

Abstract
Although multiple sclerosis (MS) is a chronic inflammatory-demyelinating disease of the white matter (WM) of the central nervous system, several pathological and magnetic resonance imaging (MRI) studies have shown that a large amount of lesions are located in the cortical and deep gray matter. The histopathological and immunological characteristics of cortical lesions differ significantly from those located in the WM, which suggests a location-dependent expression of the MS immunopathological process. More recently, the availability of not-conventional MRI sequences having higher sensitivity for the gray matter has allowed to depict in vivo a portion of such lesions. The available MRI data obtained on large cohorts of patients, having different clinical forms of the disease, indicate that cortical lesions can be detected early in the disease course, sometimes even before the appearance of WM lesions, and correlate with the severity of physical disability and cognitive impairment, and with the evolution of the disease toward the secondary progressive phase. This review provides a summary of the main histopathological and MRI findings of cortical lesions in MS and discusses their possible clinical implications.
Keywords: cortical lesions, Multiple Sclerosis, physical disability, cognitive dysfunction
Introduction
Multiple Sclerosis (MS) is an autoimmune, chronic and disabling disease of the human central nervous system, histologically characterized by multifocal areas of inflammatory demyelination within white matter (WM). For this reason MS has been traditionally considered a “pure” WM disease. However, several recent neuropathological studies disclosed a relevant, extensive and irreversible “neurodegenerative” process involving the gray matter (GM)1 and the occurrence of focal inflammatory lesions not only in the WM, but also within the cortex and deep GM.2,3
Over the past 10–15 y, quantitative magnetic resonance imaging (MRI) studies have confirmed that focal GM damage and subsequent tissue atrophy are already present at early stages of MS4 and evolve faster than WM pathology.5 In addition, GM damage has been correlated with physical disability and cognitive dysfunction more convincingly than either WM T2 hyperintense or T1 hypointense lesion load.4 So far, GM and WM damages appear to be two simultaneous components of the disease, both observable at its onset and at least partially dissociated from each other. While the relationship between WM lesions and cortical atrophy remains indefinable, it is unlikely that regional changes in cortical volume are the consequence, via retrograde degeneration, of on-going tissue axonal transection in subcortical WM lesions. On the contrary, they seem to be more depending on local (cortical) inflammation.
Focal Grey Matter Pathology: The Cortical Lesions
Classification and topographical distribution
Neuropathological studies have described a consistent number of “demyelinating” lesions in cortical and deep GM of the MS brain.6-14 In the early 1960s, Brownell and Hughes,15 in a material from 22 MS brains, described that 26% of the MS lesions affected GM and 77% of the cortical lesions (CLs) involved the subcortical WM. In a study of 60 MS cases, Lumsden16 found that 93% had a cortical involvement of varying degree, with some cases showing only a few CLs, but others having up to 465 gyral plaques. Indeed, in 10 cases, 290 lesions (59% of all hemisphere lesions) were seen to involve the cortex. Both studies showed that the demyelinating lesions involving the cortex were predominately located at the leuco-cortical junction (i.e., cortical/juxtaCL).
The currently accepted histological classification of CLs draws a distinction among four lesion subtypes (Fig. 1): Type I lesions are combined WM/GM lesions (cortical/juxta-CLs); Type II lesions are entirely located within the cerebral cortex, they are not in direct contact with subcortical WM or pia mater, and are in general small and perivascular; Type III lesions are subpial areas of demyelination, usually confined within the layers 3 and 4 of the cortex; Type IV lesions comprise the entire width of the cortex, without spreading in subcortical WM, and can extend like large sheaths over several gyri or entire lobes.6,7
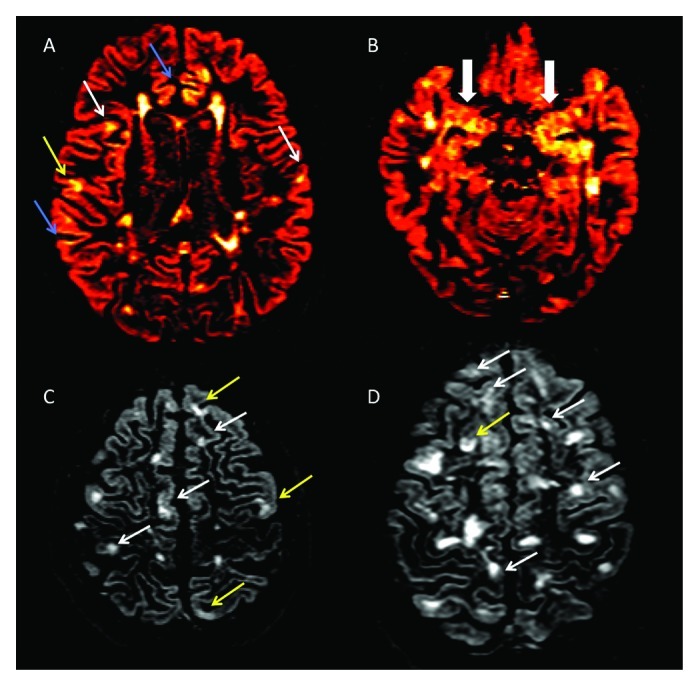
Figure 1. (A-D) Axial double inversion recovery from four patients suffering from Relapsing-Remitting Multiple Sclerosis. Several type II (white arrows), Type III (blue arrows) and Type IV (yellow arrows) are detectable (A), some of which involving the Hippocampus (B). Two patients (C and D), having a very high cortical lesion load, suffer also from epilepsy and from a significant cognitive dysfunction.
Demyelination has been reported to vary significantly among the different cortical areas, being the cingulate gyrus (up to 44%), the hippocampus (up to 30%, Figure 1B) and the temporal and frontal cortices (up to 28%) the most affected.7-9 A smaller portion of demyelinated areas was noted in other cortical areas, including the paracentral lobule (11.5%), occipital lobe (8%) and primary motor cortex (3.5%).7 Interestingly, a high percentage of demyelination was found in the cerebellar cortex,10,14 where up to 80% of the CLs are purely intracortical. Finally, focal demyelinating lesions have been described in spinal cord GM,17,18 sometimes prevailing on local WM lesions. Thus, inflammatory demyelination occurring within cortical, deep and spinal cord GM is a relevant aspect of the immunopathological process taking place in MS.
Only weak to no correlation between the extent of GM and WM demyelination has been observed,19 suggesting that the inflammatory process affecting the GM probably occurs, at least partly, independently of WM inflammation. The question whether this finding can be accounted for by a bias in the selection of the analyzed brain specimens needs to be clarified. Nevertheless, although in one study cortical demyelination was largely restricted to progressive forms of MS,9 a more recent study identified recurrent cortical demyelination with clear inflammatory profile even in the early stages of the disease.3
Immuno-histological aspects
Some significant immuno-histological differences between CLs and subcortical WM lesions have been described. Although CLs are well-demarcated areas of demyelination, with significant oligodendrocyte and axonal loss,6,11 they differ substantially from WM lesions in degree and type of inflammation, which appears for some aspects location-dependent. Compared with WM lesions, pure intracortical lesions are characterized by a lower extent of lymphocyte infiltration6 that might however depend on the patient population. In patients with long-lasting disease, perivascular infiltrates were rarely found within the cortex, and the density of infiltrating lymphocytes was very similar in pure CLs and in the so-called normal-appearing GM. Indeed, when compared with WM lesions, CLs contained 13 times fewer CD3+ lymphocytes and 6 times fewer CD68+ cells of the microglia/macrophages lineage.6 In a recent neuropathological study on brain specimens from patients with early relapsing remitting MS,3 CLs containing foamy macrophages, indicating ongoing demyelination, were detected in 66% of patients. The presence of B-cells in perivascular cortical inflammation was observed in 27% of CLs, while perivascular CD3+ T lymphocytes were observed in 82% of cortical plaques, 77% of which contained CD8+ T cells. Although leukocortical lesions were the most inflamed, the majority of intracortical and subpial plaques contained perivascular CD3+ and CD8+ T-cell infiltrates that were moderate to marked in almost 23% of subpial lesions. The authors concluded that cortical demyelination is common even in early disease stages and differs substantially from that seen in chronic multiple sclerosis. These data suggest that cortical inflammation is a frequent, transient phenomenon in early relapsing-remitting phase but rarely observed in its completeness in chronic MS patients or in very late stages of the disease.
Notes on the pathogenesis of cortical lesions
Although the pathogenesis of CL is only partially understood, several recent neuropathological studies have pointed out the role of meningeal inflammation.3,20-22
The presence of a widespread subpial demyelination, the lack of any significant leakage of plasma proteins, thus suggesting a normal blood–brain barrier function,23 and the absence of any significant signs of complement activation,24 seem to preclude that a perivascular inflammation play a major role in the pathogenesis of GM pathology. On the other hand, diffuse inflammatory cell infiltrates and ectopic lymphoid-like structures have been consistently found in the meninges, especially in the deep sulci of frontal, temporal, cingulate and insula cortex and thus topographically associated with cortical demyelination.3,20-22 Accordingly, when secondary progressive MS patients having B cell follicle-like structures in the inflamed meninges were compared with those without these structures, a substantial gradient of neuron, astrocyte and oligodendrocyte loss was found, thus suggesting the possible pathogenetic role of some soluble factors diffusing from inflamed meninges.21
Patients with B cell follicles-like structures also showed a significant increase in MHC class II+ activated microglial cells, pointing out their potential role in MS cortical pathology.21 In GM lesions, indeed, the majority of phagocytic cells were described to have the morphology of activated microglia, with only a minority having a phagocytic macrophage appearance.6 VCAM-1+ (Vascular Cell Adhesion Molecule-1+) cells in MS lesions, as defined by dual-label immunohistochemistry, were considered as derived from the monocyte/macrophage lineage and were found to be numerous at lesion edges, in coincidence with regions of oligodendrocyte injury.25 Activated microglia within CLs contained elevated levels of myeloperoxidase, indicating that reactive oxygen species may contribute to GM lesion pathogenesis.26 In a study aimed at comparing MS and Alzheimer disease cortical pathologies, profound microglia activation, determined by a wide spectrum of immunological and biochemical markers, was found in both MS and Alzheimer disease cortices, and the patterns of microglia activation were closely similar.27 However, microglia activation in MS cortices, in contrast with that in Alzheimer disease and control cortices, was clearly correlated with lymphocyte and plasmacell infiltrates in the meninges.27 Unfortunately very few studies on molecular mechanisms by which neurons degenerate within cortical lesions have been performed, yet the hypothesis of an oxygen- and nitric oxide radical-induced mitochondrial injury and energy failure as the origin of neuronal degeneration is appealing.28 Indeed the recent observation of oxidized phospholipids and DNA even in neurons within GM lesions seems to confirm the role of mitochondrial injury as a major factor driving GM tissue injury.29
Neuroimaging aspects
Although the above described pathological data suggest widespread cortical involvement, especially in patients with long-lasting disease duration, only a few studies have assessed the contribution of cortical pathology to clinical MS symptoms in vivo. This is mainly due to the fact that routinely available MRI techniques, especially T1-weighted and T2-weighted sequences, only rarely allow for identification of purely intracortical lesions. Indeed, among the four types of CLs described by pathologists, FLAIR sequence can usually detect just Type I (leuko-cortical) lesions located at the interface between cortex and WM. Several factors hamper the possibility of demonstrating CLs by means of conventional T2/FLAIR sequence, and they are mostly related to the pathophysiology of cortex and CLs. The lower inflammatory profile of CLs (compared with WM lesions),6 the absence of significant blood–brain barrier damage within CLs,23 the low myelin density in upper cortical layers, as well as technical constraints, such as partial volume effects resulting from the proximity of CLs to the cerebrospinal fluid (CSF),30-32 may help explain why in vivo CLs identification remains a challenge.
In a study comparing the number of CLs detected by histopathology and post-mortem MRI, applying dual-echo T2-weighted spin-echo images and 3D-FLAIR, detection rates of 3D-FLAIR images were of only 5% for pure CLs and 41% for leuko-cortical lesions.32 A significant improvement in the detection rate of CLs and in the delineation of GM structures was obtained by applying 3D double inversion recovery (DIR) sequences. The use of DIR imaging showed an average increase of 152% in CL detection per patient when compared with detection by means of 3D FLAIR sequence. Moreover, in comparison with T2-weighted spin echo (SE) imaging, DIR imaging showed a 500% increase in detection of CLs.33,34 The major limitation of DIR sequences is the low sensitivity in detecting CLs, especially subpial ones, when compared with histology.35 Recent comparative histological/MRI studies have however demonstrated that the “tip of the iceberg” detected by MRI and its “bulk” differ only in size, and that the number of CLs detected well correlates with their total number and with the overall percentage of cortical demyelination.36 Thus, although further improvement in CL detection shall be achieved perhaps by combining different MRI sequences as phase sensitive inversion recovery37,38 and 3D Magnetization-prepared rapid acquisition with gradient echo (MPRAGE),39 we feel that DIR-based findings can be considered an acceptable assessment of focal GM pathology.
Focal Grey Matter Pathology: Clinical implications
Cortical lesions and disability progression in patients with MS
Data from several neuroimaging studies indicate a primary role of CLs in determining clinical dysfunction and disability progression in MS. In a study conducted on a large patient population, CLs could be detected by DIR in the majority (64%) of patients with relapsing remitting (RRMS) and secondary progressive (73%) MS (SPMS), as well as in more than a third (36.8%) of patients with clinically isolated syndromes (CIS) suggestive of MS.40 In the same study, CL load modestly correlated with Expanded Disability Status Scale (EDSS) score (r = 0.48), WM T2 lesion volume (r = 0.38), and brain parenchyma fraction (r = -0.47) and patients with CLs showed higher EDSS score (p = 0.004), greater WM T2 lesion volume (p = 0.008) and smaller brain parenchyma fraction (p = 0.009) compared with patients without CLs. A three-year longitudinal follow-up study41 disclosed that CL accumulation was significantly higher in MS patients showing clinical worsening compared with those clinically stable. CL volume at baseline well correlated with baseline EDSS (r = 0.36) and even better with EDSS changes over time (r = 0.51). In the same study RRMS and SPMS patients were found to accumulate new CLs at similar rates (0.8/year in RRMS vs. 1.0/year in SPMS), suggesting that, in relapse-onset MS, the medium-term dynamics of CL evolution may not be influenced by the stage of the disease. The greater number of CLs observed in SPMS compared with RRMS patients could therefore be the consequence of the longer disease duration of the former subgroup. Indeed, post-mortem studies9 showed that GM demyelination and axonal damage are already present in the relapsing-remitting phase but become more prominent in the chronic stages. Alternatively, the fact that cortical demyelination is more extensive in progressive than in early MS could be explained by the faster and more efficient remyelination of CLs in early disease stage.11
Finally, patients with CLs show higher frequency of CSF IgG oligoclonal bands40 and higher level of intrathecal synthesis of Ig (IgG and IgM)42 compared with patients without CLs. The combination of intrathecal Ig synthesis with the presence of CLs allowed the early identification of RRMS patients having the highest risk of disease activity and the worse clinical evolution over a 3-y follow up.41 This would be in line with recent neuropathological studies3,20,22 showing association between the B-cell enriched meningeal inflammation and the subpial demyelination described in MS, thus suggesting a possible pathogenetic role of soluble factors (immunoglobulins included) diffusing from inflamed meninges into subarachnoid space. It’s interesting to notice that, although meningeal inflammation has been identified even in the early stages of the disease,3 ectopic B cell follicle-like structures have been more frequently detected in SPMS patients with an accelerated clinical course.20 These findings, along with the evidence of cortical demyelination in early MS and consequent loss of olygodendrocyte, axonal and neuronal injury in the chronic phase of the disease, suggest that disability progression could be the result of neurodegeneration running on a background of inflammation. Further studies aimed at investigating the potential role of CLs in predicting the entrance in the progressive phase of the disease could have remarkable clinical rebounds in terms of prognostic stratification of patients and optimization of available therapies.
Cortical lesions in different MS phenotypes
Additional data on possible clinical implications of CLs come from studies on specific phenotypes of MS.
CLs were observed in up to 80% of patients suffering from primary progressive form of MS (PPMS).43,44 Interestingly, when a lesion probability map was delineated, similarities rather than differences were found between RR and PPMS patients: similarities include the total number of CLs, their volume and their topographic distribution on cortical lesion probability map. In the PPMS population, as in RRMS patients, CL load correlated significantly with disease duration, EDSS score and EDSS increase after one-year follow up (Table 1).
On the contrary, patients with “benign” MS course (EDSS < 3.0 after 15 y from clinical onset and no cognitive dysfunction)45 showed significantly smaller CL load compared with early RRMS having the same degree of disability, but much shorter disease duration.46 After one-year follow-up, “benign” MS did not show an accumulation of CLs compared with early RRMS patients; again, multivariate analysis indicated that CL load at study entry and its increase over time were associated to the “benign” clinical status.47 The relative sparing of the cortex in benign MS might therefore be considered as an in vivo-feature of this disease subtype, with consequent important implications on clinical grounds: the lack of CL detection during the early RRMS stage may indeed contribute to identify MS patients with a more favorable prognosis.
MS patients with epilepsy constituted another interesting subgroup of subjects (Fig. 1C and D). As previously observed, epileptic seizures are more frequent in MS than in the general population47,48 and the association between epilepsy and MS has been demonstrated not to be mere coincidence.49,50 In a series of 20 RRMS patients who had epileptic seizures during the course of the disease, CLs were detected in 18/20 (90%) compared with only 48% of not epileptic RRMS (p = 0.001).49 Cortical pathology was quite extensive in epileptic RRMS, inasmuch as they showed five times more CLs compared with not epileptic RRMS, and had a total CL volume that was 6 times greater. Meanwhile, no significant difference between epileptic and not epileptic RRMS was observed with regard to the number and volume of juxta-cortical lesions and T2WM lesion volume. A 3-y longitudinal study on 32 RRMS patients with epilepsy revealed greater accumulation of new CLs and faster progression rate of cortical atrophy compared with 60 matched RRMS patients without epilepsy, while no difference was observed in the number of new WM or Gad + lesions.51 As expected, these patients showed a significantly worse clinical evolution, characterized by faster decline in cognition and greater EDSS increase. This suggests that these patients constitute a peculiar subgroup of MS patients, having a more pronounced and selective involvement of the GM and a severe (cortical) clinical picture.
Finally, observation of CLs even in the pediatric MS population52 supports the hypothesis that GM pathology is a very early phenomenon in MS, being strictly associated with the biologic disease onset. This result was further confirmed by a 3-y longitudinal study53 in which atrophy and CL load showed the same rate of progression in both pediatric and adult onset MS. The greater CL load characterizing adult onset MS appear to be likely the consequence of the longer interval of time between biologic and clinical onset in these patients.
Diagnostic relevance of CL
CLs have been noticed in the MS brain months/years before the appearance of inflammatory lesions in subcortical WM,54,55 strongly indicating that, at least in some patients, the pathological process underlying MS could start in the cortex. While they have been observed by DIR in more than 30% of patients presenting with CIS suggestive of MS40 as well as in asymptomatic subjects with Radiologically Isolated Syndrome (RIS),55 CLs are a frequent phenomenon in MS being present in almost 70% of RRMSpatients.40 On the basis of these observations, the predictive role of CLs in patients with CIS suggestive of MS was assessed in a recent 4-y longitudinal MRI study57 and compared with the available McDonald-Polman58,59 and Swanton60 diagnostic criteria for dissemination in space of lesions. Regression analysis showed that presence of at least one cortical (p < 0.001), one infratentorial (p = 0.03), one Gad enhancing and one spinal cord (p = 0.004) lesion were independently associated to the conversion to definite MS, while the presence of at least two of these variables resulted to be the best criterion for “dissemination in space of the lesions.” Furthermore, the combination of the intrathecal Ig synthesis with the presence of CLs identified a subgroup of CIS patients having a high risk of developing definite MS in the following three years.42 This indicates that the accuracy of MRI diagnostic criteria for MS could be increased with the integration of CLs in the diagnostic paradigm, by detection of CLs in patients suspected to be affected with MS. Interesting, cortical changes resembling CLs have been observed in other neurological disorders such as tuberous sclerosis,61 epilepsy,62,63 hepatic encephalopathy64 and neoplastic condition,65 but not in other inflammatory pathologies such as Neuromyelitis Optica (NMO)66,67 and migraine68 that may enter in differential diagnosis with MS. Therefore, CLs might represent a useful diagnostic element addressing to MS in patients with WM hyperintensities of uncertain etiology.
CL as a major substrate of cognitive dysfunction
Cortical pathology has a significant impact even on cognitive dysfunction observed early in MS course,69 with dramatic effects on personal, social and occupational functioning. Although the relationship between cognitive impairment and subcortical WM pathology remains controversial,69-72 there is increasing evidence of a primary role of cortical pathology in determining cognitive disability.73-75 Neocortical volume loss was observed to correlate with cognitive dysfunction better than whole brain atrophy.73,74 In addition, RRMS patients having cognitive dysfunctions showed widespread cortical atrophy involving almost all cortical regions, while in cognitive unimpaired RRMS patients, a thinning confined in the fronto-temporal cortical was observed.75-77
Beyond this evidence on the relationship between cognitive dysfunction and diffuse GM damage, even the focal damage has been related to the decrease in cognitive performance. In a 3 y follow up study, Roseendaal and Coll.78 found that CL number were higher in SPMS, tended to accumulate over time and was associated with a worse performance on neuropsycological measures at follow up. Furthermore, CL load was significantly heavier in cognitively impaired patients (Fig. 1C and D) compared with cognitively preserved ones77 and, together with normalized cortical volume ratio, resulted to be an independent predictor of the composite cognitive score, suggesting that focal cortical damage may be considered one of the major substrates of cognitive impairment in MS.
Conclusions
In summary, CLs appear to be quite peculiar inflammatory and demyelinating lesions. They differ from WM lesions for several quantitative and qualitative aspects of the cellular inflammatory network, for the lack of a clear evidence of local blood-brain barrier dysfunction, and for the critical role that seems to be played by reactive microglia cells, thus suggesting different underlying (immuno)pathogenetic mechanisms. Accumulating data suggest a pivotal role of CLs in physical and cognitive decline in MS, but longitudinal studies are needed to explore in detail their clinical implication. The available data, however, are particularly worth of interest and urge us to develop more sensitive MRI technologies to increase our capacity to detect and analyze cortical pathology in MS.
Table 1. Selected works studying gray matter lesions in MS and their diagnostic and prognostic relevance (physical and cognitive disability).
| Study | Patients | Follow up | Parameters Evaluated | Main outcome |
|---|---|---|---|---|
| Filippi et al. 2010 |
80 CIS |
4 y |
CLs, T2-WM lesions, Gad+ lesions |
The accuracy of MRI diagnostic criteria for MS is increased when considering the presence of CLs on baseline scans from patients at presentation with CIS suggestive of MS. |
| Calabrese et al. 2012 |
86 patients with CIS. |
3 y |
CL and WM lesion number, new MRI activity, CSF examination. |
The association of intrathecal Ig synthesis and CLs is highly predictive of an earlier CIS conversion to MS as well as of a higher disease activity. |
| Popescu et al. 2011 |
One RRMS. |
Case Report |
MRI, CSF and histological examination. |
Pathologic evidence of RRMS patient presenting with inflammatory solitary cortical enhancing lesion. |
| Calabrese et al. 2009 |
4 RRMS |
Case Report |
CLs and T2-WM lesions |
CLs were observed by DIR before the MRI evidence of inflammatory lesions in the white matter |
| Absinta et al. 2012 |
32 patients with migraine, 15 RRMS, 20 healthy controls. |
Cross-sectional |
T2-WM lesion and CL count and volume. |
No CLs identified in migraine patients. The application of DIR imaging seems to be useful in the diagnostic workup of patients with WM hyperintensities of unknown etiology, including those with migraine. |
| Giorgio et al. 2011 |
15 radiologically isolated syndrome. |
Cross-sectional |
T2-WM lesion and CL count and volume, normalized brain and cortical volume |
CLs are identified in subject with RIS (40%) and are frequent in subjects with IgOBs and dissemination in time. CLs are therefore associated with important markers of evolution to MS. |
| Popescu et al. 2011 |
19 autopsied NMO patients |
Cross-sectional |
Histological examination. |
Lack of cortical demyelination in patients with NMO is a feature that might help distinguishing NMO from MS. |
| Calabrese et al. 2012 in press |
30 NMO patients, 30 RRMS patients, 30 healthy controls. |
Cross-sectional |
CL and T2-WM lesion count, global and regional cortical thickness. |
No CLs identified in patients with NMO and no significant differences found in cortical thickness between NMO a controls. MRI analysis of the cortex may be a potential diagnostic tool, especially in ambiguous cases. |
| Absinta et al. 2011 |
24 pediatric and 15 adult MS. |
Cross-sectional |
WM lesions and CLs number and volume, GM and WM volumes. |
CL are rare in patients with MS in comparison with adult patients. |
| Calabrese et al. 2010 |
76 RRMS, 31 SPMS |
3 y |
CL and T2-WM lesion number and volume, T2-WM lesion volume, GM volume |
CL volume correlates with EDSS and EDSS changes over time and it is an independent predictor of EDSS accumulation and GM volume change in SP and RRMS patients. |
| Calabrese et al. 2009 |
48 benign MS, 96 early not disabling RRMS. |
1 y |
CL and T2-WM lesion number and volume, |
Benign MS have lower CL number compared with early RRMS patients. At one-year follow-up a significant increase of CL number and volume is observed only in early patients with RRMS. |
| Calabrese et al. 2012 |
35 pediatric and 57 adult MS |
3 y |
CL and T2-WM lesion number and volume, GM volume |
Focal (CLs) and diffuse (atrophy) GM damage are strictly associated with the biologic onset of MS, and proceed linearly and partly independently of WM pathology. |
| Calabrese et al. 2012 |
32 RRMS with epilepsy, 60 RRMS without epilepsy. |
3 y |
CL and T2-WM lesion number and volume, regional cortical thickness, new CLs and WM lesions; neuropsychological evaluation |
Cortical pathology, psysical and cognitive decline are more severe and evolving in MS patients with epilepsy compared with patients without epilepsy. |
| Roosendal et al. 2009 |
9 RRMS, 4 SPMS, 7 healthy controls |
3-y |
CL and T2-WM lesion number and volume, neuropsychological evaluation |
CLs increase significantly over a 3 y time period, are most frequent in SP patients and are associated with cognitive impairment. |
| Mike et al. 2011 |
20 RRMS, 20 SPMS |
Cross-sectional |
CLs number and volume, T2-WM volume, EDSS, cognitive testing. |
Routinely detectable cortical lesions were related to physical disability and cognitive impairment better than T2-WM lesions |
| Calabrese et al. 2009 | 70 RRMS | Cross-sectional | CL and T2-WM lesion number and volume, GM volume. neuropsychological evaluation | Cognitively impaired patients have a higher CL number and volume, decreased normalized cortical volume compared with cognitively preserved patients. |
Abbreviations: CLs, cortical lesions; CSF, Cerebro-spinal-fluid; DIR, Double inversion recovery; Gad*, gadolinium enhancing lesions; GM, gray matter; IgGOB, IgG oligoclonali bands; MRI, magnetic resonance imaging, MS, Multiple Sclerosis; NMO, Neuromyelitis optica; RIS, radiologically isolated syndrome; RRMS, relapsing remitting multiple sclerosis; SPMS, secondary progressive multiple sclerosis; T2-WM, T2 white matter; WM, white matter
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/22580
References
- 1.Pirko I, Lucchinetti CF, Sriram S, Bakshi R. Gray matter involvement in multiple sclerosis. Neurology. 2007;68:634–42. doi: 10.1212/01.wnl.0000250267.85698.7a. [DOI] [PubMed] [Google Scholar]
- 2.Popescu BF, Lucchinetti CF. Meningeal and cortical grey matter pathology in multiple sclerosis. BMC Neurol. 2012;12:11. doi: 10.1186/1471-2377-12-11. [Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–97. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Stefano N, Matthews PM, Filippi M, Agosta F, De Luca M, Bartolozzi ML, et al. Evidence of early cortical atrophy in MS: relevance to white matter changes and disability. Neurology. 2003;60:1157–62. doi: 10.1212/01.WNL.0000055926.69643.03. [DOI] [PubMed] [Google Scholar]
- 5.Chard DT, Griffin CM, Rashid W, Davies GR, Altmann DR, Kapoor R, et al. Progressive grey matter atrophy in clinically early relapsing-remitting multiple sclerosis. Mult Scler. 2004;10:387–91. doi: 10.1191/1352458504ms1050oa. [DOI] [PubMed] [Google Scholar]
- 6.Peterson JW, Bö L, Mörk SJ, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 7.Bø L, Vedeler CA, Nyland HI, Trapp BD, Mørk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol. 2003;62:723–32. doi: 10.1093/jnen/62.7.723. [DOI] [PubMed] [Google Scholar]
- 8.Geurts JJ, Bö L, Roosendaal SD, Hazes T, Daniëls R, Barkhof F, et al. Extensive hippocampal demyelination in multiple sclerosis. J Neuropathol Exp Neurol. 2007;66:819–27. doi: 10.1097/nen.0b013e3181461f54. [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulos D, Dukes S, Patel R, Nicholas R, Vora A, Reynolds R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. 2009;19:238–53. doi: 10.1111/j.1750-3639.2008.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vercellino M, Plano F, Votta B, Mutani R, Giordana MT, Cavalla P. Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol. 2005;64:1101–7. doi: 10.1097/01.jnen.0000190067.20935.42. [DOI] [PubMed] [Google Scholar]
- 11.Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 12.Gilmore CP, Donaldson I, Bö L, Owens T, Lowe JS, Evangelou N. Regional variations in the extent and pattern of grey matter demyelination in multiple sclerosis: a comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord. J Neurol Neurosurg Psychiatry. 2009;80:182–7. doi: 10.1136/jnnp.2008.148767. [DOI] [PubMed] [Google Scholar]
- 13.Albert M, Antel J, Brück W, Stadelmann C. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol. 2007;17:129–38. doi: 10.1111/j.1750-3639.2006.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kutzelnigg A, Faber-Rod JC, Bauer J, Lucchinetti CF, Sorensen PS, Laursen H, et al. Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol. 2007;17:38–44. doi: 10.1111/j.1750-3639.2006.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brownell B, Hughes JT. The distribution of plaques in the cerebrum in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1962;25:315–20. doi: 10.1136/jnnp.25.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lumsden CE. The neuropathology of multiple sclerosis. In: Vinken PJ, Brun GW, eds. Handbook of clinical neurology. Vol. 9. Amsterdam: Elsevier, 1970: 217-309. [Google Scholar]
- 17.Gilmore CP, Bö L, Owens T, Lowe J, Esiri MM, Evangelou N. Spinal cord gray matter demyelination in multiple sclerosis-a novel pattern of residual plaque morphology. Brain Pathol. 2006;16:202–8. doi: 10.1111/j.1750-3639.2006.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilmore CP, DeLuca GC, Bö L, Owens T, Lowe J, Esiri MM, et al. Spinal cord neuronal pathology in multiple sclerosis. Brain Pathol. 2009;19:642–9. doi: 10.1111/j.1750-3639.2008.00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bö L, Geurts JJ, van der Valk P, Polman C, Barkhof F. Lack of correlation between cortical demyelination and white matter pathologic changes in multiple sclerosis. Arch Neurol. 2007;64:76–80. doi: 10.1001/archneur.64.1.76. [DOI] [PubMed] [Google Scholar]
- 20.Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 21.Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–93. doi: 10.1002/ana.22230. [DOI] [PubMed] [Google Scholar]
- 22.Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–71. doi: 10.1093/brain/awr182. [DOI] [PubMed] [Google Scholar]
- 23.van Horssen J, Brink BP, de Vries HE, van der Valk P, Bø L. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol. 2007;66:321–8. doi: 10.1097/nen.0b013e318040b2de. [DOI] [PubMed] [Google Scholar]
- 24.Brink BP, Veerhuis R, Breij ECW, van der Valk P, Dijkstra CD, Bö L. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. J Neuropathol Exp Neurol. 2005;64:147–55. doi: 10.1093/jnen/64.2.147. [DOI] [PubMed] [Google Scholar]
- 25.Peterson JW, Bö L, Mörk S, Chang A, Ransohoff RM, Trapp BD. VCAM-1-positive microglia target oligodendrocytes at the border of multiple sclerosis lesions. J Neuropathol Exp Neurol. 2002;61:539–46. doi: 10.1093/jnen/61.6.539. [DOI] [PubMed] [Google Scholar]
- 26.Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008;18:86–95. doi: 10.1111/j.1750-3639.2007.00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dal Bianco A, Bradl M, Frischer J, Kutzelnigg A, Jellinger K, Lassmann H. Multiple sclerosis and Alzheimer’s disease. Ann Neurol. 2008;63:174–83. doi: 10.1002/ana.21240. [DOI] [PubMed] [Google Scholar]
- 28.Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–89. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 29.Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, Botond G, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–24. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gawne-Cain ML, O’Riordan JI, Thompson AJ, Moseley IF, Miller DH. Multiple sclerosis lesion detection in the brain: a comparison of fast fluid-attenuated inversion recovery and conventional T2-weighted dual spin echo. Neurology. 1997;49:364–70. doi: 10.1212/WNL.49.2.364. [DOI] [PubMed] [Google Scholar]
- 31.Bakshi R, Ariyaratana S, Benedict RHB, Jacobs L. Fluid-attenuated inversion recovery magnetic resonance imaging detects cortical and juxtacortical multiple sclerosis lesions. Arch Neurol. 2001;58:742–8. doi: 10.1001/archneur.58.5.742. [DOI] [PubMed] [Google Scholar]
- 32.Geurts JJ, Bö L, Pouwels PJ, Castelijns JA, Polman CH, Barkhof F. Cortical lesions in multiple sclerosis: combined postmortem MR imaging and histopathology. AJNR Am J Neuroradiol. 2005;26:572–7. [PMC free article] [PubMed] [Google Scholar]
- 33.Geurts JJ, Pouwels PJ, Uitdehaag BM, Polman CH, Barkhof F, Castelijns JA. Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging. Radiology. 2005;236:254–60. doi: 10.1148/radiol.2361040450. [DOI] [PubMed] [Google Scholar]
- 34.Pouwels PJ, Kuijer JP, Mugler JP, 3rd, Guttmann CR, Barkhof F. Human gray matter: feasibility of single-slab 3D double inversion-recovery high-spatial-resolution MR imaging. Radiology. 2006;241:873–9. doi: 10.1148/radiol.2413051182. [DOI] [PubMed] [Google Scholar]
- 35.Seewann A, Kooi EJ, Roosendaal SD, Pouwels PJ, Wattjes MP, van der Valk P, et al. Postmortem verification of MS cortical lesion detection with 3D DIR. Neurology. 2012;78:302–8. doi: 10.1212/WNL.0b013e31824528a0. [DOI] [PubMed] [Google Scholar]
- 36.Seewann A, Vrenken H, Kooi EJ, van der Valk P, Knol DL, Polman CH, et al. Imaging the tip of the iceberg: visualization of cortical lesions in multiple sclerosis. Mult Scler. 2011;17:1202–10. doi: 10.1177/1352458511406575. [DOI] [PubMed] [Google Scholar]
- 37.Nelson F, Poonawalla AH, Hou P, Huang F, Wolinsky JS, Narayana PA. Improved identification of intracortical lesions in multiple sclerosis with phase-sensitive inversion recovery in combination with fast double inversion recovery MR imaging. AJNR Am J Neuroradiol. 2007;28:1645–9. doi: 10.3174/ajnr.A0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sethi V, Yousry TA, Muhlert N, Ron M, Golay X, Wheeler-Kingshott C, et al. Improved detection of cortical MS lesions with phase-sensitive inversion recovery MRI. J Neurol Neurosurg Psychiatry. 2012;83:877–82. doi: 10.1136/jnnp-2012-303023. [DOI] [PubMed] [Google Scholar]
- 39.Nelson F, Poonawalla A, Hou P, Wolinsky JS, Narayana PA. 3D MPRAGE improves classification of cortical lesions in multiple sclerosis. Mult Scler. 2008;14:1214–9. doi: 10.1177/1352458508094644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, Barachino L, et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch Neurol. 2007;64:1416–22. doi: 10.1001/archneur.64.10.1416. [DOI] [PubMed] [Google Scholar]
- 41.Calabrese M, Rocca MA, Atzori M, Mattisi I, Favaretto A, Perini P, et al. A 3-year magnetic resonance imaging study of cortical lesions in relapse-onset multiple sclerosis. Ann Neurol. 2010;67:376–83. doi: 10.1002/ana.21906. [DOI] [PubMed] [Google Scholar]
- 42.Calabrese M, Federle L, Bernardi V, Rinaldi F, Favaretto A, Varagnolo MC, et al. The association of intrathecal immunoglobulin synthesis and cortical lesions predicts disease activity in clinically isolated syndrome and early relapsing-remitting multiple sclerosis. Mult Scler. 2012;18:174–80. doi: 10.1177/1352458511418550. [DOI] [PubMed] [Google Scholar]
- 43.Calabrese M, Rocca MA, Atzori M, Mattisi I, Bernardi V, Favaretto A, et al. Cortical lesions in primary progressive multiple sclerosis: a 2-year longitudinal MR study. Neurology. 2009;72:1330–6. doi: 10.1212/WNL.0b013e3181a0fee5. [DOI] [PubMed] [Google Scholar]
- 44.Calabrese M, Battaglini M, Giorgio A, Atzori M, Bernardi V, Mattisi I, et al. Imaging distribution and frequency of cortical lesions in patients with multiple sclerosis. Neurology. 2010;75:1234–40. doi: 10.1212/WNL.0b013e3181f5d4da. [DOI] [PubMed] [Google Scholar]
- 45.Rovaris M, Barkhof F, Calabrese M, De Stefano N, Fazekas F, Miller DH, et al. MRI features of benign multiple sclerosis: toward a new definition of this disease phenotype. Neurology. 2009;72:1693–701. doi: 10.1212/WNL.0b013e3181a55feb. [DOI] [PubMed] [Google Scholar]
- 46.Calabrese M, Filippi M, Rovaris M, Bernardi V, Atzori M, Mattisi I, et al. Evidence for relative cortical sparing in benign multiple sclerosis: a longitudinal magnetic resonance imaging study. Mult Scler. 2009;15:36–41. doi: 10.1177/1352458508096686. [DOI] [PubMed] [Google Scholar]
- 47.Spatt J, Chaix R, Mamoli B. Epileptic and non-epileptic seizures in multiple sclerosis. J Neurol. 2001;248:2–9. doi: 10.1007/s004150170262. [DOI] [PubMed] [Google Scholar]
- 48.Engelsen BA, Grønning M. Epileptic seizures in patients with multiple sclerosis. Is the prognosis of epilepsy underestimated? Seizure. 1997;6:377–82. doi: 10.1016/S1059-1311(97)80037-4. [DOI] [PubMed] [Google Scholar]
- 49.Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, Barachino L, et al. Extensive cortical inflammation is associated with epilepsy in multiple sclerosis. J Neurol. 2008;255:581–6. doi: 10.1007/s00415-008-0752-7. [DOI] [PubMed] [Google Scholar]
- 50.Catenoix H, Marignier R, Ritleng C, Dufour M, Mauguière F, Confavreux C, et al. Multiple sclerosis and epileptic seizures. Mult Scler. 2011;17:96–102. doi: 10.1177/1352458510382246. [DOI] [PubMed] [Google Scholar]
- 51.Calabrese M, Grossi P, Favaretto A, Romualdi C, Atzori M, Rinaldi F, et al. Cortical pathology in multiple sclerosis patients with epilepsy: a 3 year longitudinal study. J Neurol Neurosurg Psychiatry. 2012;83:49–54. doi: 10.1136/jnnp-2011-300414. [DOI] [PubMed] [Google Scholar]
- 52.Absinta M, Rocca MA, Moiola L, Copetti M, Milani N, Falini A, et al. Cortical lesions in children with multiple sclerosis. Neurology. 2011;76:910–3. doi: 10.1212/WNL.0b013e31820f2e69. [DOI] [PubMed] [Google Scholar]
- 53.Calabrese M, Seppi D, Romualdi C, Rinaldi F, Alessio S, Perini P, et al. Gray matter pathology in MS: a 3-year longitudinal study in a pediatric population. AJNR Am J Neuroradiol. 2012;33:1507–11. doi: 10.3174/ajnr.A3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Popescu BF, Bunyan RF, Parisi JE, Ransohoff RM, Lucchinetti CF. A case of multiple sclerosis presenting with inflammatory cortical demyelination. Neurology. 2011;76:1705–10. doi: 10.1212/WNL.0b013e31821a44f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calabrese M, Gallo P. Magnetic resonance evidence of cortical onset of multiple sclerosis. Mult Scler. 2009;15:933–41. doi: 10.1177/1352458509106510. [DOI] [PubMed] [Google Scholar]
- 56.Giorgio A, Stromillo ML, Rossi F, Battaglini M, Hakiki B, Portaccio E, et al. Cortical lesions in radiologically isolated syndrome. Neurology. 2011;77:1896–9. doi: 10.1212/WNL.0b013e318238ee9b. [DOI] [PubMed] [Google Scholar]
- 57.Filippi M, Rocca MA, Calabrese M, Sormani MP, Rinaldi F, Perini P, et al. Intracortical lesions: relevance for new MRI diagnostic criteria for multiple sclerosis. Neurology. 2010;75:1988–94. doi: 10.1212/WNL.0b013e3181ff96f6. [DOI] [PubMed] [Google Scholar]
- 58.McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–7. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 59.Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58:840–6. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 60.Swanton JK, Fernando K, Dalton CM, Miszkiel KA, Thompson AJ, Plant GT, et al. Modification of MRI criteria for multiple sclerosis in patients with clinically isolated syndromes. J Neurol Neurosurg Psychiatry. 2006;77:830–3. doi: 10.1136/jnnp.2005.073247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cotton F, Rambaud L, Hermier M. Dual inversion recovery MRI helps identifying cortical tubers in tuberous sclerosis. Epilepsia. 2006;47:1072–3. doi: 10.1111/j.1528-1167.2006.00529.x. [DOI] [PubMed] [Google Scholar]
- 62.Rugg-Gunn FJ, Boulby PA, Symms MR, Barker GJ, Duncan JS. Imaging the neocortex in epilepsy with double inversion recovery imaging. Neuroimage. 2006;31:39–50. doi: 10.1016/j.neuroimage.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 63.Grant PE, Barkovich AJ, Wald LL, Dillon WP, Laxer KD, Vigneron DB. High-resolution surface-coil MR of cortical lesions in medically refractory epilepsy: a prospective study. AJNR Am J Neuroradiol. 1997;18:291–301. [PMC free article] [PubMed] [Google Scholar]
- 64.Matsusue E, Kinoshita T, Ohama E, Ogawa T. Cerebral cortical and white matter lesions in chronic hepatic encephalopathy: MR-pathologic correlations. AJNR Am J Neuroradiol. 2005;26:347–51. [PMC free article] [PubMed] [Google Scholar]
- 65.Turetschek K, Wunderbaldinger P, Bankier AA, Zontsich T, Graf O, Mallek R, et al. Double inversion recovery imaging of the brain: initial experience and comparison with fluid attenuated inversion recovery imaging. Magn Reson Imaging. 1998;16:127–35. doi: 10.1016/S0730-725X(97)00254-3. [DOI] [PubMed] [Google Scholar]
- 66.Popescu BF, Parisi JE, Cabrera-Gómez JA, Newell K, Mandler RN, Pittock SJ, et al. Absence of cortical demyelination in neuromyelitis optica. Neurology. 2010;75:2103–9. doi: 10.1212/WNL.0b013e318200d80c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calabrese M, Oh MS, Favaretto A, Rinaldi F, Poretto V, Alessio S, et al. No MRI evidence of cortical lesions in neuromyelitis optica. Neurology. 2012;79:1671–6. doi: 10.1212/WNL.0b013e31826e9a96. [DOI] [PubMed] [Google Scholar]
- 68.Absinta M, Rocca MA, Colombo B, Copetti M, De Feo D, Falini A, et al. Patients with migraine do not have MRI-visible cortical lesions. J Neurol. 2012 doi: 10.1007/s00415-012-6571-x. In Press. [DOI] [PubMed] [Google Scholar]
- 69.Amato MP, Zipoli V, Portaccio E. Multiple sclerosis-related cognitive changes: a review of cross-sectional and longitudinal studies. J Neurol Sci. 2006;245:41–6. doi: 10.1016/j.jns.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 70.Rovaris M, Filippi M, Minicucci L, Iannucci G, Santuccio G, Possa F, et al. Cortical/subcortical disease burden and cognitive impairment in patients with multiple sclerosis. AJNR Am J Neuroradiol. 2000;21:402–8. [PMC free article] [PubMed] [Google Scholar]
- 71.Rao SM, Leo GJ, Haughton VM, St Aubin-Faubert P, Bernardin L. Correlation of magnetic resonance imaging with neuropsychological testing in multiple sclerosis. Neurology. 1989;39:161–6. doi: 10.1212/WNL.39.2.161. [DOI] [PubMed] [Google Scholar]
- 72.Benedict RH, Weinstock-Guttman B, Fishman I, Sharma J, Tjoa CW, Bakshi R. Prediction of neuropsychological impairment in multiple sclerosis: comparison of conventional magnetic resonance imaging measures of atrophy and lesion burden. Arch Neurol. 2004;61:226–30. doi: 10.1001/archneur.61.2.226. [DOI] [PubMed] [Google Scholar]
- 73.Portaccio E, Amato MP, Bartolozzi ML, Zipoli V, Mortilla M, Guidi L, et al. Neocortical volume decrease in relapsing-remitting multiple sclerosis with mild cognitive impairment. J Neurol Sci. 2006;245:195–9. doi: 10.1016/j.jns.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 74.Benedict RH, Bruce JM, Dwyer MG, Abdelrahman N, Hussein S, Weinstock-Guttman B, et al. Neocortical atrophy, third ventricular width, and cognitive dysfunction in multiple sclerosis. Arch Neurol. 2006;63:1301–6. doi: 10.1001/archneur.63.9.1301. [DOI] [PubMed] [Google Scholar]
- 75.Tekok-Kilic A, Benedict RH, Weinstock-Guttman B, Dwyer MG, Carone D, Srinivasaraghavan B, et al. Independent contributions of cortical gray matter atrophy and ventricle enlargement for predicting neuropsychological impairment in multiple sclerosis. Neuroimage. 2007;36:1294–300. doi: 10.1016/j.neuroimage.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 76.Morgen K, Sammer G, Courtney SM, Wolters T, Melchior H, Blecker CR, et al. Evidence for a direct association between cortical atrophy and cognitive impairment in relapsing-remitting MS. Neuroimage. 2006;30:891–8. doi: 10.1016/j.neuroimage.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 77.Calabrese M, Agosta F, Rinaldi F, Mattisi I, Grossi P, Favaretto A, et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch Neurol. 2009;66:1144–50. doi: 10.1001/archneurol.2009.174. [DOI] [PubMed] [Google Scholar]
- 78.Roosendaal SD, Moraal B, Pouwels PJ, Vrenken H, Castelijns JA, Barkhof F, et al. Accumulation of cortical lesions in MS: relation with cognitive impairment. Mult Scler. 2009;15:708–14. doi: 10.1177/1352458509102907. [DOI] [PubMed] [Google Scholar]