

Abstract
Disturbances in the circadian system are associated with the development of type 2 diabetes mellitus. Here, we studied the direct contribution of the suprachiasmatic nucleus (SCN), the central pacemaker in the circadian system, in the development of insulin resistance. Exclusive bilateral SCN lesions in male C57Bl/6J mice, as verified by immunochemistry, showed a small but significant increase in body weight (+17%), which was accounted for by an increase in fat mass. In contrast, mice with collateral damage to the ventromedial hypothalamus and paraventricular nucleus showed severe obesity and insulin resistance. Mice with exclusive SCN ablation revealed a loss of circadian rhythm in activity, oxygen consumption, and food intake. Hyperinsulinemic–euglycemic clamp analysis 8 weeks after lesioning showed that the glucose infusion rate was significantly lower in SCN lesioned mice compared with sham-operated mice (−63%). Although insulin potently inhibited endogenous glucose production (−84%), this was greatly reduced in SCN lesioned mice (−7%), indicating severe hepatic insulin resistance. Our data show that SCN malfunctioning plays an important role in the disturbance of energy balance and suggest that an absence of central clock activity, in a genetically intact animal, may lead to the development of insulin resistance.
Obesity and type 2 diabetes mellitus have an increasing prevalence in modern society. In the past decade, a strong and potentially causal relationship between metabolic disorders and disturbances of the circadian system has been elucidated (1). The circadian system is responsible for 24-h rhythms in a wide variety of physiological and behavioral functions (2,3). Generation of these rhythms occurs in the suprachiasmatic nuclei (SCN) of the anterior hypothalamus (2,3) and is explained by a transcriptional–translational feedback loop involving the clock genes CLOCK, BMAL1, Period (Per), and Cryptochrome (Cry) (1). The rhythms of the SCN are synchronized to the environmental 24-h cycle mainly by light–dark information perceived by the eyes. Rhythmic information is transferred from the SCN to the central nervous system and to peripheral organs of the body (4,5). This output is crucial for synchronization of many metabolic and endocrine factors such as glucose, insulin (6,7), cortisol (8), leptin, ghrelin (9,10), and neuropeptide Y (11).
Disturbances in circadian rhythms occur as a consequence of shift work and transitions in time zones, but also because of irregular sleep–wake patterns (12), aging, or neurodegenerative disorders (13). Animal studies have indicated a link between clock gene mutations and metabolic disturbances. For instance, Clock mutant mice have a greatly attenuated diurnal feeding rhythm, are hyperphagic and obese, and have development of a metabolic syndrome of hyperleptinemia, hyperlipidemia, hepatic steatosis, and hyperglycemia (14). Because clock gene mutants are not specific to the SCN, the detrimental effects of disturbed rhythms may have their origin in peripheral organs other than the SCN. It is not clear to what extent the SCN itself is involved in metabolic disorders. Given the accumulating evidence for disturbances of SCN cellular organization in aging (15), neurodegenerative disorders, and dementia (13,16), this question is also clinically relevant, as it would explain comorbidity between various disorders.
To assess the role of disturbed function of the SCN per se in the development of obesity and type 2 diabetes mellitus, we performed bilateral microlesions of the SCN in male C57Bl/6J mice. Because the SCN is anatomically surrounded by areas regulating energy homeostasis, such as the ventromedial hypothalamus (VMH) and paraventricular nucleus (PVN), great care was taken to distinguish between exclusively SCN lesioned mice and mice with collateral damage to surrounding nuclei. We show that selective ablation of the SCN results in complete loss of circadian energy metabolism and, moreover, in the development of severe hepatic insulin resistance, stressing the role of the central clock in the pathophysiology of insulin resistance.
RESEARCH DESIGN AND METHODS
Animals.
All animal experiments were approved by the Animal Ethic Committee from the Leiden University Medical Center (Leiden, the Netherlands). Male C57Bl/6J mice were housed individually in a controlled environment (21°C; 40–50% humidity) under a 12-h/12-h light/dark cycle (0700–1900 h) unless otherwise mentioned. Food (chow, RM3; Special Diet Services, Sussex, U.K.) and tap water were available ad libitum during the entire experiment. Body weight was monitored weekly for all individual mice.
SCN lesions.
Bilateral ablation of the SCN was performed in 13-week-old mice as described previously (17). Mice were anesthetized using a mixture of ketamine (100 mg/kg; Aescoket, Boxtel, the Netherlands), xylazine (10 mg/kg; Bayer AG, Leverkusen, Germany), and atropine (0.1 mg/kg; Pharmachemie, Haarlem, The Netherlands) and mounted in a stereotactic device (Digital Just for Mouse Stereotaxic Instrument; Stoelting, Wood Dale, IL). After identification of bregma, a hole was drilled through which the lesion electrode was inserted into the brain. Lesion needles were made by isolating a 0.3-mm stainless steel insect pin using isolating resin, except for 0.2 mm at the tip. The electrode tip was aimed at the SCN, 0.46 mm posterior to bregma, 0.15 mm lateral to the midline, and 5.2 mm ventral to the surface of the cortex (Paxinos Mouse Brain Atlas, Franklin 2001). Bilateral SCN lesions were made by passing a 0.6-mA current through the electrode for duration of 10 s. The sham lesioned mice underwent the same operation, but no current was passed through the electrode.
Circadian rhythm analysis.
After SCN lesioning, all mice were housed in constant dark for 10 consecutive days to determine circadian rhythm in behavioral activity of each animal using passive infrared motion detection sensors (Hygrosens, Löffingen, Germany) that were mounted underneath the lid of the cage and connected to a ClockLab data collection system (Actimetrics) that recorded the amount of sensor activation in 1-min bins. The presence of circadian rhythms was determined by F-periodogram analysis based on the algorithm of Dörrscheidt and Beck (18). Mice were included in the lesion group when no significant rhythm was present.
Positional check of the SCN lesion.
The SCN lesions were checked as described previously (10). To verify the position of the SCN lesions, brains were removed and fixed by immersion with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) at 4°C. For cryoprotection, the brain tissue was equilibrated for 48 h with 30% sucrose in 0.1 mol/L Tris-buffered saline before sectioning. Thereafter, the brain tissue was cut into 30-µm sections and divided into two equal vials for immunocytochemical staining. The two vials of brain sections were incubated overnight at 4°C with either rabbit antivasopressin or rabbit anti-vasoactive intestinal peptide primary antibodies. Sections were then rinsed in 0.1 mol/L Tris-buffered saline, incubated 1 h in biotinylated goat anti-rabbit IgG, and subsequently incubated for 1 h in avidin–biotin complex (Vector Laboratories, Burlingame, CA). The reaction product was visualized by incubation in 1% diaminobenzidine with 0.01% hydrogen peroxide for 5–7 min. Nickel ammonium sulfate (0.05%) was added to the diaminobenzidine solution to darken the reaction product (diaminobenzidine/nickel). All sections were mounted on gelatin-coated glass slides, dried, run through ethanol and xylene, and covered for observation by light microscopy. For every animal, we blindly scored the amount of damage to the SCN and surrounding hypothalamic nuclei involved in metabolism (PVN and VMH).
Indirect calorimetry and metabolic cages.
Individual measurements by indirect calorimetry were performed for a period of at least 4 consecutive days (Comprehensive Laboratory Animal Monitoring System; Columbus Instruments, Columbus, OH) (19). A period of 24 h was included at the start of the experiment to allow acclimatization of the mice to the cages. Experimental analysis started at 0900 h. Analyzed parameters included real-time energy, water intake, and activity. Oxygen consumption (energy expenditure) measurements were performed at intervals of 7 min throughout the whole period. Oxygen consumption, activity, and energy intake were analyzed separately for day and night.
Hyperinsulinemic–euglycemic clamp analysis.
Eight weeks after SCN lesioning, hyperinsulinemic–euglycemic clamp experiments were performed as previously described (20). Mice were fasted for 16 h with food withdrawn at 1700 h the day before the study. All mice ate within 60 min before the start of the fasting period, minimizing differences in fasting time. During the experiment, mice were anesthetized by intraperitoneal injection with a combination of acepromazin (6.25 mg/kg; Sanofi Sant Nutrition Animale, Libourne Cedex, France), midazolam (6.25 mg/kg; Roche, Mijdrecht, the Netherlands), and fentanyl (0.31 mg/kg; Janssen-Cilag, Tilburg, the Netherlands). Anesthesia and body temperature were maintained throughout the procedure. At the end of the basal and the hyperinsulinemic periods, hematocrit values were determined to ensure that the mice were not anemic. First, basal rates of glucose turnover were determined by administering a primed (0.8 μCi) constant (0.02 μCi/min) intravenous infusion of 3-3H-glucose (specific activity, 9.6 GBq/mmol; Amersham, Little Chalfont, U.K.) for 60 min. Glucose rate of disposal (Rd; µmol/min/kg) was determined as the rate of tracer infusion (dpm/min) divided by the plasma-specific activity of 3-3H-glucose (dpm/µmol) at both time points. Subsequently, insulin (actrapid; Novo Nordisk, Denmark) was administered intravenously by primed (4.1 mU), constant (6.8 mU/h) infusion to attain steady-state circulating insulin levels of ∼4 ng/mL together with 3-3H-glucose. A variable intravenous infusion of a 12.5% d-glucose solution was used to maintain euglycemia as determined at 10-min intervals via tail bleeding (<3 µL; Accu-chek, Sensor Comfort; Roche Diagnostics). All mice were clamped at their respective basal glucose levels, because it has been shown that alterations in basal fasting glucose levels alone are sufficient to affect insulin sensitivity (21). Seventy minutes after the start of the hyperinsulinemic period, when glucose infusion rates (GIRs) for all mice were stable for at least 30 min, blood samples were taken at 10-min intervals for 30 min to determine 3-3H-glucose. Hyperinsulinemic Rd was determined similar to the basal period. The endogenous glucose production (EGP) was calculated as the difference between Rd and the GIR. After the hyperinsulinemic–euglycemic clamp, body composition (lean vs. fat mass) was determined by DEXA using the Norland pDEXA Sabre X-Ray Bone Densitometer (Norland, Hampshire, U.K.). All data were analyzed according to the software and recommendations of the manufacturer. Subsequently, the mice were euthanized.
Plasma analysis.
Plasma insulin concentrations were measured by ELISA (Crystal Chem). For measurement of plasma 3-3H-glucose, trichloroacetic acid (final concentration 2%) was added to 10 µL plasma to precipitate proteins using centrifugation. The supernatant was dried to remove water and was resuspended in milliQ. The samples were counted using scintillation counting (Packard Instruments).
Statistical analysis.
Statistical analysis was performed using SPSS 17 for Windows (SPSS, Chicago, IL). Unpaired t tests were performed for all comparisons, with statistical significance threshold set at P = 0.05.
RESULTS
Behavioral and histological verification of SCN lesions.
Periodogram analysis of activity showed that all sham-operated mice retained a strong circadian rhythm (Fig. 1A), whereas SCN lesioned mice lost their circadian rhythm in activity after the lesion procedures (Fig. 1B). Histological analysis revealed SCN lesions without collateral damage in six mice. In addition to ablation of the SCN, four other mice had unilateral damage to the PVN, 10 other mice had bilateral damage to the PVN, and nine other mice had damage to both the PVN and the VMH. Sham-operated controls (n = 17) did not reveal damage to any of the aforementioned brain areas. Representative histological sections of the hypothalamus, illustrating the histological verification of lesion position and size, are shown in Supplementary Fig. 1; 40-µm coronal sections were alternately stained for arginine vasopressin (immunohistochemical staining, Supplementary Fig. 1A), vasoactive intestinal peptide (immunohistochemical staining, Supplementary Fig. 1B), and cresyl violet (cell nuclei staining, Supplementary Fig. 1C).
FIG. 1.

Representative double-plotted actogram analyses of sham-operated (A) and SCN lesioned (B) mice under constant dark conditions. Each line of the double-plotted actograms represents 48 h.
Selective SCN lesions
Body weight and composition.
Before the operation and after a recovery period of 8 days, the average body weight of the sham and SCN lesioned mice did not differ (operation day: sham, 26.3 ± 1.5 g, SCN lesion, 26.0 ± 1.5 g, not significant; day 8: sham, 25.9 ± 1.6 g, SCN lesion, 25.3 ± 1.6 g, not significant). Eight weeks after SCN lesion, body mass of SCN lesioned mice was 17% higher compared with that of the sham mice (24.9 ± 1.2 vs. 29.2 ± 1.9 g; P < 0.01; Fig. 2). Assessment of body composition revealed that fat mass was significantly higher in SCN lesioned mice compared with sham (5.8 ± 2.6 vs. 1.9 ± 1.0 g; P < 0.01), whereas lean mass did not differ (17.7 ± 2.2 vs. 17.2 ± 1.7 g; not significant). This indicates that SCN ablation results in only mild overweight status compared with sham mice.
FIG. 2.

Body mass and composition of sham (white bars) and SCN lesioned (black bars) mice at the time of the hyperinsulinemic–euglycemic clamp. Total body mass, lean body mass, and fat mass were determined. Data are represented as mean ± SD. **P < 0.01.
Indirect calorimetry.
Metabolic cage data on oxygen consumption, food intake, and activity were obtained over a period of a minimum of 4 consecutive days and were analyzed separately for day and night. During the night, oxygen consumption was higher compared with the day in sham mice (3,386 ± 174 vs. 2,992 ± 155 mL/kg/h; P < 0.01; Fig. 3A). This circadian rhythm in oxygen consumption was lost in SCN lesioned mice, resulting in higher oxygen consumption during the day in SCN lesioned mice compared with sham mice (3,321 ± 252 vs. 2,992 ± 155 mL/kg/h; P < 0.01). Total 24-h oxygen consumption was not different between sham and SCN lesioned mice (76,531 ± 3,854 vs. 79,518 ± 7,595 mL/kg/day, not significant). During the night, sham mice were more active compared with during the day (195 ± 68 vs. 79 ± 33 beam breaks (bb); P < 0.01; Fig. 3B). In line with the oxygen consumption rates, circadian pattern in activity was lost in SCN lesioned mice (123 ± 48 vs. 128 ± 38 bb, not significant). This loss in activity pattern in SCN lesioned mice resulted in increased activity during the day (128 ± 38 vs. 79 ± 33 bb; P < 0.01) and reduced activity during the night compared with sham mice (123 ± 48 vs. 195 ± 68 bb; P < 0.01). Total 24-h activity, however, was not different between sham and SCN lesioned mice (273 ± 96 vs. 251 ± 81 bb; not significant). Sham mice consumed 68% of their total food during the night (night vs. day: 3.2 ± 0.5 vs. 1.5 ± 0.2 g; P < 0.01; Fig. 3C), whereas SCN lesioned mice consumed only 54% during the night (night vs. day: 1.9 ± 0.3 vs. 1.6 ± 0.9 g, not significant). This resulted in reduced food intake during the night for SCN lesioned mice compared with sham mice (1.9 ± 0.3 vs. 3.2 ± 0.5 g; P < 0.01). Furthermore, total 24-h food intake also was reduced for SCN lesioned mice compared with sham mice (3.5 ± 1.2 vs. 4.7 ± 0.5 g; P < 0.01). Calculating the respiratory exchange ratio showed that SCN lesioned mice had a lower respiratory exchange ratio compared with sham mice during the day (0.86 ± 0.04 vs. 0.90 ± 0.01; P < 0.05) as well as during the night (0.87 ± 0.01 vs. 0.96 ± 0.02; P < 0.01). These data clearly show that ablation of the SCN results in a loss of circadian rhythm in respiratory metabolism and food intake.
FIG. 3.
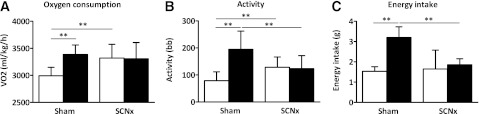
Indirect calorimetry and metabolic cage analysis of sham and SCN lesioned (SCNx) mice: oxygen consumption (A), activity (B), and energy intake (C). White bars represent the average value during the day and black bars represent the average value during the night. Data are represented as mean ± SD. **P < 0.01.
Hyperinsulinemic–euglycemic clamp studies.
Hematocrit levels were similar at the basal and the hyperinsulinemic period for sham and SCN lesioned mice, indicating that the mice were not anemic. At the start of the clamp, insulin, glucose, and free fatty acid (FFA) plasma levels were significantly higher in SCN lesioned mice compared with sham controls (Table 1). During the hyperinsulinemic period, insulin levels were increased to a similar extent for both the sham and SCN lesioned mice. At the end of the hyperinsulinemic period, circulating glucose levels were isoglycemic compared with fasting levels in sham and SCN lesioned mice, resulting in significantly higher glucose levels in the hyperinsulinemic period for SCN lesioned mice compared with sham mice (Fig. 4A). At the end of the clamp, FFA levels were decreased compared with basal plasma levels in sham and SCN lesioned mice, although this failed to reach statistical significance in SCN lesioned mice (P = 0.08). GIRs over the final 20 min of the clamp were significantly lower in SCN lesioned mice compared with sham controls (27.6 ± 19.5 vs. 74.7 ± 21.1 µmol/min/kg; P < 0.01; Fig. 4B and C). The glucose-specific activities measured at 10-min intervals indicated the presence of steady-state conditions in all groups (Supplementary Table 1).
TABLE 1.
Body mass and hyperinsulinemic–euglycemic clamp parameters of sham and suprachiasmatic nucleus lesioned (SCNx) mice
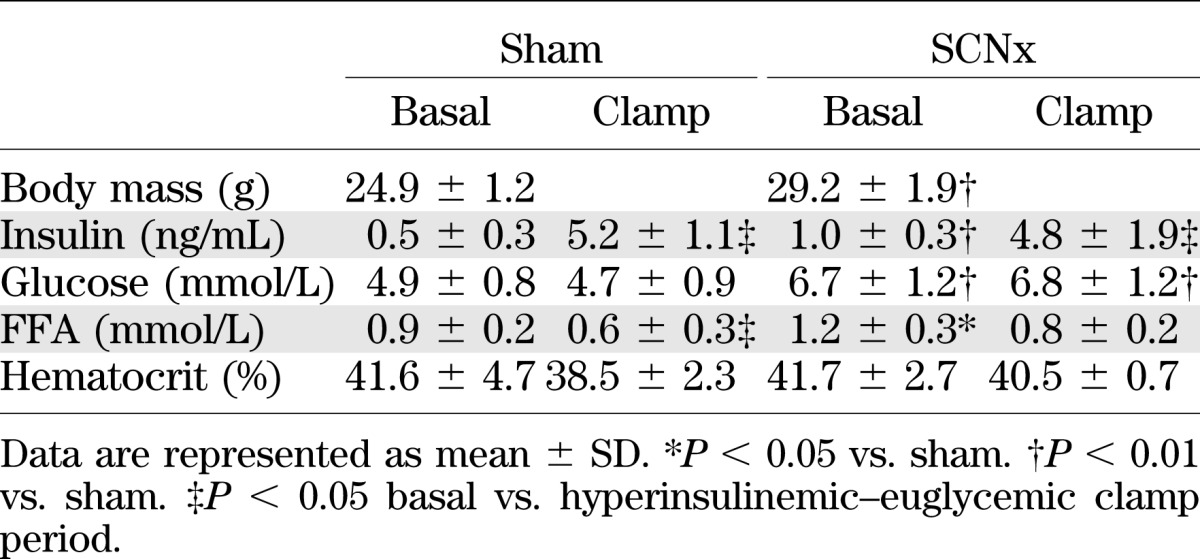
FIG. 4.
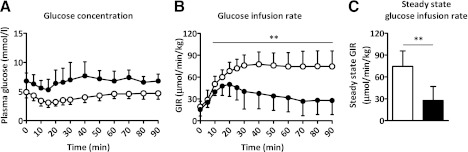
Hyperinsulinemic–euglycemic clamp analysis of sham (white) and SCN lesioned (SCNx, black) mice. Line graphs represent the plasma glucose levels (A) and GIR (B) during the hyperinsulinemic period of the clamp. Bar graph represents the clamped steady-state GIR (C). Data are represented as mean ± SD. **P < 0.01.
In the basal period, EGP, which equals Rd, was not different between sham and SCN lesioned mice (53.6 ± 16.6 vs. 65.0 ± 8.6 µmol/min/kg, not significant; Fig. 5A). In the hyperinsulinemic–euglycemic period, Rd was increased by 57% in the sham mice compared with the basal period (83.9 ± 23.8 vs. 53.6 ± 16.6 µmol/min/kg; P < 0.01) and by 27% in the SCN lesioned mice, although this failed to reach statistical significance (82.8 ± 15.9 vs. 65.0 ± 8.6 µmol/min/kg; P = 0.06). Hyperinsulinemic Rd was not significantly different between sham and SCN lesioned mice. In the hyperinsulinemic–euglycemic period, EGP was decreased by 84% in sham-operated mice compared with the basal period (8.8 ± 12.2 vs. 53.6 ± 16.6 µmol/min/kg; P < 0.01; Fig. 5B), whereas EGP was decreased by only 7% in SCN lesioned mice (60.3 ± 25.8 vs. 65.0 ± 8.6 µmol/min/kg, not significant). These data indicate that bilateral ablation of the SCN induces severe hepatic insulin resistance.
FIG. 5.

Glucose Rd (A) and EGP (B) in basal period (white bars) and hyperinsulinemic–euglycemic clamp period (black bars) of sham and SCN lesioned (SCNx) mice. Data are represented as mean ± SD. **P < 0.01.
SCN lesion with collateral hypothalamic damage.
SCN lesioned mice with collateral damage to hypothalamic nuclei involved in metabolism were analyzed separately. SCN lesioned mice with additional, but selective, unilateral damage to the PVN had modestly increased body mass compared with sham lesioned mice (29.9 ± 3.6 vs. 24.9 ± 1.2 g; P < 0.01; Fig. 6A), which was the result of increased fat mass (7.9 ± 2.5 vs. 1.9 ± 1.0 g; P < 0.01), but not lean mass (16.2 ± 1.0 vs. 17.2 ± 1.7 g, not significant). SCN lesioned mice with bilateral damage to the PVN were considerably heavier than the sham mice (42.5 ± 3.3 vs. 24.9 ± 1.2 g; P < 0.01), which was the result of increased fat mass (21.3 ± 4.8 vs. 1.9 ± 1.0 g; P < 0.01), but this also coincided with a decrease in lean mass (13.6 ± 2.2 vs. 17.2 ± 1.7 g; P < 0.01). SCN lesioned mice with collateral damage to both the PVN and the VMH had development of extreme obesity compared with sham mice (49.3 ± 5.1 vs. 24.9 ± 1.2 g; P < 0.01), which was the result of an excessive increase in fat mass (24.7 ± 3.6 vs. 1.9 ± 1.0 g; P < 0.01), whereas lean mass was not different (16.8 ± 3.3 vs. 17.2 ± 1.7 g, not significant).
FIG. 6.
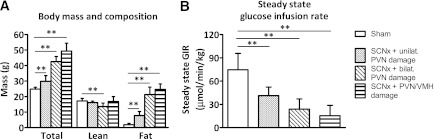
Body mass and composition (A) and hyperinsulinemic–euglycemic clamp analysis (B) of sham (white bars) and SCN lesioned (SCNx) mice with collateral damage (striped bars): unilateral PVN, bilateral PVN, and PVN plus VMH. Data are represented as mean ± SD. **P < 0.01.
Insulin sensitivity in SCN lesioned mice with collateral damage was determined by hyperinsulinemic–euglycemic clamp analysis. GIR needed to maintain euglycemia, as determined over a period of 20 min at stable plasma glucose values, was significantly lower in all SCN lesioned mice with collateral damage (SCN lesion + unilateral PVN damage: 41.3 ± 11.0 µmol/min/kg; SCN lesion + bilateral PVN damage: 23.9 ± 13.1 µmol/min/kg; SCN lesion + PVN/VMH damage: 15.4 ± 13.5 µmol/min/kg) compared with sham controls (74.7 ± 21.1 µmol/min/kg; Fig. 6B), suggesting insulin resistance. Indirect calorimetry, metabolic cage, EGP, and Rd data and insulin levels of SCN lesioned mice with collateral damage are shown in Supplementary Table 2.
DISCUSSION
This study addressed the effects of thermic, bilateral ablation of the SCN on energy metabolism and insulin sensitivity in mice. We show that exclusive lesions of the SCN disrupt the circadian pattern of energy intake, activity, and energy expenditure, as expected. However, although selective SCN ablation resulted in only mild overweight status compared with sham mice, hepatic insulin sensitivity was severely impaired. Therefore, disturbed SCN function has profound metabolic effects.
Anatomically, the SCN is connected with the VMH and the PVN through the subparaventricular zone of the hypothalamus (22). In line with previous studies, mice with collateral damage to the PVN or VMH developed severe obesity and insulin resistance (23–28). Interestingly, unilateral PVN damage in SCN lesioned mice did not result in additional overweight status, indicating that PVN damage results in obesity only in the case of bilateral damage. In light of our data, it is of utmost importance to ascertain correct and exclusive lesions of the SCN to study the role of the circadian pacemaker in the context of metabolism. However, the low success rate (∼30%) of the operation in a previous study (10) and this study (∼20%) limits the potential of this method to study the role of the SCN in metabolism, increasing the value of the current study.
The mild increase in weight gain in mice with exclusive SCN lesions compared with sham mice (+17%) is in contrast to a previous finding in rats (10). In rats, lesions of the SCN did not induce an increase in body mass, whereas the effect on body composition was not determined. This discrepancy may be attributable to the use of the well-studied obesogenic C57Bl/6J mouse strain (29–31) in our study, whereas the rat study was performed in Wistar rats. Wistar rats only become obese on hypercaloric food intake (32).
Previously, it has been shown that the SCN is involved in the regulation of energy homeostasis in mice (33) and rats (34). In the current study, indirect calorimetry and metabolic cage analysis revealed that ablation of the SCN induced a loss of circadian rhythm in oxygen consumption and activity without affecting the 24-h average levels. This loss of circadian rhythm in homeostasis is in line with previous findings, in which SCN lesions eliminated a wide range of rhythms, including leptin (10). Although total food intake over a period of 1 day and 1 night was reduced by 26%, SCN lesioned mice consumed more during the light part of the day compared with sham mice (46% vs. 32% of total food intake). Recently, it has been shown that mice and rats fed only during the day gained significantly more weight than mice fed only at night (35–37). In these studies, obesity resulted from dissociation between the timing of food intake and the intrinsic rhythm of energy expenditure and, thus, animals were eating “against their clock time.” In our study, the protocol was essentially different and animals were not forced to eat at the other part of the cycle, but rather their circadian system was impaired. Therefore, it remains unclear how the mice with selective SCN lesions developed mild overweight because they ate less than the controls and showed the same level of overall activity. Interestingly, results comparable with ours were found in continuous light–exposed mice (38). Continuous light is a strong disruptor of SCN synchrony. When applied for prolonged periods of time, mice become arrhythmic. In continuous light–induced arrhythmic mice, total activity and food intake levels did not differ from that of mice in light/dark. Even though energy balance over 24 h was similar, mice exposed to continuous light displayed impaired glucose tolerance. It has been shown that the SCN exerts excitatory effects on thermogenesis by brown adipose tissue (39), and the mild weight gain in mice with a selective SCN lesion could be the result of reduced brown adipose tissue activity. Alternatively, changes in time distribution of food intake may play a role, which remains an interesting topic for future studies.
Indirect calorimetry and metabolic cage analysis further revealed that SCN lesioned mice had lower respiratory exchange ratio during the day and during the night as compared with sham mice. This shows that SCN ablation results in lower relative carbohydrate oxidation rates and, conversely, higher fat oxidation rates compared with sham mice. These data suggest that the oxidative response to food intake is less directed to carbohydrate metabolism is lesioned, a hallmark for an impaired metabolic flexibility (40). Impaired metabolic flexibility has been associated with impaired insulin sensitivity in humans (41).
Compared with the sham-operated mice, SCN lesioned mice were hyperglycemic and hyperinsulinemic in the postabsorptive state. Furthermore, SCN lesioned mice had increased fasting FFA levels, suggesting a possible removal of inhibitory input from the SCN to the adipose tissue, thereby increasing the basal rate of lipolysis. Increased circulating levels of FFA have been implicated as a possible pathway for development of insulin resistance in obesity (42). Insulin sensitivity shows a circadian pattern in humans (43) as well as in rodents (44), with insulin sensitivity being highest during the active period and lowest during the resting period. The protocol used in this study was such that the sham mice were clamped during their resting period, when insulin sensitivity is lowest, opting for conservative conclusions. Severe hepatic insulin resistance, but not peripheral insulin resistance, was present even though body fat mass was increased only minimally. The SCN is crucial for the circadian control of glucose production and glucose uptake (45–48). Furthermore, there is a direct control of hepatic glucose metabolism resulting from cross-communication of the SCN and PVN, further mediated by innervation of the liver (49). Therefore, it is likely that the impaired hepatic insulin sensitivity found in the SCN lesioned mice is, at least in part, a direct result of the disrupted SCN-mediated control of glucose and FFA metabolism.
All SCN lesioned mice with collateral damage to PVN or VMH had development of hepatic insulin resistance. Furthermore, SCN lesioned mice with bilateral PVN damage and SCN lesioned mice with PVN or VMH damage showed peripheral insulin resistance, most likely as a result of increased fat mass in these mice.
In conclusion, we demonstrate that exclusive deletion of the SCN induces loss in circadian rhythms in energy metabolism and food intake. Although ablation of the SCN resulted in only mild overweight status, SCN lesioned mice were severely insulin resistant in the liver. Great care was taken to distinguish between exclusive SCN lesioned mice from mice that had collateral damage to the PVN and VMH areas, because the latter resulted in severe obesity. It was previously shown that mice with mutations in clock genes have altered energy homeostasis and glucose metabolism (14,50). Together, the data from several studies provide solid evidence that the SCN is crucially involved in the maintenance of energy balance and hepatic insulin sensitivity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from TI Pharma (TIP project T2-105, to J.A.R.), the Netherlands Heart Foundation (NHS project 2007B81, to J.A.R.), the Dutch Diabetes Research Foundation (DFN project 2007.00.010, to J.A.R.), the Netherlands Consortium for Systems Biology (NCSB project, to K.W.v.D.) established by The Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (NGI/NWO), NWO ZonMW (Top Go grant 81802016, to J.H.M., Top grant 91207036, to A.K.), and the Dutch Organization for Scientific Research (Clinical Fellows 90700195, to N.R.B.).
No potential conflicts of interest relevant to this article were reported.
C.P.C., S.A.A.v.d.B., E.A.L., T.H., A.C.M.P., and N.R.B. performed experiments. C.P.C., S.A.A.v.d.B., T.H., R.D.v.d.S., and A.K. researched data. C.P.C., S.A.A.v.d.B., and J.H.M. wrote the manuscript. N.R.B. and K.W.v.D. contributed to discussion. N.R.B., K.W.v.D., and J.A.R. reviewed the manuscript. J.A.R. and J.H.M. provided material. J.A.R. and J.H.M. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Hans Duindam (Department of Molecular Cell Biology), Hester van Diepen (Department of Molecular Cell Biology), Heleen Post-van Engeldorp Gastelaars (Department of Molecular Cell Biology), Rosa van den Berg (Department of Endocrinology), and Peter Stouten (Department of Molecular Cell Biology), all from Leiden University Medical Center, Leiden, the Netherlands, for excellent technical support.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0507/-/DC1
See accompanying commentary, p. 1017.
REFERENCES
- 1.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 2008;9:764–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamazaki S, Numano R, Abe M, et al. Resetting central and peripheral circadian oscillators in transgenic rats. Science 2000;288:682–685 [DOI] [PubMed] [Google Scholar]
- 3.Davidson AJ, Yamazaki S, Arble DM, Menaker M, Block GD. Resetting of central and peripheral circadian oscillators in aged rats. Neurobiol Aging 2008;29:471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalsbeek A, Buijs RM. Output pathways of the mammalian suprachiasmatic nucleus: coding circadian time by transmitter selection and specific targeting. Cell Tissue Res 2002;309:109–118 [DOI] [PubMed] [Google Scholar]
- 5.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature 2002;418:935–941 [DOI] [PubMed] [Google Scholar]
- 6.Cailotto C, La Fleur SE, Van Heijningen C, et al. The suprachiasmatic nucleus controls the daily variation of plasma glucose via the autonomic output to the liver: are the clock genes involved? Eur J Neurosci 2005;22:2531–2540 [DOI] [PubMed] [Google Scholar]
- 7.Van Cauter E, Polonsky KS, Scheen AJ. Roles of circadian rhythmicity and sleep in human glucose regulation. Endocr Rev 1997;18:716–738 [DOI] [PubMed] [Google Scholar]
- 8.Weibel L, Follenius M, Spiegel K, Ehrhart J, Brandenberger G. Comparative effect of night and daytime sleep on the 24-hour cortisol secretory profile. Sleep 1995;18:549–556 [PubMed] [Google Scholar]
- 9.Kalra SP, Bagnasco M, Otukonyong EE, Dube MG, Kalra PS. Rhythmic, reciprocal ghrelin and leptin signaling: new insight in the development of obesity. Regul Pept 2003;111:1–11 [DOI] [PubMed] [Google Scholar]
- 10.Kalsbeek A, Fliers E, Romijn JA, et al. The suprachiasmatic nucleus generates the diurnal changes in plasma leptin levels. Endocrinology 2001;142:2677–2685 [DOI] [PubMed] [Google Scholar]
- 11.Glass JD, Guinn J, Kaur G, Francl JM. On the intrinsic regulation of neuropeptide Y release in the mammalian suprachiasmatic nucleus circadian clock. Eur J Neurosci 2010;31:1117–1126 [DOI] [PubMed] [Google Scholar]
- 12.Foster RG, Roenneberg T. Human responses to the geophysical daily, annual and lunar cycles. Curr Biol 2008;18:R784–R794 [DOI] [PubMed] [Google Scholar]
- 13.Colwell CS. Linking neural activity and molecular oscillations in the SCN. Nat Rev Neurosci 2011;12:553–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005;308:1043–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura TJ, Nakamura W, Yamazaki S, et al. Age-related decline in circadian output. J Neurosci 2011;31:10201–10205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swaab DF, Fliers E, Partiman TS. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res 1985;342:37–44 [DOI] [PubMed] [Google Scholar]
- 17.Deboer T, Overeem S, Visser NA, et al. Convergence of circadian and sleep regulatory mechanisms on hypocretin-1. Neuroscience 2004;129:727–732 [DOI] [PubMed] [Google Scholar]
- 18.Dörrscheidt GJ, Beck L. Advanced methods for evaluating characteristic parameters (T, 7, p) of Circadian Rhythms. J Math Biol 1975;2:107–121 [Google Scholar]
- 19.van den Berg SA, Guigas B, Bijland S, et al. High levels of dietary stearate promote adiposity and deteriorate hepatic insulin sensitivity. Nutr Metab (Lond) 2010;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coomans CP, Biermasz NR, Geerling JJ, et al. Stimulatory effect of insulin on glucose uptake by muscle involves the central nervous system in insulin-sensitive mice. Diabetes 2011;60:3132–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley DE, Mandarino LJ. Hyperglycemia normalizes insulin-stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J Clin Invest 1990;86:1999–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieuwenhuys R, Voogd J, van Huijzen C. The Human Central Nervous System. Berlin, Springer-Verlag, 2008 [Google Scholar]
- 23.Sims JS, Lorden JF. Effect of paraventricular nucleus lesions on body weight, food intake and insulin levels. Behav Brain Res 1986;22:265–281 [DOI] [PubMed] [Google Scholar]
- 24.Touzani K, Velley L. Ibotenic acid lesion of the hypothalamic paraventricular nucleus produces weight gain but modifies neither preference nor aversion for saccharin. Physiol Behav 1992;52:673–678 [DOI] [PubMed] [Google Scholar]
- 25.Pénicaud L, Kinebanyan MF, Ferré P, et al. Development of VMH obesity: in vivo insulin secretion and tissue insulin sensitivity. Am J Physiol 1989;257:E255–E260 [DOI] [PubMed] [Google Scholar]
- 26.Grundmann SJ, Pankey EA, Cook MM, Wood AL, Rollins BL, King BM. Combination unilateral amygdaloid and ventromedial hypothalamic lesions: evidence for a feeding pathway. Am J Physiol Regul Integr Comp Physiol 2005;288:R702–R707 [DOI] [PubMed] [Google Scholar]
- 27.Kageyama A, Hirano T, Kageyama H, et al. Distinct role of adiposity and insulin resistance in glucose intolerance: studies in ventromedial hypothalamic-lesioned obese rats. Metabolism 2002;51:716–723 [DOI] [PubMed] [Google Scholar]
- 28.King BM, Phelps GR, Frohman LA. Hypothalamic obesity in female rats in absence of vagally mediated hyperinsulinemia. Am J Physiol 1980;239:E437–E441 [DOI] [PubMed] [Google Scholar]
- 29.Gallou-Kabani C, Vigé A, Gross MS, et al. C57BL/6J and A/J mice fed a high-fat diet delineate components of metabolic syndrome. Obesity (Silver Spring) 2007;15:1996–2005 [DOI] [PubMed] [Google Scholar]
- 30.Schreyer SA, Wilson DL, LeBoeuf RC. C57BL/6 mice fed high fat diets as models for diabetes-accelerated atherosclerosis. Atherosclerosis 1998;136:17–24 [DOI] [PubMed] [Google Scholar]
- 31.Parekh PI, Petro AE, Tiller JM, Feinglos MN, Surwit RS. Reversal of diet-induced obesity and diabetes in C57BL/6J mice. Metabolism 1998;47:1089–1096 [DOI] [PubMed] [Google Scholar]
- 32.Harrold JA, Widdowson PS, Clapham JC, Williams G. Individual severity of dietary obesity in unselected Wistar rats: relationship with hyperphagia. Am J Physiol Endocrinol Metab 2000;279:E340–E347 [DOI] [PubMed] [Google Scholar]
- 33.Phan Trongha X, Chan GC, Sindreu CB, Eckel-Mahan KL, Storm DR. The diurnal oscillation of MAP (mitogen-activated protein) kinase and adenylyl cyclase activities in the hippocampus depends on the suprachiasmatic nucleus. J Neurosci 2011;31:10640–10647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angeles-Castellanos M, Salgado-Delgado R, Rodriguez K, Buijs RM, Escobar C. The suprachiasmatic nucleus participates in food entrainment: a lesion study. Neuroscience 2010;165:1115–1126 [DOI] [PubMed] [Google Scholar]
- 35.Arble DM, Bass J, Laposky AD, Vitaterna MH, Turek FW. Circadian timing of food intake contributes to weight gain. Obesity (Silver Spring) 2009;17:2100–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salgado-Delgado R, Angeles-Castellanos M, Saderi N, Buijs RM, Escobar C. Food intake during the normal activity phase prevents obesity and circadian desynchrony in a rat model of night work. Endocrinology 2010;151:1019–1029 [DOI] [PubMed] [Google Scholar]
- 37.Bray MS, Tsai JY, Villegas-Montoya C, et al. Time-of-day-dependent dietary fat consumption influences multiple cardiometabolic syndrome parameters in mice. Int J Obes (Lond) 2010;34:1589–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fonken LK, Workman JL, Walton JC, et al. Light at night increases body mass by shifting the time of food intake. Proc Natl Acad Sci USA 2010;107:18664–18669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amir S, Shizgal P, Rompré PP. Glutamate injection into the suprachiasmatic nucleus stimulates brown fat thermogenesis in the rat. Brain Res 1989;498:140–144 [DOI] [PubMed] [Google Scholar]
- 40.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950 [DOI] [PubMed] [Google Scholar]
- 41.Corpeleijn E, Mensink M, Kooi ME, Roekaerts PM, Saris WH, Blaak EE. Impaired skeletal muscle substrate oxidation in glucose-intolerant men improves after weight loss. Obesity (Silver Spring) 2008;16:1025–1032 [DOI] [PubMed] [Google Scholar]
- 42.Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 2002;32(Suppl 3):14–23 [DOI] [PubMed] [Google Scholar]
- 43.Boden G, Chen X, Urbain JL. Evidence for a circadian rhythm of insulin sensitivity in patients with NIDDM caused by cyclic changes in hepatic glucose production. Diabetes 1996;45:1044–1050 [DOI] [PubMed] [Google Scholar]
- 44.La Fleur SE, Kalsbeek A, Wortel J, Fekkes ML, Buijs RM. A daily rhythm in glucose tolerance: a role for the suprachiasmatic nucleus. Diabetes 2001;50:1237–1243 [DOI] [PubMed] [Google Scholar]
- 45.Ruiter M, Buijs RM, Kalsbeek A. Hormones and the autonomic nervous system are involved in suprachiasmatic nucleus modulation of glucose homeostasis. Curr Diabetes Rev 2006;2:213–226 [DOI] [PubMed] [Google Scholar]
- 46.La Fleur SE. Daily rhythms in glucose metabolism: suprachiasmatic nucleus output to peripheral tissue. J Neuroendocrinol 2003;15:315–322 [DOI] [PubMed] [Google Scholar]
- 47.Arslanian S, Ohki Y, Becker DJ, Drash AL. Demonstration of a dawn phenomenon in normal adolescents. Horm Res 1990;34:27–32 [DOI] [PubMed] [Google Scholar]
- 48.Bolli GB, De Feo P, De Cosmo S, et al. Demonstration of a dawn phenomenon in normal human volunteers. Diabetes 1984;33:1150–1153 [DOI] [PubMed] [Google Scholar]
- 49.Kalsbeek A, La Fleur S, Van Heijningen C, Buijs RM. Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J Neurosci 2004;24:7604–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rudic RD, McNamara P, Curtis AM, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2004;2:e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.