

Abstract
Protein-based therapeutics feature large interacting surfaces. Protein folding endows structural stability to localised surface epitopes, imparting high affinity and target specificity upon interactions with binding partners. However, short synthetic peptides with sequences corresponding to such protein epitopes are unstructured in water and promiscuously bind to proteins with low affinity and specificity. Here we combine structural stability and target specificity of proteins, with low cost and rapid synthesis of small molecules, towards meeting the significant challenge of binding coiled coil proteins in transcriptional regulation. By iteratively truncating a Jun-based peptide from 37 to 22 residues, strategically incorporating i→i+4 helix-inducing constraints, and positioning unnatural amino acids, we have produced short, water-stable, α-helical peptides that bind cFos. A three-dimensional NMR-derived structure for one peptide (24) confirmed a highly stable α-helix which was resistant to proteolytic degradation in serum. These short structured peptides are entropically pre-organized for binding with high affinity and specificity to cFos, a key component of the oncogenic transcriptional regulator Activator Protein-1 (AP-1). They competitively antagonized the cJun–cFos coiled-coil interaction. Truncating a Jun-based peptide from 37 to 22 residues decreased the binding enthalpy for cJun by ∼9 kcal/mol, but this was compensated by increased conformational entropy (TΔS ≤7.5 kcal/mol). This study demonstrates that rational design of short peptides constrained by α-helical cyclic pentapeptide modules is able to retain parental high helicity, as well as high affinity and specificity for cFos. These are important steps towards small antagonists of the cJun-cFos interaction that mediates gene transcription in cancer and inflammatory diseases.
Introduction
Cellular functions are mediated by protein-protein interactions, the majority involving large interacting surface areas with binding interfaces that are shallow, hydrophilic and lack the well-defined small hydrophobic clefts that are most tractable for design of small molecule inhibitors. Driven by the need to target these larger protein surfaces, there has been renewed interest in recent years in developing larger therapeutic molecules, such as peptides and their mimetics, which can in principle combine advantages of proteins (target specificity, structural stability) with advantages of small molecules (lower cost, oral activity) [1]. One approach is to engineer small synthetic components of protein surfaces (‘protein surface mimetics’) [2] to compete for (antagonists) or mimic (agonists) protein-protein interactions that mediate disease. However, peptides have also traditionally been perceived as being problematic therapeutics as they are often considered too large, too polar and too susceptible to protease degradation in order to traverse intact across biological membranes. Conjugating small (5–12 residue) protein-transduction domains or arginine-rich peptides, such as TAT and antennapedia fragments [3], [4], [5] to peptide cargo can be used to facilitate cell penetration. However, those sequences increase peptide size and are themselves susceptible to proteolytic degradation. Rendering peptides protease resistant has been more difficult to engineer without replacing key components with non-peptidic groups, or creating N- to C-terminal ‘cyclic peptides’, to bring stability and bioavailability to the peptide (see [1], [2], [3] and references therein). More recently, constraints have been incorporated into peptide sequences to induce bioactive helix, strand or turn structural motifs that have high affinity for receptors without the need for larger sequences [6], [7], [8], [9], [10], [11]. Alpha helices have been successfully stabilized by introducing constraints in the side-chains of amino acids [12], [13], [14], [15], [16], [17], [18], [19] or within the peptide backbone using hydrogen bond surrogate approaches [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]. In the HBS approach α-helices feature a carbon−carbon bond in place of an N-terminal intramolecular hydrogen bond between the peptides i and i +4 residues. Thus organizing three consecutive amino acids into the helical orientation inherently limits the stability of short α-helices. The HBS method affords preorganized α-turns to overcome this intrinsic nucleation barrier and initiate helix formation.Other approaches include β-peptides [31], [32], without interfering with the helix surface designed to interact with the target protein, thereby conferring high helicity that confers protein-like function upon peptides that would otherwise have low or negligible biological potency. We have previously used one-turn (i→i+4) rather than two-turn (i→i+7) bridging constraints to induce α-helicity [9], [11], [33], [34], since our research supports greater per residue helicity even though this is contrary to polymer theory [35]. The approach can however be context dependent, and requires extensive further investigation to realise its promise.
The Jun-Fos Activator Protein-1 (AP-1), is a helical heterodimer and oncogenic transcriptional regulator implicated in a range of diseases that includes cancer [36], [37], [38], bone disease (e.g. osteoporosis) and inflammatory diseases such as rheumatoid arthritis and psoriasis [39], [40], [41]. A number of intracellular signalling cascades converge at AP-1, producing changes in gene expression profiles that can cause tumour formation, progression and metastasis. Here we begin with a 37 residue peptide (JunWCANDI) found to bind specifically to cFos in the presence of cJun [42] by binding to the coiled coil region that is responsible for driving AP-1 heterodimerization. In brief, the coiled coil is characterised by a repeat of seven amino acids, denoted a-g; residues a and d consist largely of hydrophobic residues, forming a stripe which associates with respective partners on the other helix. Core flanking charged residues at e and g positions form interhelical ion pairs with g and e_residues in the neighbouring helix and aid heterospecificity [43]. Core region proximity means these residues are also partially shielded from the solvent [44]. Since the Jun-Fos dimer interface is responsible for mediating key transcriptional events associated with disease induction, it may be a worthy target for therapeutic intervention. We have previously reported peptides of 33–37 amino acids, based on the coiled coil region known to control dimerization [45], [46], and flanked by N- and C-terminal capping motifs, that were able to bind and sequester either cJun or cFos to prevent Jun-Fos heterodimer formation, initiation of gene transcription, and cell differentiation and proliferation [42], [47], [48], [49]. These peptides were however too large to be useful therapeutics. By systematically truncating the 37 residue peptide ‘JunWCANDI’ [42], together with strategic introduction of helix constraints (Figure 1), we sought to establish (i) whether helix-constrained peptides could be significantly reduced in length while maintaining effective binding; (ii) whether the downsized peptides maintained high binding specificity for cFos relative to cJun; (iii) whether there was a key consensus region within the coiled coil that was essential for binding; and (iv) whether the approach required mainly enthalpic or entropic contributions to be successful. The latter is important because coiled coils typically require interactions along their entire lengths for structural stability.
Figure 1. Schematic showing sequences and constrains for all peptides.

The parental JunWCANDI sequence is shown in bold as are heptads and residue positioning within the helical wheels. Peptide constraints are shown in blue. KD values taken from ITC experiments for peptides in complex with cFos are shown in µM. Fraction helicity as measured from CD experiments are also shown. Positions of i→i+4 hydrocarbon constraints were initially placed into a JunWCANDI peptide [42] lacking capping motifs, causing a reduction in the size of the molecule from 37 residues to 32. All constraints tethered b→f or f→c residues.
We report that α-helical cyclic pentapeptide modules inserted into truncated sequences from within the JunWCANDI peptide results in much shorter water-stable α-helical peptides that retain the high affinity and specificity of the parental JunWCANDI peptide for cFos, and are stable to proteolytic degradation. Affinity for cFos is driven by a combination of interactions along most of the sequence of cJun, and we were able to pinpoint key co-facial residues that contribute to the overriding enthalpic properties that dictate peptide potency. This is an important step forward in understanding how to rationally design small transcriptional regulators.
Experimental Procedures
Peptide Synthesis and Purification
Peptide synthesis was performed as described [9], [11], [33] by Fmoc chemistry. The phenyl isopropyl ester of aspartic acid and methyl trityl group of lysine were removed from the peptide-resin with 3% TFA in dichloromethane (DCM) (5×2 min). Cyclization was effected on-resin using Benzotriazole-1-yl-oxytris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) and 1-hydroxy-7-aza-benzotriazole (HOAt), base N,N-Diisopropylethyamine (DIPEA), and DMF (1⋮1). The procedure was repeated for multiple cyclizations. Crude peptides were purified by rp-HPLC (Rt1: Vydac C18 column, 300 Å. 22×250 mm, 214 nm, Solvent A = 0.1% TFA in H2O, Solvent B = 0.1% TFA, 10% H2O in acetonitrile. Gradient: 0% B to 70% B over 35 min). Peptides were >95% purity by analytical HPLC. Correct masses were verified by electrospray mass spectrometry. Peptide masses were as follows: cFos = 4147; 1 = 3747; 2 = 3740; 3 = 3792; 4 = 3791; 5 = 2208; 6 = 2201; 7 = 2183; 8 = 2926; 9 = 2951; 10 = 2661; 11 = 2675; 12 = 2668; 13 = 2291; 15 = 2287; 16 = 2751; 17 = 2701; 18 = 2786; 19 = 2730; 20 = 2730; 21 = 2675; 22 = 2704; 23 = 2661; 24 = 2751. All synthetic peptides were N- acetylated and C-amidated. Peptide concentrations were determined by dry weight and verified via absorbance in water at 280 nm with an extinction coefficient of 1209 M−1 cm−1 [50] corresponding to a single Tyr residue inserted into a solvent-exposed b3 heptad position. The peptide concentration for 2, 6 and 7 were determined by dry weight alone since the b3 Tyr was replaced by an Lys residue that formed part of the helix constrained peptide.
NMR Spectroscopy
A sample for NMR analysis (Figure 2) was prepared by dissolving peptide 24 (2.0 mg) in 540 µL H2O and 60 µL D2O. 1D (variable temperature experiments) and 2D 1H-NMR spectra were recorded on a Bruker Avance 600 and 900 MHz spectrometers respectively. 2D 1H-spectra were recorded in phase-sensitive mode using time-proportional phase incrementation for quadrature detection in the t1 dimension. 2D experiments included TOCSY (standard Bruker mlevgpph pulse program) and NOESY (standard Bruker noesygpph pulse program) and dqf-COSY (standard Bruker dqfcosygpph pulse program). TOCSY spectra were acquired over 9920 Hz with 4096 complex data points in F2, 256 increments in F1 and 32 scans per increment. NOESY spectra were acquired over 9920 Hz with 4096 complex data points in F2, 512 increments in F1 and 32 scans per increment. TOCSY and NOESY spectra were acquired with several isotropic mixing times of 80 ms for TOCSY and 200–250 ms for NOESY. For all water suppression was achieved using modified WATERGATE and excitation sculpting sequences. For 1D 1H NMR spectra acquired in H2O/D2O (9∶1), the water resonance was suppressed by low power irradiation during the relaxation delay (1.5 to 3.0 s). Spectra were processed using Topspin (Bruker, Germany) software and NOE intensities were collected manually. The t1 dimensions of all 2D spectra were zero-filled to 1024 real data points with 90° phase-shifted QSINE bell window functions applied in both dimensions followed by Fourier transformation and fifth order polynomial baseline correction. Variable temperature NMR experiments were performed over 278–318 K. 1H chemical shifts were referenced to DSS (δ 0.00 ppm) in water. 3 J NHCHα coupling constants were measured from 1D 1H NMR and dqf-COSY spectra. The assigned 1H NMR signals for peptide 24 can be found in Table S2.
Figure 2. NMR Structure of peptide 24.

a) NOE summary diagram for peptide 24 in 90% H2O:10% D2O at 298 K. Sequential, short and medium range NOE intensities were classified as strong (upper distance constraint 2.7 Å), medium (3.5 Å), weak (5.0 Å), very weak (6.0 Å) and are proportional to bar thickness; grey bars indicate overlapping signals. 3 J NHCHαcoupling constants <6 Hz are indicated by ↓. Amide NH’s for which chemical shifts changed by <5 ppb/K are indicated by •. b) Backbone superimposition for ten lowest energy NMR-derived solution structures for Ac-cyclo-(3,7; 10,14; 17,21)-ChaR[KEIYD]LR[KKAND]LR[KHIAD]Cha-NH2 (24) in H2O:D2O (9∶1) at 298 K showing carbon atoms (green), nitrogens (blue), oxygens (red), i→i+4 hydrocarbon constraints (orange). Also for clarity, one structure is shown with its alpha helical backbone (yellow) and projecting side chains (green). N-terminus is at the top.
Structure Calculations
The distance restraints used in calculating a solution structure for 24 in water was derived from NOESY spectra recorded at 298 K or 288 K by using mixing time of 250 ms. NOE cross-peak volumes were classified manually as strong (upper distance constraint ≤ 2.7 Å), medium (≤ 3.5 Å), weak (≤ 5.0 Å) and very weak (≤ 6.0 Å) and standard pseudo-atom distance corrections were applied for non-stereospecifically assigned protons. To address the possibility of conformational averaging, intensities were classified conservatively and only upper distance limits were included in the calculations to allow the largest possible number of conformers to fit the experimental data. Backbone dihedral angle restraints were inferred from 3 J NHCHα coupling constants in 1D spectra, ϕ was restrained to –60±30° for 3 J NHCHα ≤ 6 Hz. Starting structures with randomized ϕ and ψ angles and extended side chains were generated using an ab initio simulated annealing protocol. The calculations were performed using the standard force field parameter set (PARALLHDG5.2.PRO) and topology file (TOPALLHDG5.2.PRO) in XPLOR-NIH with in house modifications to generated i→i+4 helix constraints between lysine and aspartic acid residues and unnatural amino acid Cyclohexylalanine (Cha). Refinement of structures was achieved using the conjugate gradient Powell algorithm with 2000 cycles of energy minimization and a refined force field based on the program CHARMm [51]. Structures were visualized with Pymol and analyzed for distance (>0.2 Å) and dihedral angle (>5°) violations using noe.inp and noe2emin.inp files (in Xplor). Final structures contained no distance violations (>0.2 Å) or angle violations (>5°). Corresponding NMR coordinates are available upon request.
Serum Stability
Stock solutions of 12 and 24 in both constrained forms and linear forms lacking constraints were prepared in water (1 mg/ml), 200 µL was added to human serum (800 µL) and incubated at 37°C. Aliquots (100 µL) of this diluted serum were removed at 0, 0.5, 1, 2, 4, 16, and 24 hours and a mixture of acetonitrile/water 3∶1 (300 µL) was added to each aliquot before centrifuging (17000 rpm, 15 min). Aliquots (100 µL) of the supernatant were then analysed by LC-MS-MS after passing through a 2.1×150 mm Phenomenex 300A C18 5 µm column at 10% per minute linear gradient from 0–100% acetonitrile over 12 minutes. The amount of starting material was quantified by determination of total ion counts for the m/3+ and m/4+ ion for each peptide (Figure 3).
Figure 3. Serum stability of peptides 12 and 24.

Shown are the effects of helix-inducing constraints (• and ▪) versus the linear sequences (▾ and ▴) in human serum at 37°C.
Circular Dichroism
Circular dichroism (CD) spectroscopy was conducted with an Applied Photophysics Chirascan CD spectroploarimeter (Leatherhead, U.K.) using a 200 µL sample in a CD cell with a 1 mm path length (Hellma, Müllheim Germany). Samples contained 150 µM total peptide concentration suspended in 10 mM potassium phosphate and 100 mM potassium fluoride at pH 7. The CD spectra of the homodimeric and heterodimeric complexes were scanned between 300 nm and 190 nm at 20°C (for both pre- and post-melt samples to check for reversibility of unfolding) and at −8°C to assess helical levels and coiled-coil structure (see Figure 4). All data have been converted from raw ellipticity to molar residue ellipticity according to the equation:
| (1) |
Where θ is the CD signal of the sample in millidegrees, l is the pathlength of the cell in centimetres, r is the number of residues in the peptide, and c is the total peptide molar concentration of the sample.
Figure 4. Raw thermal melting data for all homo and heterodimeric complexes.

Data have been collected by measuring the level of helicity at 222 nm in an applied photophysics chirascan Circular Dichroism (CD) Spectrometer. Data have been converted from raw ellipticity to Molar Residue Ellipticity (MRE) according to equation 1 to take account of the different peptide lengths. Thermal melting data for cFos is shown in black, data for the constrained peptide in isolation is shown in blue and the cFos/constrained peptide mixture is shown in red. Also shown is the average of the cFos and constrained peptide (black dotted line). Where possible data have been fitted to equation 2 to generate thermal melting (Tm) values (e.g. for cFos-1 and cFos-2) and in such instances it is clear from an increase in the averaged homomeric Tm values that an interaction is occurring (e.g. 1: −1+50 = 49/2 = 24.5<55). However, some data were unable to be fitted owing to the lack of a melting transition, or of a lower baseline (e.g. cFos-3 and cFos-4), indicating that an interaction is not occurring. CD spectra for these pairs are given in Figure S3.
Thermal Melts
Spectra and thermal melts were performed on 150 µM peptides in 10 mM potassium phosphate, 100 mM potassium fluoride, pH 7, using an Applied Photophysics Chirascan CD instrument (Leatherhead, U.K.). The temperature ramp was set to stepping mode using 1°C increments and paused for 30 seconds at each temperature before measuring ellipticity at 222 nm. For all temperature denaturation experiments data collection was started at −8°C, and at this temperature the peptide solutions remained aqueous. Data points for thermal denaturation profiles represent the averaged signal after 4 s of data collection. Samples were identical in composition to the CD buffer samples. Melting profiles (see Figure 4) were ≥95% reversible with equilibrium denaturation curves fitted to a two-state model (see [52]) to yield the melting temperature (Tm):
| (2) |
where ΔH is the change in enthalpy, TA is the reference temperature in Kelvin; R is the ideal gas constant (1.9872 cal⋅mol–1⋅K–1); Pt the total peptide concentration (150 µM); and ΔCp the change in heat capacity [47]. Helix heterodimerisation is inferred in cases where melting profiles for heterodimers are clearly distinct from averages of constituent homodimeric melts (Shown in Figure 4 and via dimer exchange in Figure S3). The cooperative nature of the melting profiles suggests an apparent two-state process. Tm values were determined by least-squares fitting of the denaturation assuming a two-state folding model that is widely used for coiled coils and provided an excellent fit to our data.
Inspection of Homo and Heterotypic CD Data
Thermal denaturation data and spectra for the cFos homotypic complex (black), helix constrained peptide (blue), and heteromeric complex (red) were recorded using 200 µL of sample at 150µM total peptide concentration (Figure 4, Figure S1). Next, 100 µL of each solution was mixed and the spectra taken such that the final total peptide concentration was also 150 µM. More experimental procedures can be found in File S1.
Results
Design Rational and Evolution of the Helix-constrained Peptide Sequences
Our first aim was to systematically introduce helix-inducing constraints into JunWCANDI which has been shown to be specific for cFos in the presence of cJun [42]. All helix-constrained peptides lack five residues that served as N-terminal and C-terminal capping motifs in the JunWCANDI parent peptide [42], [47], [48]. This led us to examine the effect on helix induction of one or more such constraints placed at different positions within the sequence, while concomitantly truncating the sequence from either terminus. The goal was to decrease the peptide length while maintaining the peptide helicity using one or more helix-inducing constraints. The process was iterative, generating 24 peptides. We find that introduction of helical constraints leads to a stable heterodimeric peptide with cFos for some but not all of the constructs developed, indicating that induction of helicity alone is insufficient to achieve binding:
Peptides 1–4
Our initial series of four constructs (Figure 1) were based on the JunWCANDI scaffold (4.5 heptads in length), but lacked the N- and C-terminal capping motifs. Thus at 32 residues these peptides were five residues shorter than this ‘Protein-fragment complementation assay’ (PCA) derived parent peptide [42], [47], [48], but retained all regions of the coiled coil. The first four peptides synthesized (1–4, Figure 1) each had one i→i+4 helix constraint formed by inserting Lys and Asp in place of solvent exposed residues between positions b and f in one of the four heptads of the coiled coil. Circular dichroism spectra were used to measure the relative extent of α-helix induction and showed that we were successful in our design process (Table S1, Figure S1). Constraints in either of the two C-terminal heptads were more effective than in the two N-terminal heptads at inducing α-helicity (1, 63%; 2, 30%; 3, 14%; 4, 15%; Table S1), suggesting an important effect of the sequence environment surrounding the constraint which was most helix-inducing when placed in the most C-terminal heptad (e.g. 1). This led to two peptides that bound (1 and 2– constrained at the C-terminal end of the molecule; see Figure 1) and two that did not (3 and 4– constrained at the N-terminal end). Our interpretation of this result was that the first two heptads are less important in binding cFos and that the latter two heptads were crucial. Indeed, cFos-1 displays a Tm of 55°C; 11°C higher than the cFos-JunWCANDI [42], while cFos-2 displays a Tm of 53°C, amounting to KD values of approximately 1 µM as verified by ITC (see Table 1). In contrast, cFos-3 and cFos-4 did not generate a thermal denaturation profile that could be fitted to equation 2; rather the profile lacked a lower baseline and entered partway through the unfolding transition (see Figure 4). The data also shows that 3 or 4 in solution with cFos results in much lower helicity than cFos with 1 or 2 (cFos-1, 65%; cFos-2, 39%), compared to 3 or 4 (cFos-3, 17%; cFos-4, 26%). For this reason, thermal denaturation data could only be fitted and normalised to a fraction unfolded where satisfactory denaturation profiles, lower baselines and overall fit to equation 2 could be attained. We interpreted cFos-1 stability as being attributable to helical propensity resulting from a constraint at the C-terminus, where less helical structure in the parent protein might be assumed. The same outcome is not observed for the N-terminal constrained peptides 3 and 4, which are significantly less helical. We therefore inferred that the N-terminal region does not play a substantial role in binding, or that these constraints cause steric interference at this end of the molecule. All of these peptides retained ten core a/d residues per helix for hydrophobic interactions and six electrostatic e/g interactions (Figure 1).
Table 1. Thermodynamics of binding of cJun analogues to cFos. Columns (from left to right) show i) Tm values from thermal denaturation analysis ii) calculated % helicity for each respective pair calculated from circular dichroism spectra and iii) KD values calculated from thermal denaturation data.
| Peptide complex | Tm °C | % Helical | KD(20) (Thermal) µM | *KD(20) M−1 (ITC) µM | N | ΔG(20) kcal/mol (ITC) | ΔH kcal/mol | #TΔS kcal/mol [ΔH – ΔG] |
| cFos-cFos | −1 | 20% | 325 | N/D | N/D | N/D | N/D | N/D |
| cFos-JunWCANDI | 44 | 42% | 0.45 | 1.2±0.05 | 0.8±0.01 | −7.9±0.03 | −14.8±0.3 | −6.9±0.3 |
| cFos-1 | 55 | 65% | 0.50 | 0.39±0.03 | 0.8±0.01 | −8.6±0.04 | −11.6±0.1 | −3.0±0.1 |
| cFos-2 | 53 | 39% | 0.19 | 1.44±0.09 | 1.2±0.01 | −7.8±0.04 | −7.1±0.1 | +0.7±0.1 |
| cFos-8 | 32 | 38% | 21.5 | 55±12.45 | 1.0±0.35 | −5.7±0.13 | −9.5±0.4 | −3.8±0.4 |
| cFos-9 | 38 | 47% | 9.2 | N/A | N/A | N/A | N/A | N/A |
| cFos-10 | 49 | 52% | 1.7 | 5.8±0.78 | 0.8±0.01 | −7.0±0.08 | −8.4±0.9 | −1.4±0.9 |
| cFos-11 | 47 | 55% | 5.8 | 5.4±0.28 | 0.7±0.01 | −7.0±0.03 | −7.7±0.2 | −0.7±0.2 |
| cFos-12 | 52 | 40% | 1.2 | 7.6±0.51 | 1.0±0.02 | −6.8±0.04 | −6.1±0.2 | 0.7±0.2 |
| cFos-17 | 44 | 40% | 7.8 | 11.4±0.88 | 1.0±0.03 | −6.6±.0.05 | −5.4±0.3 | 1.2±0.3 |
| cFos-20 | 48 | 49% | 3.8 | 11.2±0.82 | 1.1±0.03 | −6.6±0.04 | −7.5±0.3 | −0.9±0.3 |
| cFos-22 | 51 | 40% | 1.4 | 11.9±1.16 | 1.4±0.04 | −6.6±0.06 | −5.0±0.2 | 1.6±0.2 |
| cFos-24 | 58 | 52% | 7.2 | 7.25±0.64 | 1.1±0.03 | −6.9±0.05 | −8.8±0.4 | −1.9±0.4 |
The remaining three columns give stoichiometry of binding and thermodynamic data calculated from ITC, with TΔS calculated according to the Gibbs Helmholtz equation.
data calculated using the midpoint of the transition from thermal denaturation profiles (and fit as temperature as a function of lnKD, with the fit lnKD = aT+C where a is the gradient, T is the temperature in Celsius and C is the intercept) and calculated at 20°C.
Calculated according to TΔS = ΔH−ΔG.
Peptides 5–7
Using data from the first four peptides, our next group of synthetic constrained peptides were synthesized. These were truncated by two heptads at the N-terminus of the peptide (NΔ14, 18 residues) and a single (5 and 6) or double (bicyclic –7) constraint introduced. Much shorter peptides 5 and 6 had a single helix constraint but were not much more helical (34% and 30%) than the expected statistical ratio of just making 5 of the 18 residues (27%) helical due to their presence in the cyclic pentapeptide component, while there was no additive effect of combining both helix-inducing constraints within the same sequence (e.g. as for peptide 8 ). Unfortunately, none of these peptides resulted in a stable interaction with cFos. Again, no lower baseline for thermal denaturation profiles was observed and therefore no fit could be given in estimation of the Tm (see Figure 4 for raw data). The MRE at θ222 was approximately −12000 at 20°C, indicating a comparatively low level of helicity ([26–29% helical], Table S1) compared to cFos-1 or cFos-2. In addition, peptides 5–7 were all more helical in isolation than as heteromeric mixtures with cFos, with all transitions unable to produce a thermal denaturation profile indicative of a stable interaction. Peptides 5–7 had six core residues per helix for hydrophobic interaction (four less than peptides 1–4) and only three electrostatic e/g interactions (three less than peptides 1–4), which may account for the loss in activity.
Peptides 8–11
Data taken from peptides 1–7 was used in the design of four further peptides 8–11 (Figure 1). In this group, 8 contained the same sequence as 1 but with an additional seven residues truncated from the C-terminus (CΔ7, 25 residues). The constraint also resides closer to the C-terminus of the molecule relative to 1. The bi-cyclic 9 contained the same sequence and constraint as 8 but with the additional N-terminal constraint found in the unsuccessful peptide 4; its insertion resulted in a modest helicity gain over 8 (θ222 at 20°C = −21000 versus. −14000). The logic behind this was that by constraining both the N-and C-terminus of the peptide, helicity would be propagated and maintained across the entire molecule. Interestingly, cFos-8 (which contains one f→c constraint, 32°C) performed comparably to cFos-9 (one N-terminal b→f constraint and one C-terminal f→c constraint, Tm 38°C), indicating that the inclusion of the N-terminal constraint in this bicyclic peptide had only a modest influence on the interaction stability. Taken together with data for 4, this reinforces the argument that inclusion/constraint of this N-terminal region is less important for interaction with cFos. The heterodimeric stability for these two peptides was almost 20°C less than for cFos-1 or cFos-2, therefore indicating that at least part of the CΔ7 deletion is an important binding determinant. Given the low affinity of the N-terminally truncated constrained peptides 5–7 for cFos, and the modest binding affinity afforded by peptides 8–9, two bi-cyclic peptides 10 and 11 were created to make less severe N-terminal truncations than peptides 5–7 (NΔ10 versus NΔ14, see Figure 1). Importantly, this more modest truncation incorporated the hydrophobic ‘d’ residue of the second heptad with the constraint inserted in close proximity, ensuring that the helical integrity of this region was maintained entirely to the helical termini. In addition, 10 and 11 retained the complete C-terminal region with a second constraint either close to (11) or at (10) the C-terminus. 10 contains an f2→c3 and an f4→c5 constraint whereas peptide 11 contains an f2→c3 and a b4→f4 constraint. These peptides increased the Tm with cFos by 9–17°C relative to peptides 8–9, and within 4–8°C of peptides 1 and 2 (and with comparable levels of helicity) despite being 10 residues shorter. Reassuringly, peptides 8–11 all give thermal denaturation profiles that can be fit by equation 2. It is also worth noting that although the position of the constraint is always between b→f or f→c (and therefore always tethers i→i+4 and is positioned away from the a/d/e/g residues associated with the dimeric coiled coil interface), it cannot be ruled out that the precise position of the constraint, and the residues that become replaced by the Lys-Asp pair, do not affect binding affinity in the resulting peptide. In addition, although some peptides are very helical in isolation (as would be expected for such conformationally restricted chains), others exhibit no unfolding transition upon thermal melting, with interaction clearly observed via a cooperative unfolding transition upon mixing with cFos. Lastly, peptides 8–11 had eight (for 8–9) or seven (for 10–11) core residues per helix for hydrophobic interaction (two or three respectively less than for 1–4) and five electrostatic e/g interactions (one less than 1–4). Despite the fact that more core residues are present in 8–9, more stability is observed for cFos in complex with 10–11, indicating that reduced core packing is compensated for in these constrained variants. From 9 onward, all peptides contained two or three helix-inducing constraints, with only the 25-mer 9 (52%), and 22-mers 10 (57%), 11 (63%), 20 (51%) and 24 (69%) displaying about twice the helicity of peptides 5–8 (30–35%).
Peptides 12–17
Based on the above findings, six constrained peptide variants were synthesised: 12–17. 12 contained the three combined constraints of peptides 9 and 10 based on the scaffold of 10 and produced a comparable Tm of 52°C. 13 was the same as 11 but truncated by four residues from the C-terminus, and resulting in severely diminished binding and helicity. 15 was truncated by the same four residues as 13 with a similar bi-cyclic constraint; it was also unsuccessful and similarly displayed reduced helicity and poor thermal denaturation profile. 16 was identical to 11 with an a4 changed from Ala to Phe. This was due to a hydrophobic pocket in the crystal structure that was anticipated to be occupied by the exchange, but this design was also unsuccessful. 17 was also based on 11 with the d3 Leu changed to Cyclohexylalanine (Cha) as an alternative attempt to add hydrophobic bulk to the core in this central region. In particular, we wanted to increase the hydrophobicity in this part of the leucine zipper, since both 13 and 15 had failed in being truncated by four Leu residues relative to 11. 12 and 17 both represent NΔ10 truncations relative to JunWCANDI and maintains seven of ten a/d core residues and five of six electrostatic e/g interactions.
Peptides 18–24
From the success of 12 and 17 in the previous cohort, six further peptides were synthesised; all contained the same f2→c3 and b4→f4 constraint. 18 contained four Cha sidechains – one at each d position (see Figure 1). 19 contained two Cha residues at the central two d positions of the same template, with 20 containing two Cha residues at the outermost d positions of the same template. The final three peptides were identical to 11 but contained point mutations; Ile→Asn at a3 to provide a partner for a3 Lys in cFos, Lys→Arg at e2 to provide an enhanced electrostatic contact with a g’1 Glu in cFos, and a Lys→Leu at g3 for a potentially enhanced hydrophobic effect with e’4 Leu in cFos. These changes were made in 21–23 respectively. Of these six, only 20 and 22 gave profiles consistent with a strong interaction affinity for cFos. For 20, placement of bulky hydrophobic groups at the outermost d positions helps to stabilise the dimer, possibly by also helping to constrain the helix to its target and aid in maintaining overall helicity. For 22 enhancing this electrostatic interaction would be predicted to add around 0.3 kcal/mol of stability [53], but clearly makes a large difference since 21 has a much lower affinity. Lastly, peptide 24 was synthesized to incorporate all of the changes introduced into 12, 20, and 22 - the three shortest and most effective peptides studied. This, the most helical peptide, was fifteen residues shorter than the JunWCANDI parent peptide, ten residues shorter from the N-terminus than peptide 1, and more helical than both (Figure 1). Indeed, 24 was three and a half times more helical than JunWCANDI (69% vs 19%) based on CD spectra. Lastly, conventional substitutions were made, as well as substituting leucine with the bulkier unnatural amino acid cyclohexylalanine (Cha) in 17–20 and 24 to increase both hydrophobicity and potentially the core packing in this part of the coiled coil. Unnatural amino acids at the ends of a peptide can potentially confer proteolytic stability to degradation by carboxy- or amino- peptidases. The CD data for peptides 17–24 suggest that placement of Cha groups at the temini enhances helicity. The two triply bridged peptides 12 (43%) and 24 (69%) differed only in the N-terminal Leu-Lys and C-terminal Leu in 12 being replaced by Cha-Arg and Cha respectively in 24. The Cha residues contribute to this helix increase (cf. 20, 51% vs. 12, 43%) but there is an even larger helix induction through the Lys to Arg change (20 vs 24).
Binding Affinity of Helix-constrained Peptides for cFos
Having identified a number of peptides able to adopt highly helical conformations upon introduction of the constraint, we sought to measure their binding affinities for cFos and explore relationships between affinity and helicity. Peptides 1, 2, 8, 9, 10, 11, 12, 17, 20, 22, and 2 4 are compared in Figure 4 for their capacity to interact with cFos based on thermal denaturation experiments. Although there was no quantitative linear correlation between peptide helicity and affinity for cFos, there was a qualitative relationship. No peptides with <30% helicity were able to form a stable interaction with cFos and most peptides with high helicity also had high affinity for cFos, the mean helicity of interacting peptides being 49% versus 32% for non-interacting peptides. Despite truncating 15 residues from JunWCANDI (>40% of all residues) and 10 residues from 1 (>30% of the molecule), peptide 24 had the highest Tm of 58°C. Similarly, most of the more helical peptides (e.g. 1, 2, 10, 11, 12, 20, 22) had a Tm around 50°C, resulting in KD values of ∼1–11 µM for binding to cFos (Table 1).
In addition to thermal denaturation profiles, isothermal calorimetric data could be obtained for all but one of the peptides in complex with cFos. To verify interactions between constrained peptides and cFos, we compared spectra for cFos alone, constrained peptide alone, and for the mixture of the two (Figure S1). For certain cFos-constrained peptide mixtures (e.g. <30% helicity), the observed spectra matched the average of the previous two spectra, indicating that no exchange of dimer had occurred. For some mixtures of cFos with a constrained peptide, the observed signal from the spectra exceeded that of the averaged homodimeric spectra, indicating binding. All peptides that perfomed well in thermal denaturation studies generated spectra that exceeded the average of component homodimers.
Specificity of Helix-constrained Peptides for cFos
To establish if the truncated and constrained peptides retained the interaction specificity of the JunWCANDI parent peptide, peptides capable of interacting with cFos were also incubated at equimolar concentrations with cJun and monitored via CD spectra. In addition, thermal denaturation was used to establish if an interaction took place, and therefore if the specificity exhibited by the JunWCANDI parent was retained for constrained and truncated sequences. None of the peptides capable of forming an interaction with cFos were found to interact with cJun in these experiments (Figure S2, S3, S4), indicating specificity of the truncated helix constrained, peptides. In addition, a dimer exchange experiment was performed for the most helical peptide 24. Spectra for a cJun-peptide solution and a cFos solution should, upon mixing, generate an averaged spectrum if there was no dimer exchange. However, helical spectra exceeded the average of the two component spectra, indicating that dimer exchange did occur. In contrast, for a cFos-peptide mixed with cJun, the average of the two component spectra was observed, indicating that no change in binding partner for peptide had occurred. Spectra for both mixtures can be superimposed (Figure S4).
Thermodynamic Parameters for cFos Binding
To provide more insight to the origin of the binding affinity (KD) between helix-constrained peptides and cFos, we decided to dissect out the relative contributions of enthalpy versus entropy to the affinities. Isothermal Titration Calorimetry (ITC) experiments were conducted, enabling the free energy of binding to be split into entropic and enthalpic components (Figure 5 and Table 1), while also providing a stoichiometric measure of binding. Owing to the relative stabilities of the interacting pairs, only complexes formed between cFos and peptide 1 , 2 , 8, 10 , 11 , 12 , 17 , 20 and 22 and 24 were able to be characterised (see Table 1), with weaker associations formed by 9 unable to be assessed. For these and other pairs the thermodynamic parameters were unable to be determined by this technique owing to the large quantities of peptide required in both the cell and the syringe.
Figure 5. Isothermal Titration Calorimetry (ITC) analysis of leucine zipper domain interactions between constrained peptides and cFos.

Shown are isotherms for all ten measureable heterodimers (1, 2, 8, 10, 11, 12, 17, 20, 22, and 24) injected into a cell containinginto cFos. The top and bottom panels show, respectively, raw data after baseline correction. During ITC experiments, approximately 200–600 µM of peptide A was injected in 30–40×5 µl batches from the injection syringe into the cell, which contained 10–40 µM cFos. Both partners were in a 10 mM Potassium Phosphate buffer, 100 mM Potassium Fluoride at pH 7. Experiments were undertaken at 20°C. The solid lines represent the fit of the data to the function based on the binding of a ligand to a macromolecule using the Microcal (GE Healthcare) Origin software [57].
Thermodynamic parameters determined from ITC measurements on the above peptides indicate that the free energy of binding is predominantly driven by a favourable enthalpic term (ΔH −5.0 to −11.6 kcal/mol) with the entropic component either favourable or weakly opposing (Table 1). The free energy of binding for the longer parental JunWCANDI peptide is driven by an even stronger enthalpic component (ΔH = −14.8 kcal/mol) but retarded by an unfavourable entropy term (TΔS = −6.9 kcal/mol). This is precisely as expected from the increased conformational entropy in the denatured state, relative to the more ordered helix-constrained peptides, that is lost upon binding to cFos [8]. Thus, truncation of the peptides has significantly reduced the enthalpic contribution to ΔG, but this is in part compensated for by a loss in the entropic barrier due to the helix-constraints. This is consistent with the helix-constraints pre-organizing the peptides into the receptor-binding conformation.
Structure and Stability of 24
2D proton NMR spectra were obtained for peptide 24. The NOE summary diagram (Figure 2) reveals small 3JNHHα coupling constants, low temperature coefficients Δδ/T, and many dαN(i, i+3) NOEs which collectively indicate that peptide 24 is heavily populated with helical structures in water. Strong d αN(i,i+4) and d αN(i,i+3), but only a few d αβ(i,i+2), NOEs reveal an alpha-, rather than 310-, helical structure. A structure calculation using these NMR restraints produced a highly convergent set of 20 lowest energy structures (RMSD = 1.87 Å over backbone heavy atoms) without any violations in distance restraints (>0.2 Å) or dihedral angles (>5°). The NMR-derived structures for peptide 24 are clearly in an alpha helical conformation (Figure 2), with an averaged backbone RMSD deviation from an idealized alpha helix of only 1.27 Å. However, the family of structures are somewhat flexible at the N- and C-terminal Cha residues. Figure 2 shows that thee three i→i+4 helix constraints are located on a helix face away from the face exposing the hydrophobic side chains (Cha1, I5, L8, A12, L15, I19, Cha22) for interaction with Fos peptide in a leucine zipper. This is consistent with the design intent to rigidly constrain the hydrophobic residues at desired positions of the heptad repeat for optimal exposure to cFos. Peptide 24 was also fairly stable in human serum over 4 h at 37°C, whereas the same sequences lacking the constraint were undetectable after 4 hours due to proteolytic cleavage (Figure 3).
Discussion
One, two, or three i→i+4 helix constraints have been introduced into truncated peptide derived from a 37 residue peptide sequence from cJun (JunWCANDI), itself a weak antagonist of cJun binding to cFos. The aim was to investigate whether (a) much shorter helix-constrained peptides could maintain high affinity binding to cFos by virtue of pre-organizing the sequence in a conformation ideal for interaction with cFos [42], (b) key side chains in the Jun-based peptide represented sequence “hot spots” which were responsible for most of the energetic contributions to binding with cFos, (c) enthalpy or entropy contributions were most influential for binding, and (d) the downsized, helix-constrained, peptides retained high specificity for cFos, competing with cJun.
Helix Induction
The use of i→i+4 helix-inducing constraints was successful in producing highly alpha helical peptides, allowing truncation of the sequence from 37 to 22 residues (but no shorter) without loss of helicity. A constraint near the C-terminus of the peptides (heptads 3 and 4) was more effective in inducing helicity and enhancing binding to cFos than a constraint introduced near the N-terminus (heptads 1 and 2). It was found that although CΔ7 truncation preserved some of the binding properties (8–9: Tm 32–38°C), NΔ10 deletion peptides (10–24: Tm 44–52°C) were much improved over NΔ14 deletions (5–7), suggesting a key role for residues d2-g2. In addition to constraint insertion, other amino acid substitutions were made to enhance core hydrophobic and electrostatic contacts. The addition of non-natural Cha sidechains to core residues within NΔ10 bicyclic peptides resulted in stabilisation with cFos for replacement of the two outermost d sidechains, suggesting that these act to constrain the helix to its target while maintaining overall helicity. In arriving at 24 from JunWCANDI over 40% of the molecule was removed while still increasing overall helicity from 19 to 69%.
Stability and Specificity
While constraining heptads 3 or 4 at the C-terminus was most effective in promoting binding to cFos, truncating both heptads 1 and 2 did impact the binding suggesting that at least part of the second heptad from the N-terminus is required for effective binding. Subsequent designs incorporated residues d2-g2, since the hydrophobic core appeared to be compromised if residue d2 was missing, and thus binding affinity is broadly distributed along the length of the peptide from residues d2-d5 (Figure 6). Qualitatively similar affinities were observed for all helix-constrained peptides, regardless of the size of the molecule. Importantly, all peptides capable of binding to cFos also retained specificity in the presence of cJun that was displayed by the CANDI-PCA selected parent peptide [42]. This was shown by dimer exchange experiments and thermal melt comparisons of equimolar solutions of peptide-cFos or peptide-cJun (Figures S2, S3, S4). Finally, the most helical peptide by CD measurements was 24, which was examined by 2D-NMR spectroscopy and an alpha helical structure was verified along its length (Figure 2). This peptide was much more stable in human serum at 37° than the same peptide lacking the constraints, due to a consequence of the helix constraints and unnatural amino acids at each end (Figure 3). Degradative proteolytic enzymes in serum are known to recognize only denatured or extended strand peptide conformations in their active sites, with helical structures being too large and protective of peptide bonds to be cleaved by proteases [54], [55]. In addition to serum stability hydrocarbon restraints have additionally been speculated to facilitate cell membrane permeability, with several studies demonstrating this directly [7], [15].
Figure 6. Coiled coil structure of the cJun-cFos interaction.
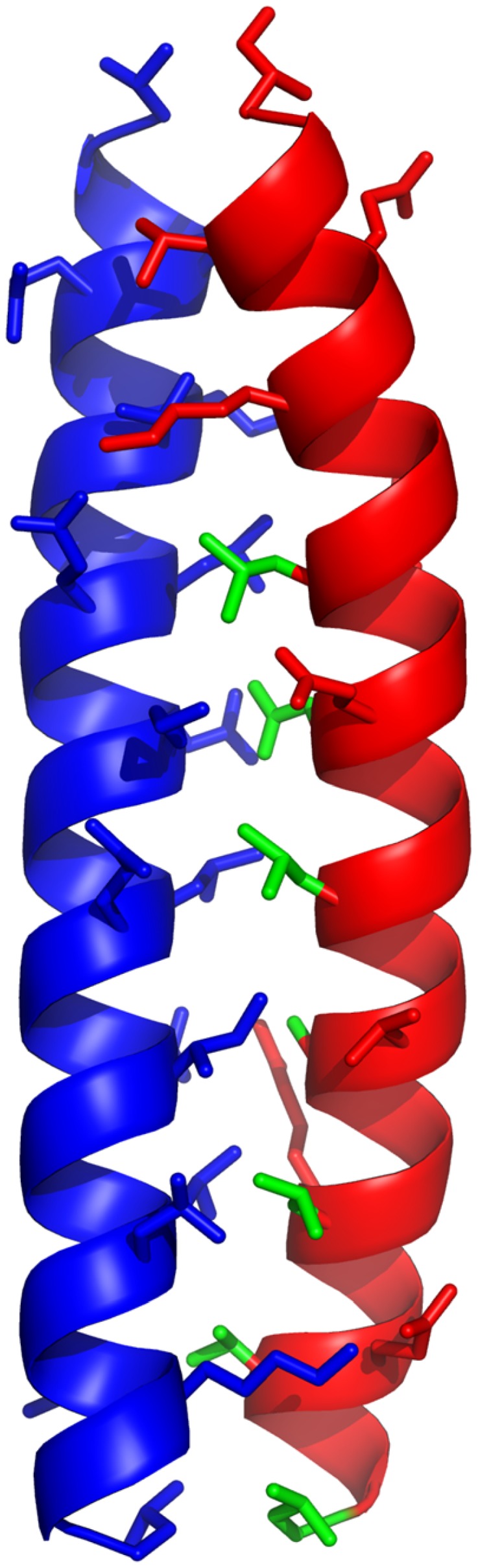
Shown are the coiled coil regions of cJun (red) and cFos (blue) interaction (PDB coordinates: 1FOS [58]). Side-chains for interfacial ‘ a’ ‘d’ ‘e’ and ‘g’ residues are shown and highlight the fact that the interactions between them are distributed broadly across the molecule. Key hydrophobic interfacial side-chains within cJun that are predicted to be required for effective binding to cFos are shown in green (d2, a3, d3, a4, d4, a5, d5, top to bottom: L, N, L, A, L, V, L).
Thermodynamics
Consistent with a reduction in conformational freedom imposed by the helix constraints, there is a measurable increase in the entropic contribution to cFos binding. TΔS was −6.9 kcal/mol for cFos-JunWCANDI versus −3.8 to +1.6 kcal/mol for a cFos-constrained peptide pair, with the triply bridged helical peptide 12 exhibiting a 7.5 kcal/mol more positive TΔS than for JunWCANDI. This contrasts with the weaker binding, but more helical, 24 where the Cha-Arg at the N-terminus and Cha at the C-terminus appear to disfavour binding. Thus favourable free energy changes on binding to cFos stem from a combination of enthalpic contributions, reduced upon peptide truncation, and more importantly, increased conformational entropy that arises from introduction of the helix-inducing constraints. This contrasts with the cFos-JunWCANDI interaction which was driven primarily by a larger enthalpy than for the much shorter constrained peptides. Because the coiled coil requires interactions along its entire length for thermodynamic stability, truncation of the peptide causes a large enough loss in enthalpy to compensate for the conformational entropy advantage that is conferred by the helix constraints. Thus the extent of sequence truncation has been limited here to splicing ∼40% off the sequence and this balancing act, between focussed entropic targeting of hot spots in a coiled coil (such as helical heptads at the C-terminus in this case) and losing enthalpy due to removal of many of the interactions that stabilize the coiled coil [56], may be more difficult to achieve than for other helical protein surfaces where there are more pronounced hot spots. In particular for 24, hydrophobic residues d2, a3, d3, a4, d4, a5 and d5 (Cha, I, L, A L, I, Cha) are predicted to make large enthalpic contributions to coiled-coil binding potency.
In summary, the affinity of cJun for cFos is dependent upon interactions along the entire length of these two helical coils, with the two C-terminal heptads contributing a little more enthalpy to the interaction energy than the two N-terminal heptads. Thus, shortening the sequence by over 40% from JunWCANDI to peptide 24 has correspondingly and proportionately reduced the free energy and enthalpy of interaction, which has been compensated for to some extent by pre-organizing the peptide in a stable alpha helical conformation. This has led to peptides that bind to cFos in the µM affinity range. In the future it may be possible to retain or further improve the affinity of peptide interactions with cFos by using further iterations of truncating the sequence, with alternative positioning of helix-conferring constraints, such as these or others, combined with non-natural amino acids, HBS approaches or the use of β-peptides to generate even smaller peptidomimetics of AP-1 components. However, the large surface area of interaction, the shallow binding pockets, and especially the coiled coil nature of the heterodimer make this and other transcription factors more challenging than many other protein alpha helices to mimic using smaller helix-constrained peptides. The extent to which helix-constrained transcription factors can be shortened is uncertain but this does represent a promising approach to generating smaller transcriptional regulators. This research is particularly timely given the rapid increase in knowledge from proteomics and interactomics studies on transcriptional regulators in signalling pathways.
Supporting Information
CD spectra for all constraints in this study. These are shown both in isolation and as a mixture with cFos. Data have been collected by measuring the level of helicity at 222 nm in an applied photophysics chirascan Circular Dichroism (CD) Spectrometer. Data have been converted from raw ellipticity to Molar Residue Ellipticity (MRE). From these raw data it is possible to see which heterodimers constitute an increase over the average of the homodimeric components (black dotted line). Any increase in the helical signal that exceeds the average of cFos (black) and the constrained peptide (blue), that would be anticipated for a non-interacting pair, is clearly observed in the heterodimeric profiles (red) and therefore strongly indicates the presence of an interaction.
(TIFF)
Raw thermal melting data for homo and heterodimeric complexes with cJun for constrained peptides 1 , 2 , 8 , 9, 10 , 11, 12, 17, 20, 22, and 24 . Shown are raw thermal melting data for all homo and heterodimeric complexes. Data have been collected by measuring the level of helicity at 222 nm in an applied photophysics chirascan Circular Dichroism (CD) Spectrometer. Data have been converted from raw ellipticity to Molar Residue Ellipticity (MRE) according to equation 1 to take account of the different peptide lengths. Thermal melting data for cJun is shown in black, data for the constrained peptide in isolation is shown in blue, the average of these two as a black dotted line, with the cJun/constrained peptide mixture is shown in red. It is clear that none of the peptides form a stable interaction with cJun.
(TIFF)
CD spectra as MRE both in isolation and as a mixture with cJun. From these raw data it is also clear that no interaction is occurring between constrained peptides and cJun. Rather, specta appear as averages of their homodimeric components (i.e. superimpose with the homomeric averages). We observe no heteromeric helical signal (red) that exceeds the average (black dotted line) of cJun (black) and the constrained peptide (blue), that would be anticipated for a non-interacting pair.
(TIFF)
Dimer exchange experiments between cJun, cFos and constrained peptide 24 . a) Equimolar mixures of cJun-cJun and cFos-24 are mixed and the observed signal closely resembles the average of the two constituent spectra, indicating no change has occurred. b) Equimolar mixtures of cFos-cFos and cJun-24 are mixed and the observed spectra greatly exceeds the average of the two constituent spectra, indicating that dimer exchange has occurred. c) Mixtures from a) and b) superimpose, indicating that the same species is populated in both cases.
(TIFF)
Helical Data obtained via Circular Dichroism. Shown are A) homo and B) heteromeric samples. Column 1 displays the 222/208 ratio which can be used as an indication of the presence of coiled coils. A ratio higher than 1 is generally indicated evidence that a coiled coil has formed, while a ratio of less than 0.9 is taken to indicate the presence of isolated helices. Shown in column two are the calculated fractional helicities taken from the Molar Residue Ellipticity at 22 nm. Fraction helicity (ƒH) can be calculated as ƒH = (θ222−θc)/(θ222∞−θc) where θ222∞ = (−44000+250*T)*(1−k/Nr) and θc = 2220–(53*T). In these equations the wavelength dependent constant k = 2.4 (at 222 nm), Nr = the number of residues, and T = 20 degrees Celsius (293 K). The 222/208 ratio is used to provide evidence on whether the helices are monomeric or are adopting a quaternary structure. Measured helicity for the original JunWCANDI peptide and for the template on which the hydrocarbon constraints have been introduced. These data take into account the constrained variants which have been shown to introduce significant helicity into the molecule. The N- and C-capping motifs have been removed for the constrained since they are considered to be largely redundant upon their introduction.
(DOC)
Assigned 1H NMR signals for peptide 24 in H2O:D2O (9∶1) at 298 K.
(DOC)
Supporting Information.
(DOC)
Acknowledgments
The authors also wish to thank Dr Miao Yu for technical assistance.
Funding Statement
JMM gratefully acknowledges support from the Wellcome Trust (WT090184MA) and Cancer Research UK for a career establishment award (DBB3800). DPF acknowledges the Australian Research Council (FF668733, DP00770936) and National Health and Medical Research Council (511194, APP1028423, APP1027369) for grant support and for ARC Federation and NHMRC Senior Principal Research Fellowships. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Mason JM (2010) Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med Chem 2: 1813–1822. [DOI] [PubMed] [Google Scholar]
- 2. Fairlie DP, West ML, Wong AK (1998) Towards protein surface mimetics. Curr Med Chem 5: 29–62. [PubMed] [Google Scholar]
- 3. Derossi D, Joliot AH, Chassaing G, Prochiantz A (1994) The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 269: 10444–10450. [PubMed] [Google Scholar]
- 4. Fuchs SM, Raines RT (2006) Internalization of cationic peptides: the road less (or more?) traveled. Cell Mol Life Sci 63: 1819–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vives E, Brodin P, Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 272: 16010–16017. [DOI] [PubMed] [Google Scholar]
- 6. Judice JK, Tom JY, Huang W, Wrin T, Vennari J, et al. (1997) Inhibition of HIV type 1 infectivity by constrained alpha-helical peptides: implications for the viral fusion mechanism. Proc Natl Acad Sci U S A 94: 13426–13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, et al. (2004) Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305: 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kutchukian PS, Yang JS, Verdine GL, Shakhnovich EI (2009) All-atom model for stabilization of alpha-helical structure in peptides by hydrocarbon staples. J Am Chem Soc 131: 4622–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harrison RS, Shepherd NE, Hoang HN, Ruiz-Gomez G, Hill TA, et al. (2010) Downsizing human, bacterial, and viral proteins to short water-stable alpha helices that maintain biological potency. Proc Natl Acad Sci U S A 107: 11686–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, et al. (2010) Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc Natl Acad Sci U S A 107: 14093–14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shepherd NE, Hoang HN, Abbenante G, Fairlie DP (2005) Single turn peptide alpha helices with exceptional stability in water. J Am Chem Soc 127: 2974–2983. [DOI] [PubMed] [Google Scholar]
- 12. Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL (2007) Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc 129: 2456–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, et al. (2006) A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 24: 199–210. [DOI] [PubMed] [Google Scholar]
- 14. Bird GH, Bernal F, Pitter K, Walensky LD (2008) Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzymol 446: 369–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, et al. (2009) Direct inhibition of the NOTCH transcription factor complex. Nature 462: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim YW, Verdine GL (2009) Stereochemical effects of all-hydrocarbon tethers in i,i+4 stapled peptides. Bioorg Med Chem Lett 19: 2533–2536. [DOI] [PubMed] [Google Scholar]
- 17. Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, et al. (2010) A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 18: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim YW, Grossmann TN, Verdine GL (2011) Synthesis of all-hydrocarbon stapled alpha-helical peptides by ring-closing olefin metathesis. Nat Protoc 6: 761–771. [DOI] [PubMed] [Google Scholar]
- 19. Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, et al. (2012) Structure of the stapled p53 peptide bound to Mdm2. J Am Chem Soc 134: 103–106. [DOI] [PubMed] [Google Scholar]
- 20. Chapman RN, Dimartino G, Arora PS (2004) A highly stable short alpha-helix constrained by a main-chain hydrogen-bond surrogate. J Am Chem Soc 126: 12252–12253. [DOI] [PubMed] [Google Scholar]
- 21. Wang D, Liao W, Arora PS (2005) Enhanced metabolic stability and protein-binding properties of artificial alpha helices derived from a hydrogen-bond surrogate: application to Bcl-xL. Angew Chem Int Ed Engl 44: 6525–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang D, Chen K, Kulp Iii JL, Arora PS (2006) Evaluation of biologically relevant short alpha-helices stabilized by a main-chain hydrogen-bond surrogate. J Am Chem Soc 128: 9248–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang D, Chen K, Dimartino G, Arora PS (2006) Nucleation and stability of hydrogen-bond surrogate-based alpha-helices. Org Biomol Chem 4: 4074–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patgiri A, Jochim AL, Arora PS (2008) A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation. Acc Chem Res 41: 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chapman R, Kulp JL 3rd, Patgiri A, Kallenbach NR, Bracken C, et al (2008) Trapping a folding intermediate of the alpha-helix: stabilization of the pi-helix. Biochemistry 47: 4189–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu J, Wang D, Zheng Q, Lu M, Arora PS (2008) Atomic structure of a short alpha-helix stabilized by a main chain hydrogen-bond surrogate. J Am Chem Soc 130: 4334–4337. [DOI] [PubMed] [Google Scholar]
- 27. Wang D, Lu M, Arora PS (2008) Inhibition of HIV-1 fusion by hydrogen-bond-surrogate-based alpha helices. Angew Chem Int Ed Engl 47: 1879–1882. [DOI] [PubMed] [Google Scholar]
- 28. Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, et al. (2010) Inhibition of hypoxia inducible factor 1-transcription coactivator interaction by a hydrogen bond surrogate alpha-helix. J Am Chem Soc 132: 941–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guarracino DA, Bullock BN, Arora PS (2011) Mini review: protein-protein interactions in transcription: a fertile ground for helix mimetics. Biopolymers 95: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bullock BN, Jochim AL, Arora PS (2011) Assessing helical protein interfaces for inhibitor design. J Am Chem Soc 133: 14220–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Appella DH, Christianson LA, Klein DA, Powell DR, Huang X, et al. (1997) Residue-based control of helix shape in beta-peptide oligomers. Nature 387: 381–384. [DOI] [PubMed] [Google Scholar]
- 32. Seebach D, Gardiner J (2008) Beta-peptidic peptidomimetics. Acc Chem Res 41: 1366–1375. [DOI] [PubMed] [Google Scholar]
- 33. Shepherd NE, Abbenante G, Fairlie DP (2004) Consecutive cyclic pentapeptide modules form short alpha-helices that are very stable to water and denaturants. Angew Chem Int Ed Engl 43: 2687–2690. [DOI] [PubMed] [Google Scholar]
- 34. Shepherd NE, Hoang HN, Desai VS, Letouze E, Young PR, et al. (2006) Modular alpha-helical mimetics with antiviral activity against respiratory syncitial virus. J Am Chem Soc 128: 13284–13289. [DOI] [PubMed] [Google Scholar]
- 35. Pace CN, Grimsley GR, Thomson JA, Barnett BJ (1988) Conformational stability and activity of ribonuclease T1 with zero, one, and two intact disulfide bonds. J Biol Chem 263: 11820–11825. [PubMed] [Google Scholar]
- 36. Ozanne BW, Spence HJ, McGarry LC, Hennigan RF (2007) Transcription factors control invasion: AP-1 the first among equals. Oncogene 26: 1–10. [DOI] [PubMed] [Google Scholar]
- 37. Eferl R, Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859–868. [DOI] [PubMed] [Google Scholar]
- 38. Libermann TA, Zerbini LF (2006) Targeting transcription factors for cancer gene therapy. Curr Gene Ther 6: 17–33. [DOI] [PubMed] [Google Scholar]
- 39. Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, et al. (2008) Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther 10: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner EF, Eferl R (2005) Fos/AP-1 proteins in bone and the immune system. Immunol Rev 208: 126–140. [DOI] [PubMed] [Google Scholar]
- 41. Aud D, Peng SL (2006) Mechanisms of disease: Transcription factors in inflammatory arthritis. Nat Clin Pract Rheumatol 2: 434–442. [DOI] [PubMed] [Google Scholar]
- 42. Mason JM, Muller KM, Arndt KM (2007) Positive aspects of negative design: simultaneous selection of specificity and interaction stability. Biochemistry 46: 4804–4814. [DOI] [PubMed] [Google Scholar]
- 43. Lavigne P, Sonnichsen FD, Kay CM, Hodges RS (1996) Interhelical salt bridges, coiled-coil stability, and specificity of dimerization. Science 271: 1136–1138. [DOI] [PubMed] [Google Scholar]
- 44. O’Shea EK, Lumb KJ, Kim PS (1993) Peptide ‘Velcro’: design of a heterodimeric coiled coil. Curr Biol 3: 658–667. [DOI] [PubMed] [Google Scholar]
- 45. O’Shea EK, Rutkowski R, Kim PS (1989) Evidence that the leucine zipper is a coiled coil. Science 243: 538–542. [DOI] [PubMed] [Google Scholar]
- 46. O’Shea EK, Rutkowski R, Kim PS (1992) Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell 68: 699–708. [DOI] [PubMed] [Google Scholar]
- 47. Mason JM, Schmitz MA, Muller KM, Arndt KM (2006) Semirational design of Jun-Fos coiled coils with increased affinity: Universal implications for leucine zipper prediction and design. Proc Natl Acad Sci U S A 103: 8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mason JM, Hagemann UB, Arndt KM (2009) Role of hydrophobic and electrostatic interactions in coiled coil stability and specificity. Biochemistry 48: 10380–10388. [DOI] [PubMed] [Google Scholar]
- 49. Crooks RO, Rao T, Mason JM (2011) Truncation, Randomization, and Selection: Generation of a Reduced Length c-Jun Antagonist That Retains High Interaction Stability. J Biol Chem 286: 29470–29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dixon JM, Taniguchi M, Lindsey JS (2005) PhotochemCAD 2: a refined program with accompanying spectral databases for photochemical calculations. Photochem Photobiol 81: 212–213. [DOI] [PubMed] [Google Scholar]
- 51. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, et al. (1983) CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4: 187–217. [Google Scholar]
- 52. Ibarra-Molero B, Makhatadze GI, Matthews CR (2001) Mapping the energy surface for the folding reaction of the coiled-coil peptide GCN4-p1. Biochemistry 40: 719–731. [DOI] [PubMed] [Google Scholar]
- 53. Krylov D, Barchi J, Vinson C (1998) Inter-helical interactions in the leucine zipper coiled coil dimer: pH and salt dependence of coupling energy between charged amino acids. J Mol Biol 279: 959–972. [DOI] [PubMed] [Google Scholar]
- 54. Fairlie DP, Tyndall JD, Reid RC, Wong AK, Abbenante G, et al. (2000) Conformational selection of inhibitors and substrates by proteolytic enzymes: implications for drug design and polypeptide processing. J Med Chem 43: 1271–1281. [DOI] [PubMed] [Google Scholar]
- 55. Tyndall JD, Nall T, Fairlie DP (2005) Proteases universally recognize beta strands in their active sites. Chem Rev 105: 973–999. [DOI] [PubMed] [Google Scholar]
- 56. Johnson LM, Horne WS, Gellman SH (2011) Broad distribution of energetically important contacts across an extended protein interface. J Am Chem Soc 133: 10038–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wiseman T, Williston S, Brandts JF, Lin LN (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem 179: 131–137. [DOI] [PubMed] [Google Scholar]
- 58. Glover JN, Harrison SC (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 373: 257–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CD spectra for all constraints in this study. These are shown both in isolation and as a mixture with cFos. Data have been collected by measuring the level of helicity at 222 nm in an applied photophysics chirascan Circular Dichroism (CD) Spectrometer. Data have been converted from raw ellipticity to Molar Residue Ellipticity (MRE). From these raw data it is possible to see which heterodimers constitute an increase over the average of the homodimeric components (black dotted line). Any increase in the helical signal that exceeds the average of cFos (black) and the constrained peptide (blue), that would be anticipated for a non-interacting pair, is clearly observed in the heterodimeric profiles (red) and therefore strongly indicates the presence of an interaction.
(TIFF)
Raw thermal melting data for homo and heterodimeric complexes with cJun for constrained peptides 1 , 2 , 8 , 9, 10 , 11, 12, 17, 20, 22, and 24 . Shown are raw thermal melting data for all homo and heterodimeric complexes. Data have been collected by measuring the level of helicity at 222 nm in an applied photophysics chirascan Circular Dichroism (CD) Spectrometer. Data have been converted from raw ellipticity to Molar Residue Ellipticity (MRE) according to equation 1 to take account of the different peptide lengths. Thermal melting data for cJun is shown in black, data for the constrained peptide in isolation is shown in blue, the average of these two as a black dotted line, with the cJun/constrained peptide mixture is shown in red. It is clear that none of the peptides form a stable interaction with cJun.
(TIFF)
CD spectra as MRE both in isolation and as a mixture with cJun. From these raw data it is also clear that no interaction is occurring between constrained peptides and cJun. Rather, specta appear as averages of their homodimeric components (i.e. superimpose with the homomeric averages). We observe no heteromeric helical signal (red) that exceeds the average (black dotted line) of cJun (black) and the constrained peptide (blue), that would be anticipated for a non-interacting pair.
(TIFF)
Dimer exchange experiments between cJun, cFos and constrained peptide 24 . a) Equimolar mixures of cJun-cJun and cFos-24 are mixed and the observed signal closely resembles the average of the two constituent spectra, indicating no change has occurred. b) Equimolar mixtures of cFos-cFos and cJun-24 are mixed and the observed spectra greatly exceeds the average of the two constituent spectra, indicating that dimer exchange has occurred. c) Mixtures from a) and b) superimpose, indicating that the same species is populated in both cases.
(TIFF)
Helical Data obtained via Circular Dichroism. Shown are A) homo and B) heteromeric samples. Column 1 displays the 222/208 ratio which can be used as an indication of the presence of coiled coils. A ratio higher than 1 is generally indicated evidence that a coiled coil has formed, while a ratio of less than 0.9 is taken to indicate the presence of isolated helices. Shown in column two are the calculated fractional helicities taken from the Molar Residue Ellipticity at 22 nm. Fraction helicity (ƒH) can be calculated as ƒH = (θ222−θc)/(θ222∞−θc) where θ222∞ = (−44000+250*T)*(1−k/Nr) and θc = 2220–(53*T). In these equations the wavelength dependent constant k = 2.4 (at 222 nm), Nr = the number of residues, and T = 20 degrees Celsius (293 K). The 222/208 ratio is used to provide evidence on whether the helices are monomeric or are adopting a quaternary structure. Measured helicity for the original JunWCANDI peptide and for the template on which the hydrocarbon constraints have been introduced. These data take into account the constrained variants which have been shown to introduce significant helicity into the molecule. The N- and C-capping motifs have been removed for the constrained since they are considered to be largely redundant upon their introduction.
(DOC)
Assigned 1H NMR signals for peptide 24 in H2O:D2O (9∶1) at 298 K.
(DOC)
Supporting Information.
(DOC)