

Abstract
Type 2 diabetes is a strong risk factor for stroke. Linagliptin is a dipeptidyl peptidase-4 (DPP-4) inhibitor in clinical use against type 2 diabetes. The aim of this study was to determine the potential antistroke efficacy of linagliptin in type 2 diabetic mice. To understand whether efficacy was mediated by glycemia regulation, a comparison with the sulfonylurea glimepiride was done. To determine whether linagliptin-mediated efficacy was dependent on a diabetic background, experiments in nondiabetic mice were performed. Type 2 diabetes was induced by feeding the mice a high-fat diet for 32 weeks. Mice were treated with linagliptin/glimepiride for 7 weeks. Stroke was induced at 4 weeks into the treatment by transient middle cerebral artery occlusion. Blood DPP-4 activity, glucagon-like peptide-1 (GLP-1) levels, glucose, body weight, and food intake were assessed throughout the experiments. Ischemic brain damage was measured by determining stroke volume and by stereologic quantifications of surviving neurons in the striatum/cortex. We show pronounced antistroke efficacy of linagliptin in type 2 diabetic and normal mice, whereas glimepiride proved efficacious against stroke in normal mice only. These results indicate a linagliptin-mediated neuroprotection that is glucose-independent and likely involves GLP-1. The findings may provide an impetus for the development of DPP-4 inhibitors for the prevention and treatment of stroke in diabetic patients.
Type 2 diabetes is a strong risk factor for severe stroke. In addition, stroke patients with type 2 diabetes show higher stroke recurrence and mortality compared with nondiabetic stroke patients (1–4). Finally, a prediabetic state with impaired glucose tolerance is often detected in stroke patients after hospital admission, and such patients generally exhibit a poor prognosis (5,6).
Glucagon-like peptide-1 receptor (GLP-1R) agonists are novel treatments in clinical use against type 2 diabetes (7). They specifically bind G-protein–coupled GLP-1R, enhancing insulin secretion and decreasing glucagon production in a glucose-dependent manner (8). Besides its glucoregulatory action, the activation of GLP-1R by the specific ligand exendin-4 is efficacious against stroke in diabetic and nondiabetic animal models (9–13). In addition, GLP-1R activation by exendin-4 has proven beneficial in other animal models for neurodegenerative diseases such as Parkinson’s (14–16), Alzheimer’s (17–19), and Huntington’s (20). Finally, anti-inflammatory (15,21) and neurogenic (14,22,23) actions mediated by GLP-1R activation have been recently reported. Whether all effects of GLP-1 and its mimetics are mediated by the known GLP-1R is not yet completely clear because GLP-1R–independent activation pathways have only recently been reported (24).
In addition to GLP-1R agonists, GLP-1R activation can also be achieved through the prolongation of the short half-life of the endogenous GLP-1 by inhibition of the enzyme dipeptidyl peptidase-4 (DPP-4) (25). Upon food ingestion, intestinal endocrine L cells secrete GLP-1. However, GLP-1 is rapidly degraded by the enzyme DPP-4, which proteolytically removes two amino acids from the N-terminal end of GLP-1, thereby abolishing its interaction with GLP-1R. Thus, GLP-1 as such has no clinical use. This limitation has been overcome by the development of specific DPP-4 inhibitors (26). In addition to GLP-1, DPP-4 has many other substrates, including peptides with potential neurotrophic or neuroprotective effects (27).
Linagliptin is a recently approved DPP-4 inhibitor for the treatment of type 2 diabetes in monotherapy or combined with other antidiabetic drugs (28). Furthermore, some studies have suggested beneficial effects of linagliptin on secondary cardiovascular end points such as stroke (29,30). The aim of this study was to determine the potential efficacy of linagliptin against stroke in diabetic and normal mice by using a drug administration paradigm and a dose that mimics a type 2 diabetic and obese patient receiving DPP-4 inhibitor therapy. As a glycemic comparator, we used the sulfonylurea glimepiride.
RESEARCH DESIGN AND METHODS
Animals and experimental groups.
The stroke experiments used 44 male C57Bl mice. In the first set of experiments, 21 8-week-old mice were exposed to a high-fat diet (HFD; Research Diets, Inc., New Brunswick, NJ) for 32 weeks (Fig. 1). Body weight was measured every fifth week. The intraperitoneal glucose tolerance test (IPGTT) and IP insulin tolerance test (IPinsTT) were carried out before and 12 weeks after the HFD treatment. When IPGTT and IPinsTT verified the animals’ diabetic state, the drug treatment was not started for an additional 13 weeks to mimic the clinical situation of an overtly diabetic patient who later suffers a stroke. We thus wanted to allow for metabolic toxicity of hyperglycemia and other diabetes manifestations to affect the body and the central nervous system.
FIG. 1.
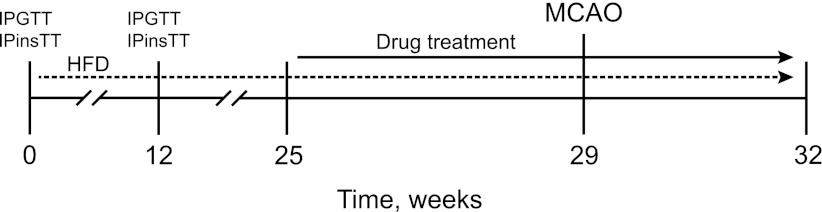
Experimental design and drug-treatment paradigm.
Before the start of the linagliptin (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany) and glimepiride (Sigma Aldrich, Stockholm, Sweden) treatments at week 25, baseline fasting blood glucose concentrations were measured and the animals assigned to the different treatment groups so that mean blood glucose values were equalized. The treatment groups thus created were tested for normality using the Shapiro-Wilk normality test. Starting from week 25, all HFD-fed mice received oral administration of 10 mg/kg/body weight (bw) linagliptin daily (n = 7), 2 mg/kg/bw glimepiride daily (n = 7), or vehicle (n = 7) for 4 weeks before being subjected to stroke at week 29 (Fig. 1). The glimepiride and linagliptin treatments were continued 3 weeks until the animals were killed (Fig. 1).
In a second set of experiments, 23 10-week-old mice fed a normal diet were treated, as mice in the first experiment, for 4 weeks with 10 mg/kg/bw linagliptin daily (n = 7), 2 mg/kg/bw glimepiride daily (n = 7), or vehicle (n = 9). After 4 weeks of drug treatment, all mice were subjected to stroke, and the treatments were continued for an additional 3 weeks until they were killed.
All experiments were conducted according to the “Guide for the Care and Use of Laboratory Animals” published by U.S. National Institutes of Health (NIH publication #85–23, revised 1985) and approved by the regional ethics committee for animal experimentation.
IPGTT and IPinsTT.
IPGTT and IPinsTT were carried out before the HFD treatment began and at week 12 (Fig. 1). The mice were fasted for 5 h, and intraperitoneal injections of 3 g/kg/bw glucose or 1 unit/kg/bw insulin were given. Blood was drawn from the tail vein, and glycemia was measured using a One-Touch Ultra 2 glucometer (LifeScan, Milpitas, CA) immediately before (time 0) and at 5, 10, 30, 60, and 120 min after the injection.
Transient middle cerebral artery occlusion.
The intraluminal filament model of focal ischemia was used (31). All animals received linagliptin, glimepiride, or vehicle treatments 1 h before surgery. Anesthesia was induced by 3% isoflurane and continued during surgery with 1.5% isoflurane using a snout mask. Briefly, the carotid arteries on the left side were exposed, the external carotid was ligated, and temporary sutures were placed over the common carotid artery. Through a small incision in the external carotid artery, a 7-0 monofilament coated with silicone was advanced through the internal carotid artery until it blocked the origin of the middle cerebral artery. When the filament had been positioned, wounds were closed and anesthesia was discontinued. After 30 min of occlusion, the mice were anesthetized again, the filament was withdrawn, and the ligatures were removed from the common carotid artery. Body temperature was maintained between 36 and 38°C with a heat lamp during surgery and ischemia. The mice were transferred to a heated box where they regained wakefulness and were kept for 2 h. The surgeon performing the operation was blinded to the treatment groups.
Measurements of fasting and fed blood glucose levels.
Fasting blood glucose levels were measured after 4 weeks of drug treatment. To do so, animals were given linagliptin, glimepiride, or vehicle and fasted for 5 h. Fed blood glucose levels were measured 1 h after drug treatment immediately before middle cerebral artery occlusion (MCAO), under anesthesia. Blood was drawn from the tail vein, and glycemia was measured using the One-Touch Ultra 2 glucometer.
Measurements of DPP-4 activity and active GLP-1 levels.
To measure the DPP-4 enzymatic activity and levels of active GLP-1, all animals received linagliptin, glimepiride, or vehicle treatments, and blood was collected 1 h thereafter on the day they were killed. Plasma total active GLP-1 concentrations were determined by means of a GLP-1 assay kit (Meso Scale Discovery, Gaithersburg, MD). DPP-4 activity was detected, as recently published by our group (32).
Brain immunocytochemistry.
Animals were deeply anesthetized and perfused transcardially with 4% paraformaldehyde. The brains were extracted, postfixed in 4% paraformaldehyde overnight at 4°C, and submersed in 20% sucrose in phosphate buffer until they sank. A sliding microtome was used to cut 40-µm-thick coronal sections, which were stained as free-floating sections. The primary antibody anti-NeuN (1:100; Millipore Corp., Billerica, MA) was used to stain surviving neurons in striatum and cerebral cortex. Sections were incubated with the primary antibody for 36 h at 4°C in phosphate buffer containing 3% normal horse serum and 0.25% Triton-X. Primary antibody was detected using biotin-conjugated anti-mouse (Vector Laboratories, Burlingame, CA) secondary antibody (1:200). Sections were incubated with secondary antibody for 2 h at room temperature in phosphate buffer containing 3% normal horse serum and 0.25% Triton-X. For chromogenic visualization, avidin-biotin complex (ABC kit, Vector Laboratories) and diaminobenzidine were used. For GLP-1R/NeuN staining, the sections were first microwave-boiled in citrate buffer for 10 min and then incubated with NeuN (1:100; Millipore) and GLP-1R (1:1,000; Abcam, Cambridge, U.K.) antibodies at 4°C overnight. The primary antibodies were visualized using Alexa Fluor 488 and Alexa Fluor 594 conjugated secondary antibodies (1:200; Invitrogen, Paisley, U.K.) for 2 h at room temperature in phosphate buffer containing 3% normal donkey serum and 0.25% Triton-X. The sections were counterstained with DAPI.
Brain infarct volume and cell quantifications.
An investigator blinded to the experimental groups performed tissue damage quantification and cell counting. For tissue damage evaluation, the NeuN-labeled tissue sections were displayed live on the computer monitor, and the area of contralateral hemisphere and the area of the intact ipsilateral tissue were measured in every section containing stroke damage using NewCast software (Visiopharm, Hoersholm, Denmark). To compensate for the stroke-induced morphologic tissue changes, the infarct volume was calculated by subtracting the volume of remaining intact tissue in the ipsilateral hemisphere from the volume of the contralateral hemisphere. The stroke volume in linagliptin and glimepiride groups has been normalized to its own respective vehicle-treated group (HFD and normal).
Immunoreactive cells were counted using a computerized nonbiased setup for stereology, driven by NewCast software. The number of neurons was quantified using the optical fractionator method (33,34). Briefly, brain sections were displayed live on the computer monitor and the striatum and cortex delineated at low magnification. Quantifications were performed using an oil immersion lens (original magnification ×100) with a numeric aperture of 1.30. Ten evenly spaced sections in parallel-cut series through the entire striatum were included. Random sampling was carried out using the counting frame, which systematically was moved at predefined intervals so that ∼300 immunoreactive cells were counted. The total number of cells was estimated according to the optical fractionator formula (33,34).
Statistics.
Statistical analyses were performed using the Student unpaired t test or one-way ANOVA, followed by the Bonferroni post hoc test. Differences between groups were considered statistically significant when P < 0.05. Data are presented as means ± SEM.
RESULTS
HFD exposure leads to insulin resistance, glucose intolerance, and hyperglycemia.
After 25 weeks of HFD treatment, the mice exhibited ∼200% weight gain (Fig. 2A). As previously described (35), the HFD feeding led to insulin resistance, glucose intolerance, and hyperglycemia. The HFD-fed mice developed these metabolic derangements already after 12 weeks on this diet (Fig. 2B–D).
FIG. 2.

Metabolic phenotype of HFD feeding. A: Body weight gain after HFD treatment. B: IPGTT before and 12 weeks into the HFD. C: IPinsTT before and 12 weeks into the HFD. D: Fasted blood glucose levels before and 12 weeks into the HFD. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 for a chance difference vs. controls using the Student unpaired t test.
Linagliptin inhibits DPP-4 activity, raises blood GLP-1 levels, and regulates glycemia.
Seven weeks of linagliptin treatment in HFD-fed mice, as well as in normal mice, significantly inhibited DPP-4 activity (Fig. 3A and E), leading to a 20- to 40-fold increase of blood GLP-1 levels (Fig. 3B and F), whereas glimepiride had no effect on these parameters. The results also show that linagliptin and glimepiride treatment decreased fed and fasting blood glucose levels in HFD-fed mice (Fig. 3C and D), while—as expected—in normal diet–fed mice, only glimepiride reduced glycemia (both fed and fasting; Fig. 3G and H).
FIG. 3.
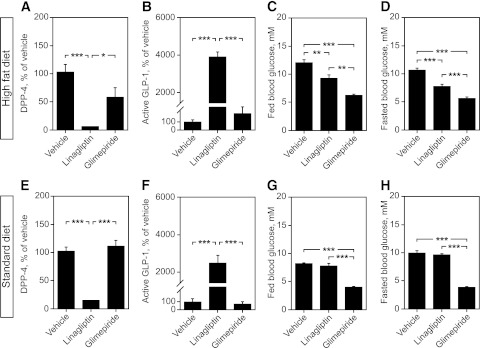
Effects of linagliptin and glimepiride on DPP-4 activity, GLP-1 levels, and blood glucose in HFD-fed vs. normal mice. HFD-fed mice: DPP-4 activity (A), GLP-1 levels (B), fed glucose levels (C), and fasted blood glucose levels (5 h) at 1 h after drug administration (D). Normal mice: DPP-4 activity (E), GLP-1 levels (F), fed glucose levels (G), and fasted blood glucose levels (5 h) at 1 h after drug administration (H). Bars represent means ± SEM. One-way ANOVA, followed by Bonferroni post hoc tests was used. *P < 0.05, **P < 0.01, and ***P < 0.001.
Linagliptin decreases ischemic brain damage.
To determine the potential antistroke efficacy mediated by linagliptin in HFD-treated mice, the brain infarct volume was assessed at 3 weeks after stroke. This measurement revealed that linagliptin treatment showed a noticeable, albeit not statistically significant, trend toward reduction of ischemic tissue damage, whereas glimepiride did not (Fig. 4A). We previously show that stereological counting of surviving neurons in stroke-damaged striatum and cortex provides a highly accurate method, considerably more sensitive than merely estimating infarction volume, to quantify the antistroke efficacy of candidate drugs (10). Thus, to further and in greater detail assess the neuroprotective effect of linagliptin and glimepiride after stroke, NeuN-positive neurons were quantified in both stroke-damaged striatum and cortex using the optical fractionator method (see research design and methods). Consistent with the volume measurements, no increase of neuronal survival in the cortex and/or striatum was evident in glimepiride-treated mice (Fig. 4B–D). In contrast, in linagliptin-treated animals the cortex contained ∼30% more surviving neurons, a statistically significant effect above both vehicle and glimepiride (Fig. 4C). This effect remained statistically significant when data from the cortex and striatum were pooled (Fig. 4D).
FIG. 4.

Neuroprotective effects of linagliptin and glimepiride treatments. A: Ischemic volume (mm3) after 30 min of MCAO in HFD-fed mice. Number of surviving neurons in stroke-damaged striatum (B), cortex (C), and striatum and cortex combined (D) in HFD-fed mice. E: Ischemic volume (mm3) after 30 min of MCAO in nondiabetic mice. Number of surviving neurons in stroke-damaged striatum (F), cortex (G), and striatum and cortex combined (H) in nondiabetic mice. The dashed lines in B, C, F, G, represent the average number of neurons in the brain areas of naïve animals (no stroke) where the neuronal quantification was performed. Bars represent means ± SEM. One-way ANOVA, followed by Bonferoni post hoc tests, was used. *P < 0.05, **P < 0.01. K: An illustration of typical brain damage in our stroke model. I and J: Photomicrographs of the area of the cortex illustrated in L on the contralateral, nondamaged, side of the brain, show normal neuronal density. Photomicrographs of the area (L) of the stroke-damaged cortex in HFD (M–O) and normal diet (P–R) illustrating the changes in neuronal density in vehicle, linagliptin, and glimepiride-treated mice, respectively. All photomicrographs have been enhanced with high-contrast monochromatic adjustment for better visual representation on small images.
To determine whether a diabetic background influences the linagliptin-mediated antistroke efficacy, the same type of experiment was performed in normal nondiabetic mice. The results show that linagliptin was also efficacious against stroke in nondiabetic mice and significantly reduced infarct volume (Fig. 4E), which was more pronounced than in the HFD-fed mice. The results also indicate that glimepiride showed a strong, albeit not statistically significant, trend toward reduction of ischemic tissue damage (Fig. 4E). When counting the total number of surviving NeuN-positive neurons in cortex and striatum, we observed a similar neuroprotective effect of linagliptin mainly limited to cerebral cortex (Fig. 3G and H), resembling the findings in diabetic conditions (Fig. 4C and D). In addition and contrarily to the findings in the diabetic mice (Fig. 4C and D), the results show that glimepiride induced a statistically significant neuroprotective effect, similar to that of linagliptin, in nondiabetic mice (Fig. 4G and H).
GLP-1R is expressed in mouse brain neurons.
To determine GLP-1R expression in the brain, we performed immunohistochemical staining in the cortex/striatum of HFD-treated mice without stroke. Double staining with GLP-1R/NeuN revealed that GLP-1R was expressed exclusively in neurons, with the strongest expression levels in cortical pyramidal neurons. Virtually no cell that was negative for NeuN was positive for GLP-1R (Fig. 5).
FIG. 5.
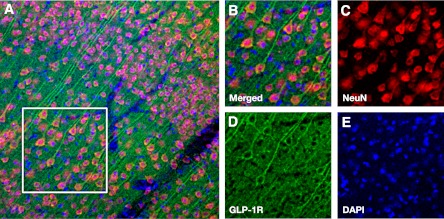
GLP-1R expression in the mouse cerebral cortex. A: Low magnification image of GLP-1R expression in cortex. B: High magnification image corresponding to the white borders square within panel A. Split channel images show immunoreactivity for NeuN (C), GLP-1R (D), and DAPI (E).
DISCUSSION
Most preclinical studies that aim to prove the antistroke efficacy of candidate drugs are performed in experimental settings bearing little—if any—resemblance to clinical reality, which is a possible reason for several neuroprotective drug failures (36,37). Potential causes of this lack of success include use of preclinical drug administration paradigms not achievable at the clinic level (e.g., drug administration before or very shortly after stroke, intracerebroventricular injections, and too-high doses of the candidate drugs (38)), efficacy experiments performed using animal models that lack common comorbidities of stroke patients, such as diabetes and hypertension (36), and finally, nearly all rodent stroke studies are performed in young animals, whereas most stroke patients are elderly (39).
Our goal in the current study was to determine the potential antistroke efficacy of a DPP-4 inhibitor therapy by mimicking the likely clinical scenario of an obese type 2 diabetic patient receiving this treatment suffering a stroke. To this end, we used middle-aged obese and diabetic mice and a drug administration route and dosages resembling a type 2 diabetic patient receiving chronic linagliptin treatment. Linagliptin is a recently approved DPP-4 inhibitor for the treatment of type 2 diabetes (28). Our results show a significant antistroke efficacy mediated by linagliptin treatment. To understand whether the neuroprotective efficacy by linagliptin was direct or rather secondary to its glycemic effects, we used two strategies: 1) We determined whether linagliptin showed antistroke efficacy also in nondiabetic mice, and 2) we performed a head-to-head comparison of linagliptin with the sulfonylurea glimepiride, which does not affect the incretin system.
By comparing the linagliptin antistroke effects in type 2 diabetic versus normal mice, our results show that linagliptin is strongly efficacious against stroke in both phenotypes. The effect was even stronger in nondiabetic mice. They also point to an effect that occurs mainly in the ischemic penumbra (peri-infarct cortex). Our stroke model results in ischemic damage that originates in the striatum and then spreads across the overlaying cortex, depending on the duration of the MCAO. In contrast to the striatal damage, the cortical damage in our model is typically limited to a general decrease in neuronal density, often without clearly defined borders from the primary stroke-damaged areas. This prevents the accurate estimation of ischemic damage by only using volume measurements. On the contrary, stereologic quantifications of neurons can accurately identify the differences within such infarct areas, and therefore, is less likely to overlook the neuroprotective effects of a potential treatment. A typical change in neuronal density in the cortex after MCAO and the effect of drug treatments is illustrated in Fig. 4I–R. Thus, the cortex in our model contains mostly ischemic penumbra, where a neuroprotective intervention can be more effective, and that is exactly where we found most of the antistroke effect mediated by linagliptin.
Glimepiride treatment showed a stronger effect than linagliptin in decreasing glycemia in type 2 diabetic and nondiabetic mice. As expected in the latter, no changes in blood glucose levels were observed after linagliptin treatment. Despite this, linagliptin treatment led to decreased DPP-4 activity, increased levels of blood GLP-1, and neuroprotection. Collectively, our results strongly suggest that the neuroprotective effect by linagliptin is unrelated to its glycemic actions. Because it mimicked the neuroprotective effects by the GLP-1R agonist exendin-4 previously reported by us and others (as noted earlier), this likely occurs by the increased GLP-1 levels observed. This is further supported by the fact that GLP-1 has been shown to pass the blood–brain barrier (40), whereas linagliptin does not (41). Our results also indicate the linagliptin-mediated neuroprotection occurs directly at the neuronal level because we found GLP-1R expression exclusively in neurons, with the strongest expression (based on immunohistochemistry) in cortical pyramidal neurons (Fig. 5). However, other peptides and substrates of DPP-4 (27,42) with reported neuroprotective and neurogenic actions, such as pituitary adenylate cyclase-activating polypeptide (43), glucose-dependent insulinotropic peptide (44), and stromal cell-derived factor 1α (45) may also be involved in the neuroprotective action mediated by linagliptin.
The results obtained by comparing linagliptin and glimepiride are intriguing because linagliptin was efficacious against stroke in both nondiabetic and diabetic mice, whereas glimepiride was efficacious only in nondiabetic mice. Because insulin has been suggested to have direct (nonglycemic) neuroprotective effects in the brain (46), we hypothesize that glimepiride-mediated efficacy against stroke in nondiabetic mice results from a direct neurotrophic effect mediated by increased insulin secretion and that this effect cannot be fully achieved in diabetic mice. Indeed, a nutritional regimen based on an HFD has been shown to render the brain insulin-resistant (47,48), thus potentially decreasing the neuroprotective actions mediated by insulin against stroke (46). In support of this hypothesis are also the inconclusive results from clinical trials aimed at assessing the role of tight glucose control against stroke in type 2 diabetes by using insulin as well as the sulfonylurea glimepiride (3,49). In line with our observed effects of linagliptin and glimepiride in diabetic animals are the recently observed results from a phase 3 trial in type 2 diabetic patients showing reduced incidences of stroke in linagliptin- versus glimepiride-treated patients (30).
The results obtained in our study have two implications of potential clinical relevance: First, they could be pertinent to type 2 diabetic patients receiving chronic linagliptin treatment. DPP-4 inhibition in these patients could decrease the risk of developing severe brain damage after a stroke while at the same time provide glycemic control without hypoglycemic side effects (6).
Second, given the glucose-independent effects of linagliptin, they advocate the use of DPP-4 inhibition as secondary prevention in nondiabetic and type 2 diabetic patients to minimize the damaging effects of recurrent stroke. Individuals who suffer a stroke or transient ischemic attack, in particular diabetic subjects, are at very high risk for another cardiovascular event (50). Thus, these patients could be prescribed DPP-4 inhibition therapy (safe and with minimal side effects) (28) aiming at reducing these types of complications. To test the feasibility of this hypothesis, we encourage preclinical and also clinical efforts in future work.
In conclusion, by using an experimental paradigm applicable to the clinical situation, we report the efficacy of linagliptin against stroke that is essentially glucose-independent and likely involves GLP-1. Furthermore, we demonstrate that linagliptin mediates neuroprotection in both type 2 diabetic and normal mice. Finally, we show significant differences between the linagliptin and glimepiride neuroprotective effects in normal versus diabetic background underlining the importance of performing this type of study in view of designing clinically suitable strategies. We believe that these findings provide an impetus for the further development of incretin-based drugs for the prevention and treatment of stroke in both diabetic and nondiabetic high-risk patients.
ACKNOWLEDGMENTS
Financial support was provided by Boehringer Ingelheim Pharma GmbH & Co. KG; by the regional agreement on medical training and clinical research (A.L.F.) between Stockholm County Council and the Karolinska Institutet; by grants from AFA Insurance, Diabetes Research & Wellness Foundation; a European Foundation for the Study of Diabetes/sanofi-aventis grant; by Magnus Bergvalls Stiftelse, Fredrik and Ingrid Thuring’s Foundation, Axel and Signe Lagerman’s Donation Foundation, Loo and Hans Osterman’s Foundation, Stohne’s stiftelse, Åhlén-stiftelsen, and STROKE-Riksförbundet stiftelser och fonder; and by Karolinska Institutet.
Å.S. has received research grants, consultancy fees, lecture honoraria, and fees for expert testimony from Eli Lilly, Novo Nordisk, Merck, Boehringer Ingelheim, AstraZeneca, Novartis, and sanofi-aventis, and is on the national/Nordic/European/global advisory boards of Eli Lilly, Merck, Boehringer Ingelheim, AstraZeneca, sanofi-aventis, and Novartis. T.K. is an employee of Boehringer Ingelheim Pharma GmbH & Co. No other potential conflicts of interest relevant to this article were reported.
V.D. designed and performed the stroke experiments, part of the immunohistochemistry studies, and stereology analysis; acquired and processed images and figures; contributed to discussion; and wrote the manuscript. H.O. planned and performed bioactivity studies and contributed to discussion. A.O. performed NeuN/GLP-1R immunohistological staining and acquired and processed images. E.D. performed part of NeuN staining and quantification. P.W. performed bioactivity studies and drug administrations. T.N. provided expertise in GLP-1R detection and contributed to discussion. T.K. conceived the research plan, provided expertise in DPP-4 inhibitors and GLP-1R, coordinated GLP-1 and DPP-4 inhibition activity assays, contributed to discussion, and edited the manuscript. Å.S. conceived the hypothesis and the research plan, provided expertise in diabetes and the HFD mice, contributed to discussion, and edited the manuscript. C.P. conceived, designed, and coordinated the research plan, contributed to discussion and to the stroke experiments, and wrote and edited the manuscript. V.D. and C.P. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in poster form at the 72nd Scientific Sessions of the American Diabetes Association, Philadelphia, Pennsylvania, 8–12 June 2012.
The authors thank Richelle Fall and Diana Rydholm (Södersjukhuset AB) for skilled animal technical assistance, Dr. Hans Pettersson and Lina Benson (Karolinska Institutet) for help with statistical analyses, and Jeannette Lundblad Magnusson (Södersjukhuset AB) and Julia Dennenmoser (Boehringer-Ingelheim) for laboratory technical assistance.
Footnotes
See accompanying commentary, p. 1029.
REFERENCES
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414:782–787 [DOI] [PubMed] [Google Scholar]
- 2.Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003;348:383–393 [DOI] [PubMed] [Google Scholar]
- 3.Sander D, Kearney MT. Reducing the risk of stroke in type 2 diabetes: pathophysiological and therapeutic perspectives. J Neurol 2009;256:1603–1619 [DOI] [PubMed] [Google Scholar]
- 4.Harmsen P, Lappas G, Rosengren A, Wilhelmsen L. Long-term risk factors for stroke: twenty-eight years of follow-up of 7457 middle-aged men in Göteborg, Sweden. Stroke 2006;37:1663–1667 [DOI] [PubMed] [Google Scholar]
- 5.Haratz S, Tanne D. Diabetes, hyperglycemia and the management of cerebrovascular disease. Curr Opin Neurol 2011;24:81–88 [DOI] [PubMed] [Google Scholar]
- 6.Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol 2012;11:261–271 [DOI] [PubMed] [Google Scholar]
- 7.Perry T, Greig NH. The glucagon-like peptides: a double-edged therapeutic sword? Trends Pharmacol Sci 2003;24:377–383 [DOI] [PubMed] [Google Scholar]
- 8.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006;368:1696–1705 [DOI] [PubMed] [Google Scholar]
- 9.Briyal S, Gulati K, Gulati A. Repeated administration of exendin-4 reduces focal cerebral ischemia-induced infarction in rats. Brain Res 2012;1427:23–34 [DOI] [PubMed] [Google Scholar]
- 10.Darsalia V, Mansouri S, Ortsäter H, et al. Glucagon-like peptide-1 receptor activation reduces ischaemic brain damage following stroke in Type 2 diabetic rats. Clin Sci (Lond) 2012;122:473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CH, Yan B, Yoo KY, et al. Ischemia-induced changes in glucagon-like peptide-1 receptor and neuroprotective effect of its agonist, exendin-4, in experimental transient cerebral ischemia. J Neurosci Res 2011;89:1103–1113 [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Perry T, Kindy MS, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A 2009;106:1285–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teramoto S, Miyamoto N, Yatomi K, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, provides neuroprotection in mice transient focal cerebral ischemia. J Cereb Blood Flow Metab 2011;31:1696–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertilsson G, Patrone C, Zachrisson O, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res 2008;86:326–338 [DOI] [PubMed] [Google Scholar]
- 15.Kim S, Moon M, Park S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J Endocrinol 2009;202:431–439 [DOI] [PubMed] [Google Scholar]
- 16.Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation 2008;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry T, Lahiri DK, Sambamurti K, et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res 2003;72:603–612 [DOI] [PubMed] [Google Scholar]
- 18.Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J Clin Invest 2012;122:1339–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holscher C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Patents CNS Drug Discov 2010;5:109–117 [DOI] [PubMed] [Google Scholar]
- 20.Martin B, Golden E, Carlson OD, et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes 2009;58:318–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pugazhenthi U, Velmurugan K, Tran A, Mahaffey G, Pugazhenthi S. Anti-inflammatory action of exendin-4 in human islets is enhanced by phosphodiesterase inhibitors: potential therapeutic benefits in diabetic patients. Diabetologia 2010;53:2357–2368 [DOI] [PubMed] [Google Scholar]
- 22.Hunter K, Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci 2012;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isacson R, Nielsen E, Dannaeus K, et al. The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. Eur J Pharmacol 2011;650:249–255 [DOI] [PubMed] [Google Scholar]
- 24.Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008;117:2340–2350 [DOI] [PubMed] [Google Scholar]
- 25.Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 1993;214:829–835 [DOI] [PubMed] [Google Scholar]
- 26.Deacon CF. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes Obes Metab 2011;13:7–18 [DOI] [PubMed] [Google Scholar]
- 27.Ahrén B, Hughes TE. Inhibition of dipeptidyl peptidase-4 augments insulin secretion in response to exogenously administered glucagon-like peptide-1, glucose-dependent insulinotropic polypeptide, pituitary adenylate cyclase-activating polypeptide, and gastrin-releasing peptide in mice. Endocrinology 2005;146:2055–2059 [DOI] [PubMed] [Google Scholar]
- 28.Barnett AH. Linagliptin: a novel dipeptidyl peptidase 4 inhibitor with a unique place in therapy. Adv Ther 2011;28:447–459 [DOI] [PubMed] [Google Scholar]
- 29.Johansen OE, Neubacher D, von Eynatten M, Patel S, Woerle HJ. Cardiovascular safety with linagliptin in patients with type 2 diabetes mellitus: a pre-specified, prospective, and adjudicated meta-analysis of a phase 3 programme. Cardiovasc Diabetol 2012;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallwitz B, Rosenstock J, Rauch T, et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: a randomised, double-blind, non-inferiority trial. Lancet 2012;380:475–483 [DOI] [PubMed] [Google Scholar]
- 31.Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab 1996;16:605–611 [DOI] [PubMed] [Google Scholar]
- 32.Klein T, Niessen HG, Ittrich C, et al. Evaluation of body fat composition after linagliptin treatment in a rat model of diet-induced obesity: a magnetic resonance spectroscopy study in comparison with sibutramine. Diabetes Obes Metab 2012;14:1050–1053 [DOI] [PubMed] [Google Scholar]
- 33.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec 1991;231:482–497 [DOI] [PubMed] [Google Scholar]
- 34.West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 1999;22:51–61 [DOI] [PubMed] [Google Scholar]
- 35.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988;37:1163–1167 [DOI] [PubMed] [Google Scholar]
- 36.Sena E, van der Worp HB, Howells D, Macleod M. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci 2007;30:433–439 [DOI] [PubMed] [Google Scholar]
- 37.Legos JJ, Tuma RF, Barone FC. Pharmacological interventions for stroke: failures and future. Expert Opin Investig Drugs 2002;11:603–614 [DOI] [PubMed] [Google Scholar]
- 38.Wahlgren NG, Ahmed N. Neuroprotection in cerebral ischaemia: facts and fancies—the need for new approaches. Cerebrovasc Dis 2004;17(Suppl. 1):153–166 [DOI] [PubMed] [Google Scholar]
- 39.Marini C, Triggiani L, Cimini N, et al. Proportion of older people in the community as a predictor of increasing stroke incidence. Neuroepidemiology 2001;20:91–95 [DOI] [PubMed] [Google Scholar]
- 40.Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci 2002;18:7–14 [DOI] [PubMed] [Google Scholar]
- 41.Fuchs H, Binder R, Greischel A. Tissue distribution of the novel DPP-4 inhibitor BI 1356 is dominated by saturable binding to its target in rats. Biopharm Drug Dispos 2009;30:229–240 [DOI] [PubMed] [Google Scholar]
- 42.Jungraithmayr W, De Meester I, Matheeussen V, Baerts L, Arni S, Weder W. CD26/DPP-4 inhibition recruits regenerative stem cells via stromal cell-derived factor-1 and beneficially influences ischaemia-reperfusion injury in mouse lung transplantation. Eur J Cardiothorac Surg 2012;41:1166–1173. [DOI] [PubMed]
- 43.Reglodi D, Somogyvari-Vigh A, Vigh S, Kozicz T, Arimura A. Delayed systemic administration of PACAP38 is neuroprotective in transient middle cerebral artery occlusion in the rat. Stroke 2000;31:1411–1417 [DOI] [PubMed] [Google Scholar]
- 44.Figueiredo CP, Pamplona FA, Mazzuco TL, Aguiar AS, Jr, Walz R, Prediger RD. Role of the glucose-dependent insulinotropic polypeptide and its receptor in the central nervous system: therapeutic potential in neurological diseases. Behav Pharmacol 2010;21:394–408 [DOI] [PubMed] [Google Scholar]
- 45.Yoo J, Seo JJ, Eom JH, Hwang DY. Effects of stromal cell-derived factor 1α delivered at different phases of transient focal ischemia in rats. Neuroscience 2012;209:171–186 [DOI] [PubMed] [Google Scholar]
- 46.Auer RN. Insulin, blood glucose levels, and ischemic brain damage. Neurology 1998;51(Suppl. 3):S39–S43 [DOI] [PubMed] [Google Scholar]
- 47.Yue JT, Lam TK. Lipid sensing and insulin resistance in the brain. Cell Metab 2012;15:646–655 [DOI] [PubMed] [Google Scholar]
- 48.Kim B, Sullivan KA, Backus C, Feldman EL. Cortical neurons develop insulin resistance and blunted Akt signaling: a potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid Redox Signal 2011;14:1829–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Favilla CG, Mullen MT, Ali M, Higgins P, Kasner SE, Virtual International Stroke Trials Archive (VISTA) Collaboration Sulfonylurea use before stroke does not influence outcome. Stroke 2011;42:710–715 [DOI] [PubMed] [Google Scholar]
- 50.Sacco RL, Adams R, Albers G, et al. American Heart Association. American Stroke Association Council on Stroke. Council on Cardiovascular Radiology and Intervention. American Academy of Neurology Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Stroke 2006;37:577–617 [DOI] [PubMed] [Google Scholar]