

Abstract
We investigated whether HLA-A*24 typing complements screening for HLA-DQ and for antibodies (Abs) against insulin, GAD, IA-2 (IA-2A), and zinc transporter-8 (ZnT8A) for prediction of rapid progression to type 1 diabetes (T1D). Persistently Ab+ siblings/offspring (n = 288; aged 0–39 years) of T1D patients were genotyped for HLA-DQA1-DQB1 and HLA-A*24 and monitored for development of diabetes within 5 years of first Ab+. HLA-A*24 (P = 0.009), HLA-DQ2/DQ8 (P = 0.001), and positivity for IA-2A ± ZnT8A (P < 0.001) were associated with development of T1D in multivariate analysis. The 5-year risk increased with the number of the above three markers present (n = 0: 6%; n = 1: 18%; n = 2: 46%; n = 3: 100%). Positivity for one or more markers identified a subgroup of 171 (59%) containing 88% of rapid progressors. The combined presence of HLA-A*24 and IA-2A+ ± ZnT8A+ defined a subgroup of 18 (6%) with an 82% diabetes risk. Among IA-2A+ ± ZnT8A+ relatives, identification of HLA-A*24 carriers in addition to HLA-DQ2/DQ8 carriers increased screening sensitivity for relatives at high Ab- and HLA-inferred risk (64% progression; P = 0.002). In conclusion, HLA-A*24 independently predicts rapid progression to T1D in Ab+ relatives and complements IA-2A, ZnT8A, and HLA-DQ2/DQ8 for identifying participants in immunointervention trials.
Autoimmune type 1 diabetes (T1D) results from the T-cell–mediated destruction of pancreatic insulin-producing β-cells, preferentially in individuals carrying HLA class II susceptibility haplotypes (1). Immunointerventions with anti-CD3 antibodies (Abs) (2,3), rituximab (4), and cytotoxic T-lymphocyte antigen-4-immunoglobulin fusion protein (5) transiently preserved β-cell function, preferentially in a patient subgroup with younger age at diagnosis and with relatively preserved residual functional β-cell mass (3), hereby opening perspectives for future trials at the preclinical stage (3,6). To limit numbers needed to treat and the time to reach significant conclusions, such trials require identification of individuals at high risk to develop diabetes short-term (7,8). Abs against IA-2 (IA-2A) and zinc transporter-8 (ZnT8A) generally appear later than Abs against insulin (IAA) or GAD (GADA) in pre-T1D and have been associated with more rapid disease progression in first-degree relatives (FDRs) (9–12).
Genetic factors can further modulate diabetes risk in Ab+ relatives; for example, the presence of HLA-DQA1*0501-DQB1*0201(DQ2)/DQA1*0301-DQB1*0302(DQ8) identifies a subgroup of IA-2A+ or multiple Ab+ relatives with impending diabetes (13–15). The HLA class I region has also been shown to contribute to T1D risk (16–18). The presence of HLA-A*24 has been associated with progression from autoimmunity to T1D (19) and with early and complete β-cell destruction after diagnosis (20). It is, however, still unclear to what extent genotyping for HLA class I can usefully complement screening for Ab- and HLA-DQ–inferred risk in identifying relatives with impending diabetes. We investigated in a large representative group of Ab+ FDRs aged younger than 40 years whether 1) the presence of HLA-A*24 affects progression from autoimmunity to T1D independently from other risk markers; 2) HLA-A*24 interacts with HLA-DQ and/or auto-Ab-inferred risk of diabetes; and 3) HLA-A*24 typing can refine the sensitivity or specificity of screening to identify rapid progressors.
RESEARCH DESIGN AND METHODS
Study population.
Offspring or siblings of T1D probands were Caucasians (aged 0–39 years) consecutively recruited by the Belgian Diabetes Registry (BDR), as previously described (13). At entry and during yearly follow-up, blood sampling and completion of a short questionnaire were performed after written informed consent. Only relatives with at least two contacts during follow-up, the last being at diagnosis of diabetes in case of progression to clinically overt disease, were included in this study. Blood, serum, and plasma samples were stored as aliquots at −80°C until analyzed. Screening for auto-Abs against insulin (IAA), GAD 65 kDa (GADA), IA-2 (IA-2A), and ZnT8 (ZnT8A) identified 288 persistently positive FDRs (i.e., ≥1 molecular Abs in ≥2 consecutive samples) with available DNA for HLA-DQ and HLA-A*24 typing (flowchart in Supplementary Fig. 1). At baseline (first Ab+ sample), the median age (interquartile range [IQR]) of FDRs was 12 (6–19) years. Because we focused in the current study on rapid progression to diabetes, follow-up was truncated at 60 months. The median (IQR) follow-up time was 60 (36–60) months (73% of FDRs completed the 5-year follow-up). Diabetes was diagnosed according to the American Diabetes Association criteria (21). Diagnosis was ascertained through repeated contacts with Belgian diabetologists, self-reporting through yearly questionnaires, and a link to the BDR patient database, where newly diagnosed patients are registered.
The study was carried out according to The Helsinki Declaration as revised in 2008 (http://www.wma.net/en/30publications/10policies/b3/, accessed on 18 April 2012), and the protocol was approved by the ethics committees of the BDR and participating university hospitals.
Analytical methods.
Diabetes auto-Abs and HLA-DQ genotyping were performed as reported previously (9,13,22). HLA-A*24 typing was performed by the PCR sequence-specific oligonucleotide dot-blot method. Briefly, genomic DNA was amplified by HLA-A*24 group-specific primers (23), followed by DNA dot-blot hybridization (22) with a panel of three sequence-specific oligonucleotide probes (probe 1: 5′-GAG-GAG-ACA-GGG-AAA-3′; probe 2: 5′-CGC-TCT-TGG-ACC-GCG-3′; and probe 3: 5′-GCG-GCC-CGT-GTG-GCG-3′), and the signals were subsequently detected with a Digoxigenin Luminescent Detection Kit (Roche Molecular Biochemicals, Mannheim, Germany). Our probe set identified all 255 HLA-A*24 alleles except HLA-A*240217/0227/0249/08/24/29/31/42/67/77/89/129/145/156191.
Statistical methods.
Statistical differences in proportions between groups were tested by χ2 with Yates continuity correction for independent samples or by McNemar test for paired samples. For continuous variables, comparisons were performed by Mann-Whitney U test. Follow-up of relatives started at the time of the first Ab+ sample and ended at the last contact with the relative or at clinical onset, whichever came first. For the identification of rapid progressors, follow-up was truncated after 5 years. Kaplan-Meier survival analysis and log-rank test were used to compare differences in diabetes-free survival between groups. Cox regression univariate analysis (enter method) was used to select potential predictors (P < 0.05) to be tested in a multivariate model (forward stepwise method) for identifying independent predictors of impending diabetes and calculation of their hazard ratio. Statistical analyses were performed two-tailed by EpiInfo 6.0 (USD Inc., Stone Mountain, GA), IBM SPSS Statistics 20.0 (IBM Corporation, Armonk, NY), or by GraphPad Prism 5 (GraphPad, San Diego, CA) software. Significance was defined as P < 0.05 or P < 0.05/k in case of multiple comparisons (Bonferroni adjustment).
RESULTS
HLA-A*24 as an independent predictor of progression to diabetes.
We identified 288 persistently IAA+, GADA+, IA-2A+ and/or ZnT8A+ relatives at baseline who were tested for the presence or absence of HLA-DQ2, -DQ8 and -A*24. Within 5 years after the first Ab+, 50 relatives (17%) developed diabetes after a median (IQR) follow-up time of 29 (12–42) months. Their baseline characteristics are reported in Table 1. Compared with nonprogressors, rapid progressors were younger and more frequently IA-2A+, ZnT8A+, multiple Ab+, and/or HLA-DQ2/DQ8+. They tended to be less frequently offspring of a diabetic mother and to carry more often HLA-DQ2, -DQ8, and -A*24.
TABLE 1.
Baseline characteristics of Ab+ FDRs** according to 5-year diabetes status

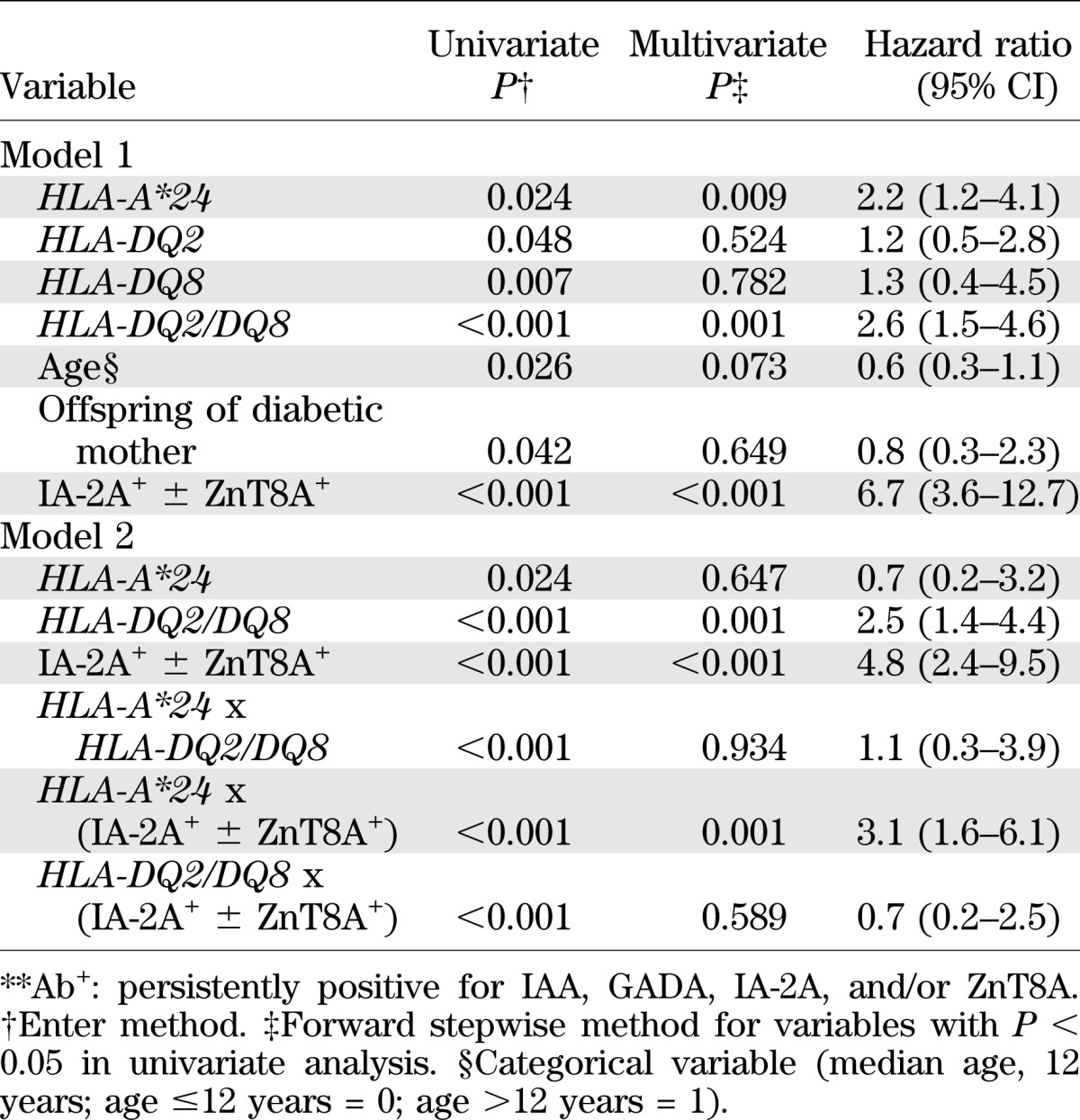
Cox regression analysis (Table 2) identified HLA-A*24 as an independent predictor of rapid progression to diabetes in addition to the established high-risk markers IA-2A+, ZnT8A+, and HLA-DQ2/DQ8. When positivity for two or more Abs was included in the first model, it also came out as an independent predictor (P = 0.024) in addition to HLA-A*24 (P = 0.015), HLA-DQ2/DQ8 (P = 0.001), and IA-2A+ ± ZnT8A+ (P = 0.006; data not shown). A second model looking at interactions between independent predictors revealed a significant interaction between the presence of HLA-A*24 and IA-2A+ ± ZnT8A+ (Table 2). Similar results were obtained when considering 10-year progression to diabetes (Supplementary Table 1). Except for a tendency toward a higher prevalence of GADA+ in carriers of HLA-DQ2/DQ8 (P = 0.062 vs. absence of HLA-DQ2/DQ8) or HLA-A*24 (P = 0.077 vs. absence of HLA-A*24), Ab frequency did not differ significantly according to HLA-DQ2/DQ8 and -A*24 status (data not shown).
TABLE 2.
Cox regression analysis for 5-year progression to diabetes in 288 Ab+ FDRs**
Kaplan-Meier survival analysis.
Survival analysis confirmed a more rapid progression to diabetes in presence of HLA-A*24 (Fig. 1A), HLA-DQ2/DQ8 (Fig. 1B), and IA-2A ± ZnT8A (Fig. 1C), than in their respective absence (Fig. 1A–C). Positivity for HLA-A*24 selectively and significantly increased the 5-year progression rate to diabetes in presence of HLA-DQ2/DQ8 (Fig. 1D) or IA-2A ± ZnT8A (Fig. 1E) but not in their absence (Fig. 1D and E). Similarly, HLA-DQ2/DQ8 carrier status increased the 5-year diabetes risk more in relatives with IA-2A+ ± ZnT8A+ than in those without (Fig. 1F). Table 3 lists the numbers of relatives presenting various combinations of the three markers (IA-2A+ ± ZnT8A+, HLA-A*24, HLA-DQ2/DQ8) at baseline, their 5-year progression rate, and the fraction of diabetes cases they identify.
FIG. 1.
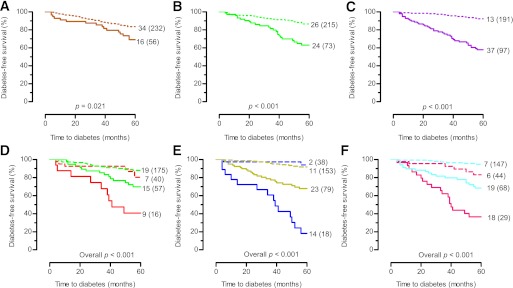
Diabetes-free survival in persistently Ab+ FDRs at baseline (n = 288) stratified according to HLA-A*24, HLA-DQ2/DQ8, and IA-2A/ZnT8A status: presence of HLA-A*24, solid orange line; absence of HLA-A*24, broken orange line (A); presence of HLA-DQ2/DQ8, solid green line; absence of HLA-DQ2/DQ8, broken green line (B); presence of IA-2A and/or ZnT8A, solid purple line; absence of IA-2A and ZnT8A, broken purple line (C); presence of HLA-A*24 and HLA-DQ2/DQ8, solid red line; presence of HLA-DQ2/DQ8 and absence of HLA-A*24, solid green line; presence of HLA-A*24 and absence of HLA-DQ2/DQ8, broken red line; absence of HLA-DQ2/DQ8 and of HLA-A*24, broken green line (D); presence of HLA-A*24 and (IA-2A and/or ZnT8A), solid blue line; presence of (IA-2A and/or ZnT8A) and absence of HLA-A*24, solid yellow line; presence of HLA-A*24 and absence of (IA-2A and ZnT8A), broken blue line; absence of IA-2A, ZnT8A and of HLA-A*24, broken yellow line (E); presence of IA-2A and/or ZnT8A and HLA-DQ2/DQ8, solid fuchsia line; presence of IA-2A and/or ZnT8A and absence of HLA-DQ2/DQ8, solid cyan line; presence of HLA-DQ2/DQ8 and absence of IA-2A and ZnT8A, broken fuchsia line; absence of IA-2A, ZnT8A, and HLA-DQ2/DQ8, broken cyan line (F). The numbers in each panel indicate the number of events for each arm (total number at study entry). P by log-rank test.
TABLE 3.
Five-year progression to type 1 diabetes according to biomarker profile
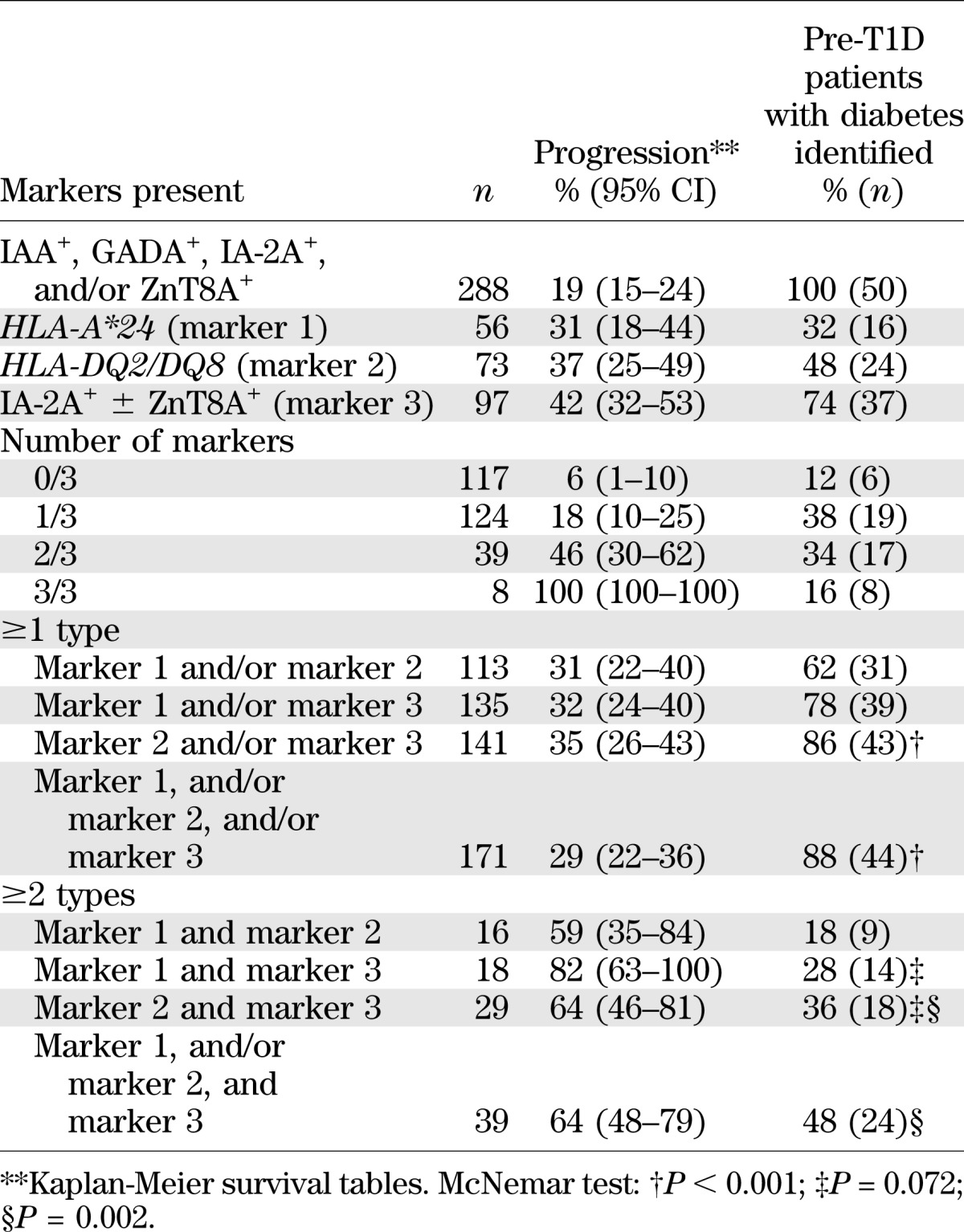
The 5-year risk of diabetes and diagnostic sensitivity were higher in IA-2A+ ± ZnT8A+ relatives than when HLA-DQ2/DQ8 or HLA-A*24 carrier status alone was examined (Table 3). The 5-year progression rate increased according to the number of independent risk markers (IA-2A+ ± ZnT8A+, HLA-A*24, HLA-DQ2/DQ8), reaching 100% in presence of all three markers but with 72% of the future patients belonging to the groups with one or two markers (Table 3).
Positivity for at least one of the three high-risk markers identified a subgroup of 171 relatives (57%) harboring 44 of the 50 (88%) rapid progressors (Table 3). Typing for HLA-A*24 in addition to HLA-DQ and IA-2A ± ZnT8A only marginally increased screening sensitivity in this group. However, the presence of at least two of the three risk markers (IA-2A+ ± ZnT8A+, HLA-A*24, HLA-DQ2/DQ8) was associated with 59–82% 5-year progression but at the expense of a low sensitivity between 18% and 48%, depending on the marker combinations (Table 3). The combined presence of HLA-A*24 and IA-2A+ ± ZnT8A+ (regardless of HLA-DQ2/DQ8 genotype) conferred the highest 5-year risk (82%) of diabetes but identified only 28% of the rapid progressors (Table 3). The combination of IA-2A+ ± ZnT8A+ and HLA-DQ2/DQ8 was more sensitive (36%), with a high progression rate (64%), and an additional search for HLA-A*24 carriers among IA-2A+ ± ZnT8A+ relatives further increased sensitivity (48%) without affecting overall 5-year progression rate. When 10-year progression to diabetes was considered (Supplementary Table 2), results were similar to the observations in Table 3 except for a slight decrease in the fraction of diabetic patients identified and an overall increase in progression rate, as expected.
DISCUSSION
The current study confirms the association of HLA-A*24 with progression from autoimmunity to T1D (16–18). The identification of this allele as an independent predictor of clinical onset within 5 years and interacting with the presence of HLA-DQ2/DQ8 and IA-2A+ ± ZnT8A+ constitutes the major finding of our study. Our observation has practical implications for screening strategies in preparation of secondary prevention trials with immune intervention. The joint presence of IA-2A+ ± ZnT8A+ and HLA-A*24 identifies a small subgroup at extremely high diabetes risk (82% within 5 years). In IA-2A+ ± ZnT8A+ FDRs, however, detection of HLA-A*24 carrier status significantly increases screening sensitivity for individuals at high Ab- and HLA-inferred 5-year risk (64% progression to diabetes) as a useful complement to HLA-DQ typing. This selected group of relatives qualifies for precise assessment of β-cell function with standardized tests (e.g., hyperglycemic clamp test) to identify individuals with already decreased stimulated hormone release in the perspective of composing homogeneous groups of high-risk participants in immunointervention trials with an acceptable risk-to-benefit ratio in the light of the potentially harmful drugs used (e.g., 80% 3-year diabetes risk) (7,10,13).
Strengths of this study include its registry-based nature, the broad age range of recruited FDRs, its longitudinal nature, with 73% of nonprogressors followed for 5 years, and the completeness of the dataset for all variables. Given the broad age range, most relatives were not monitored from birth, which is the best approach to study the natural history of diabetes. However, Vermeulen et al. (9) showed that relatives who seroconverted to Ab+ during follow-up did not differ in baseline characteristics and progression rate toward diabetes from relatives who were Ab+ at the first sampling. Because we focused on rapid progressors, the number of events is relatively limited. However, our cohort constitutes one of the largest and most representative groups of relatives without preselection on the basis of an initial screening.
The mechanisms by which polymorphic HLA class II and I molecules confer T1D risk are incompletely understood but inferred to involve thymic selection of T-cell repertoire and peripheral selection of peptide repertoire (1). Tait et al. (19) suggested that HLA class II and class I, respectively, mediate initiation and progression of β-cell autoimmunity, although observations by us and others also suggest a major role for DQ2-DR3/DQ8-DR4 in disease progression (13,24). Recent findings by Lipponen et al. (18) showed a strong effect of HLA-B*39, but not of HLA-A*24, on progression to T1D after seroconversion to persistent Ab+, but only in individuals carrying HLA-DR3/DR4. In contrast, Tait et al. (19) found that disease progression was associated with HLA-A*24, -A*30, and -B*18 in Ab+ relatives of a diabetic proband. Our observation of an accelerating role of HLA-A*24 on β-cell loss is in accordance with the results of Tait et al. (19) and with previous reports on its association with early-onset T1D and subsequent complete β-cell destruction (17,20) but at variance with Lipponen et al. (18). In line with the latter Finnish study (18), however, the disease-promoting effect of HLA class I alleles was restricted to individuals carrying the HLA-DQ2/DQ8 high-risk genotype.
Several reasons may explain this discrepancy between Finnish and Belgian studies, including differences in the background population (Finland vs. Belgium), familial history (general population vs. FDRs), age at inclusion (neonates monitored from birth vs. age 0–39 years), and initial Ab screening (ICA vs. four molecular Abs). Our results do not allow us to reach firm conclusions about the exact nature of the mechanisms involved in the disease-promoting effects of HLA-A*24 and in its interactions with other genetic and immune risk markers. Nevertheless, the accelerating effect of HLA-A*24 on the underlying process and its interaction with immune markers of impending diabetes are compatible with a role in efficient HLA class I–mediated presentation of antigenic self-epitopes to cytotoxic CD8+ lymphocytes (25). Our data reiterate the need to stratify according to closely linked HLA class II alleles when looking for class I effects (17,18). Finally and more importantly, they demonstrate for the first time how HLA class I typing may contribute in practice to the refinement of screening strategies to select suitable candidates for participation in immune interventions in pre-T1D.
In conclusion, we demonstrate that HLA-A*24 is an independent predictor of 5-year progression to diabetes in Ab+ FDRs of T1D patients. It interacts significantly with the presence of HLA-DQ2/DQ8 and, in particular, IA-2A+ ± ZnT8A+. Depending on whether the screening goals intend to favor specificity or sensitivity, HLA-A*24 can help select a subgroup at extremely high risk or increase sensitivity of detecting relatives with high combined Ab- and HLA-inferred risk.
Supplementary Material
ACKNOWLEDGMENTS
The study was supported by grants from the Juvenile Diabetes Research Foundation (JDRF Center Grant 4-2005-1327), the European Union (FP-7 Project No. 241833), the Belgian Fund for Scientific Research (FWO Vlaanderen Projects G.0319.01, G.0514.04, G.0311.07, G.0374.08, and G.0868.11, senior clinical research fellowships for I.W.), the Research Council of the Brussels Free University (research fellowship to E.M.), and the Willy Gepts Fund (projects 3-2005 and 3/22-2007, University Hospital Brussels–UZ Brussel). J.C.H. acknowledges Diabetes and Endocrinology Research Center (DERC) National Institutes of Health (NIH) grants (P30-DK57516 and R01-DK052068), and JDRF Grant 4-2007-1056.
The BDR was sponsored by the Belgian National Lottery, the ministries of Public Health of the Flemish and French Communities of Belgium, Hippo & Friends, Weight Watchers, Ortho-Clinical Diagnostics, Novo Nordisk Pharma, LifeScan, Roche Diagnostics, Bayer, and Eli Lilly. No other potential conflicts of interest relevant to this article were reported.
E.M. designed research; acquired data; undertook statistical analysis and interpretation of the results; wrote, reviewed, and edited the manuscript; and approved the final version of the manuscript. B.J.V.d.A. designed research, obtained funding, supervised data collection, interpreted data, reviewed and edited the manuscript, and approved the final version of the manuscript. I.V., S.D., A.V.D., and E.V.B. analyzed and interpreted data, participated in discussions, reviewed and edited the manuscript, and approved the final version of the manuscript. S.V.A., L.D., H.D., and J.D.S. designed the study, recruited FDRs, reviewed and edited the manuscript, and approved the final version of the manuscript. C.v.S. designed the study, participated in discussion, analyzed and interpreted data, reviewed and edited the manuscript, and approved the final version of the manuscript. J.M.W. and J.C.H. contributed new reagents and analytical tools, analyzed and interpreted data, contributed to discussion, reviewed and edited the manuscript, and approved the final version of the manuscript. D.P. designed research, analyzed and interpreted data, contributed to discussion, reviewed and edited the manuscript, and approved the final version of the manuscript. I.W. designed research, recruited FDRs, supervised statistical analysis, interpreted data, reviewed and edited the manuscript, and approved the final version of the manuscript. F.K.G. designed research; obtained funding; supervised the study; analyzed and interpreted data; drafted, reviewed, and edited the manuscript; and approved the final version of the manuscript. F.K.G. is the guarantor of this work, and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors gratefully acknowledge the expert technical assistance of coworkers at the central unit of the BDR (P. Goubert, C. Groven) and at the reference laboratory of the BDR (V. Baeten, T. De Mesmaeker, H. Dewinter, N. Diependaele, S. Exterbille, T. Glorieux, T. Haulet, A. Ivens, D. Kesler, F. Lebleu, M. Van Molle, S. Vanderstraeten, K. Verhaeghen, and A. Walgrave from the Department of Clinical Chemistry and Radio-immunology, University Hospital Brussels Free University-UZ Brussel, Brussels, Belgium, and G. De Block, E. Quartier, G. Schoonjans from the Brussels Free University-VUB, Brussels). The authors also thank the various university teams of coworkers for their excellent assistance in collecting samples and organizing the fieldwork in Antwerp (L. Van Gaal, C. De Block, R. Braspenning, J. Michiels, J. Van Elven, and J. Vertommen, University Hospital Antwerp), in Brussels (B. Keymeulen, K. Decochez, E. Vandemeulebroucke, and U. Van de Velde, University Hospital Brussels Free University-UZ Brussel), in Ghent (J.M. Kaufman, J. Ruige, A. Hutse, A. Rawoens, and N. Steyaert, University Hospital Ghent), and in Leuven (C. Mathieu, P. Gillard, M. Carpentier, M. Robijn, K. Rouffe, A. Schoonis, and H. Morobé, University Hospital Leuven), all in Belgium. The authors sincerely thank all members of the BDR who contributed to the recruitment of relatives for the current study (list of names in the Supplementary Data).
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0747/-/DC1.
A complete list of the members of the Belgian Diabetes Registry can be found in the Supplementary Data.
See accompanying commentary, p. 1020.
REFERENCES
- 1.Todd JA. Etiology of type 1 diabetes. Immunity 2010;32:457–467 [DOI] [PubMed] [Google Scholar]
- 2.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692–1698 [DOI] [PubMed] [Google Scholar]
- 3.Keymeulen B, Walter M, Mathieu C, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 2010;53:614–623 [DOI] [PubMed] [Google Scholar]
- 4.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orban T, Bundy B, Becker DJ, et al. Type 1 Diabetes TrialNet Abatacept Study Group Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011;378:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skyler JS, Ricordi C. Stopping type 1 diabetes: attempts to prevent or cure type 1 diabetes in man. Diabetes 2011;60:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandemeulebroucke E, Keymeulen B, Decochez K, et al. Belgian Diabetes Registry Hyperglycaemic clamp test for diabetes risk assessment in IA-2-antibody-positive relatives of type 1 diabetic patients. Diabetologia 2010;53:36–44 [DOI] [PubMed] [Google Scholar]
- 8.Mahon JL, Dupré J. The limitations of clinical trials for prevention of IDDM. Diabetes Care 1997;20:1027–1033 [DOI] [PubMed] [Google Scholar]
- 9.Vermeulen I, Weets I, Costa O, et al. Belgian Diabetes Registry An important minority of prediabetic first-degree relatives of type 1 diabetic patients derives from seroconversion to persistent autoantibody positivity after 10 years of age. Diabetologia 2012;55:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Grijse J, Asanghanwa M, Nouthé B, et al. Belgian Diabetes Registry Predictive power of screening for antibodies against insulinoma-associated protein 2 beta (IA-2beta) and zinc transporter-8 to select first-degree relatives of type 1 diabetic patients with risk of rapid progression to clinical onset of the disease: implications for prevention trials. Diabetologia 2010;53:517–524 [DOI] [PubMed] [Google Scholar]
- 11.Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 2007;104:17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu L, Boulware DC, Beam CA, et al. Type 1 Diabetes TrialNet Study Group Zinc transporter-8 autoantibodies improve prediction of type 1 diabetes in relatives positive for the standard biochemical autoantibodies. Diabetes Care 2012;35:1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decochez K, Truyen I, van der Auwera B, et al. Belgian Diabetes Registry Combined positivity for HLA DQ2/DQ8 and IA-2 antibodies defines population at high risk of developing type 1 diabetes. Diabetologia 2005;48:687–694 [DOI] [PubMed] [Google Scholar]
- 14.Verge CF, Gianani R, Kawasaki E, et al. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996;45:926–933 [DOI] [PubMed] [Google Scholar]
- 15.Kulmala P, Savola K, Reijonen H, et al. Childhood Diabetes in Finland Study Group Genetic markers, humoral autoimmunity, and prediction of type 1 diabetes in siblings of affected children. Diabetes 2000;49:48–58 [DOI] [PubMed] [Google Scholar]
- 16.Noble JA, Valdes AM, Varney MD, et al. Type 1 Diabetes Genetics Consortium HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes 2010;59:2972–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nejentsev S, Howson JM, Walker NM, et al. Wellcome Trust Case Control Consortium Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007;450:887–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipponen K, Gombos Z, Kiviniemi M, et al. Effect of HLA class I and class II alleles on progression from autoantibody positivity to overt type 1 diabetes in children with risk-associated class II genotypes. Diabetes 2010;59:3253–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tait BD, Colman PG, Morahan G, et al. HLA genes associated with autoimmunity and progression to disease in type 1 diabetes. Tissue Antigens 2003;61:146–153 [DOI] [PubMed] [Google Scholar]
- 20.Nakanishi K, Inoko H. Combination of HLA-A24, -DQA1*03, and -DR9 contributes to acute-onset and early complete beta-cell destruction in type 1 diabetes: longitudinal study of residual beta-cell function. Diabetes 2006;55:1862–1868 [DOI] [PubMed] [Google Scholar]
- 21.The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 1997;20:1183–1197 [DOI] [PubMed] [Google Scholar]
- 22.van der Auwera B, Schuit F, Lyaruu I, et al. Genetic susceptibility for insulin-dependent diabetes mellitus in Caucasians revisited: the importance of diabetes registries in disclosing interactions between HLA-DQ- and insulin gene-linked risk. Belgian Diabetes Registry. J Clin Endocrinol Metab 1995;80:2567–2573 [DOI] [PubMed] [Google Scholar]
- 23.Williams F, Meenagh A, Maxwell AP, Middleton D. Allele resolution of HLA-A using oligonucleotide probes in a two-stage typing strategy. Tissue Antigens 1999;54:59–68 [DOI] [PubMed] [Google Scholar]
- 24.Krolewski AS, Warram JH, Rand LI, Kahn CR. Epidemiologic approach to the etiology of type I diabetes mellitus and its complications. N Engl J Med 1987;317:1390–1398 [DOI] [PubMed] [Google Scholar]
- 25.Kronenberg D, Knight RR, Estorninho M, et al. Circulating preproinsulin signal peptide-specific CD8 T cells restricted by the susceptibility molecule HLA-A24 are expanded at onset of type 1 diabetes and kill β-cells. Diabetes 2012;61:1752–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.