

Abstract
Human effector memory (EM) CD4 T cells may be recruited from the blood into a site of inflammation in response either to inflammatory chemokines displayed on or specific antigen presented by venular endothelial cells (ECs), designated as chemokine-driven or TCR-driven transendothelial migration (TEM), respectively. We have previously described differences in the morphological appearance of transmigrating T cells as well as in the molecules that mediate T cell-EC interactions distinguishing these two pathways. Here we report that TCR-driven TEM requires ZAP-70-dependent activation of a pathway involving Vav, Rac and myosin IIA. Chemokine-driven TEM also utilizes ZAP-70, albeit in a quantitatively and spatially different manner of activation, and is independent of Vav, Rac and mysosin IIA, depending instead on an as yet unidentified GTP exchange factor that activates Cdc42. The differential use of small Rho family GTPases to activate the cytoskeleton is consistent with the morphological differences observed in T cells that undergo TEM in response to these distinct recruitment signals.
Keywords: Endothelial cells, T cells, Inflammation, Signal Transduction
Introduction
A critical step in the inflammatory response is the recruitment of circulating leukocytes across activated post-capillary venular endothelium into the peripheral tissues. The multi-step paradigm of myeloid leukocyte recruitment starts with sequential tethering, rolling, firm attachment, and intra-luminal crawling on the endothelial cell (EC) surface until the leukocyte is positioned above or near an inter-endothelial cell junction. The process concludes with cytoplasmic probing and cell process penetration of and ultimately transmigration across the endothelial monolayer (1). Activated venular ECs express leukocyte adhesion molecules that mediate tethering, rolling and firm adhesion and display chemokines that interact with chemokine receptors on the rolling leukocytes. Leukocyte responses to chemokines mediate all but the initial tethering and rolling steps in this cascade so that recruitment may be said to be chemokine-driven. T cells in the circulation may be divided into various subsets including naïve and central memory T cells that recirculate to secondary lymphoid organs and effector memory (EM) T cells (identified as CCR7−, CD62L−, CD45RA−, CD45RO+) that can be recruited to peripheral sites of inflammation (2, 3). EM T cells may follow the same general paradigm as myeloid leukocytes and will transmigrate in response to chemokines bound to luminal proteoglycans on the EC surface (4). In the presence of shear stress comparable to that present in post-capillary venules, EM CD4 T cells can complete the process of TEM across a microvascular EC monolayer in vitro within 10–15 minutes. The requirement for shear stress as an inducer of rapid TEM is unique to T cells (5). Primary resting human T cells crawling on the EC in the presence of shear stress are polarized, displaying a leading edge and a trailing uropod. Shear stress has been proposed to allow mechanical stretching of T cell LFA-1 molecules attached to EC ICAM-1, leading to a greater increase in LFA-1 affinity than that produced by chemokine signaling alone (6). As the T cell approaches an inter-endothelial junction, it extends sub-micron ventral adhesive and invasive filipodia into the EC surface, and subsequently into the junction between ECs, creating a gap through which TEM occurs (7).
EM T cells may alternatively undergo TEM by a process that is independent of chemokines. Human venular ECs in peripheral tissues basally express both MHC Class I and Class II molecules, enabling them to present antigens and thus signal through the TCR of a rolling EM CD8 or CD4 T cell, respectively. Since TCRs are clonally expressed, very few T cells actually respond to any particular antigen. In vivo, this is not a problem because the circulatory system constantly delivers fresh EM T cells to sample the antigens displayed by the venular ECs and those rare EM T cells that recognize their cognate antigen may serve as “pioneer cells,” initiating a recall response (8). Experimentally, the number of T cells capable of being activated through their TCR can be increased (and thus studied in vitro) by presentation of a superantigen, such as toxic shock syndrome toxin 1 (TSST-1), that can be recognized by 5–20% of peripheral blood EM T cells. Surprisingly, the activation of TCR signaling in EM CD4 T cells blocks TEM in response to inflammatory chemokines (9). Instead, TCR-activated EM CD4 T cells round up on the EC surface and extrude a long (up to 20 μm) cytoplasmic protrusion that crosses and tunnels beneath the EC monolayer; we have designated these structures as transendothelial protrusions (TEPs). In experiments using microvascular ECs, the T cell body eventually follows the TEP in a second step that depends upon EC expression of fractalkine (10). In addition to the morphological differences from chemokine-driven TEM, TCR-driven TEM is slower (requiring about 50 minutes) but similarly requires shear stress and utilizes LFA-1. However, TCR-driven TEM involves several EC junctional molecules, such as platelet-endothelial cell adhesion molecule-1 (CD31), CD99, and polio virus receptor (CD155) or nectin-2 (CD112), engaged by their cognate receptors on the T cell that are not required for chemokine-driven TEM (10–12).
Since changes in cell shape, exemplified by TEP formation, are generally controlled by changes in the actin cytoskeleton and since TEP formation is only observed in TCR-driven TEM, we reasoned that the cytoskeleton of EM CD4 T cells must undergo different forms of reorganization following TCR- vs. chemokine-signaling. Some of the pathways by which TCR or chemokine receptors can modulate the cytoskeleton are well described (13, 14). An early step in TCR signaling involves phosphorylation of tyrosine residues of several immunoreceptor tyrosine activation motifs (ITAMs) located within the cytoplasmic portions of the TCR-associated CD3 protein subunits, including the zeta chains, by src family kinases such as lck or fyn. These phosphorylated ITAMs then serve as binding sites for ZAP-70, a syk family cytosolic tyrosine kinase, that is, in turn, activated through phosphorylation by src-family kinases. Activated ZAP-70 then phosphorylates TCR-associated adaptor proteins such as LAT and SLP76, forming a complex that serves as a scaffold for the recruitment and activation of Vav, a GTP exchange factor (GEF) that activates Rac, a small Rho family GTP-binding (G) protein. Among other actions, Rac can reorganize the actin cytoskeleton in a manner that leads to polarized outgrowths of cell protrusions such as lamellipodia (15). Chemokine receptors are G-protein coupled receptors that serve as GEFs for trimeric G protein family members, and chemokine binding results in trimeric G protein activation. Through processes that are less well understood, activated trimeric G proteins may activate both cytosolic tyrosine kinases (such as ZAP-70 in T cells) and small Rho family G proteins such as Rho, Rac and/or Cdc42. Rho and Cdc42, like Rac, can reorganize the actin cytoskelton, but Rho typically mediates cellular retraction and Cdc42 commonly promotes the formation of filopodia (15). Thus, based on morphological difference between TCR-driven TEM (such as TEP formation) and chemokine-driven TEM (such as filopodia formation) it seemed likely that TCR and chemokine signaling would result in distinct patterns of small G protein activation. In addition to causing cell shape change, rearrangements of the actin cytoskeleton also modulates T cell attachment to other cells by regulating integrin affinity. The principal integrin expressed by T cells that mediates attachment to endothelial cells is LFA-1 (CD11a/CD18) and the control of LFA-1 affinity for its ligands on the endothelium is mediated through proteins that link actin filaments to the intracellular tails of the LFA-1 β chain, namely talin and kindlin 3, the latter being associated with the scaffold protein RACK1 (16, 17). Binding of these linker proteins to the intracellular domains of the integrin β chain causes the extracellular domains of both the LFA-1 α and β chains to extend and bind more tightly to ICAM-1. The quantity and distribution of extended forms of LFA-1 as well as the linker proteins associated with the cytoplasmic tail of LFA-1 may well change in a different manner in response to TCR vs. chemokine signaling.
In the present study, we have used a variety of approaches to analyze signaling events of the distinct processes of TEM that we have observed with a focus on small G proteins and the cytoskeleton. By means of pharmacological inhibition, we show that ZAP-70 activation is necessary for TCR-driven TEP formation and TEM as well as for chemokine-driven TEM. However, as detected by immunofluorescence microscopy, these stimuli activate ZAP-70 and LAT in a quantitatively and spatially distinct manner. Similarly, the cellular distribution of activated LFA-1, talin, and RACK1 reflect the morphological changes attendant with TCR-driven TEM. By transducing primary EM CD4 T cells with membrane-permeable recombinant proteins, we show that Vav and its target small G protein Rac are required for TCR-driven TEM, while the small G protein Cdc42 controls chemokine-driven TEM in a manner that is independent of Vav and Rac. Furthermore, pharmacological inhibition suggests that myosin IIA is necessary for TCR-driven TEM but dispensable for TEP formation and for chemokine-driven TEM. These differences in the manner by which TCR vs. chemokine receptors affect the cytoskeleton further highlight differences between these two modes of T cell recruitment to sites of inflammation.
Materials and Methods
Cells and reagents
All human materials were obtained from de-identified blood or tissue donors under protocols approved by the Yale Human Investigation Committee. CIITA-transduced human dermal microvascular EC (HDMEC) were generated using a retroviral vector and characterized as described (10). Prior to flow experiments, CIITA HDMEC were incubated in the presence of 10 ng/ml recombinant human TNF (rhTNFα, R&D Systems) for 18–28 hours to upregulate adhesion molecules and inflammatory chemokines, then overlaid with 100 ng/ml recombinant TSST-1 (Toxin Technology, Inc.) for 30 minutes to allow TCR-mediated activation of all T cells that utilize a Vβ2 gene segment to form their TCR, approximately 5–20% of the circulating T cells in most donors.
Leukapheresis was performed on healthy adult donors and PBMCs were enriched by Ficoll-Hypaque density gradient centrifugation prior to cryopreservation of aliquots. Total peripheral blood CD4 T cells were isolated from the cryopreserved samples by positive selection with CD4 Dynabeads magnetic beads and released with Detachabead (Dynal). Memory (CD4+CD45RA−) T cells were enriched by depletion of CD45RA cells from CD4 T cells using anti-CD45RA mAb (eBiosciences) and pan-mouse IgG beads (Dynal). EM CD4 T cells were further purified from the total CD4 memory pool by depletion of central memory markers with anti-CCR7 mAb (R&D Systems) and anti-CD62L (eBioscience) and pan-mouse IgG beads. Approximately 90% of the initial T cell population as well as essentially all other leukocyte types were removed by these manipulations. The remaining 10% of CD4 T cells, highly enriched for EM CD4 T cells, was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and penicillin/streptomycin (all from Invitrogen) overnight prior to assays. In experiments in which we used transduction of dominant negative proteins, the EM CD4 T cells were transduced with recombinant proteins for 2h and then washed prior to the overnight incubation. Where indicated, EM CD4 T cells were treated with piceatannol (Calbiochem) and Syk inhibitor II (Calbiochem) to inhibit ZAP-70, with, blebbistatin (Sigma) to inhibit myosin IIA, ML-7 (Calbiochem) to inhibit myosin light chain kinase (MLCK), EHop-016 (Calbiochem) to inhibit Rac and Cdc42, 6-Thio-GTP (Jena Bioscience) to inhibit Vav, or vehicle control (DMSO) and then washed prior to flow TEM assays.
Antibodies used to stain the samples are as follows: anti-Vβ2TCR mAb (Meridian Life Science), anti-NFAT mAb (Becton Dickenson), anti-Phospho-ZAP-70(Tyr319) rabbit polyclonal antibody (Cell Signaling), anti-CD18 mAb, clone MEM-148 (Enzo Life Sciences), anti-Phospho LAT (Tyr191) rabbit polyclonal antibody (Cell Signaling), anti-talin mAb clone 8d4 (Sigma), anti-RACK1 mAb (Santa Cruz Biotechnology). Phalloidin-488 (Invitrogen) was used to stain F-actin, and DAPI in the mounting media (ProLong Gold, Invitrogen) to stain nuclei.
Preparation of cell-permeable recombinant proteins
Bacterial expression constructs encoding His-tagged Hph1-EGFP-Rac1 and Hph-1-EGFP-Cdc42 wild type (wt) and dominant negative (DN) proteins were made by subcloning the products of PCR using EGFP-Rac1 and EGFP-Cdc42 plasmids (Addgene plasmids 12980, 12982, 12599, and12601 corresponding to Rac1 wt, Rac1 DN. Cdc42 wt, and Cdc42 DN, respectively, provided by Gary Bokoch (Scripps Research Institute, Lo Jolla, CA)(18) and Klaus Hahn (University of North Carolina School of Medicine, Chapel Hill, North Carolina)(19)) as templates with 5′-GGATCCGTATGCGCGCGTGCGCCGTCGCGGCCCGCGTCGCGGTGGTGGTATG GTGAGCAAGGGCGAGGAGCTG-3′ and 5′-TAGAAGGCACAGTCGAAGG-3′ as primers into the BamHI and XhoI sites of pET28b (Novagen); PCR products were cloned into pCR 2.1-TOPO vector (Invitrogen) and sequenced prior to subcloning into pET-28b. Similarly, His-tagged Hph1-EGFP and Hph-1-EGFP-Vav DN were made using pEGFP-c1 (Clontech) and pNM117 (a plasmid kindly provided by Xose Bustelo, University of Salamanca, Salamanca, Spain, that encodes EGFP-Vav SH3-SH2-SH3 (20)) as templates. However, due to the presence of a BamHI site in the 3′ UTR of the Vav1 cDNA in pNM117, the Vav DN construct was subcloned into the BamHI site only of pET-28b. Recombinant proteins were purified from BL21 (DE3) bacteria containing pET-28b constructs induced with 1 mM IPTG for 5–6 hours at 30°C using Talon metal affinity resin as described by the manufacturer (Clontech) using imidazole for elution from the resin, and then concentrated with Amicon centrifugal filters.
FACS analysis
Flow Cytometry Standard (FCS) files of EM CD4 T cells transduced (or not) with GFP fusion recombinant proteins were acquired using an LSRII flow cytometer (Becton Dickenson) with FACSDiva software (Becton Dickenson) and analyzed using Flowjo software (Tree Star).
TEM assays
CIITA HDMEC were grown to confluence on 20 μg/ml human plasma fibronectin-coated 35 mm coverglasses, treated with TNF and loaded with TSST-1 as described, washed twice with RPMI/10% FBS, and a parallel plate flow chamber apparatus (Glycotech) using the 0.01 inch height, 5 mm wide slit gasket provided by the manufacturer was mounted on the coverglass. On a 37°C heating surface, CD4+CD45RA−CCR7lowCD62Llow (EM) human T cells (106 cells/500 μl) suspended in the same medium were loaded onto the EC monolayer at 0.75 dyne/cm2 for 2 minutes, followed by washing with medium only at 1 dyne/cm2 for 5, 15, 30 or 50 minutes. Samples were then fixed with 3.7% formaldehyde in PBS, stained with anti-Vβ2TCR mAb, followed by Alexafluor 546, 568, or 488-conjugated donkey or goat anti-mouse IgG, mounted on slides using mounting medium containing DAPI (Prolong Gold, Invitrogen), and examined by microscopy with a Zeiss Axiovert 200M microscope. A FITC filter was used to detect FITC or Alexafluor 488-stained cells, a TRITC filter was used to detect PE and Alexafluor 546 and 568-stained cells, a DAPI filter was used to detect DAPI-stained nuclei, and a Cy5 filter was used to detect Alexafluor 647 stained cells. A 40X/0.60 korr Ph2 objective and phase contrast optics were used to determine whether CD4 T cells were either on top or underneath the HDMEC monolayer. T cells that were captured on top of the monolayer but not spread were round and bright when viewed under phase contrast. CD4 T cells that were spread, but still on top of the HDMEC monolayer were surrounded by a bright corona of light in contrast to those that had transmigrated, that were dark without a corona of light. The percentage of transmigrated CD4 T cells was calculated for 100–200 cells per sample by analyzing five to ten groups of 20 cells each, calculating the percentage for each group, and calculating the mean and s.e.m. for the groups. For NFAT staining, anti-NFAT mAb was pre-complexed with Zenon-647 (Invitrogen) and incubated with Triton X-100-permeabilized samples after anti-Vβ2TCR mAb and anti-mouse IgG staining. Activated ZAP-70 and P-LAT was detected by using anti-Vβ2TCR mAb and anti-Phospho(Tyr319)ZAP-70 and anti-Phospho(Tyr191)LAT rabbit polyclonal antibodies followed by Alexafluor-488 or-546-conjugated donkey anti-mouse IgG and Alexafluor 647-conjugated chicken anti-rabbit IgG (Invitrogen), and mounted on slides using mounting medium containing DAPI. Staining was quantified with Image J.
Confocal Microscopy
Images were captured with a Leica TCS SP5 Spectral Confocal Microscope, 405UV using a 63X oil immersion objective and sequential scanning with 405 Diode, argon and He/Ne laser excitation lines of 405 nm, 488 nm, 543 nm, and 633 nm.
Statistics
For experiments in which more than two groups were compared, statistical significance was determined by one-way ANOVA using a 95% confidence interval and the Tukey post-test (Prism 4.0 for Macintosh). Statistical error is expressed as s.e.m. For experiments in which two groups were compared, a t-test was used.
Results
The aim of this study is to identify proteins in EM CD4 T cells that couple TCR to transendothelial migration (TEM), focusing on signals that could potentially reorganize the cytoskeleton to form the novel TEPs we have observed during this process. We are particularly interested in signaling pathways that distinguish TCR-driven TEM from chemokine-driven TEM. To simultaneously analyze chemokine- and TCR-driven TEM, both of which are shear-stress-dependent, we use TNF-treated CIITA-transduced HDMEC monolayers overlaid with superantigen TSST-1, as described previously (10, 12), and analyze the morphology and location of isolated human peripheral blood EM CD4 T cells allowed to interact with the EC monolayer in a flow chamber apparatus that applies venular levels of shear stress. In this system, those EM CD4 T cells that express a TCR containing a Vβ2 gene segment (Vβ2TCR, comprising 5–20% of the T cells in a normal adult) will interact with and become activated by TSST-1 bound to MHC Class II molecules expressed on the EC as a result of CIITA transduction. (EC lose MHC Class II expression in culture and transduction with CIITA was used to restore expression.) As reported previously, T cells that do not express a Vβ2-containing TCR (Vβ2TCR−, the other 80–95% T cells in a normal adult) are not activated by TSST-1 and transmigrate within 15 minutes in response to TNF-induced chemokines displayed on the surface of the EC. Vβ2TCR+ T cells respond similarly to Vβ2TCR− T cells in the absence of TSST-1. However, essentially all of the Vβ2TCR+ cells activated by TSST-1 no longer respond to inflammatory chemokines displayed by the EC but instead round up, and a proportion of these cells form TEPs that penetrate and tunnel beneath the EC monolayer, and subsequently transmigrate within 50 minutes. We have previously shown that pharmacological inhibition of src-family kinases reverses TCR inhibition of chemokine-driven TEM (9), likely by preventing the recruitment and activation of ZAP-70 to the TCR/CD3 complex.
To analyze the role of ZAP-70 in TEM, we treated the EM CD4 T cells with two different ZAP-70 inhibitors, namely piceatannol and Syk Inhibitor II, and analyzed samples after 15 and 50 minutes flow. If ZAP-70 only contributed to TCR-driven TEM but not chemokine-driven TEM, there would be a reversal of the TCR “stop” signal, and an increase in TEM of the Vβ2TCR+ cells at 15 minutes. Treatment of EM CD4 T cells with ZAP-70 inhibitors did not reverse the TCR “stop” signals at 15 minute (data not shown), since it abrogated both chemokine-and TCR-driven TEM (Figure 1A). To assess specificity, we stained the samples for NFAT, since TCR-mediated activation of ZAP-70 leads to activation of NFAT, detectable by immunofluorescence as translocation of this transcription factor from the cytosol to the nucleus. The effects of ZAP-70 inhibitors on TEM were observed at concentrations that corresponded with the inhibition of NFAT translocation to the nucleus in Vβ2TCR+ cells (Figure 1B and data not shown), consistent with the interpretation that the effect of the inhibitors on TEM was due to the inhibition of ZAP-70.
Figure 1.

T cell ZAP-70 activity is required for both chemokine- and TCR-driven TEM. A. TEM assays of EM CD4 T cells treated with 0, 10, 30 and 100 μg/ml piceatannol (pic) for 30 minutes or 0 and 40 μM Syk Inhibitor II for 60 minutes. Graphs show %TEM of cells lacking the Vβ2TCR (VB2−, which do not interact with TSST-1 and transmigrate in response to chemokines on the EC; chemokine-driven TEM) at 15 min flow (pic) or 50 min flow (Syk Inhib. II) and % TEM of cells expressing Vβ2TCR (VB2+, those that interact with TSST-1 presented by the EC; TCR-driven TEM) after 50 min flow. Graphs display combined data from 2 separate experiments (pic) or one representative experiment of two (Syk inhib. II). *, p<0.001, **, p<0.0001. B. Samples from TEM experiments were stained for Vβ2TCR, NFAT, and nuclei (DAPI). Shown are samples from T cells treated with 30 and 100 μg/ml pic. Arrows point to NFAT staining in Vβ2TCR+ T cells. Note that NFAT is translocated to the nucleus in the Vβ2TCR+ T cell at 30 μg/ml pic, but is excluded from the nucleus in all Vβ2TCR− cells as well as the Vβ2TCR+ cell at 100 μg/ml pic.
In addition to its well-established role in TCR signaling, previous studies have reported that ZAP-70 is also involved in chemokine receptor responses (21–24). We directly assessed ZAP-70 activation by immunofluorescence microscopy using an antibody to a phosphorylated form of the enzyme. By this approach, we observed that all EM CD4 T cells bound to the ECs activate ZAP-70, but that there are marked differences in the extent and localization of ZAP-70 activation in the EM CD4 T cells depending on whether or not they encounter antigen presented by the EC under flow. Within a few minutes of contacting the EC surface, the cells unable to recognize TSST-1 (the Vβ2TCR− cells) adopt the well-characterized polarized morphology associated with flattened crawling T cells, whereas the Vβ2TCR+ cells appear circular and rounded-up. Activated ZAP-70 is abundant in the Vβ2TCR+ cells compared to the Vβ2TCR− cells and persists throughout the time course used in these studies (up to 50 minutes, Figure 2 and data not shown), and is located uniformly around the plasma membrane, including the TEPs, with no obvious preference for one site or another. LAT, a target of ZAP-70 kinase activity in the TCR signaling pathway, is also abundantly phosphorylated in the Vβ2TCR+ cells compared to the Vβ2TCR− cells, but appears to be more intense at the T cell-EC interface, and similarly distributed in the TEPs (Figure 3).
Figure 2.

Activated ZAP-70 in EM CD4 T cells undergoing chemokine- and TCR-driven TEM. A. EM CD4 T cells on TNF-treated CIITA HDMEC overlaid with TSST-1 after 5 minutes of flow stained for DAPI (blue), Vβ2TCR (VB2, green), and Phospho(Tyr319)ZAP-70 (P-ZAP-70, white) were imaged with a confocal microscope at different Z sections corresponding to the T cell-EC interface and above, as indicated by the numbers to the right (in μm). Z sections of the two T cells in the top panels are shown, one Vβ2TCR− (left panels) and the other Vβ2TCR+ (right panels). Note the strong symmetrical P-ZAP-70 staining pattern in the VB2+ cell at all layers, and the focal dots in the VB2− cell apparent mostly at the T cell-EC interface. B. Quantification of P-ZAP-70 staining of Vβ2TCR− (VB2−) and Vβ2TCR+ (VB2+) T cells at 5 minute flow. N=22, *, p<0.0001. C. Confocal images of a Vβ2TCR+ T cell (VB2 in red) at 30 minutes flow with a TEP that is underneath the EC monolayer (top panel), the cell body near the apical surface of the EC (middle panels) and the top of the T cell (bottom panels).
Figure 3.

Phospho-LAT staining of EM CD4 T cells at 5 and 30 minutes flow. A. 5 minute samples stained with DAPI (blue), phalloidin (green), Vβ2TCR (VB2, red), and Phospho-LAT (Tyr191)(P-LAT, white). Left panels show one Z section of a Vβ2TCR− T cell at the EC interface, and the right panels show two Z sections, one at the EC interface and another 1.26 μm above. B. Quantification of P-LAT staining of Vβ2TCR− (VB2−) and Vβ2TCR+ (VB2+) T cells at 5 minute flow. N=20, *, p<0.0001. C. Confocal images of a Vβ2TCR+ T cell at 30 minutes flow with a TEP that is underneath the EC monolayer (top panels), the cell body near the apical surface of the EC (middle panels) and further above the EC interface (bottom panels). Numbers indicate μm above the TEP.
ZAP-70 has been reported to constitutively associate with LFA-1 and become activated upon LFA-1 binding to ICAM-1 (25), whereas focal dots of high-affinity LFA-1-ICAM-1 have been described in primary T cells crawling along EC under flow, although these focal dots seemed to be devoid of phosphotyrosine (7). In EM CD4 T cells under flow, activated ZAP-70 in the cells responding to chemokine appears to colocalize with activated LFA-1 in some, but not all, areas of the cells undergoing chemokine-driven TEM, whereas activated LFA-1 broadly colocalized with P-ZAP-70 everywhere in the Vβ2TCR+ cells (Figure 4).
Figure 4.

Activated LFA-1 and P-ZAP-70 staining of chemokine- and TCR-driven TEM at 5 minutes. Left four panels: Chemokine-driven TEM. Z sections showing DAPI, P-ZAP-70 (red), activated LFA-1 (white), and an averlay of P-ZAP-70 and activated LFA-1. Right four panels: TCR-driven TEM. Z sections showing DAPI, Vβ2TCR (green), P-ZAP-70 (red), and activated LFA-1 (white).
In T cells, LFA-1 is activated by inside-out signals originating from chemokine receptors or the TCR. Binding of activated LFA-1 to ICAM-1 not only serves to attach the T cell to the EC, but may also provide signals as well, so called outside-in signaling. Talin and kindlin-3 control the activation and function of LFA-1 in T cells (16, 26, 27). Talin does not appear to be localized to any particular area in either Vβ2TCR+ or Vβ2TCR− cells (Figure 5). Staining for kindlin-3 was not successful (unpublished data, TDM), but staining for the scaffolding protein RACK1, which forms a ternary complex with kindlin-3 and LFA-1 (17), showed an exclusion from zones underneath the nucleus in Vβ2TCR+ cells, whereas some dots were apparent on the ventral surface underneath the nucleus of the Vβ2TCR− cells as well as at the leading edge and especially at the ventral surface of the uropod (Figure 5).
Figure 5.
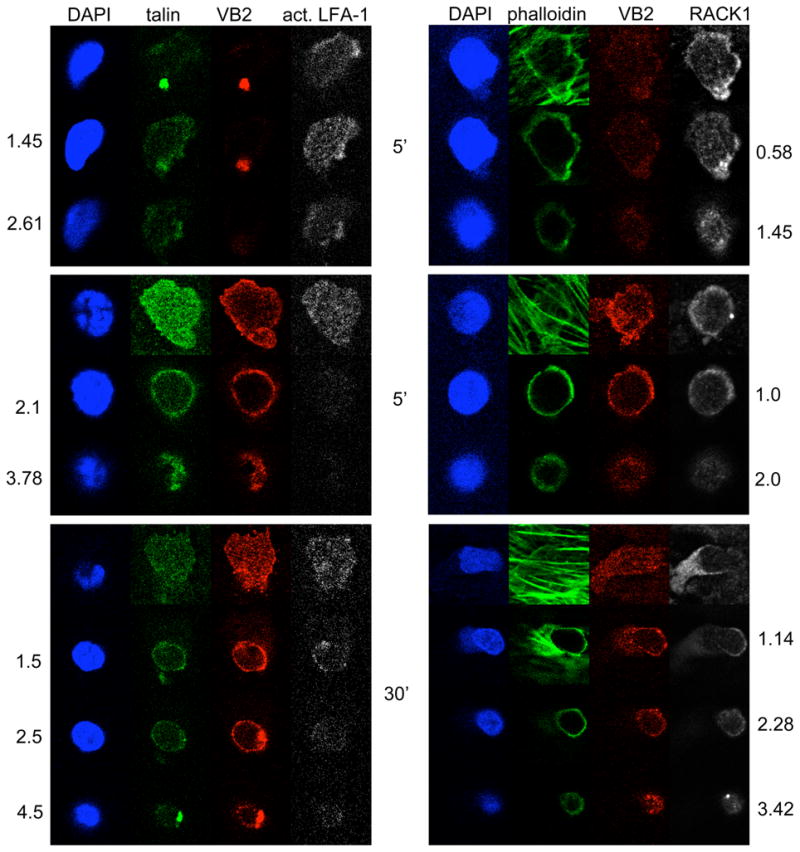
Confocal analysis of talin and RACK1 in EM CD4 T cells undergoing chemokine- and TCR-driven TEM. Left panels show Z slices of samples stained for nuclei (DAPI, blue), talin (green, Vβ2TCR (VB2, red) and activated LFA-1 (white). The right panels show Z slices of samples stained for nuclei (DAPI, blue), F-actin (phalloidin, green), Vβ2TCR (VB2, red), and RACK1 (white). The top set of panels represent Vβ2TCR− T cells at 5 min flow, the middle panels represent Vβ2TCR+ T cells at 5 min flow, and the bottom panels represent Vβ2TCR+ T cells at 30 min flow. The top panels of the 5 min flow samples are at the T cell EC interface, with the middle and bottom panels taken above, as the numbers indicate in μm. The top panels of the 30 min samples are underneath the EC monolayer, with the next three rows taken at layers above that plane, in the numbers indicated in μm.
In response to TCR signaling, ZAP-70 activates Vav once it is recruited to the phosphorylated LAT signalosome. T cells treated with the Vav inhibitor 6-thio-GTP reduced TCR-but not chemokine-driven TEM (Figure 6A). To support these findings, and since primary resting EM CD4 T cells are not amenable to genetic manipulations such as knockdown by siRNA or overexpression of proteins from genes introduced by transfection or viral transduction (TDM, unpublished observations), we adopted protein transduction of recombinant dominant negative proteins as an alternative approach to examine cellular signaling. By using recombinant proteins fused to both a protein transduction domain (Hph-1), shown to be effective for introducing functional protein into primary T cells (28, 29), and to EGFP as a marker, we introduced similar levels of dominant negative Vav1 (Vav DN) (20, 30) or Hph-1 fused directly to EGFP as a control into EM CD4 T cells (Figure 6B) and examined the transduced T cells in TEM assays. Vav DN reduced TCR- but not chemokine-driven TEM (Figure 6C).
Figure 6.

Vav is required for TCR-driven TEM. A. TEM assays of EM CD4 T cells treated with vehicle and 5 μM 6-Thio GTP for 44 hours. Graphs show %TEM of cells lacking the Vβ2TCR (VB2−, which do not interact with TSST-1 and transmigrate in response to chemokines on the EC; chemokine-driven TEM) and cells with Vβ2TCR (VB2+, those that interact with TSST-1 presented by the EC; TCR-driven TEM) after 50 min flow. Graphs display data combined from 3 separate experiments. *, p<0.001. B. FACS analysis of EM CD4+ T cells transduced with recombinant fusion proteins. Filled peak denotes nontransduced cells, and lines depict cells transduced with Hph-EGFP (GFP) and Hph-EGFP-Vav dominant negative (Vav DN). C. TEM assays of EM CD4+ T cells transduced with recombinant proteins. Graphs show %TEM of cells lacking the Vβ2TCR (VB2−)at 15 min flow and cells with Vβ2TCR (VB2+) after 50 min flow. Graphs display data combined from 3 separate experiments. *, p<0.001.
Vav has been described to act primarily as a GEF for the Rho-family GTPase Rac, but may also act on Cdc42, another Rho-family GTPase (31). Using the protein transduction approach described above for Vav, Rac1 and Cdc42 wild type and dominant negative proteins were transduced into EM CD4+ T cells (Figure 7A) and their effects tested in TEM assays. Dominant negative Cdc42 (Cdc42 DN) impaired chemokine-driven TEM, but had no effect on TCR-driven TEM (Figure 7B). Conversely, dominant negative Rac1 (Rac DN) had no effect on chemokine driven TEM, but reduced TCR-driven TEM (Figure 7B). Wild type versions of Rac and Cdc42 had no effect on either process compared to nontransduced cells. Treatment of EM CD4 T cells with EHop-016 reduced TCR-driven TEM at 1 μM and chemokine-driven TEM at 4 μM, consistent with the IC50 of this inhibitor for Rac and Cdc42, respectively (Figure 7C) (32).
Figure 7.

Chemokine-driven TEM requires Cdc42 and TCR-driven TEM requires Rac. A. FACS analysis of EM CD4 T cells transduced with recombinant fusion proteins. Filled peak denotes nontransduced cells, and lines depict cells transduced with Hph-GFP-Rac1 wild type (Rac wt), Hph-GFP-Rac dominant negative (Rac DN), Hph-GFP-Cdc42 wild type (Cdc42 wt), and Hph-GFP-Cdc42 dominant negative (Cdc42 DN). B. TEM assays of EM CD4 T cells transduced with recombinant proteins. Graphs show %TEM of cells lacking the Vβ2TCR (VB2−, which do not interact with TSST-1 and transmigrate in response to chemokines on the EC; chemokine-driven TEM) at 15 min flow and cells with Vβ2TCR (VB2+, those that interact with TSST-1 presented by the EC; TCR-driven TEM) after 50 min flow. Graphs display data from one representative experiment of at least 3. *, p<0.001. C. TEM assays of EM CD4 T cells treated with 0, 1, and 4 μM EHop-016 20 hours, flow 50 minutes. Graphs display data combined from three experiments. *, p<0.0001.
The organization of the cytoskeleton in non-muscle cells may be controlled either by regulating actin filament assembly or disassembly or by regulating the enzymatic activity of myosin. Myosin IIA is the only myosin II isoform in T lymphocytes (33). To investigate myosin IIA function in TEM of EM CD4 T cells under flow, we treated the T cells with blebbistatin, a myosin IIA-selective inhibitor, and tested them in the flow assay. Surprisingly, chemokine-driven TEM was not affected, but TCR-driven TEM was blocked by blebbistatin and was accompanied by an increase in the number of TEPs (Figure 8A), many of which became quite extensive (Figure 8B, C). Blebbistatin had no effect on ZAP-70 activation in Vβ2TCR+ EM CD4 T cells detectable within the first few minutes and all time points thereafter (up to 50 minutes, Supplemental Figure 1). Similarly, treatment of T cells with myosin light chain kinase inhibitor ML-7 had no effect on chemokine-driven TEM, but reduced TCR-driven TEM while augmenting the number of TEPs (Figure 8D). These data suggest that the primary role of myosin in TCR-driven TEM is to allow the cell body to follow the TEP across the EC monolayer rather than contribute to upstream signaling.
Figure 8.

Myosin IIA activity is required for TCR-driven TEM at a step after TEP formation, but not chemokine-driven TEM. A. TEM assays of EM CD4 T cells treated with blebbistatin. Graphs show %TEM of cells lacking the Vβ2TCR (VB2−, which do not interact with TSST-1 and transmigrate in response to chemokines on the EC; chemokine-driven TEM, left graph) at 15 min flow and cells with Vβ2TCR (VB2+, those that interact with TSST-1 presented by the EC; TCR-driven TEM, middle graph) after 50 min flow, as well as %TEPs of the VB2+ cells (right graph). Graphs display data from one representative of three separate experiments. *, p<0.001. B. EM CD4 T cells treated with vehicle or blebbistatin 30 minutes on TNF-treated CIITA HDMEC overlaid with TSST-1 after 30 minutes of flow stained for DAPI (blue) and Vβ2TCR (VB2, green) were imaged with a confocal microscope at different Z sections corresponding to below the EC monolayer, first row, and the T cell body 1.5 μm above on the apical side of the EC (second row). Note the exaggerated TEP (the entire VB2 staining underneath the EC monolayer, first row) in the blebbistatin treated sample compared to vehicle. Scalebar = 20 μm. C. Quantification of TEP length in vehicle and blebbistatin treated samples at 30 min flow. N=26, *p<0.0001. D. TEM assays of EM CD4 T cells treated with vehicle or 10 μM ML-7 for 30 minutes, 50 minute flow. Graphs display data combined from three experiments. *, p=0.0005, **, p<0.0001.
Discussion
In this report we provide evidence that TCR-driven TEM of EM CD4 T cells is controlled by Vav, Rac, and myosin IIA, each of which is dispensable for chemokine-driven TEM. This process is initiated by ZAP-70, but this kinase appears to affect chemokine-driven TEM as well. This is in agreement with previous studies reporting that ZAP-70 is involved in chemokine receptor responses (21–24), and explains why we did not see a reversal of the TCR “stop” signal, as was seen with lck inhibitors (9). Since ZAP-70 is constitutively associated with LFA-1 and becomes activated upon LFA-1 binding to ICAM-1 (25), it may be postulated that activated ZAP-70 may colocalize to the sites of high-affinity LFA-1-ICAM-1 described in primary T cells crawling along EC under flow, even though staining with anti-phosphtyrosine antibodies indicated that the focal dots did not contain phosphotyrosine (7). Staining of EM CD4 T cells undergoing chemokine-driven TEM for activated ZAP-70, although very weak, and activated LFA-1 indicated that there may be some colocalization of these two molecules. Since ZAP-70 levels are elevated in memory compared to naïve T cells (34), this may explain why it was not detected previously. In contrast, activation of ZAP-70 in TCR-stimulated T cells is much more robust and distributed throughout the entire plasma membrane as revealed by immunofluorescence microscopy. Furthermore, a substrate of ZAP-70, LAT, is negligibly phosphorylated in T cells undergoing chemokine-driven TEM as compared to TCR-driven TEM. The differences in the site and extent of ZAP-70 activation likely explains how the same enzyme can activate distinct downstream signaling pathways. Indeed, the confocal analysis of all molecules examined here, including activated LFA-1, talin, and RACK1, support the assertion that a major difference between chemokine- and TCR-driven TEM is the early change in cell shape and polarity, a process controlled by polarity proteins, which are required for conformational changes triggered by TCR signaling (35). It should be noted that, while essentially all of the TCR-activated cells have activated ZAP-70 and adopt a different morphology as compared to the chemokine-driven transmigrating cells, not all of these will form TEPs and transmigrate in the time course analyzed, indicating that while the “stop” signals are shared between all EM CD4 T cells, there may be some other signals that are necessary for TCR-driven TEM besides those from the TCR, or that our analysis did not extend long enough. The ability to follow TCR-driven TEM for longer time points is hampered by the downregulation of the TCR on the TSST-1-interacting T cells after about 90 minutes of flow, and hence the inability to detect and analyze those cells with the anti-Vβ2TCR antibody at time points after that (TDM, unpublished observations). By analyzing cells by immunofluorescence microscopy at the 30 minute time point, we were able to examine cells in the later stages of TCR-driven TEM; many had TEPs, and some even showed nuclei in transit through the monolayer. It appears that P-ZAP-70 and P-LAT (and SLP-76, not shown) are located not just at the T cell-EC interface at early time points, but also in the TEPs, suggesting that TEPs are a site of active signaling. TEPs also contain activated LFA-1 and talin, as well as RACK1, a molecule that associates with both kindlin-3 and LFA-1 and may regulate LFA-1 affinity and outside-in signaling (17). Of all these proteins, only RACK1 displayed any sort of selective distribution, being excluded from zones underneath nuclei. The significance of this particular localization is unclear.
In TCR signaling, src-family kinase-phosphorylated ITAM motifs of the CD3 zeta chain at the plasma membrane recruit and activate ZAP-70, which in turn phosphorylates LAT and SLP-76, leading to recruitment and activation of Vav, as well as other downstream mediators. We expected that the activation of Vav would provide a direct link between TCR signaling and the reorganization of the cytoskeleton that is required for TCR-driven TEM, and reduction of TCR-driven TEM of T cells treated with a pharmacological inhibitor of Vav, 6-thio-GTP, supports that hypothesis. However, since pharmacological inhibitors have off-target effects, and another pharmacological inhibitor of Vav is not available, we sought to support these findings using transduction of a recombinat dominant negative protein. This approach was required since genetic manipulation of EM CD4 T cells, such as overexpression from a transfected or transduced vector or knock down by siRNA, requires time to exert its effect and prolonged culture of primary human peripheral blood T cells changes the characteristics of the cells such that they are no longer truly resting EM T cells. Significantly, EM CD4 T cells lose expression of receptors necessary for TCR-driven TEM after a few days in culture (TDM, unpublished observations). Therefore, in order to investigate Vav function in EM CD4 T cells, we exploited a technique used successfully by others to modify intracellular events in primary T cells, namely introducing functional proteins via fusion to a protein transduction domain peptide called Hph (28). Hph-fusion proteins enter the cell within a one hour incubation, and are functional after overnight incubation, or perhaps sooner. While the levels of transduction for all proteins used in these studies were concentration-dependent, the maximum amount of any given protein that could be transduced into an EM T cell varied with the identity of the protein domains fused to the Hph transduction domain. Unexpectedly, lower levels of control Hph-EGFP could be introduced into the cell than the other proteins used in this study, all of which are fused to EGFP. Therefore it was fortunate that relatively low levels of Hph-EGFP-Vav DN (similar to levels of control EGFP) were able to produce an effect since we could not control for off target or non-specific effects following higher levels of transduction. Consistent with the observations reported here for human EM CD4 T cells, genetic knock out of Vav in mice inhibits antigen-specific but not chemokine-driven recruitment of primed T cells in peripheral tissues (36).
Vav is the primary Rac GEF in TCR signaling, and, indeed, Rac appears to control TCR-driven TEM. Pharmacological inhibition of Cdc42 has been reported to inhibit chemokine-driven TEM of primary human CD4 cells under flow (7), and our results using cells transduced with a Cdc42 DN protein and treated with another pharmacological inhibitor support those findings. It is perhaps surprising that Rac DN did not inhibit chemokine-driven TEM or adhesion to the EC, as mouse Rac1 and Rac2 double knockout T cells are defective in secondary lymph node homing in vivo as well as adhesion to and TEM across CCL21-overlaid EC in vitro under flow (37). Furthermore, transduction of total primary human peripheral blood T cell populations with Rac DN protein inhibited binding to ICAM-1 in response to CXCL12 in vitro as well as arrest on mouse high endothelial venules in vivo (38). However, it should be noted that the T cells used in those studies are either all (in the mouse model) or substantially (total T cells from human blood) naïve, responding to homeostatic chemokines, whereas our studies used purified EM human T cells (typically less than 10% of total T cells) responding to inflammatory chemokines. In other words, different chemokine receptors may activate distinct signaling pathways. Indeed, Rac1 has been shown to associate with and control CXCR4, the receptor for CXCL12, but not other related chemokine receptors (39). Consistent with these findings, two Rac-specific GEFs, namely DOCK2 and Tiam, control homeostatic recirculation through lymphoid organs (40–42). Thus while Rac may play a role in homeostatic recirculation of naïve T cells, it may not be involved in the response of EM T cells to inflammatory chemokines.
Although inhibition of Vav and Rac both reduced TCR-driven TEM of human EM CD4 T cells, they did not significantly affect TEP formation. This was a surprise since we had hypothesized that TEPs were related to lamellipodia, a cellular protrusion that is controlled by Rac. Our new findings suggest that TEPs are a unique cellular structure and further investigation will be required to understand their formation.
In our study, we identified a role for myosin IIA in allowing the T cell body to follow TEP formation and lead to TEM. Myosin IIA activity has also been reported to be necessary for maturation of microclusters and sustained TCR signaling at the immunological synapse, affecting ZAP-70 activation by src kinases in T cells interacting with professional APCs or planar bilayers in shear-free conditions (43). In our system, i.e., EM CD4 T cells interacting with superantigen presented by EC under conditions of flow, blebbistatin did not affect ZAP-70 activation. Rather, myosin IIA appears to be acting downstream of ZAP-70, since TEP formation is prevented by piceatannol, but not by blebbistatin. TEPs are dynamic, undergoing both extension and retraction. The concomitant increase in TEPs (both in number and size) and decrease in TEM in samples treated with blebbistatin suggests that myosin IIA may be controlling two events: the retraction of the TEP as it probes the subendothelial surface and the release of the nucleus into the TEP to complete TEM. Both processes may involve de-adhesion of the T cell or its TEP and myosin IIA has been associated with de-adhesion of the T cell uropod in a previous study (44). Interestingly, in our previous work (10), we had observed that fractalkine signals were needed for completion of TCR-driven TEM but not for TEP formation. It is therefore possible that myosin IIA activation, which appears to play a similar role, may be downstream of fractalkine signaling.
The unique intracellular signaling pathway that controls TCR-driven TEM as opposed to chemokine-driven TEM may have significant therapeutic implications. EM CD4 T cells are present in the circulation of every normal adult, and produce inflammatory cytokines (TNF and IFN-γ) rapidly after TCR stimulation (2). They are perfectly positioned, then, to act as “pioneers” in an inflammatory response, where a small number of antigen-specific EM T cells are activated and recruited by antigen presenting EC and, via production of TNF and IFN-γ, initiate a cascade of antigen-independent recruitment of other leukocytes (8, 45). In this scenario, interfering with the recruitment of the pioneers is an attractive approach to prevent the onset of an unwanted inflammatory response such as allograft rejection. Evidence in support of this concept has been provided by studies of the effects of azathioprine, a widely used immunosuppressant whose mode of action has been elucidated relatively recently; azathioprine is metabolized to 6-Thio-GTP and inhibits Vav GEF-mediated activation of Rac in T cells (46). An interpretation of these findings has been that azathioprine inhibits activation of naïve T cells. An alternative, but not mutually exclusive, possibility is that azathioprine may inhibit TCR-driven recruitment of EM CD4 T cells.
Supplementary Material
Acknowledgments
Grant Support: This work is funded by National Institute of Health grant R01-HL051014.
We thank Je-Min Choi for advice on the Hph protein transduction technology and Xose Bustelo for the Vav DN plasmid. We also thank Louise Benson, Gwendoline Davis, and Lisa Gras for excellent assistance in cell culture.
Abbreviations
- EC
endothelial cell
- EM
effector memory
- GEF
GTP exchange factor
- HDMEC
human dermal microvascular endothelial cells
- TEM
transendothelial migration
- TEP
transendothelial protrusion
- TSST-1
toxic shock syndrome toxin 1
Footnotes
The authors have no conflicting financial interests.
References
- 1.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 2.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 3.Masopust D, Picker LJ. Hidden memories: frontline memory T cells and early pathogen interception. J Immunol. 2012;188:5811–5817. doi: 10.4049/jimmunol.1102695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manes TD, Pober JS, Kluger MS. Endothelial cell-T lymphocyte interactions: IP-10 stimulates rapid transendothelial migration of human effector but not central memory CD4+ T cells. Requirements for shear stress and adhesion molecules. Transplantation. 2006;82:S9–14. doi: 10.1097/01.tp.0000231356.57576.82. [DOI] [PubMed] [Google Scholar]
- 5.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–522. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- 6.Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 2007;26:17–27. doi: 10.1016/j.immuni.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, Montresor A, Bolomini-Vittori M, Feigelson SW, Kirchhausen T, Laudanna C, Shakhar G, Alon R. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity. 2009;30:384–396. doi: 10.1016/j.immuni.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghani S, Feuerer M, Doebis C, Lauer U, Loddenkemper C, Huehn J, Hamann A, Syrbe U. T cells as pioneers: antigen-specific T cells condition inflamed sites for high-rate antigen-non-specific effector cell recruitment. Immunology. 2009;128:e870–880. doi: 10.1111/j.1365-2567.2009.03096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manes TD, Shiao SL, Dengler TJ, Pober JS. TCR signaling antagonizes rapid IP-10-mediated transendothelial migration of effector memory CD4+ T cells. J Immunol. 2007;178:3237–3243. doi: 10.4049/jimmunol.178.5.3237. [DOI] [PubMed] [Google Scholar]
- 10.Manes TD, Pober JS. Antigen Presentation by Human Microvascular Endothelial Cells Triggers ICAM-1-Dependent Transendothelial Protrusion by, and Fractalkine-Dependent Transendothelial Migration of, Effector Memory CD4+ T Cells. J Immunol. 2008;180:8386–8392. doi: 10.4049/jimmunol.180.12.8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manes TD, Hoer S, Muller WA, Lehner PJ, Pober JS. Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. J Immunol. 2010;184:5186–5192. doi: 10.4049/jimmunol.0902938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manes TD, Pober JS. Identification of endothelial cell junctional proteins and lymphocyte receptors involved in transendothelial migration of human effector memory CD4+ T cells. J Immunol. 2011;186:1763–1768. doi: 10.4049/jimmunol.1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol. 2007;7:131–143. doi: 10.1038/nri2021. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Madrid F, del Pozo MA. Leukocyte polarization in cell migration and immune interactions. Embo J. 1999;18:501–511. doi: 10.1093/emboj/18.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. 2009;9:630–644. doi: 10.1038/nri2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alon R, Feigelson SW. Chemokine-triggered leukocyte arrest: force-regulated bi-directional integrin activation in quantal adhesive contacts. Curr Opin Cell Biol. 2012;24:670–676. doi: 10.1016/j.ceb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Feng C, Li YF, Yau YH, Lee HS, Tang XY, Xue ZH, Zhou YC, Lim WM, Cornvik TC, Ruedl C, Shochat SG, Tan SM. Kindlin-3 mediates integrin alphaLbeta2 outside-in signaling, and it interacts with scaffold protein receptor for activated-C kinase 1 (RACK1) J Biol Chem. 2012;287:10714–10726. doi: 10.1074/jbc.M111.299594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subauste MC, Von Herrath M, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem. 2000;275:9725–9733. doi: 10.1074/jbc.275.13.9725. [DOI] [PubMed] [Google Scholar]
- 19.Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM. Activation of endogenous Cdc42 visualized in living cells. Science. 2004;305:1615–1619. doi: 10.1126/science.1100367. [DOI] [PubMed] [Google Scholar]
- 20.Zugaza JL, Lopez-Lago MA, Caloca MJ, Dosil M, Movilla N, Bustelo XR. Structural determinants for the biological activity of Vav proteins. J Biol Chem. 2002;277:45377–45392. doi: 10.1074/jbc.M208039200. [DOI] [PubMed] [Google Scholar]
- 21.Inngjerdingen M, Torgersen KM, Maghazachi AA. Lck is required for stromal cell-derived factor 1 alpha (CXCL12)-induced lymphoid cell chemotaxis. Blood. 2002;99:4318–4325. doi: 10.1182/blood.v99.12.4318. [DOI] [PubMed] [Google Scholar]
- 22.Ottoson NC, Pribila JT, Chan AS, Shimizu Y. Cutting edge: T cell migration regulated by CXCR4 chemokine receptor signaling to ZAP-70 tyrosine kinase. J Immunol. 2001;167:1857–1861. doi: 10.4049/jimmunol.167.4.1857. [DOI] [PubMed] [Google Scholar]
- 23.Ticchioni M, Charvet C, Noraz N, Lamy L, Steinberg M, Bernard A, Deckert M. Signaling through ZAP-70 is required for CXCL12-mediated T-cell transendothelial migration. Blood. 2002;99:3111–3118. doi: 10.1182/blood.v99.9.3111. [DOI] [PubMed] [Google Scholar]
- 24.Dar WA, Knechtle SJ. CXCR3-mediated T-cell chemotaxis involves ZAP-70 and is regulated by signalling through the T-cell receptor. Immunology. 2007;120:467–485. doi: 10.1111/j.1365-2567.2006.02534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans R, Lellouch AC, Svensson L, McDowall A, Hogg N. The integrin LFA-1 signals through ZAP-70 to regulate expression of high-affinity LFA-1 on T lymphocytes. Blood. 2011;117:3331–3342. doi: 10.1182/blood-2010-06-289140. [DOI] [PubMed] [Google Scholar]
- 26.Feigelson SW, Grabovsky V, Manevich-Mendelson E, Pasvolsky R, Shulman Z, Shinder V, Klein E, Etzioni A, Aker M, Alon R. Kindlin-3 is required for the stabilization of TCR-stimulated LFA-1:ICAM-1 bonds critical for lymphocyte arrest and spreading on dendritic cells. Blood. 2011;117:7042–7052. doi: 10.1182/blood-2010-12-322859. [DOI] [PubMed] [Google Scholar]
- 27.Simonson WT, Franco SJ, Huttenlocher A. Talin1 regulates TCR-mediated LFA-1 function. J Immunol. 2006;177:7707–7714. doi: 10.4049/jimmunol.177.11.7707. [DOI] [PubMed] [Google Scholar]
- 28.Choi JM, Ahn MH, Chae WJ, Jung YG, Park JC, Song HM, Kim YE, Shin JA, Park CS, Park JW, Park TK, Lee JH, Seo BF, Kim KD, Kim ES, Lee DH, Lee SK. Intranasal delivery of the cytoplasmic domain of CTLA-4 using a novel protein transduction domain prevents allergic inflammation. Nat Med. 2006;12:574–579. doi: 10.1038/nm1385. [DOI] [PubMed] [Google Scholar]
- 29.Choi JM, Kim SH, Shin JH, Gibson T, Yoon BS, Lee DH, Lee SK, Bothwell AL, Lim JS. Transduction of the cytoplasmic domain of CTLA-4 inhibits TcR-specific activation signals and prevents collagen-induced arthritis. Proc Natl Acad Sci U S A. 2008;105:19875–19880. doi: 10.1073/pnas.0805198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Martin L, Sanchez-Sanchez N, Gutierrez-Lopez MD, Rojo AI, Vicente-Manzanares M, Perez-Alvarez MJ, Sanchez-Mateos P, Bustelo XR, Cuadrado A, Sanchez-Madrid F, Rodriguez-Fernandez JL, Cabanas C. Signaling through the leukocyte integrin LFA-1 in T cells induces a transient activation of Rac-1 that is regulated by Vav and PI3K/Akt-1. J Biol Chem. 2004;279:16194–16205. doi: 10.1074/jbc.M400905200. [DOI] [PubMed] [Google Scholar]
- 31.Zeng R, Cannon JL, Abraham RT, Way M, Billadeau DD, Bubeck-Wardenberg J, Burkhardt JK. SLP-76 coordinates Nck-dependent Wiskott-Aldrich syndrome protein recruitment with Vav-1/Cdc42-dependent Wiskott-Aldrich syndrome protein activation at the T cell-APC contact site. J Immunol. 2003;171:1360–1368. doi: 10.4049/jimmunol.171.3.1360. [DOI] [PubMed] [Google Scholar]
- 32.Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, Vlaar CP, Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem. 287:13228–13238. doi: 10.1074/jbc.M111.334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobelli J, Chmura SA, Buxton DB, Davis MM, Krummel MF. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat Immunol. 2004;5:531–538. doi: 10.1038/ni1065. [DOI] [PubMed] [Google Scholar]
- 34.Farber DL. Biochemical signaling pathways for memory T cell recall. Semin Immunol. 2009;21:84–91. doi: 10.1016/j.smim.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ludford-Menting MJ, Oliaro J, Sacirbegovic F, Cheah ET, Pedersen N, Thomas SJ, Pasam A, Iazzolino R, Dow LE, Waterhouse NJ, Murphy A, Ellis S, Smyth MJ, Kershaw MH, Darcy PK, Humbert PO, Russell SM. A network of PDZ-containing proteins regulates T cell polarity and morphology during migration and immunological synapse formation. Immunity. 2005;22:737–748. doi: 10.1016/j.immuni.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 36.David R, Ma L, Ivetic A, Takesono A, Ridley AJ, Chai JG, Tybulewicz VL, Marelli-Berg FM. T-cell receptor- and CD28-induced Vav1 activity is required for the accumulation of primed T cells into antigenic tissue. Blood. 2009;113:3696–3705. doi: 10.1182/blood-2008-09-176511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faroudi M, Hons M, Zachacz A, Dumont C, Lyck R, Stein JV, Tybulewicz VL. Critical roles for Rac GTPases in T-cell migration to and within lymph nodes. Blood. 2010;116:5536–5547. doi: 10.1182/blood-2010-08-299438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolomini-Vittori M, Montresor A, Giagulli C, Staunton D, Rossi B, Martinello M, Constantin G, Laudanna C. Regulation of conformer-specific activation of the integrin LFA-1 by a chemokine-triggered Rho signaling module. Nat Immunol. 2009;10:185–194. doi: 10.1038/ni.1691. [DOI] [PubMed] [Google Scholar]
- 39.Zoughlami Y, Voermans C, Brussen K, van Dort KA, Kootstra NA, Maussang D, Smit MJ, Hordijk PL, van Hennik PB. Regulation of CXCR4 conformation by the small GTPase Rac1: implications for HIV infection. Blood. 2012;119:2024–2032. doi: 10.1182/blood-2011-06-364828. [DOI] [PubMed] [Google Scholar]
- 40.Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, Sasazuki T. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001;412:826–831. doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- 41.Nombela-Arrieta C, Lacalle RA, Montoya MC, Kunisaki Y, Megias D, Marques M, Carrera AC, Manes S, Fukui Y, Martinez AC, Stein JV. Differential requirements for DOCK2 and phosphoinositide-3-kinase gamma during T and B lymphocyte homing. Immunity. 2004;21:429–441. doi: 10.1016/j.immuni.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 42.Gerard A, van der Kammen RA, Janssen H, Ellenbroek SI, Collard JG. The Rac activator Tiam1 controls efficient T-cell trafficking and route of transendothelial migration. Blood. 2009;113:6138–6147. doi: 10.1182/blood-2008-07-167668. [DOI] [PubMed] [Google Scholar]
- 43.Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morin NA, Oakes PW, Hyun YM, Lee D, Chin YE, King MR, Springer TA, Shimaoka M, Tang JX, Reichner JS, Kim M. Nonmuscle myosin heavy chain IIA mediates integrin LFA-1 de-adhesion during T lymphocyte migration. J Exp Med. 2008;205:195–205. doi: 10.1084/jem.20071543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pober JS, Tellides G. Participation of blood vessel cells in human adaptive immune responses. Trends Immunol. 2012;33:49–57. doi: 10.1016/j.it.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poppe D, Tiede I, Fritz G, Becker C, Bartsch B, Wirtz S, Strand D, Tanaka S, Galle PR, Bustelo XR, Neurath MF. Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J Immunol. 2006;176:640–651. doi: 10.4049/jimmunol.176.1.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.