 776 cm−1; 1H NMR (400 MHz, CDCl3): δ 6.96 (dt, J = 15.6, 1H), 5.82 (dt, J = 15.6, 1.6 Hz, 1H), 4.18 (q, J = 7.2 Hz, 2H), 3.69-3.58 (m, 2H), 2.29-2.13 (m, 2H), 1.66-1.43 (m, 3H), 1.38-1.24 (m, 2H), 1.28 (t, J = 7.2 Hz, 3H), 0.895 (s, 9H), 0.894 (d, J = 6.4 Hz, 3H), 0.45 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 167.0, 149.7, 121.4, 61.4, 60.3, 39.9, 35.4, 29.9, 29.2, 26.2, 19.6, 18.5, 14.5, −5.1; EI-MS m/z (%): 299 (M – CH3, 3), 257 (M – C4H9, 100), 211 (16); HRMS (EI) calcd for C13H25O3Si [M – C4H9]+ = 257.1573, found 257.1580.
(E)-t-Butyl 8-acetoxy-6-methyloct-2-enoate (Table 5, entry 9, R = tBu)
The representative procedure above was followed using 3,7-dimethyloct-6-en-1-yl acetate43 (99 mg, 0.50 mmol), t-butyl acrylate (128 mg, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 6% EtOAc/hexanes) afforded the product as a colorless oil (108 mg, 80%). IR (neat): 2973, 2931, 2874, 1740, 1715, 1653, 1458, 1391, 1367, 1290, 1236, 1157, 1125, 1049, 984, 851 cm-1; 1H NMR (400 MHz, CDCl3):δ 6.82 (dt, J = 15.6, 6.8 Hz, 1H), 5.72 (dt, J = 15.9, 1.6 Hz, 1H), 4.13-4.02 (m, 2H), 2.25-2.08 (m, 2H), 2.02 (s, 3H), 1.69-1.38 (m, 3H), 1.45 (s, 9H), 1.36-1.23 (m, 2H), 0.90 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 171.3, 166.2, 147.9, 123.2, 80.2, 62.8, 35.4, 35.2, 29.6, 29.5, 28.3, 21.2, 19.3; ESI-MS m/z: 293 (M + Na); HRESIMS calcd for C15H26O4Na [M + Na]+ = 293.1723, found 293.1730.
(E)-2-Ethylhexyl 8-acetoxy-6-methyloct-2-enoate (Table 5, entry 9, R = 2-ethylhexyl)
The representative procedure above was followed using 3,7-dimethyloct-6-en-1-yl acetate43 (99 mg, 0.50 mmol), 2-ethylhexyl acrylate (0.21 mL, 1.00 mmol) and Grubbs-2 catalyst (12.8 mg, 0.015 mmol). Column chromatography on silica gel (eluting with 6% EtOAc/hexanes) afforded the product as a colorless oil (127 mg, 78%). IR (neat): 2959, 2930, 2873, 2861, 1741, 1721, 1655, 1462, 1382, 1366, 1309, 1239, 1171, 1125, 1047, 985 cm-1; 1H NMR (400 MHz, CDCl3): δ 6.92 (dt, J = 15.6, 6.8 Hz, 1H), 5.81 (dt, J = 15.9, 1.6 Hz, 1H), 4.13-4.04 (m, 2H), 4.03-3.98 (m, 2H), 2.27-2.12 (m, 2H), 2.02 (s, 3H), 1.70-1.27 (m, 14H), 0.92-0.86 (m, 9H); 13C NMR (100 MHz, CDCl3): δ 171.3, 167.0, 149.1, 121.6, 66.8, 62.8, 38.9, 35.4, 35.2, 30.6, 29.7, 29.5, 29.1, 24.0, 23.1, 21.2, 19.3, 14.2, 11.2; ESI-MS m/z: 349 (M + Na), 327 (M + H); HRESIMS calcd for C19H34O4Na [M + Na]+ = 349.2349, found 349.2354.
(E)-12-(1,3-Dioxolan-2-yl)dodec-3-en-2-one
The representative procedure above was followed using 2-(dec-9-en-1-yl)-1,3-dioxolane (22) (106 mg, 0.50 mmol), methyl vinyl ketone (106 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (114 mg, 90%). IR (neat): 2926, 2855, 1697, 1675, 1626, 1465, 1434, 1361, 1253, 1139, 1036, 980 cm−1; 1H NMR (400 MHz, CDCl3): δ 6.78 (dt, J = 16.0, 7.2 Hz, 1H), 6.04 (dt, J = 16.0, 1.2 Hz, 1H), 4.81 (t, J = 4.8 Hz, 1H), 3.98-3.90 (m, 2H), 3.87-3.78 (m, 2H), 2.22 (s, 3H), 2.19 (qd, J = 7.2, 1.2 Hz, 2H), 1.63 (td, J = 7.2, 4.8 Hz, 2H), 1.46-1.28 (m, 12H); 13C NMR (100 MHz, CDCl3): δ 198.8, 148.7, 131.4, 104.8, 65.0, 34.0, 32.6, 29.6, 29.5, 29.4, 29.3, 28.2, 27.0, 24.2; ESI-MS m/z: 393 (M + K), 277 (M + Na); HRESIMS calcd for C15H26O3Na [M + Na]+ = 277.1780, found 277.1774.
(E)-6-(2-(t-Butyldimethylsilyloxy)phenyl)hex-4-en-3-one (23)
The representative procedure above was followed using t-butyl(2-allylphenoxy)dimethylsilane44 (20) (124 mg, 0.50 mmol), ethyl vinyl ketone (126 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (134 mg, 88%). IR (neat): 3063, 3035, 2956, 2897, 2857, 1697, 1682, 1632, 1599, 1583, 1495, 1454, 1451, 1361, 1258, 1201, 1123, 1107, 1043, 982, 934, 838, 781, 756, 704, 663 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.14 (td, J = 7.6, 1.6 Hz, 1H), 7.10 (dd, J = 7.2, 1.6 Hz, 1H), 6.98 (dt, J = 16.0, 6.8 Hz, 1H), 6.91 (td, J = 7.2, 1.2 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 6.05 (dt, J = 16.0, 1.6 Hz, 1H), 3.53 (dd, J = 6.8, 1.6 Hz, 2H), 2.56 (q, J = 7.6 Hz, 2H), 1.09 (t, J = 7.6 Hz, 3H), 1.01 (s, 9H), 0.25 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 201.2, 153.7, 145.3, 130.9, 130.7, 128.6, 128.0, 121.4, 118.6, 33.6, 33.2, 26.0, 18.4, 8.3, −3.9; ESI-MS m/z: 327 (M + Na), 305 (M + H); HRESIMS calcd for C18H28O2SiNa [M + Na]+ = 327.1756, found 327.1758.
(E)-6-(2-(t-Butyldimethylsilyloxy)phenyl)-1-phenylhex-4-en-3-one (24)
The representative procedure above was followed using t-butyl(2-allylphenoxy)dimethylsilane44 (20) (124 mg, 0.50 mmol), 5-phenylpent-1-en-3-one (240 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (154 mg, 81%). IR (neat): 3062, 3028, 2955, 2929, 2896, 2858, 1697, 1674, 1628, 1600, 1582, 1492, 1453, 1362, 1256, 1105, 981, 928, 839, 781, 756, 699 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.30-7.26 (m, 2H), 7.20-7.18 (m, 3H), 7.14 (td, J = 7.2, 1.2 Hz, 1H), 7.08 (dd, J = 7.2, 1.2 Hz, 1H), 6.98 (dt, J = 16.0, 6.4 Hz, 1H), 6.91 (t, J = 7.2 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.05 (d, J = 16.0 Hz, 1H), 3.52 (d, J = 6.4 Hz, 2H), 2.96-2.92 (m, 2H), 2.88-2.84 (m, 2H), 1.00 (s, 9H), 0.25 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 199.7, 153.7, 146.0, 141.5, 131.1, 130.7, 128.7, 128.54, 128.49, 128.1, 126.2, 121.5, 118.7, 41.7, 33.6, 30.2, 26.0, 18.4, −3.9; ESI-MS m/z: 403 (M + Na), 381 (M + H); HRESIMS calcd for C24H32O2SiNa [M + Na]+ = 403.2069, found 403.2078.
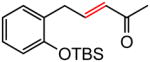
(E)-5-(2-(t-Butyldimethylsilyloxy)phenyl)pent-3-en-2-one (Table 7, entry 1)
The representative procedure above was followed using t-butyl(2-allylphenoxy)dimethylsilane44 (20) (124 mg, 0.50 mmol), methyl vinyl ketone (106 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (132 mg, 91%). IR (neat): 3062, 3034, 2932, 2894, 2859, 1699, 1676, 1626, 1599, 1582, 1492, 1472, 1452, 1422, 1390, 1361, 1254, 1182, 1108, 1043, 982, 929 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.15 (td, J = 7.6, 1.6 Hz, 1H), 7.11 (dd, J = 7.6, 1.6 Hz, 1H), 6.95(dt, J = 16.0, 6.4 Hz, 1H), 6.92 (td, J = 7.6, 1.2 Hz, 1H), 6.84 (dd, J = 7.6, 1.2 Hz, 1H), 6.03 (dt, J = 16.0, 1.6 Hz, 1H), 3.54 (dd, J = 6.4, 1.6 Hz, 2H), 2.24 (s, 3H), 1.01 (s, 9H), 0.26 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 198.8, 153.7, 146.8, 132.0, 130.7, 128.4, 128.1, 121.4, 118.6, 33.7, 26.9, 25.9, 18.4, −4.0; EI-MS m/z (%): 275 (M – CH3+, 2), 233 (M – C4H9+, 100), 215 (20), 151 (8), 75 (42); HRMS(EI) calcd for C13H17O2Si [M – C4H9]+ = 233.0998, found 233.1006.
4-(2-(t-Butyldimethylsilyloxy)phenyl)but-2-enenitrile (Table 7, entry 2)
The representative procedure above was followed using t-butyl(2-allylphenoxy)dimethylsilane44 (20) (124 mg, 0.50 mmol), acrylonitrile (80 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (41 mg, 30%). IR (neat): 3067, 3035, 2956, 2931, 2896, 2859, 2222, 1683, 1620, 1599, 1583, 1492, 1472, 1454, 1390, 1362, 1258, 1184, 1109, 1045, 1108, 928, 838 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.17-7.14 (m, 2H), 6.93 (t, J = 7.6 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 6.65 (dt, J = 11.2, 7.2 Hz, 1H), 5.38 (d, J = 11.2 Hz, 1H), 3.74 (d, J = 7.2 Hz, 2H), 1.03 (s, 9H), 0.27 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 153.3, 130.8, 130.4, 128.6, 128.4, 121.6, 118.7, 116.3, 99.7, 34.4, 33.2, 26.0, −3.9; FI-MS m/z: 273 (M), 216 (M – C4H9); HRFIMS calcd for C16H23NOSi [M]+ = 273.1549, found 273.1557.
(E)-4-(2-(t-Butyldimethylsilyloxy)phenyl)but-2-enal (Table 7, entry 3)
The representative procedure above was followed using t-butyl(2-allylphenoxy)dimethylsilane44 (20) (124 mg, 0.50 mmol), acrolein (84 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 3% EtOAc/hexanes) afforded the product as a colorless oil (97 mg, 70%). IR (neat): 3365, 3064, 3035, 2932, 2895, 2859, 2810, 2739, 1686, 1635, 1599, 1582, 1491, 1471, 1452, 1413, 1390, 1361, 1254, 1183, 1128, 1095, 1043, 1008, 978, 927, 838, 782, 757, 734, 704 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.53 (d, J = 7.6 Hz, 1H), 7.16 (td, J = 7.6, 1.6 Hz, 1H), 7.11 (dd, J = 7.6, 1.6 Hz, 1H), 6.99 (dt, J = 15.6, 6.4 Hz, 1H), 6.93 (td, J = 7.6, 1.6 Hz, 1H), 6.84 (dd, J = 7.6, 1.6 Hz, 1H), 6.07 (ddt, J = 15.6, 7.6, 1.6 Hz, 1H), 3.64 (dd, J = 6.4, 1.6 Hz, 2H), 1.00 (s, 9H), 0.26 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 194.1, 157.1, 153.7, 133.5, 130.7, 128.4, 127.9, 121.6, 118.7, 34.0, 26.0, 18.4, −3.9; ESI-MS m/z: 315 (M + K), 299 (M + Na); HRESIMS calcd for C16H24O2SiNa [M + Na]+ = 299.1443, found 299.1445.
(E)-Diethyl (3-(2-(t-butyldimethylsilyloxy)phenyl)prop-1-en-1-yl)phosphonate (Table 7, entry 4)
The representative procedure above was followed using t-butyl(2-allylphenoxy)dimethylsilane44 (20) (124 mg, 0.50 mmol), diethyl vinylphosphonate (164 mg, 1.00 mmol) and Grubbs-2 catalyst (17 mg, 0.02 mmol). Column chromatography on silica gel (eluting with 25% EtOAc/hexanes) afforded the product as a colorless oil (125 mg, 65%). IR (neat): 3063, 2931, 2900, 2859, 1631, 1599, 1583, 1493, 1472, 1453, 1391, 1362, 1250, 1164, 1104, 1023, 964 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.12 (td, J = 7.6, 1.6 Hz, 1H), 7.08 (dd, J = 7.6, 1.6 Hz, 1H), 6.94 (dt, J = 17.2, 6.0 Hz, 1H), 6.92-6.85 (m, 1H), 6.80 (d, J = 8.0 Hz, 1H), 5.59-5.49 (m, 1H), 4.08-3.99 (m, 4H), 3.53-3.51 (m, 2H), 1.29 (t, J = 7.2 Hz, 6H), 0.99 (s, 9H), 0.22 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 153.7, 151.83, 151.78, 130.8, 128.2, 128.0, 121.4, 118.6, 116.7, 61.73, 61.67, 35.1, 34.9, 25.9, 18.4, 16.54, 16.47, −4.0; ESI-MS m/z: 407 (M + Na), 385 (M + H); HRESIMS calcd for C19H33O4SiPNa [M + Na]+ = 407.1783, found 407.1783.
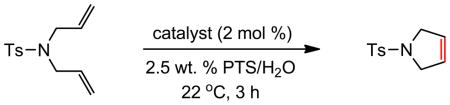
1-Tosyl-2,3,4,7-tetrahydro-1H-azepine (Table 8)
The representative procedure above was followed using N-allyl-4-methyl-N-(pent-4-en-1-yl)benzenesulfonamide45 (56 mg, 0.20 mmol) and Grubbs-2 catalyst (3.4 mg, 0.004 mmol). Column chromatography on silica gel (eluting with 4% EtOAc/hexanes) afforded the product as a colorless oil (46 mg, 92%). 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 5.79-5.74 (m, 1H), 5.67-5.62 (m, 1H), 3.83-3.82 (m, 2H), 3.38 (t, J = 6.0 Hz, 2H), 2.42 (s, 3H), 2.20-2.15 (m, 2H), 1.82-1.76 (m, 2H).45
(E)-t-Butyl 6-(1-phenyl-1H-tetrazol-5-ylthio)-2-hexenoate (Table 12, entry 1)
The representative procedure above was followed using 5-(pent-4-en-1-ylthio)-1-phenyl-1H-tetrazole16a (123 mg, 0.50 mmol), t-butyl acrylate (217 μL, 1.50 mmol), CuI (7.2 mg, 0.038 mmol) and Grubbs-2 catalyst (21.2 mg, 0.025 mmol). Column chromatography on silica gel (eluting with 10% EtOAc/hexanes) afforded the product as colorless oil (138 mg, 80%). 1H NMR (400 MHz, CDCl3): δ 7.59-7.55 (m, 5H), 6.82 (dt, J = 15.6, 7.2 Hz, 1H), 5.79 (dt, J = 15.6, 1.6 Hz, 1H), 3.41 (t, J = 7.2 Hz, 2H), 2.36 (qd, J = 7.2, 1.6 Hz, 2H), 2.03 (quintet, J = 7.2 Hz, 2H), 1.48 (s, 9H).16a
(E)-t-Butyl 3-(2,4-dimethylphenyl)-2-propenoate (Table 12, entry 3)
The representative procedure above was followed using 2,4-dimethylstyrene (74 μL, 0.50 mmol), t-butyl acrylate (145 μL, 1.00 mmol), CuI (2.9 mg, 0.015 mmol) and Grubbs second-generation catalyst (8.5 mg, 0.010 mmol). Column chromatography on silica gel (eluting with 2% EtOAc/hexanes) afforded the product as colorless oil (94 mg, 81%). 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 15.6 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.02-7.00 (m, 2H), 6.28 (d, J = 15.6 Hz, 1H), 2.40 (s, 3H), 2.33 (s, 3H), 1.55 (s, 9H).16a
(E)-7-(4-Nitrophenoxy)-7-phenylhept-4-en-3-one (26)
The representative procedure above was followed using 1-nitro-4-(1-phenylbut-3-en-1-yloxy)benzene (25)21a (135 mg, 0.50 mmol), ethyl vinyl ketone (126 mg, 1.50 mmol) and Grubbs-2 catalyst (8.5 mg, 0.01 mmol). Column chromatography on silica gel (eluting with 15% EtOAc/hexanes) afforded the product as a pale yellow liquid (122 mg, 75%). 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 9.2 Hz, 2H), 7.38–7.27 (m, 5H), 6.89 (d, J = 9.2 Hz, 2H), 6.83 (dt, J = 16.0, 7.2 Hz, 1H), 6.17 (d, J = 16.0 Hz, 1H), 5.34 (dd, J = 7.6, 4.8 Hz, 1H), 2.97–2.89 (m, 1H), 2.83–2.76 (m, 1H), 2.54 (q, J = 7.2 Hz, 2H), 1.07 (t, J = 7.2 Hz, 3H).21a
t-Butyldimethyl(11-methyldodec-10-en-1-yloxy)silane
TBSCl (0.29 g, 1.92 mmol) and imidazole (0.17 g, 2.50 mmol) were dissolved in dry CH2Cl2 (6 mL) in a round bottom flask. 11-methyldodec-10-en-1-ol46 (0.25 g, 1.26 mmol) was then added dropwise and the resulting solution was allowed to warm to rt and left to stir for another 12 h, after which it was quenched with water. The aqueous layer was extracted with CH2Cl2 and the organic extracts were washed with water and dried over Na2SO4. The volatiles were removed in vacuo to afford the crude product which was subsequently purified by column chromatography on silica gel (eluting with 5% EtOAc/hexanes) to yield the product (0.38 g, 96%) as a colorless oil. IR (neat): 2851, 1464, 1386, 1361, 1253, 1100, 1007, 984, 938, 840, 775 cm−1; 1H NMR (400 MHz, CDCl3): δ 5.12 (t, J = 7.2 Hz, 1H), 3.60 (t, J = 6.8 Hz, 2H), 1.96 (q, J = 6.8 Hz, 2H), 1.69 (s, 3H), 1.61 (s, 3H), 1.51 (quint, J = 6.8 Hz, 2H), 1.28 (br s, 12H), 0.91 (s, 9H), 0.06 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 131.3, 125.2, 63.6, 33.1, 30.1, 29.9, 29.8, 29.7, 29.6, 29.6, 28.3, 26.2, 26.0, 18.6, 17.9, −5.0; EI-MS m/z (%): 255 (M – C4H9, 70), 75 (100); HRMS (EI) calcd for C15H31OSi [M – C4H9]+ = 255.2144, found 255.2146.
(R)-11-Methyldodec-10-en-1-yl 2-(benzyloxycarbonylamino)-3-phenylpropanoate
To a 50 mL round-bottomed-flask containing a stir bar, was added CH2Cl2 (12 mL) which was cooled to 0 oC. N-carbobenzyloxy-L-phenylalanine (0.45 g, 1.51 mmol), 11-methyldodec-10-en-1-ol46 (0.25 g, 1.26 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.33 g, 2.14 mmol), and 4-dimethylaminopyridine (0.015 g, 0.13 mmol) were then added. The resulting solution was allowed to warm to rt over a 20 h period, after which it was quenched by the addition of H2O. The reaction mixture was extracted with CH2Cl2, and the organic layer washed with aqueous NaHCO3, water and dried over anhydrous Na2SO4. Removal of the volatiles in vacuo afforded the crude residue, which was purified by column chromatography on silica gel (eluting with 10% EtOAc/hexanes) to yield the product (0.58 g, 96%) as a colorless liquid. IR (neat): 3348, 3087, 3063, 3031, 2925, 2854, 1720, 1604, 1499, 1455, 1395, 1376, 1346, 1254, 1208, 1081, 1057, 1029 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.39-7.22 (m, 8H), 7.11 (dd, J = 7.6, 1.6 Hz, 2H), 5.25 (d, J = 8.4 Hz, 1H), 5.14-5.07 (m, 3H), 4.66 (dt, J = 8.4, 5.6 Hz, 1H), 4.15-4.05 (m, 2H), 3.14 (dd, J = 13.6, 5.6 Hz, 1H), 3.10 (dd, J = 13.6, 5.6 Hz, 1H), 1.99-1.95 (m, 2H), 1.70 (s, 3H), 1.61 (s, 3H), 1.60-1.58 (m, 2H), 1.28 (br s, 12H); 13C NMR (100 MHz, CDCl3): δ 171.6, 155.7, 136.4, 135.9, 131.2, 129.4, 128.61, 128.58, 128.2, 128.1, 127.1, 125.0, 67.0, 65.8, 55.0, 38.5, 30.1, 29.68, 29.65, 29.5, 29.4, 28.6, 28.2, 26.0, 25.9, 17.9; ESI-MS m/z: 502 (M + Na); HRESIMS calcd for C30H41NO4Na [M + Na]+ = 502.2933, found 502.2909.
(R)-11-Methyldodec-10-en-1-yl 2-(benzyloxycarbonylamino)-3-phenylpropanoate
To a 50 mL round-bottomed-flask containing a stir bar, was added CH2Cl2 (12 mL) which was cooled to 0 oC. N-carbobenzyloxy-L-phenylalanine (0.45 g, 1.51 mmol), 11-methyldodec-10-en-1-ol46 (0.25 g, 1.26 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.33 g, 2.14 mmol), and 4-dimethylaminopyridine (0.015 g, 0.13 mmol) were then added. The resulting solution was allowed to warm to rt over a 20 h period, after which it was quenched by the addition of H2O. The reaction mixture was extracted with CH2Cl2, and the organic layer washed with aqueous NaHCO3, water and dried over anhydrous Na2SO4. Removal of the volatiles in vacuo afforded the crude residue, which was purified by column chromatography on silica gel (eluting with 10% EtOAc/hexanes) to yield the product (0.58 g, 96%) as a colorless liquid. IR (neat): 3348, 3087, 3063, 3031, 2925, 2854, 1720, 1604, 1499, 1455, 1395, 1376, 1346, 1254, 1208, 1081, 1057, 1029 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.39-7.22 (m, 8H), 7.11 (dd, J = 7.6, 1.6 Hz, 2H), 5.25 (d, J = 8.4 Hz, 1H), 5.14-5.07 (m, 3H), 4.66 (dt, J = 8.4, 5.6 Hz, 1H), 4.15-4.05 (m, 2H), 3.14 (dd, J = 13.6, 5.6 Hz, 1H), 3.10 (dd, J = 13.6, 5.6 Hz, 1H), 1.99-1.95 (m, 2H), 1.70 (s, 3H), 1.61 (s, 3H), 1.60-1.58 (m, 2H), 1.28 (br s, 12H); 13C NMR (100 MHz, CDCl3): δ 171.6, 155.7, 136.4, 135.9, 131.2, 129.4, 128.61, 128.58, 128.2, 128.1, 127.1, 125.0, 67.0, 65.8, 55.0, 38.5, 30.1, 29.68, 29.65, 29.5, 29.4, 28.6, 28.2, 26.0, 25.9, 17.9; ESI-MS m/z: 502 (M + Na); HRESIMS calcd for C30H41NO4Na [M + Na]+ = 502.2933, found 502.2909.
2-(Dec-9-en-1-yl)-1,3-dioxolane (22)
A round bottom flask, equipped with a stirrer bar and a Dean Stark apparatus, was charged with Undec-10-enal (1.34 g, 8.00 mmol), ethylene glycol (2.7 mL, 48.00 mmol), pTSA.H2O (0.06 g, 0.32 mmol), and benzene (15 mL) and the reaction mixture heated at reflux for 8 h. After usual work-up the pure acetal (1.52 g, 90%) was obtained by distillation in vacuo as a colorless oil. IR (neat): 3076, 2976, 2926, 2855, 1640, 1465, 1438, 1411, 1361, 1212, 1142, 1037, 993, 944, 910 cm−1; 1H NMR (400 MHz, CDCl3): δ 5.87-5.76 (m, 1H), 5.02-4.92 (m, 2H), 4.85 (t, J = 4.8 Hz, 1H), 4.01-3.93 (m, 2H), 3.90-3.81 (m, 2H), 2.04 (q, J = 7.2 Hz, 2H), 1.68-1.63 (m, 2H), 1.45-1.29 (m, 12H); 13C NMR (100 MHz, CDCl3): δ 139.3, 114.2, 104.8, 65.0, 34.1, 34.0, 29.8, 29.7, 29.6, 29.3, 29.1, 24.3; EI-MS m/z (%): 211 (M – H, 2), 73 (100); HRMS (EI) calcd for C13H23O2 [M – H]+ = 211.1698, found 211.1693.
Recycling of PTS
N,N′-Diallyl-4-methylbenzenesulfonamide (25 mg, 0.10 mmol) and Grubbs-2 catalyst (1.7 mg, 0.002 mmol) were both added into a Teflon-coated-stir-bar-containing Biotage 5 mL microwave reactor vial at rt, and sealed with a septum. An aliquot of PTS/H2O (1.0 mL; 2.5% PTS by weight) was added, via syringe, and the resulting solution was allowed to stir at rt for 2 h. Et2O (3 mL) was then added to the reaction mixture and stirred for 10 s. The reaction mixture was then allowed to separate and the upper (Et2O) layer was removed by pipette. The aqueous layer was successively washed with Et2O (3 × 3 mL). The combined Et2O extracts layers were evaporated in vacuo to afford the crude product, which was examined by 400 MHz 1H NMR spectroscopy to reveal complete conversion of diene and clean formation of the corresponding cyclized product. 1H NMR (400 MHz, CDCl3):δ 7.72 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 5.65 (t, J = 4.4 Hz, 2H), 4.11 (t, J = 4.4 Hz, 2H), 2.42 (s, 3H).47 For the second run, the diene (25 mg, 0.10 mmol) and Grubbs-2 catalyst (1.7 mg, 0.002 mmol) were both added again to the same reaction vessel and stirred at rt for another 2 h. The workup was conducted in exactly the same way as described for the first cycle. This reaction was repeated eight more times, each using the above diene (25 mg, 0.10 mmol) and Grubbs-2 catalyst (1.7 mg, 0.002 mmol).
Supplementary Material
Acknowledgments
Financial support provided by the NIH (GM 86485) is warmly acknowledged. We also thank Dr. Thomas J. Colacot (Johnson Matthey), Dr. Richard Pederson (Materia) and Dr. Oliver Briel (Umicore) for supplying the palladium, Grubbs, and Neolyst catalysts, respectively, and to Zannan Pharma Ltd. for catalysts RC-303 and RC-304, used in this study. We are also grateful to the Materials Research Lab Central Facilities at UCSB, supported by the MRSEC Program of the National Science Foundation (DMR05-20415), for cryo-TEM experiments.
Footnotes
Supporting Information Available: Synthesis of starting materials and copies of 1H and 13C NMR spectra of all new compounds and copies of 1H NMR spectra of all known compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Bruce H. Lipshutz, Email: lipshutz@chem.ucsb.edu, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106.
Subir Ghorai, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106.
Wendy Wen Yi Leong, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106.
Benjamin R. Taft, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106
Daniel V. Krogstad, Materials Department, University of California, Santa Barbara, CA 93106
References
- 1.(a) Dwars T, Paetzold E, Oehme G. Angew Chem, Int Ed. 2005;44:7174–7199. doi: 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]; (b) Khan MN. Micellar Catalysis. CRC Press; Boca Raton, FL: 2006. [Google Scholar]
- 2.(a) Borowy-Borowski H, Sikorska-Walker M, Walker PR. U.S. Patent 6,045,826. Water-Soluble Compositions of Bioactive Lipophilic Compounds. 2000 Apr 4;; (b) Borowy-Borowski H, Sikorska-Walker M, Walker PR. U.S. Patent 6,191,172. Water-Soluble Compositions of Bioactive Lipophilic Compounds. 2001 Feb 20;; (c) Borowy-Borowski H, Sikorska-Walker M, Walker PR. U.S. Patent 6,632,443. Water-Soluble Compositions of Bioactive Lipophilic Compounds. 2003 Oct 14;
- 3.Lipshutz BH, Ghorai S. Aldrichimica Acta. 2008;41:59–72. [PMC free article] [PubMed] [Google Scholar]
- 4.Huang S, Voigtritter KR, Unger JB, Lipshutz BH. Synlett. 2010:2041–2044. [Google Scholar]
- 5.(a) Schott H. J Colloid Interface Sci. 1995;173:265–277. [Google Scholar]; (b) Nishikido N, Matuura R. Bul Chem Soc Jap. 1977;50:1690–1694. [Google Scholar]
- 6.(a) Knowles JP, Whiting A. Org Biomol Chem. 2007;5:31–44. doi: 10.1039/b611547k. [DOI] [PubMed] [Google Scholar]; (b) Phan NTS, van der Sluys M, Jones CW. Adv Synth Catal. 2006;348:609–679. [Google Scholar]; (c) Alonso F, Beletskaya IP, Yus M. Tetrahedron. 2005;61:11771–11835. [Google Scholar]; (d) Bhattacharya S, Srivastava A, Sengupta S. Tetrahedron Lett. 2005;46:3557–3560. [Google Scholar]; (e) Beletskaya IP, Cheprakov AV. Aqueous Palladium Catalysis. In: Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Part X.1. Vol. 2. Wiley-Interscience; New York, NY: 2002. pp. 2957–3006. [Google Scholar]; (f) Stambuli JP, Stauffer SR, Shaughnessy KH, Hartwig JF. J Am Chem Soc. 2001;123:2677–2678. doi: 10.1021/ja0058435. [DOI] [PubMed] [Google Scholar]; (g) Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 7.Significant rate accelerations were also observed in Suzuki-Miyaura couplings with aryl chloride.
- 8.Lipshutz BH, Taft BR. Org Lett. 2008;10:1329–1332. doi: 10.1021/ol702755g. [DOI] [PubMed] [Google Scholar]
- 9.For applications and properties of BASF Solutol HS 15 (CAS Registry No. 70142-34-6), see [accessed Jan., 2011]; http://www.basf-korea.co.kr/02_products/04_finechemicals/document/pharma/techinfo/ti/down.asp?file=Solutol%20HS%2015.pdf.
- 10.For more information on Cremophor_ EL (CAS Registry No. 61791-12-6), see [accessed Jan., 2011]; http://www.pharma-ingredients.basf.com/Statements/Technical%20Informations/EN/Pharma%20Solutions/EMP%20030711e_Cremophor%20EL.pdf.
- 11.For applications and properties of Eastman Vitamin E TPGS NF, see [accessed Jan., 2011]; http://www.eastman.com/Literature_Center/P/PCI102.pdf.
- 12.Narayan S, Muldoon J, Finn MG, Fokin VV, Kolib HC, Sharpless KB. Angew Chem, Int Ed. 2005;44:3275–3279. doi: 10.1002/anie.200462883.See also: Blackmond DG, Armstrong A, Coombe V, Wells A. Angew Chem, Int Ed. 2007;46:3798–3800. doi: 10.1002/anie.200604952.
- 13.(a) Krasovskiy A, Duplais C, Lipshutz BH. J Am Chem Soc. 2009;131:15592–15593. doi: 10.1021/ja906803t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nishikata T, Lipshutz BH. J Am Chem Soc. 2009;131:12103–12105. doi: 10.1021/ja905082c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nishikata T, Abela AR, Lipshutz BH. Angew Chem, Int Ed. 2010;49:781–784. doi: 10.1002/anie.200905967. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Moser R, Nishikata T, Lipshutz BH. Org Lett. 2010;12:28–31. doi: 10.1021/ol9023908. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nishikata T, Lipshutz BH. Org Lett. 2010;12:1972–1975. doi: 10.1021/ol100331h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nishikata T, Lipshutz BH. Chem Commun. 2009:6472–6474. doi: 10.1039/b914982a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lipshutz BH, Chung DW, Rich B. Org Lett. 2008;10:3793–3796. doi: 10.1021/ol801471f. [DOI] [PubMed] [Google Scholar]
- 14.(a) Carrow BP, Hartwig JF. J Am Chem Soc. 2010;132:79–81. doi: 10.1021/ja909306f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Littke AF, Fu GC. J Am Chem Soc. 2001;123:6989–7000. doi: 10.1021/ja010988c. [DOI] [PubMed] [Google Scholar]
- 15.(a) Grubbs RH, Chang S. Tetrahedron. 1998;54:4413–4450. [Google Scholar]; (b) Fürstner A. Angew Chem, Int Ed. 2000;39:3012–3043. [PubMed] [Google Scholar]; (c) Trnka TM, Grubbs RH. Acc Chem Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; (d) Schrock RR, Hoveyda AH. Angew Chem, Int Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (e) Connon SJ, Blechert S. Angew Chem, Int Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]; (f) Grubbs RH, editor. Handbook of Metathesis. Wiley-VCH; Weinheim, Germany: 2003. (three-volume set) [Google Scholar]; (g) Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; (h) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]; (i) Gradillas A, Pérez-Castells J. Angew Chem, Int Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]; (j) Schrodi Y, Pederson RL. Aldrichimica Acta. 2007;40:45–52. [Google Scholar]
- 16.(a) Lipshutz BH, Aguinaldo GT, Ghorai S, Voigtritter K. Org Lett. 2008;10:1325–1328. doi: 10.1021/ol800028x. [DOI] [PubMed] [Google Scholar]; (b) Lipshutz BH, Ghorai S, Aguinaldo GT. Adv Synth Catal. 2008;350:953–956. [Google Scholar]
- 17.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 18.(a) Chatterjee AK, Morgan JP, Scoll M, Grubbs RH. J Am Chem Soc. 2000;122:3783–3784. [Google Scholar]; (b) Lipshutz BH, Ghorai S, Leong WWY. J Org Chem. 2009;74:2854–2857. doi: 10.1021/jo900012z. [DOI] [PubMed] [Google Scholar]; (c) Ettari R, Micale N. J Organomet Chem. 2007;692:3574–3576. [Google Scholar]; (d) Michrowska A, Bujok R, Harutyunyan S, Sashuk V, Dolgonos G, Grela K. J Am Chem Soc. 2004;126:9318–9325. doi: 10.1021/ja048794v. [DOI] [PubMed] [Google Scholar]; (e) Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem, Int Ed. 2002;41:4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 19.(a) Hong SH, Grubbs RH. J Am Chem Soc. 2006;128:3508–3509. doi: 10.1021/ja058451c. [DOI] [PubMed] [Google Scholar]; (b) Dunbar MA, Balof SL, Roberts AN, Valente EJ, Schanz HJ. Organometallics. 2011;30:199–203. [Google Scholar]; (c) Gallivan JP, Jordan JP, Grubbs RH. Tetrahedron Lett. 2005;46:2577–2580. [Google Scholar]; (d) P’Pool SJ, Schanz HJ. J Am Chem Soc. 2007;129:14200–14212. doi: 10.1021/ja071938w. [DOI] [PubMed] [Google Scholar]; (e) Lynn DM, Mohr B, Grubbs RH, Henling LM, Day MW. J Am Chem Soc. 2000;122:6601–6609. [Google Scholar]; (f) Lynn DM, Mohr B, Grubbs RH. J Am Chem Soc. 1998;120:1627–1628. [Google Scholar]
- 20.(a) Rivard M, Blechert S. Eur J Org Chem. 2003:2225–2228. [Google Scholar]; (b) Wright DL, Usher LC, Esterella-Jimenez M. Org Lett. 2001;3:4275–4277. doi: 10.1021/ol016936+. [DOI] [PubMed] [Google Scholar]; (c) Gessler S, Randl S, Blechert S. Tetrahedron Lett. 2000;41:9973–9976. [Google Scholar]
- 21.(a) Lipshutz BH, Ghorai S, Bošković ŽV. Tetrahedron. 2008;64:6949–6954. [Google Scholar]; (b) BouzBouz S, Cossy J. Org Lett. 2001;3:1451–1454. doi: 10.1021/ol0157095. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee AK, Choi TL, Grubbs RH. Synlett. 2001:1034–1037. [Google Scholar]
- 23.(a) Thayer AM. Chem Eng News. 2007;85:37. [Google Scholar]; (b) Jafarpour L, Schanz HJ, Stevens ED, Nolan SP. Organometallics. 1999;18:5416–5419. [Google Scholar]; (c) Furstner A, Grabowski J, Lehmann CW. J Org Chem. 1999;64:8275–8280. doi: 10.1021/jo991021i. [DOI] [PubMed] [Google Scholar]; (d) Furstner A, Thiel OR, Ackermann L, Schanz HJ, Nolan SP. J Org Chem. 2000;65:2204–2207. doi: 10.1021/jo9918504. [DOI] [PubMed] [Google Scholar]; (e) Furstner A, Guth O, Duffels A, Seidel G, Liebl M, Gabor B, Mynott R. Chem Eur J. 2001;7:4811–4820. doi: 10.1002/1521-3765(20011119)7:22<4811::aid-chem4811>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]; (f) Dragutan V, Dragutan I. Platinum Met Rev. 2005;49:33–40. [Google Scholar]; (g) Schanz HJ, Jafarpour L, Stevens ED, Nolan SP. Organometallics. 1999;18:5187–5190. [Google Scholar]
- 24.Paquette LA, Dong S, Parker GD. J Org Chem. 2007;72:7135–7147. doi: 10.1021/jo070862j.For more information on Zhan catalyst (CAS Registry No. for RC-303: 918870-76-5 and for RC-304: 918871-44-0), see [accessed Jan., 2011]; http://www.zannanpharma.com/Zannan/Zannan/Zhan%20Catalysts%20&%20Metathesis.pdf.
- 25.(a) Lipshutz BH, Ghorai S. Org Lett. 2009;11:705–708. doi: 10.1021/ol8027829. [DOI] [PubMed] [Google Scholar]; (b) Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 26.(a) Morgan JP, Grubbs RH. Org Lett. 2000;2:3153–3155. doi: 10.1021/ol0063510. [DOI] [PubMed] [Google Scholar]; (b) Dias EL, Nguyen ST, Grubbs RH. J Am Chem Soc. 1997;119:3887–3897. [Google Scholar]; (c) Sanford MS, Henling LM, Grubbs RH. Organometallics. 1998;17:5384–5389. [Google Scholar]
- 27.Voigtritter K, Ghorai S, Lipshutz BH. J Org Chem. 2011 doi: 10.1021/jo200360s. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Choi TL, Chatterjee AK, Grubbs RH. Angew Chem, Int Ed. 2001;40:1277–1279. doi: 10.1002/1521-3773(20010401)40:7<1277::aid-anie1277>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (b) Goldberg SD, Grubbs RH. Angew Chem, Int Ed. 2002;41:807–810. doi: 10.1002/1521-3773(20020301)41:5<807::aid-anie807>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; (c) Chatterjee AK, Toste FD, Choi TL, Grubbs RH. Adv Synth Catal. 2002;344:634–637. [Google Scholar]
- 29.Donohoe TJ, Bower JF. PNAS. 2010;107:3373–3376. doi: 10.1073/pnas.0913466107.Randl S, Connon SJ, Blechert S. Chem Comm. 2001:1796–1797.See also ref. 17, 18a, 28a.
- 30.Tian X, Rychnovsky SD. Org Lett. 2007;9:4955–4958. doi: 10.1021/ol702200t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The importance of sample viscosity in DLS measurements, see [accessed Jan., 2011]; http://www.rci.rutgers.edu/~moghe/Viscosity%20in%20DLS%20meas.pdf.
- 32.(a) Friedrich H, Frederik PM, de With G, Sommerdijk NAJM. Angew Chem, Int Ed. 2010;49:7850–7858. doi: 10.1002/anie.201001493. [DOI] [PubMed] [Google Scholar]; (b) González YI, Kaler EW. Curr Opin Colloid Interface Sci. 2005;10:256–260. [Google Scholar]
- 33.Menger FM, Keiper JS. Angew Chem, Int Ed. 2000;39:1906–1920. doi: 10.1002/1521-3773(20000602)39:11<1906::aid-anie1906>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 34.(a) Bhattacharya S, Kumar VP. Langmuir. 2005;21:71–78. doi: 10.1021/la048858f. [DOI] [PubMed] [Google Scholar]; (b) Bhattacharya S, Kumar VP. J Org Chem. 2004;69:559–562. doi: 10.1021/jo034745+. [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Yu Y, Xia W, Song Y, Huang Z. J Org Chem. 2002;67:3096–3103. doi: 10.1021/jo025586h. [DOI] [PubMed] [Google Scholar]
- 36.Davies SG, Mulvaney AW, Russell AJ, Smith AD. Tetrahedron: Asymmetry. 2007;18:1554–1566. [Google Scholar]
- 37.Wu Y, Zhao J, Chen J, Pan C, Li L, Zhang H. Org Lett. 2009;11:597–600. doi: 10.1021/ol8026208. [DOI] [PubMed] [Google Scholar]
- 38.Caron S, Vazquez E. J Org Chem. 2003;68:4104–4107. doi: 10.1021/jo034274r. [DOI] [PubMed] [Google Scholar]
- 39.Lipshutz BH, Ghorai S. Tetrahedron. 2010;66:1057–1063. [Google Scholar]
- 40.Henderson WH, Check CT, Proust N, Stambuli JP. Org Lett. 2010;12:824–827. doi: 10.1021/ol902905w. [DOI] [PubMed] [Google Scholar]
- 41.Bunnage ME, Davies SG, Goodwin CJ, Ichihara O. Tetrahedron. 1994;50:3975–3986. [Google Scholar]
- 42.Oh K, Knabe WE. Tetrahedron. 2009;65:2966–2974. [Google Scholar]
- 43.Yadav JS, Narsaiah AV, Reddy BVS, Basak AK, Nagaiah K. J Mol Cat, A: Chem. 2005;230:107–111. [Google Scholar]
- 44.Ikawa T, Hattori K, Sajiki H, Hirota K. Tetrahedron. 2004;60:6901–6911. [Google Scholar]
- 45.Audic N, Clavier H, Mauduit M, Guillemin JC. J Am Chem Soc. 2003;125:9248–9249. doi: 10.1021/ja021484x. [DOI] [PubMed] [Google Scholar]
- 46.Michrowska A, Bujok R, Harutyunyan S, Sashuk V, Dolgonosb G, Grela K. J Am Chem Soc. 2004;126:9318–9325. doi: 10.1021/ja048794v. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama R, Kobayashi S. Angew Chem, Int Ed. 2002;41:2602–2604. doi: 10.1002/1521-3773(20020715)41:14<2602::AID-ANIE2602>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.