

Abstract
In the search for the most efficacious antisense oligonucleotides (AOs) aimed at inducing SMN2 exon 7 inclusion, we systematically assessed three AOs, PMO25 (−10, −34), PMO18 (−10, −27), and PMO20 (−10, −29), complementary to the SMN2 intron 7 splicing silencer (ISS-N1). PMO25 was the most efficacious in augmenting exon 7 inclusion in vitro in spinal muscular atrophy (SMA) patient fibroblasts and in vitro splicing assays. PMO25 and PMO18 were compared further in a mouse model of severe SMA. After a single intracerebroventricular (ICV) injection in neonatal mice, PMO25 increased the life span of severe SMA mice up to 30-fold, with average survival greater by 3-fold compared with PMO18 at a dose of 20 μg/g and 2-fold at 40 μg/g. Exon 7 inclusion was increased in the CNS but not in peripheral tissues. Systemic delivery of PMO25 at birth achieved a similar outcome and produced increased exon 7 inclusion both in the CNS and peripherally. Systemic administration of a 10-μg/g concentration of PMO25 conjugated to an octaguanidine dendrimer (VMO25) increased the life span only 2-fold in neonatal type I SMA mice, although it prevented tail necrosis in mild SMA mice. Higher doses and ICV injection of VMO25 were associated with toxicity. We conclude that (1) the 25-mer AO is more efficient than the 18-mer and 20-mer in modifying SMN2 splicing in vitro; (2) it is more efficient in prolonging survival in SMA mice; and (3) naked Morpholino oligomers are more efficient and safer than the Vivo-Morpholino and have potential for future SMA clinical applications.
Zhou and colleagues systematically examine the efficacy of three different antisense oligonucleotides (AOs) with a morpholino backbone targeting the ISS-N1 sequence of SMN. Of the three AOs examined, they find that a single intracerebral ventricular injection of the 25-mer AO PMO25 increases the life span of severe spinal muscular atrophy (SMA) mice up to 30-fold.
Introduction
Spinal muscular atrophy (SMA) is a recessively inherited neuromuscular disease characterized by the selective loss of spinal motor neurons and paralysis of skeletal muscle in the trunk and limbs. The carrier frequency in the general population is 1 in 35 and the prevalence is 1 in 6000 live births. It is the most common genetic cause of infant mortality (Pearn, 1980; Feldkotter et al., 2002). The genetic defect in patients with SMA is the loss of survival of motor neuron (SMN) protein, caused in 95% of cases by homozygous deletions of the survival of motor neuron 1 (SMN1) gene and, rarely, by missense mutations (Lefebvre et al., 1995; Feldkotter et al., 2002; Monani, 2005). A neighboring gene, SMN2, is intact in all patients with SMA. SMN2 copy number is polymorphic in the general population, and in SMA, there is an inverse correlation between the number of copies of SMN2 and the severity of the disease. Patients with severe disease carry one or two copies of SMN2, whereas patients with milder disease carry three or more copies (McAndrew et al., 1997; Taylor et al., 1998; Feldkotter et al., 2002). The SMN2 gene cannot fully compensate for the loss of the SMN1 gene, as only 10% of its transcripts correctly splice exon 7, whereas in SMN1 the exon is constitutive and its inclusion is essential for the production of a functional SMN protein (Lorson et al., 1999; Monani et al., 1999; Cartegni and Krainer, 2002; Kashima and Manley, 2003). As SMN2 is intact in all patients with SMA, it represents an attractive therapeutic target.
SMA is currently incurable. However, significant progress in the development of therapeutic strategies has been achieved. These include the following: (1) introducing the exogenous SMN1 gene by viral vectors (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011); (2) increasing the SMN2 transcript and SMN protein by small-molecule drugs, such as histone deacetylase (HDAC) inhibitors (Kernochan et al., 2005; Avila et al., 2007; Garbes et al., 2009; Hauke et al., 2009); (3) transplantation of motor neuron stem cells (Corti et al., 2010); and (4) redirecting SMN2 splicing and increasing the full-length SMN2 transcript by antisense oligonucleotides (AOs) (Skordis et al., 2003; Hua et al., 2007, 2008, 2010, 2011; Baughan et al., 2009; Williams et al., 2009; Owen et al., 2011; Passini et al., 2011; Osman et al., 2012; Porensky et al., 2012).
The application of AOs that modify pre-messenger RNA splicing has great potential for treating genetic diseases. The strategy used to redirect splicing for therapeutic purpose involves the use of AOs complementary to splice motifs or enhancer sequences, including exonic and intronic splicing enhancers (ESEs/ISEs) (Dominski and Kole, 1993; Dunckley et al., 1998; Aartsma-Rus et al., 2010). One approach is to block the binding of essential splicing factors at a specific exon, causing it to be skipped from the mature pre-mRNA. This strategy has been proved to be effective, and promising results have emerged from clinical trials of AOs that promote exon skipping in Duchenne muscular dystrophy (DMD) (van Deutekom et al., 2007; Kinali et al., 2009; Cirak et al., 2011; Goemans et al., 2011). In addition to enhancer sequences, silencer sequences can also be targeted by AOs, such as exonic and intronic splicing silencers (ESSs/ISSs), which are binding sites for splicing repressors (Singh et al., 2006). These have become important targets for the treatment of SMA.
Alternative splicing of the SMN2 gene transcript is due to a C>T single-nucleotide substitution in exon 7 that, without altering the amino acid encoded, disrupts an ESE in that exon and causes it to be excluded from the mature SMN2 transcript during splicing (Cartegni and Krainer, 2002). Two strategies have emerged for stimulating the inclusion of exon 7: (1) the use of AOs to block silencer motifs (ESSs/ISSs) or (2) the use of bifunctional AOs to provide trans-acting ESEs in exon 7. The bifunctional AOs were designed with one domain that was intended to anneal to the target exon and a second (tail) domain that contained sequences (i.e., the GGA motif) to which activator proteins, such as the serine/arginine (SR) proteins, would bind. We previously demonstrated that one such AO stimulated the splicing of SMN2 exon 7 in nuclear extracts of a cell-free model and that it stimulated both splicing and SMN protein expression in fibroblasts derived from patients with SMA. This method was termed “targeted oligonucleotide enhancers of splicing” (TOES) (Skordis et al., 2003; Owen et al., 2011). A similar strategy, exon-specific silencing enhancement by small chimeric effectors (ESSENCE), which comprises a minimal synthetic RS domain to emulate the function of SR proteins, has also shown efficacy in redirecting exon 7 spicing in vitro (Cartegni and Krainer, 2003).
In the SMN2 gene, a few regulatory elements have been shown to modulate the alternative splicing of exon 7. These include element 1 in intron 6, an SF2/ASF (alternative splicing factor/splicing factor-2) or hnRNP (heterogeneous nuclear ribonucleoprotein) A1-binding site in exon 7 (the site containing the C>T substitution), the Tra2-β1-binding site in exon 7, the 3′ cluster at the 3′ end of exon 7, the splicing silencer N1 in intron 7 (ISS-N1), and hnRNP A1-binding sites in intron 7 (Fig. 1) (Hofmann et al., 2000; Lim and Hertel, 2001; Cartegni and Krainer, 2002; Miyajima et al., 2002; Kashima and Manley, 2003; Singh et al., 2004, 2006; Kashima et al., 2007). Some AOs complementary to these regulatory sites, including conventional and bifunctional AOs, have been designed and validated in vitro and in vivo, as shown in Fig. 1. Among them, AOs targeting the ISS-N1 element in intron 7 have been intensively investigated and found to be more efficacious than AOs targeting other elements when evaluated in various SMA mouse models (Baughan et al., 2009; Williams et al., 2009; Hua et al., 2010, 2011; Passini et al., 2011; Porensky et al., 2012).
FIG. 1.
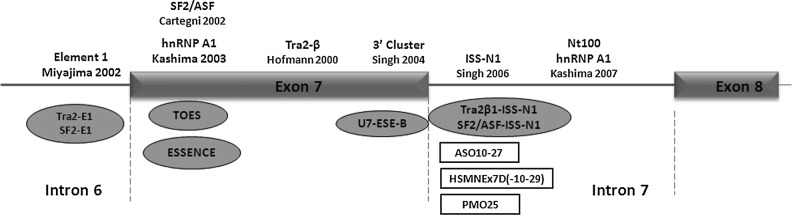
Schematic representation of SMN2 exon 7 splicing regulatory elements. The functional elements regulating exon 7 splicing in SMN2 are depicted along the top. Antisense oligonucleotides (AOs) targeting these regulatory elements are listed at the bottom, including bifunctional AOs (gray ovals) and conventional AOs (open rectangles).
AOs are designed to bind to specific nucleotide sequences by Watson–Crick base paring. This reduces the risk of off-target side effects when compared with small-molecule drugs. The efficiency of a splice-switching AO depends on many factors: the particular sequence motifs targeted, their length, base composition and the backbone chemical modification used (Harding et al., 2007; Owen et al., 2011). In this study, we have investigated the efficacy of three Morpholino AOs of various lengths complementary to ISS-N1. The in vitro efficacy was validated in an in vitro splicing assay and in cultured SMA patient fibroblasts. The in vivo validation was conducted in a standard SMA mouse model that produces both mice with severe type I SMA and mice with mild SMA (Hsieh-Li et al., 2000; Hua et al., 2010, 2011). Single intracerebroventricular (ICV) injections of phosphoroamidate morpholino oligomers (PMOs) were administered to neonatal SMA mice. Systemic delivery in neonatal SMA mice was also investigated to exploit the early incomplete closure of the blood–brain barrier (BBB) and to determine the efficacy of the combined targeting of both the peripheral tissues and motor neurons. Last, we investigated the outcomes in transgenic SMA mice of adding an octaguanidine dendrimer to the PMO backbone. Our comprehensive analyses identified a promising AO sequence that should be considered in future experimental studies for SMA.
Materials and Methods
SMA mouse model with human SMN2 transgene
All mouse experiments were performed according to protocols approved by University College London (London, UK) Biological Services and U.K. Home Office under the Animals (Scientific Procedures) Act 1986. The initial breeding strain of the SMA transgenic mice, FVB.Cg-Tg(SMN2)2Hung Smn1tm1Hung/J, originally created by Hsieh-Li and colleagues (2000), was purchased from Jackson Laboratory (TJL005058; Jackson Laboratory, Bar Harbor, ME). Various breeding schemes were followed as previously described (Gogliotti et al., 2009).
Morpholino antisense oligonucleotides
All Morpholinos and the Vivo-Morpholino were synthesized and purified by Gene Tools (Philomath, OR). They were dissolved to standard concentrations and stored according to the manufacturer's instructions.
Animal procedures
Newborn SMA mice on postnatal day (PND) 0 were cryoanesthetized, followed by either ICV injections or facial vein injections. Injections were performed with a glass capillary (10 μl) with a fine tip. For ICV injections, the glass capillary was inserted to an approximate depth of 1 mm, the injection site being 1 mm lateral and before the intersection of the coronal and sagittal cranial sutures (Passini et al., 2011; Porensky et al., 2012). Intravenous injections in newborn mice were performed via the facial vein on PND0 with the aid of a microscope. Intravenous injections in adult mice were performed via the tail vein. Other systemic deliveries used in this study included subcutaneous and intraperitoneal injections, using protocols in accordance with the Laboratory Animal Science Association (LASA) guidelines (www.lasa.co.uk).
In vitro splicing assay
Radiolabeled SMN2 transcript, prepared as described previously (Skordis et al., 2003; Owen et al., 2011), was annealed in silanized tubes to various concentrations of Morpholinos (PMO18, PMO20, and PMO25) at 30°C for 10 min in 5 mM Tris, 0.5 mM EDTA, pH 8. Splicing reactions were done as described previously (Skordis et al., 2003; Owen et al., 2011). Quantification of pre-mRNA and both mRNA products was done with Optiquant software (PerkinElmer, Waltham, MA), and intensities were adjusted to account for the number of radioactive guanosines in each molecular species.
Transfection of SMA fibroblasts
Skin fibroblast cells, derived from a patient with type III SMA who has a homozygous deletion of the SMN1 gene and three copies of the SMN2 gene, were cultured as previously described (Owen et al., 2011). Cells were plated into 30-mm diameter plates at a concentration of ∼2×105 cells per plate, which gave 90% confluence by the next day. PMOs were complexed with Lipofectamine 2000 (Invitrogen/Life Technologies, Carlsbad, CA) in Opti-MEM (Invitrogen/Life Technologies) according to the manufacturer's instructions, and added to cells. Cells were harvested 48 hr after transfection for RNA and protein extraction. All experiments were repeated at least three times for each PMO.
Reverse transcription PCR and real-time RT-PCR analyses
Total RNA was extracted from cultured fibroblasts or homogenized mouse tissues, using an RNeasy kit (Qiagen, Chatsworth, CA). Total RNA (500 ng) was used for first-strand cDNA synthesis, using a SuperScript III reverse transcription kit (Invitrogen/Life Technologies). Human SMN-specific primers (forward, 5′-CTC CCA TAT GTC CAG ATT CTC TT-3′; reverse, 5′-CTA CAA CAC CCT TCT CAC AG-3′) were used to amplify SMN2 and gave two PCR products, the full-length SMN2 (505 bp) and Δ7 SMN2 (451 bp). Quantitative real-time PCR was done with a MESA Blue qPCR kit (Eurogentec, Seraing, Belgium). Samples were incubated in a 25-μl reaction mix according to the manufacturer's instructions. The same cDNA products as described previously were used as templates for real-time PCR. Human-specific full-length SMN2 primers (forward, 5′-ATA CTG GCT ATT ATA TGG GTT TT-3′; reverse, 5′-TCC AGA TCT GTC TGA TCG TTT C-3′ [133 bp]) and human-specific SMN Δ7 primers [forward, 5′-TGG ACC ACC AAT AAT TCC CC-3′; reverse, 5′-ATG CCA GCA TTT CCA TAT AAT AGC C-3′ [125 bp]) were used. Quantitative real-time PCR was performed with an Applied Biosystems/Life Technologies (Carlsbad, CA) fast 7500 real-time PCR system as described previously (Owen et al., 2011). The program includes activation at 95°C for 5 min, 40 cycles of 95°C for 3 sec, and 60°C for 1 min. Quantification was based on concurrent standard curves produced from serial dilutions of cDNA from the fibroblasts of an untreated patient with SMA. The cycle at which the amount of fluorescence was above the threshold (Ct) was detected. The ratios of full-length SMN2 to Δ7 SMN2 of the treated samples were normalized, taking the ratio of the untreated sample, or saline control of mouse tissues, as 1.0.
Western blotting
Fibroblasts or mouse brain was lysed in buffer containing 75 mM Tris-HCl (pH 6.8) and 0.25% sodium dodecyl sulfate (SDS) supplemented with protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Protein concentration was analyzed with a bicinchoninic acid (BCA) kit (Pierce Biotechnology/Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's instructions. Five micrograms of total protein extracted from fibroblasts, or 50 μg of total protein extracted from mouse brain, was loaded into 10% NuPAGE Bis-Tris precast gels (Invitrogen/Life Technologies). SMN protein was blotted as previously described (Owen et al., 2011). The primary anti-SMN antibody used in SMA fibroblasts was BD Transduction Laboratories mouse anti-SMN monoclonal antibody (diluted 1:2000; BD Biosciences, San Jose, CA); the human-specific SMN antibody used in mouse brain was mouse anti-SMN 4F11 monoclonal antibody (diluted 1:1000; from C. Lorson, University of Missouri, Columbia, MO) (Mattis et al., 2008). β-Tubulin was used as protein loading control and was detected with mouse anti-β-tubulin monoclonal antibody (1:5000; Sigma-Aldrich, St. Louis, MO). Membranes were blocked overnight in 10% semiskimmed milk in phosphate-buffered saline (PBS)–0.1% Tween (PBST). After incubation for 1 hr at room temperature with various primary antibodies, membranes were washed in PBST, followed by administration of secondary horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody (GE Healthcare Life Sciences, Piscataway, NJ) at a dilution of 1:50,000 in PBST. Blots were developed with an enhanced chemiluminescence detection kit (GE Healthcare Life Sciences). Quantification of band intensity was analyzed with ImageJ software (Rasband, 1997–2012).
Statistical analysis
Student t tests were used to determine statistical significance. Data are displayed as means±standard error (means±SE). Kaplan–Meier survival curves were generated to analyze the survival data, followed by a log-rank test for statistical significance. GraphPad Prism 5.0 software (GraphPad, San Diego, CA) was used for statistical analysis and graph design.
Results
ISS-N1 is an intronic splicing silencer of 15 nucleotides located in the 5′ region of intron 7 in SMN1 and SMN2 genes (Singh et al., 2006). It has been shown previously that AOs complementary to ISS-N1 from intron nucleotide 10 (−10) onward produced efficient exon 7 inclusion and rescued SMA transgenic mice (Hua et al., 2010, 2011; Passini et al., 2011; Porensky et al., 2012). These AOs comprised an 18-mer based on 2′-O-methoxyethyl chemistry and a 20-mer phosphorodiamidate morpholino antisense oligonucleotide (PMO). To test whether length was a factor influencing their splice-switching ability, Morpholino AOs complementary to 18 (−10, −27; PMO18), 20 (−10, −29; PMO20), and 25 (−10, −34; PMO25) bases of ISS-N1 were tested, all of which extended from nucleotide 10 of the intron 7 (Fig. 2).
FIG. 2.

Diagrammatic representation of the positions of AOs targeting the ISS-N1 region. The sequence of exon 7 is shown in upper case and that of intron 7 in lower case. The sequence of ISS-N1 and the annealing sites for the 18-mer ASO-10–27, 20-mer HSMNEx7D, and 25-mer PMO25 are highlighted. Note: In the Morpholinos the uridine (U) is replaced by thymine (T).
Effects of PMOs on SMN2 alternative splicing in cell-free in vitro splicing assay
An SMN2 cell-free in vitro splicing assay was used to compare the efficiencies of PMO18, PMO20, and PMO25. The PMOs were incubated with the pre-mRNA substrate and then mixed with a splicing reaction mixture (Skordis et al., 2003; Owen et al., 2011). All three PMOs gave rise to substantial exon 7 inclusion at concentrations of 250 and 500 nM (Fig. 3A). This demonstrates that their primary effect is directly on pre-mRNA splicing rather than on other processes, such as RNA stability. When comparing the intensities of the bands representing exon 7 inclusion with the exon 7 exclusion product, PMO25 was more efficient at inducing exon 7 inclusion than PMO18 and PMO20 at 250 nM (p<0.0001; Fig. 3B).
FIG. 3.

PMO25 promotes exon 7 inclusion in vitro. (A) Cell-free in vitro splicing assay using radiolabeled SMN2 transcripts combined with PMO18, PMO20, or PMO25 at concentrations between 0 and 500 nM. (B) Graph showing ratio of SMN2 exon 7 inclusion to exclusion with increasing concentrations of PMOs. The relative ratio labeled on the y axis indicates that the readings have been normalized against the reading obtained for the no-PMO (0 nM) sample. (C) A representative reverse transcription-PCR image shows the effect of PMOs on SMN2 exon 7 inclusion in cultured SMA fibroblasts. Cells were treated at 100 and 500 nM in each PMO group. (D) Quantitative real-time PCR assay of the relative expression of full-length SMN2 transcript against the Δ7 SMN2 transcript in PMO-treated SMA fibroblasts. (E) Western blotting of SMN protein in PMO-treated SMA fibroblasts. (F) Semiquantitative analysis of SMN protein expression normalized to tubulin. *p<0.05; **p<0.01; ***p<0.001.
Comparative analysis of PMO18, PMO20, and PMO25 in SMA patient fibroblasts
PMO18, PMO20, and PMO25 were transfected into SMA type III patient fibroblasts at concentrations of 100 and 500 nM. Reverse transcription-PCR revealed clear differences in the splicing patterns of cells treated with the three PMOs (Fig. 3C). Exon 7 inclusion in PMO-treated fibroblasts was further quantified by measuring the ratio of full-length SMN2 transcript to Δ7 SMN2 transcript by quantitative real-time PCR (Owen et al., 2011). At 100 nM, there was no difference between PMO18 and PMO20, but a significant increase in exon 7 inclusion was identified in PMO25-treated compared with PMO18- and PMO20-treated fibroblasts (p=0.0033 and p=0.0022, respectively). At 500 nM, PMO25-treated fibroblasts showed the strongest exon 7 inclusion compared with both PMO18 and PMO20 (p<0.0001 and p=0.0003, respectively). The exon 7 inclusion induced by PMO25 was 2.6- and 1.8-fold greater than that induced with PMOs 18 and 20, respectively (Fig. 3D).
The restoration of SMN protein consequent to the increased exon 7 inclusion in transfected fibroblasts was examined by Western blotting. PMOs 18, 20, and 25 were transfected into SMA type III patient fibroblasts at concentrations of 500 and 1000 nM. At both concentrations, PMO25 gave rise to significantly more SMN protein than did PMOs 18 and 20 (p<0.0001 and p=0.0007 at 500 nM, p=0.0015 and p=0.0013 at 1000 nM, respectively), resulting in 2- and 1.6-fold, respectively, higher levels of protein expression (Fig. 3E and F).
SMN2 exon 7 inclusion and SMN protein expression induced by PMOs in SMA mice
Because PMO25 was significantly more effective than PMO18 and PMO20 in correcting SMN2 exon 7 splicing in vitro, whereas PMO18 and PMO20 showed a similar effect, the efficacy of PMO25 was then compared with PMO18 in an SMA mouse model.
Litters born from the breeding pairs of mice with genotype (SMN2)2+/+; smn–/– crossed with the genotype smn+/– have 50% wild-type “heterozygous” progeny with genotype (SMN2)2+/–; smn+/– and 50% severe type I SMA progeny with genotype (SMN2)2+/–; smn–/–. Newborn (PND0) mice, of both genotypes, were given a single ICV injection of PMO18 or PMO25 at 20 μg/g. Injected mice were genotyped to identify heterozygous and type I SMA mice. The heterozygotes [(SMN2)2+/–; smn+/–] were used in the analysis of SMN2 exon 7 inclusion and SMN protein by using human SMN2-specific primers and a human-specific SMN antibody. Brain and spinal cord tissues were collected at intervals of 10 days, starting from 10 days postinjection until 40 days. The ratio of human full-length SMN2 transcript to human Δ7 SMN2 transcript was measured by quantitative real-time PCR with primers specific for human SMN2 transcripts (Fig. 4A). Ten days after PMO administration, RNA isolated from brain and spinal cord showed prominent exon 7 inclusion in mice treated with both PMO18 and PMO25 (Fig. 4A). No significant difference in exon 7 inclusion was observed between the two PMOs at this time point. Twenty days after injection, exon 7 inclusion remained at high levels in brain and spinal cord from PMO25-treated mice, but it was dramatically reduced in PMO18-treated mice (Fig. 4A). Analyzed by quantitative real-time PCR, the ratio of human full-length SMN2 transcript to human Δ7 SMN2 transcript induced by PMO25 was more than 2-fold greater than that induced by PMO18 in both brain (p=0.0297) and spinal cord (p=0.0055; Fig. 4B). The effect on exon 7 inclusion in peripheral tissues was also measured after ICV injection in PMO18- and PMO25-treated heterozygous [(SMN2)2+/–; smn+/–] mice. Tibialis anterior (TA) muscle, heart, liver, and kidney were collected 10 days after injection (PND10). Real-time PCR showed that ICV injection produced no change in exon 7 inclusion in these peripheral tissues, either in PMO18- or PMO25-treated mice (data not shown).
FIG. 4.

Effects of PMO18 and PMO25 on exon 7 inclusion and human SMN protein expression in SMA transgenic mice. (A) Newborn SMA transgenic mice that carried two copies of the human SMN2 gene were administered, by intracerebroventricular (ICV) injection, PMO18 or PMO25 at 20 μg/g on postnatal day (PND) 01. Exon 7 inclusion in brain and spinal cord was analyzed by reverse transcription-PCR 10 and 20 days postinjection. Representative RT-PCR gel images are displayed. (B) Quantitative real-time PCR analyses of exon 7 inclusion in brain and spinal cord, 10 and 20 days after a single ICV injection of saline or either PMO18 or PMO25 at 20 μg/g on PND0. *p<0.05; **p<0.01. (C) Quantitative real-time PCR analyses of exon 7 inclusion in brain and spinal cord, examined from PND10 to PND40 at 10-day intervals, after a single ICV injection of PMO25 (20 μg/g) on PND0. (D) Newborn SMA transgenic mice that carried two copies of the human SMN2 gene were given PMO18 or PMO25 at 40 μg/g by ICV injection on PND0. Quantitative real-time PCR analyses of exon 7 inclusion in brain and spinal cord were performed 20 days later. *p<0.05. (E) Western blotting of human SMN protein expressed in brain from PMO-treated SMA transgenic mice. The expressed protein was probed by a human-specific SMN antibody in samples extracted from mice treated by ICV injection of PMO25 or PMO18 (20 μg/g) on PND0. (F) Semiquantitative analysis of human SMN protein expression normalized to tubulin. *p<0.05; **p<0.01; ***p<0.001.
To assess the persistence of the effect from a single administration of PMO25 (20 μg/g) in mouse brain and spinal cord, SMN2 exon 7 inclusion was measured at 10-day intervals until PND40. Three treated mice per time point were collected. The ratio of human full-length SMN2 transcript to human Δ7 SMN2 transcript declined on PND30 and nearly reached baseline levels on PND40 (Fig. 4C).
The dose was increased to 40 μg/g, again administered in a single ICV injection to newborn mice on PND0. Twenty days after injection, the ratio of human full-length SMN2 transcript to human Δ7 SMN2 transcript induced by PMO25 was about 2-fold greater than that induced by PMO18 in brain (p=0.0033), and 1.6-fold in spinal cord (p=0.0496; Fig. 4D).
The level of human SMN protein expression after the administration of PMOs was measured by Western blotting with a human-specific SMN antibody (Mattis et al., 2008). A significant increase in the level of expression was detected 10 days after PMO administration in the brains of mice treated with PMO18 and PMO25 at 20 μg/g. Expression was markedly higher in PMO25-treated mice than in PMO18-treated mice (p=0.0186; Fig. 4E and F).
The survival of severe SMA mice treated by single ICV injection of PMO25 or PMO18
Survival curve studies were performed on type I SMA mice [(SMN2)2+/–; smn–/–] given a single ICV injection on PND0 of either PMO25 (20 μg/g, n=9 or 40 μg/g, n=8) or PMO18 (20 μg/g, n=6 or 40 μg/g, n=6), or saline control (n=6). Mice were weighed daily and assessed for tail and ear necrosis and mortality. Mice injected with saline survived for an average of 9.5 days. However, mice treated with PMO18 (20 μg/g) survived for an average of 12 days, with a maximal life span of 25 days. Mice treated with PMO25 survived for an average of more than 43 days, the longest survival being 185 days (p=0.01 for the comparison of PMO25 and PMO18; Fig. 5A). Increasing the single ICV injection on PND0 to 40 μg/g further improved the average survival to 32 days with PMO18 and 85.5 days with PMO25, with the longest survival being 298 days (Fig. 5B). The survival advantage was significant for both PMO18- and PMO25-treated groups when compared with saline control mice (p<0.0001). There was also a significant difference between the PMO25 and PMO18 groups, in favor of PMO25 (p=0.0111).
FIG. 5.

Survival curves and weight gain of type I SMA mice treated with PMO18 and PMO25 by ICV injection. (A) Survival curves of type I SMA mice treated with a single ICV injection of PMO18 or PMO25 at 20 μg/g on PND0. (B) Survival curves of type I SMA mice treated with a single ICV injection of PMO18 or PMO25 at 40 μg/g on PND0. (C) Body weight gain up to 30 days in type I mice treated with PMOs at 20 μg/g. Less weight gain was observed in PMO18-treated mice, corresponding to their poor survival. (D) Body weight gain up to 30 days in type I mice treated with PMOs at 40 μg/g. Saline-injected type I mice and heterozygous control mice [(SMN2)2+/–; smn+/–] were used as body weight controls.
In addition to the prolongation of life span, a single ICV injection of PMOs into newborn type I mice had a significant effect on body weight (Fig. 5C and D). At 40 μg/g, mice treated with both PMO18 and PMO25 showed a steady increase in weight, which was similar to the gain in body mass induced by PMO25 at 20 μg/g. The low weight gain observed in mice given PMO18 at 20 μg/g might be related to their poor survival. All treated type I mice had short tails, with tail necrosis commencing between 60 and 80 days of age, leading to complete tail loss between 80 and 100 days. Ear necrosis started between 40 and 50 days. No difference in tail and ear necrosis was observed between PMO18- and PMO25-treated type I mice.
Efficacies of systemically delivered PMOs in type I SMA mice
The efficacies of delivery routes other than ICV injection were assessed in type I SMA mice. For the assessment of a single systemic application, type I mice were injected with PMO25 (40 μg/g) intravenously, via the facial vein, on PND0 (n=8); for repeat systemic deliveries, intravenous injection on PND0 was followed by a second intraperitoneal or subcutaneous injection of PMO25 (40 μg/g) on PND3 (n=6). Mice were then weighed daily and assessed for survival. The average survival of the single intravenous-injected group has not yet been determined, as five of eight mice are still alive at the time of submission of this manuscript (beyond 230 days). Nevertheless, the predicted average survival of the single intravenous-injected group, beyond 230 days, is longer than the average survival of the repeat systemic group, which was 93.5 days. This suggests that repeated injections do not confer an advantage to severe SMA mice. The average survival in the repeated systemically injected group is similar to the average 85-day survival of mice given a single 40-μg/g ICV injection (Fig. 6A).
FIG. 6.

Efficacies of various delivery routes in type I SMA mice. Type I SMA mice were administered PMO25 (40 μg/g) by a single ICV injection on PND0, or by single systemic delivery (via the facial vein) on PND0, or by repeat systemic deliveries on PND0 (via the facial vein) and PND3 (subcutaneously or intraperitoneally). (A) Survival curves of type I SMA mice treated with PMO25 via different delivery routes at 40 μg/g. (B) Body weight gain in type I mice treated with PMO25 at 40 μg/g by single ICV, single intravenous, or repeated systemic injections. (C) Representative reverse transcription-PCR gel images show exon 7 inclusion in CNS tissues (brain and spinal cord) and peripheral tissues (TA muscle, heart, liver, and kidney) 10 days after the repeat systemic administration. (D) Quantitative analyses of exon 7 inclusion by real-time PCR in CNS and peripheral tissues after systemic administrations. *p<0.05; **p<0.01; ***p<0.001.
The three different delivery routes showed a similar ascending trend of body weight gain over times. At about 50 days of age, the ICV-treated type I SMA mice (n=3) increased body weight to 77% of control heterozygotes [(SMN2)2+/–; smn+/–, n=5]; the single intravenous-injected type I SMA mice (n=6) gained body weight to 80% of heterozygous controls; and the repeat systemically injected type I SMA mice (n=3) gained 70% of the body weight of heterozygous controls. At 140 days of age, there were three type I mice still alive in the ICV 40-μg/g treated group, with an average body weight of 19.3 g, which was equivalent to 83% of the average body weight of heterozygous controls. The average body weight increased to 86% of the heterozygous controls at 160 days of age (Fig. 6B).
In addition, we examined exon 7 inclusion in the CNS and peripheral tissues in heterozygous [(SMN2)2+/–; smn+/–] mice after repeat systemic deliveries. Tissues from CNS (brain and spinal cord) and other organs (heart, TA muscle, liver, and kidney) were collected on PND10 after PND0 (intravenous) and PND3 (subcutaneous) systemic injections. The ratio of full-length SMN2 to Δ7 SMN2 was measured by SMN2-specific real-time PCR. Compared with the saline-treated control, there was an approximately 2-fold increase in exon 7 inclusion in brain (p=0.0014) and spinal cord (p=0.0003) after systemic injection of PMO25 at 40 μg/g on PND0. Similar increases were also observed in the liver (p=0.0129), but not in muscle, heart, and kidney (Fig. 6C and D). This suggests that PMO25 administered systemically to neonatal mice may reach the CNS because of the immaturity of the BBB barrier in very young mice. Our data imply that the correction of exon 7 inclusion in the CNS, induced by systemic PMO administration in the neonatal period, contributes in a major way to extending the life span of type I SMA mice.
Efficacy of Vivo-Morpholino in SMA mice
To investigate whether an octaguanidine moiety can improve the efficacy of the Morpholino AO in SMA mice, PMO25 was conjugated with a dendrimeric octaguanidine during synthesis. The product, 25-mer Vivo-Morpholino AO (VMO25), was administered to both severe type I and mild SMA mice.
Type I SMA mice were treated by an intravenous injection of VMO25 (7 μg/g) via the facial vein on PND0, followed by an intraperitoneal injection of the same dose on PND3. The average survival of VMO25-treated type I SMA mice (n=7) was 16 days, compared with 10 days in saline-treated type I mice (n=7; p=0.0039). Increasing the dose to 10 μg/g did not improve the efficacy. The survival of VMO25-treated type I mice was significantly lower than that of mice treated by systemic delivery of naked PMO25 at 40 μg/g, which was 93.5 days (n=6) (p=0.0085; Fig. 7A).
FIG. 7.

Efficacy of VMO25 in type I and mild SMA mice. (A) Survival curves of type I mice after systemic delivery of VMO25 or PMO25 on PND0. (B) Tail rescue by VMO25 in mild SMA mice. The genotype of mouse 1 was (SMN2)2+/+; smn+/–, used as wild-type control; the genotype of mice 2 and 3 was (SMN2)2+/+; smn–/–, that is, mild SMA mice. Single facial vein injections of saline into mice 1 and 2, and of VMO25 (10 μg/g) into mouse 3, were conducted on PND0. Images of the mice were captured at ages of 3 weeks, 4 weeks, 8 weeks, and 9 months. Tail necrosis in mouse 2 (arrowed) had clearly progressed with time. VMO25-treated mild SMA mouse 3 (asterisked) had a slightly shorter but normal-looking tail compared with wild-type mouse 1. (C) Effect of systemically delivered VMO25 in young adult mice on exon 7 inclusion in CNS and peripheral tissues. VMO25 (10 or 5 μg/g) was injected into young adult heterozygous SMA mice on PND21 via the tail vein. Brain, spinal cord, skeletal muscle (TA), heart, liver, and kidney were collected 10 days after the injection. Exon 7 inclusion in these tissues was analyzed by reverse transcription-PCR and quantified by real-time PCR. Representative reverse transcription-PCR pictures are displayed. (D) The ratio of full-length SMN2 transcript to Δ7 SMN2 transcript was quantified by real-time PCR (*p<0.05; ***p<0.001). (E) Survival curves of type I SMA mice treated with a single ICV injection of VMO25 at 4 or 2 μg/g.
The mild SMA mice [(SMN2)2+/+; smn–/–] carry four copies of the human SMN2 gene. These mice have a normal life span, body weight, and fertility, but nevertheless exhibit tail and ear necrosis (Hsieh-Li et al., 2000). The onset of tail necrosis typically starts between 2 and 3 weeks of age, leading to complete loss of the tail by 8 weeks. Mice were injected with saline or VMO25 at 10 μg/g via the facial vein on PND0. In saline-treated mice, tail necrosis started after 3 weeks, part of the tail was lost by 4 weeks, and the tail had nearly completely disappeared by 8 weeks of age (Fig. 7B). In contrast, the tails of mild SMA mice treated with VMO25 appeared normal at 9 months of age, although they were slightly short in comparison with those of wild-type mice (Fig. 7B). Slow progress of tail necrosis started at about 10 months of age in VMO25-treated mild SMA mice. Unconjugated PMO25 was also tested at 20 μg/g by ICV injection into neonatal mild SMA mice. The onset of tail necrosis started at about 8 weeks of age, significantly delayed compared with 3 weeks in saline control, but still less impressive than that of VMO25.
Because Vivo-Morpholino has shown significant efficacy on exon skipping in skeletal and cardiac muscle in mdx mice (Wu et al., 2009), a model of human DMD, we then examined the target organs of VMO25 in SMA mice and its ability to cross the BBB in adult mice. Three-week-old heterozygous mice with the genotype of (SMN2)2+/–; smn+/– were injected with VMO25 at a dose of 5 or 10 μg/g via the tail vein. After 10 days, tissues were collected from brain, spinal cord, TA muscle, heart, liver, and kidney. Exon 7 inclusion was analyzed by reverse transcription-PCR and quantified by real-time PCR. Whereas prominent exon 7 inclusion was observed in peripheral tissues such as skeletal muscle and kidney, there was no evidence of inclusion in brain and spinal cord (Fig. 7C). This observation was confirmed by quantitative real-time PCR. Striking exon 7 inclusion was obtained in the TA muscle and kidney, followed by liver, but not in heart, brain, and spinal cord (Fig. 7D). Hence, the 25-mer Vivo-Morpholino was inefficient in inducing exon 7 inclusion in the CNS after its systemic delivery in adult mice.
The administration of VMO25 by ICV injection was also examined in newborn SMA mice. After administration of 10 μg/g, all injected pups, including both type I and heterozygous mice, died overnight. When the dose was reduced to 4 μg/g, half of the injected mice died overnight. We monitored the survival and general condition of the remaining VMO25-injected mice. Three type I mice survived until PND15–17. Two injected heterozygous mice lived until PND25 and had to be culled because of severe hydrocephalus. After reducing the dose to 2 μg/g, no mice died on the second day, but no significant improvement in survival was observed in type I SMA mice. The injected type I mice died between PND7 and PND12, with survival similar to that of saline-treated type I SMA mice (Fig. 7E).
Discussion
Manipulating SMN2 pre-mRNA splicing by AOs to induce the inclusion of exon 7 in the mature transcript, hence increasing the amount of SMN protein, is a promising therapeutic strategy for SMA. Nine AOs, including bifunctional AOs, have been previously investigated in in vitro and in vivo experimental systems (Fig. 1). The most impressive outcomes in SMA transgenic mice are from two AOs binding to the ISS-N1 in intron 7, the 18-mer ASO 10–27 (−10, −27) and 20-mer HSMN2Ex7D (−10, −29) (Hua et al., 2011; Porensky et al., 2012). Our aim was to refine the design of an AO binding to ISS-N1 and explore the feasibility of obtaining an even more potent AO, so as to reduce the frequency of repeated administrations in future clinical trials in patients with SMA. For this purpose, an AO that can safely induce sustained and pronounced full-length SMN2 expression when applied at a low dose is required.
In this study, we conducted a systematic comparison of the efficacies of three AOs with a Morpholino backbone, all targeting the ISS-N1 core sequence. These had lengths of 18 (PMO18), 20 (PMO20), and 25 (PMO25) bases, respectively. The efficiencies of the PMOs were assessed by previously validated and widely used in vitro and in vivo assays, that is, by in vitro cell-free splicing and measurements of exon 7 inclusion and SMN protein expression in transfected SMA fibroblasts (Owen et al., 2011); and, more importantly, by their efficacy in an SMA transgenic mouse model. Consistent results were obtained in all the in vitro assays: PMO25 is more efficient than PMO18 and PMO20 in inducing exon 7 inclusion in splicing in vitro and in SMA fibroblasts. The superiority of PMO25 to PMO18 was further confirmed in SMA transgenic mice after administrations in newborn type I SMA mice.
Porensky and colleagues administered a single ICV injection of Morpholino HSMN2Ex7D AO, which is the same as PMO20, to newborn severely affected SMNΔ7 mice. The AO increased survival at all doses (27, 54, and 81 μg in total), with an increase in the average survival to 83 to 112 days, compared with 15 days for the untreated control (Porensky et al., 2012). This outcome is similar to our survival data obtained from ICV or intravenous injection of PMO25 at 40 μg/g. However, the experiments were conducted in a different mouse model of SMA, SMNΔ7 mice (SMN2+/+; Smn–/–; SMNΔ7+/+), developed by Le and colleagues (2005), which lacks mouse Smn and carries two copies of human SMN2 and SMNΔ7. Despite the fact that these mice display a phenotype similar to the type I SMA mouse model used here, which carries truncated mouse Smn lacking exon 7 and two copies of human SMN2, developed by Hsieh-Li and colleagues (2000), SMNΔ7 mice have a longer median survival (∼15 days) than the type I SMA mice (∼10 days). The difference in baseline survival time between these two SMA mouse models obviously affects survival after AO administration.
The most common AO chemistries used at present are 2′-O-methyl (2′-OMe) phosphorothioate, 2′-O-2-methoxyethyl (MOE) phosphorothioate, and PMO. They have all been widely used in preclinical studies and clinical trials. However, the therapeutic application of 2′-OMe and MOE AOs is limited by liver toxicity when using MOE AOs, and liver and kidney toxicity when using 2′-OMe AOs (Kling, 2010; Raal et al., 2010; Akdim et al., 2011; Goemans et al., 2011). In contrast, the PMO backbone has been demonstrated to be well tolerated with no drug-related adverse effect in two completed clinical trials in which systemic administration of a PMO at doses up to 20 and 50 mg/kg, respectively, was done in boys with DMD (Cirak et al., 2011; and http://investorrelations.avibio.com/phoenix.zhtml?c=64231&p=irol-newsArticle&ID=1678924&highlight=).
Preclinical studies have indicated no drug-related adverse events in primates receiving repeated administrations of the maximal feasible dose (320 mg), or in mice receiving long-term (>1 year) PMO at doses of 1000 μg/g (Sazani et al., 2011). The absence of PMO toxicity in CNS was confirmed in a study using ICV injection of the PMO20 (HSMN2Ex7D) in neonatal SMAΔ7 mice, where doses as high as 405 μg showed no evidence of toxicity (Porensky et al., 2012).
In the present study, we showed that ICV injection of PMO18 at 40 μg/g significantly increased the average survival of type I SMA mice from 10 to 43 days. A similar 20-mer antisense sequence but with a 2′-OMe backbone did not improve survival of SMNΔ7 mice, after repeated ICV injections (Williams et al., 2009). Furthermore, a proinflammatory effect in the brain and spinal cord was detected when the 20-mer 2′-OMe was administered into the CNS, in the absence of any effect on SMN2 exon 7 inclusion (Hua et al., 2010). Modification of the 2′-OMe backbone to MOE chemistry, by using ASO-10–27, only extended the life span of SMNΔ7 mice from 15 to 26 days after ICV injection of 4 μg into neonatal mice (Passini et al., 2011). Increasing the dose to 16 μg resulted in a dramatic death rate within 24 hr of ICV injection (Passini et al., 2011). In contrast, a subsequent study using systemic administration of the 18-mer MOE ASO-10–27 at a high dose of 160 μg/g extended the median life span of type I SMA mice from 10 days to more than 200 days. A possible explanation of these results might be that there is some MOE toxicity in direct CNS administration. In that study, although SMN2 splicing in the CNS was corrected at a similar efficiency as in other peripheral tissues, it was concluded that peripheral administration of MOE was essential for longer survival and required for the rescue of the SMA phenotype. On the other hand, scAAV8-SMN CNS delivery and transgenic approaches limiting SMN expression to the CNS or skeletal muscle suggest that rescue of SMN expression in the CNS is essential for effective treatment of SMA in mice (Gavrilina et al., 2008). Moreover, both the study on neonatal ICV injection of the 20-mer Morpholino and our present study show dramatic rescue of the SMA phenotype but no peripheral modulation of SMN2 splicing, in keeping with the poor crossing of the BBB by the PMOs (Porensky et al., 2012). Our current results and those of Porensky and colleagues show that there is no significant difference in survival between the delivery routes when comparing ICV with systemic Morpholino administrations in neonatal SMA mice.
Novel approaches to improve systemic AO delivery have been intensively investigated with the aim of permitting lower doses of AOs to be used, compatible with acceptable safety and toxicity profiles, and to facilitate the efficiency of therapeutic AOs in target tissues. Two principal delivery moieties have been conjugated to the uncharged PMO: short arginine-rich cell-penetrating peptide-conjugated PMOs (PPMOs) and nonpeptide dendrimeric octaguanidine (Vivo-Morpholinos). These delivery moieties have been intensively investigated in mdx mice, and showed enhanced delivery of Morpholino and improved expression of dystrophin in skeletal and cardiac muscle after systemic delivery (Wu et al., 2008, 2009; Yin et al., 2008; Moulton and Jiang, 2009; Wood et al., 2010). A Vivo-Morpholino targeting mouse dystrophin exon 23 (Vivo-ME23) showed significantly improved efficacy in inducing exon skipping in mdx mice without any noticeable toxicity (Wu et al., 2009). However, the conjugation of the octaguanidine moiety with PMO25 (VMO25) in the present study in SMA mice gave equivocal results. VMO25 was much less effective than PMO25 in rescuing severe type I SMA mice after systemic delivery (average survival, 16 vs. 93.5 days). In addition, the CNS toxicity on direct delivery of VMO25 by ICV injection was noticeable. Lowering the dose diminished the toxicity but failed to rescue the mice. These data therefore highlight limitations of the octaguanidine Morpholino as a potential therapy for SMA or other CNS conditions in humans. Nevertheless, systemic delivery of VMO25 in newborn mild SMA mice showed striking success in rescuing tail and ear necrosis. Tail and ear necrosis is a typical feature in SMA mice with a mild phenotype (Hsieh-Li et al., 2000; Tsai et al., 2006), which could be reminiscent of the digit necrosis observed in some severe cases of type I SMA in children (Araujo et al., 2009; Rudnik-Schoneborn et al., 2010). Although the mechanism of tail necrosis in SMA mice is still unclear, a possibility is that it could be related to vasculopathy induced by skeletal muscle denervation or autonomic nervous system dysfunction (Tsai et al., 2006; Araujo et al., 2009; Hua et al., 2010; Rudnik-Schoneborn et al., 2010). The tail rescue observed in mild SMA mice is more impressive in VMO25-treated than PMO25-treated mice. When comparing the target organs, whereas both AOs showed efficacy in the brain and spinal cord, only VMO25 showed an effect on SMN2 exon 7 inclusion in skeletal muscle. This result may indicate that skeletal muscle or other peripheral tissues are closely involved in the scenario of vasculopathy in SMA.
Our study has shown that the naked Morpholino is more efficient than any of the other AOs studied, with no apparent toxicity after both CNS and systemic administrations. There is clear evidence that CNS restoration of SMN protein is essential in rescuing SMA phenotypes. Although we and others have shown that AOs can cross the BBB and exert their action in the CNS when administered systemically at high dose in newborn mice, it is unlikely they are capable of BBB penetration in humans, even in infants, as the BBB is fully developed before birth in humans. Therefore direct CNS delivery, for example, ICV or intrathecal delivery, will be necessary in clinical trials, unless efficient brain targeting after systemic delivery becomes feasible. Thus, the use of an AO with no CNS toxicity will be vital. Our data provide clear evidence that PMO25 is superior, both in terms of potency at low dose and in terms of longevity of action, compared with PMO18. From a therapeutic point of view it would be preferable to use AOs at the lowest possible concentration for safety reasons as well as to minimize the repeated administration and cost of treatment, and in light of the excellent safety track record of PMOs, PMO25 should be considered for future clinical applications.
Acknowledgments
The authors thank Professor Christian Lorson (University of Missouri, Columbia, MO) for the generous gift of human-specific anti-SMN antibody. This work was supported by the UCL Therapeutic Innovation Fund (to F.M. and H.Z.) provided by the Wellcome Trust Institutional Strategic Support Fund (grant reference 097815/Z/11/Z), the UCL Biomedical Research Centre (grant reference CBRC 161), and a Wellcome Trust Value in People award grant to H.Z. (grant code FPEG). Great Ormond Street Hospital Children's Charity is gratefully acknowledged for its support of F.M. J.E.M. is supported by a Wellcome Trust University award. K.A. is supported by an AFM program grant.
Author Disclosure Statement
The authors declare that they have no conflict of interest.
References
- Aartsma-Rus A. Houlleberghs H. van Deutekom J.C., et al. Exonic sequences provide better targets for antisense oligonucleotides than splice site sequences in the modulation of Duchenne muscular dystrophy splicing. Oligonucleotides. 2010;20:69–77. doi: 10.1089/oli.2009.0215. [DOI] [PubMed] [Google Scholar]
- Akdim F. Tribble D.L. Flaim J.D., et al. Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur. Heart J. 2011;32:2650–2659. doi: 10.1093/eurheartj/ehr148. [DOI] [PubMed] [Google Scholar]
- Araujo A.Q. Araujo M. Swoboda K.J. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J. Pediatr. 2009;155:292–294. doi: 10.1016/j.jpeds.2009.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila A.M. Burnett B.G. Taye A.A., et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughan T.D. Dickson A. Osman E.Y. Lorson C.L. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum. Mol. Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L. Krainer A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- Cartegni L. Krainer A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- Cirak S. Arechavala-Gomeza V. Guglieri M., et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti S. Nizzardo M. Nardini M., et al. Embryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in mice. Brain. 2010;133:465–481. doi: 10.1093/brain/awp318. [DOI] [PubMed] [Google Scholar]
- Dominguez E. Marais T. Chatauret N., et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- Dominski Z. Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley M.G. Manoharan M. Villiet P., et al. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 1998;7:1083–1090. doi: 10.1093/hmg/7.7.1083. [DOI] [PubMed] [Google Scholar]
- Feldkotter M. Schwarzer V. Wirth R., et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust K.D. Wang X. McGovern V.L., et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Garbes L. Riessland M. Holker I., et al. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum. Mol. Genet. 2009;18:3645–3658. doi: 10.1093/hmg/ddp313. [DOI] [PubMed] [Google Scholar]
- Gavrilina T.O. McGovern V.L. Workman E., et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans N.M. Tulinius M. van den Akker J.T., et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N. Engl. J. Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- Gogliotti R.G. Hammond S.M. Lutz C. DiDonato C.J. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2009;39:517–522. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding P.L. Fall A.M. Honeyman K., et al. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol. Ther. 2007;15:157–166. doi: 10.1038/sj.mt.6300006. [DOI] [PubMed] [Google Scholar]
- Hauke J. Riessland M. Lunke S., et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet. 2009;18:304–317. doi: 10.1093/hmg/ddn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann Y. Lorson C.L. Stamm S., et al. Htra2-β1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc. Natl. Acad. Sci. U.S.A. 2000;97:9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li H.M. Chang J.G. Jong Y.J., et al. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Hua Y. Vickers T.A. Baker B.F. Bennett C.F, et al. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y. Vickers T.A. Okunola H.L. Bennett C.F., et al. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y. Sahashi K. Hung G., et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y. Sahashi K. Rigo F., et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima T. Manley J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- Kashima T. Rao N. Manley J.L. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 2007;104:3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernochan L.E. Russo M.L. Woodling N.S., et al. The role of histone acetylation in SMN gene expression. Hum. Mol. Genet. 2005;14:1171–1182. doi: 10.1093/hmg/ddi130. [DOI] [PubMed] [Google Scholar]
- Kinali M. Arechavala-Gomeza V. Feng L., et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling J. Safety signal dampens reception for mipomersen antisense. Nat. Biotechnol. 2010;28:295–297. doi: 10.1038/nbt0410-295. [DOI] [PubMed] [Google Scholar]
- Le T.T. Pham L.T. Butchbach M.E., et al. SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S. Burglen L. Reboullet S., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lim S.R. Hertel K.J. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J. Biol. Chem. 2001;276:45476–45483. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- Lorson C.L. Hahnen E. Androphy E.J. Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis V.B. Butchbach M.E. Lorson C.L. Detection of human survival motor neuron (SMN) protein in mice containing the SMN2 transgene: Applicability to preclinical therapy development for spinal muscular atrophy. J. Neurosci. Methods. 2008;175:36–43. doi: 10.1016/j.jneumeth.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew P.E. Parsons D.W. Simard L.R., et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima H. Miyaso H. Okumura M., et al. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J. Biol. Chem. 2002;277:23271–23277. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- Monani U.R. Spinal muscular atrophy: A deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Monani U.R. Lorson C.L. Parsons D.W., et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- Moulton J.D. Jiang S. Gene knockdowns in adult animals: PPMOs and Vivo-Morpholinos. Molecules. 2009;14:1304–1323. doi: 10.3390/molecules14031304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman E.Y. Yen P.F. Lorson C.L. Bifunctional RNAs targeting the intronic splicing silencer N1 increase SMN levels and reduce disease severity in an animal model of spinal muscular atrophy. Mol. Ther. 2012;20:119–126. doi: 10.1038/mt.2011.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen N. Zhou H. Malygin A.A., et al. Design principles for bifunctional targeted oligonucleotide enhancers of splicing. Nucleic Acids Res. 2011;39:7194–7208. doi: 10.1093/nar/gkr152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini M.A. Bu J. Roskelley E.M., et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini M.A. Bu J. Richards A.M., et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- Porensky P.N. Mitrpant C. McGovern V.L., et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raal F.J. Santos R.D. Blom D.J., et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- Rasband W.S. U.S. National Institutes of Health; Bethesda, MD: 1997–2012. ImageJ. [Google Scholar]
- Rudnik-Schoneborn S. Vogelgesang S. Armbrust S., et al. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve. 2010;42:144–147. doi: 10.1002/mus.21654. [DOI] [PubMed] [Google Scholar]
- Sazani P. Ness K.P. Weller D.L., et al. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of Duchenne muscular dystrophy. Int. J. Toxicol. 2011;30:313–321. doi: 10.1177/1091581811403505. [DOI] [PubMed] [Google Scholar]
- Singh N.K. Singh N.N. Androphy E.J. Singh R.N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.N. Androphy E.J. Singh R.N. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10:1291–1305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skordis L.A. Dunckley M.G. Yue B., et al. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.E. Thomas N.H. Lewis C.M., et al. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur. J. Hum. Genet. 1998;6:467–474. doi: 10.1038/sj.ejhg.5200210. [DOI] [PubMed] [Google Scholar]
- Tsai L.K. Tsai M.S. Lin T.B., et al. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol. Dis. 2006;24:286–295. doi: 10.1016/j.nbd.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Valori C.F. Ning K. Wyles M., et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- van Deutekom J.C. Janson A.A. Ginjaar I.B., et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- Williams J.H. Schray R.C. Patterson C.A., et al. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M.J. Gait M.J. Yin H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain. 2010;133:957–972. doi: 10.1093/brain/awq002. [DOI] [PubMed] [Google Scholar]
- Wu B. Moulton H.M. Iversen P.L., et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B. Li Y. Morcos P.A., et al. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol. Ther. 2009;17:864–871. doi: 10.1038/mt.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H. Moulton H.M. Seow Y., et al. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008;17:3909–3918. doi: 10.1093/hmg/ddn293. [DOI] [PMC free article] [PubMed] [Google Scholar]