

Abstract
Melanocytes undergo extensive genetic changes during transformation into aggressive melanomas. These changes deregulate genes whose aberrant activity promotes the development of this disease. The PI3K (phosphoinositide 3-kinase) and MAP (mitogen-activated protein) kinase pathways are two key signaling cascades that have been found to play prominent roles in melanoma development. These pathways relay extra-cellular signals via an ordered series of consecutive phosphorylation events from cell surface throughout the cytoplasm and nucleus regulating diverse cellular processes including proliferation, survival, invasion and angiogenesis. It is generally accepted that therapeutic agents would need to target these two pathways to be an effective therapy for the long-term treatment of advanced-stage melanoma patients. This review provides an overview of the PI3 kinase pathway focusing specifically on two members of the pathway, called PTEN and Akt3, which play important roles in melanoma development. Mechanisms leading to deregulation of these two proteins and therapeutic implications of targeting this signaling cascade to treat melanoma are detailed in this review.
Keywords: melanoma, Akt3, PTEN, oncogene, tumorigenesis, apoptosis
1. INTRODUCTION
Identification of signaling pathways and proteins involved in melanoma progression and drug resistance has the potential to lead to the development of novel, more effective targeted therapies. Two of these pathways are the PI3 (phosphoinositide 3-kinase) and MAP (mitogen-activated protein) kinase signaling cascades involved in melanoma development by relaying extra-cellular signals to regulate diverse cellular processes including proliferation, survival, invasion and angiogenesis (Smalley and Herlyn, 2005, Hsu et al., 2002, Chudnovsky et al., 2005). It is generally accepted in the melanoma research and clinical communities that therapeutic agents targeting these two pathways will be effective for the long-term treatment of advanced-stage patients; but which members of the cascades to target remain to be established. This review provides an overview of the PI3 kinase pathway in melanoma development detailing mechanisms leading to deregulation of PTEN and Akt3 in this cascade, cellular processes regulated by these proteins and therapeutic implications of targeting these proteins to treat this deadly disease. Finally, key issues that remain to be answered concerning this important signaling cascade are discussed.
1.1 Introduction to PTEN as a cancer suppressor gene
The PTEN (phosphatase and tensin homologue deleted from chromosome 10) gene, which is also known as MMAC1 (mutated in multiple advanced cancers) and TEP1 (TGF-β regulated and epithelial cell-enriched phosphatase) is an important tumor suppressor gene located at 10q23-24 (Wu et al., 2003, Robertson et al., 1998a). PTEN is a unique 55 kDa dual specificity phosphatase dephosphorylating phosphoserine and phosphotyrosine residues in proteins (Lee et al., 1999, Maehama and Dixon, 1999, Waite and Eng, 2002). It is also a lipid phosphatase hydrolyzing the secondary messenger inositol trisphosphates (PIP3s) (Maehama andDixon, 1999, Gericke et al., 2006). Structurally, it is the N-terminal phosphatase domain of PTEN from amino acids 22 to 185 that catalyzes the hydrolysis of phospholipids. The central C2 lipid binding domain from amino acids 190 – 351 and c-terminal PDZ ligand sequence from amino acids 401 – 403 are associated with binding to the lipid (Miller et al., 2002, Gericke et al., 2006, Tamguney and Stokoe, 2007). PTEN also contains the characteristic protein phosphatase signature sequence “HCKAGKGR” spanning amino acids 123 – 130 (Fig. 1A) (Maehama et al., 2004). This signature sequence also contains two lysine residues that are key for establishing interactions with negatively charged PIPs (Maehama et al., 2004).
Figure 1. Structure of the PTEN and Akt3 proteins.

1A PTEN, a tumor suppressor protein and lipid phosphatase, is a 55 kDa enzyme containing an N-terminal phosphatase domain, a central C2 domain, and a C-terminal tail. Each domain has sites for post-translational modifications that could involve: acetylation, oxidation, ubiquitination, or phosphorylation, which have potential to regulate PTEN activity. 1B. Structural analysis of Akt3 reveals that this oncogenic survival kinase increasingly active during melanoma progression consists of an N-terminal PH domain, a central kinase domain and a C-terminal regulatory motif. Complete activation of this kinase occurs only when the threonine 308 (T308) and serine 472 (S472) gets phosphorylated by PDK1 and a PDK2.
Although, PTEN exhibits both protein and lipid phosphatase activities, it is the lipid phosphatase activity that is generally thought to be the major function of this phosphatase (Maehama and Dixon, 1999, Waite and Eng, 2002). The PTEN active site is structurally designed to facilitate electrostatic interactions with phosphatidylinositol 3,4,5-trisphosphate (PIP3) for binding and hydrolysis (Lee et al., 1999). As a proteinphosphatase, PTEN can dephosphorylate: (a) focal adhesion kinase, regulating cell-to-cell adhesion, and: (b) the Src homologous and collagen gene (an adaptor protein) to disrupt cell spreading and migration. PTEN can also suppress MAPK signaling activity to modulate cell-to-cell adhesion and migration (Li and Ross, 2007, Waite and Eng, 2002, Wu et al., 2003). In its role as a lipid phosphatase, PTEN removes a phosphate group at the 3rd position from cellular PIP3 and phosphatidylinositol 3,4-bisphosphate (PIP2) thereby decreasing the activities ofseveral proteins or pathways whose activities are regulated by this lipid thereby modulating cell proliferation, survival and apoptosis (Wu et al., 2003, Mikhail et al., 2005).
PTEN-null embryonic fibroblasts have elevated levels of PIP3 leading to constitutive Akt activity, whichdemonstrates that PTEN regulates the Akt pathway (Wu et al., 1998a, Dahia et al., 1999). PTEN is essential for murine embryonic development beyond day 7.5 (Di Cristofano et al., 1998) and loss of a single PTEN allele leads to hyperplasia and dysplasia in the skin, gastrointestinal tract, and prostate, as well as tumor formation (Di Cristofano et al., 1998). PTEN protein is also absent or functionally inactivated in many different tumor types causing increased Akt activation (Wu et al., 1998b, Dahia et al., 1999). PTEN expression in cells can suppress colony formation, growth in soft agar, and tumor formation in nude mice (Cheney et al., 1998).
1.2 Regulation of PTEN activity
PTEN expression and activity are regulated at transcriptional and post-translational levels (Robertson et al., 1998a, Mirmohammadsadegh et al., 2006, Gericke et al., 2006). Alterations in PTEN promoter activity have been shown to modulate cellular protein levels in response to positive and negative regulators (Gericke et al., 2006, Maehama et al., 2004). Some of the positive regulators of PTENpromoter activity include EGR-1 (early growth regulated transcription factor), PPARγ (peroxisome proliferator-activated receptors) and p53 (Gericke et al., 2006). Negative regulators such as MKK-4 (mitogen activated protein kinase kinase 4), NFκB (nuclear factor kappa-light-chain-enhancer of activated B-cells), TGF-β (transforming growth factor beta) and c-JUN have been shown to inhibit PTEN protein expression in several cancers (Wu et al., 2003, Lopez-Bergami et al., 2008, Gericke et al., 2006). Although, these pathways have been demonstrated to regulate melanoma development, detailed mechanisms by which they inhibit PTEN activity remain to be established.
PTEN activity is also modulated by interaction with lipids and proteins. Recently, PICT-1 (protein interacting with the C-tail-1) has been shown to interact with the C-tail of PTEN thereby regulating its turnover (Gericke et al., 2006, Tamguney and Stokoe, 2007). SiRNA mediated down regulation of PICT-1 protein levels led to destabilization of PTEN and decreased protein levels. Aberrant PICT-1 mRNA levels have been reported in human gliomas, neuroblastomas and ovarian carcinomas (Tamguney and Stokoe, 2007). However, its expression and activity patterns in melanomas are yet to be studied.
Post-translational modifications such as phosphorylation, oxidation, acetylation and ubiquitination can also regulate the activity of this phosphatase (Fig. 1A) (Tamguney and Stokoe, 2007, Miller et al., 2002, Gericke et al., 2006). The oxidation of cysteine residues C71 and C124 by reactive oxygen species inhibits the phosphatase activity of PTEN. Acetylation is another mechanism by which PTEN activity is negatively regulated in many cancer cell types. The histone acetyltransferase PCAF (p300/CBP-associated factor) acetylates lysine residues 125 and 128 (located within the catalytic cleft) of PTENthereby decreasing affinity towards PIP3s. PCAF regulates melanoma development by acetylation, thereby stabilizing NFκB in the nucleus (Ueda et al., 2007). Regulation of PTEN activity by PCAF inmelanomas warrants investigation.
Phosphorylation of the C-terminal tail of PTEN has been found to regulate PTEN activity. The C-terminal tail of PTEN contains multiple serine and threonine residues (S362, T366, S370, S380, T382, T383, S385), which can be phosphorylated (Fig. 1A) (Miller et al., 2002, Tamguney and Stokoe, 2007). Amino acids S370 and S385 are phosphorylated by CK2 (casein kinase 2). Recently, other kinases suchas LKB1, Src, GSK3β have also been shown to play a role in PTEN phosphorylation (Tamguney andStokoe, 2007, Gericke et al., 2006). Phosphorylated PTEN is stable but less active compared to unphosphorylated PTEN, which is less stable (Gericke et al., 2006, Dahia, 2000). C-tail mutants of PTEN, including deletions and missense mutations, tend to be short lived. A deletion of the C-terminal 18 amino acids has been shown to completely destabilize PTEN protein. Furthermore, amino acid substitutions at L345 or T348 to L345Q and T348I, respectively, produce highly unstable proteins, contributing to ~20% of known tumor-associated PTEN mutations (Dahia, 2000, Tamguney and Stokoe, 2007, Maehama et al., 2004). It is uncertain whether these occur in melanoma.
The sub-cellular distribution of PTEN is a further key to its activity and may play a role in cancer development (Wu et al., 2003, Gericke et al., 2006). C-tail phosphorylation can influence translocation ofPTEN to the cell membrane. Kinases such as CK2 and GSK3β (glycogen synthase kinase) have been shown to phosphorylate PTEN thereby decreasing its association with the cell membrane (Al-Khouri et al., 2005, Gericke et al., 2006). Cytosolic PTEN is inactive but highly stable. PTEN has intrinsic membrane translocation signals in the phosphatase and C2 domains. Mutations in these regions impair its ability to translocate to the plasma membrane. Additionally, PTEN localization and stability were regulated by interaction with other proteins (Gericke et al., 2006, Dahia, 2000, Tamguney and Stokoe, 2007). For example monoubiquitination of PTEN promotes nuclear localization whereas polyubiquitination causes the protein to remain in the cytosol. Ubiquitination of PTEN is catalyzed by NEDD4-1 (neural precursor cell expressed, developmentally down-regulated 4) (Kim et al., 2008, Wang et al., 2007b). However, whether similar ubiquitination patterns occur in melanomas need investigation.
Finally, genetic and epigenetic mechanisms such as promoter methylation, mutations, chromosomal deletions, and expression of endogenous microRNAs have also been shown to modulate the expression and activity of this important phosphatase (Waite and Eng, 2002, Gericke et al., 2006). Genetic mechanisms regulating PTEN expression are: (a) loss of the whole chromosome, (b) homozygous deletion, (c) frameshift or point mutations, and (d) inframe deletion and truncation (Yin and Shen, 2008, Li and Ross, 2007). Loss of chromosome 10, which contains the PTEN gene, has been reported in several cancers, including ~30–60% of non-inherited melanomas (Stahl et al., 2003). Furthermore, mutations in the PTEN gene have been shown to be associated with ~28% of glioblastomas, ~34% endometrial carcinomas, ~11.8% prostate cancer and ~12% melanomas (Yin and Shen, 2008). Mechanistically, loss or inactivation of PTEN leads to Akt activation, which upon induction leads to the phosphorylation and monoubiquitination of DNA damage checkpoint kinase, Chk1, causing genomic instability, double stranded DNA breaks and ultimately, cancer development (Yin and Shen, 2008). Epigenetic modifications of PTEN arise by intrinsic (development, differentiation and cancer) or extrinsic (environmental) factors and can be reversed by chemical or environmental treatment modalities. CpG rich regions of DNA, also known as CpG islands, which are distributed throughout the whole genome, have been shown to undergo methylation in several cancers including melanoma (Costello et al., 2000, Furuta et al., 2004, Wu et al., 2003). DNA methylation is a well-studied epigenetic mechanism for silencing of PTEN in melanoma (Furuta et al., 2004, Mirmohammadsadegh et al., 2006, Liu et al., 2008).
MicroRNAs are small non-coding RNAs of ~22 nucleotides long (Wang and Lee, 2009). Recent studies have shown that microRNAs can negatively regulate gene expression at the post-transcriptional and/or translational level in different cell types (Wang and Lee, 2009). MicroRNAs targeting PTEN gene expression have been identified in several cancers (Huang et al., 2009, Yang et al., 2008, Pezzolesi et al., 2008). miR-214 has been shown to regulate PTEN translation by binding to its 3′-UTR, which in turn leads to Akt activation and chemoresistance in human ovarian cancers (Yang et al., 2008). In a different study, miR-21 has been demonstrated to cause hepato-cellular carcinoma by inhibiting PTEN expression (Meng et al., 2007). However, microRNAs regulating PTEN expression in melanoma have not yet been identified. Since melanomas are highly resistant to chemotherapies and currently no treatment options are available for combating the advanced stage disease, identification of microRNAs regulating PTEN expression has significant potential to lead to novel therapeutic interventions.
1.3 The Akt family as oncogenes
The Akt kinase family consists of three protein kinases Akt1 (PKBα), Akt2 (PKBβ) and Akt3 (PKBγ) (Brazil et al., 2002, Nicholson and Anderson, 2002). Akt isoforms share >80% amino acid homology, containing three structurally distinct functional domains (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002) (Akt3 shown in Fig. 1B). The N-terminal pleckstrin homology domain spans amino acids 1 to 107, mediating protein-protein and protein-lipid interactions (Lietzke et al., 2000, Ferguson et al., 2000). The central catalytic domain contains a threonine residue (T305) whose phosphorylation is essential for activating this protein (Jones et al., 1991, Andjelkovic et al., 1995). The carboxy terminal tail region, known as the regulatory domain, contains a second phosphorylation site serine (S472). Phosphorylation of this second site is required for complete Akt3 activation (Fig. 1B). In response to growth factors or other extracellular stimuli both sites become phosphorylated, resulting in the complete activation of Akt isoforms (Alessi et al., 1996). Other possible phosphorylation sites may also be important and research in this area continues (Alessi et al., 1996). Splice variants of Akt3 lacking serine 472 have been identified but the significance of this form remains unknown (Konishi et al., 1995, Brodbeck et al., 2001).
1.4 Regulation of Akt activity
Activity of the Akt isoforms is regulated through similar mechanisms, but activity of a particular isoform is cell type dependent (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002). In normal cells, growth factors bind to a cell surface receptor or through G-protein-coupled receptors, activating PI3K as shown in Fig. 2 (Burgering and Coffer, 1995, Franke et al., 1995). Active PI3K then phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) on the 3-OH group generating thesecond messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) (Vanhaesebroeck and Alessi, 2000). PIP3 does not activate Akt directly, but rather promotes Akt translocation to the plasma membrane, where altered conformation of the protein mediated by PIP3 binding allows subsequent phosphorylation by phosphoinositide-dependent kinase-1 (PDK-1).
Figure 2. PI3K and MAPK signaling interactions in melanoma.

PI3 and MAP kinase signaling cascades regulate cell senescence, proliferation and apoptosis. Whereas the Ras/Raf/Mek/Erk signaling arm is primarily involved in controlling proliferation, it also induce cell senescence in benign nevi to inhibit further progression into melanomas. The survival kinase Akt3 inhibits apoptosis and induces proliferation by phosphorylation of substrate proteins. Recently, Akt3 has been shown to inhibit V600EB-Raf activity by phosphorylation thereby promoting early melanoma development.
Akt3 activity is regulated by phosphorylation at threonine 305 and serine 472 (Figs. 1B and 2). Phosphorylation at threonine 305, which is mediated by PDK1, partially activates Akt3, but full activation also requires phosphorylation of serine 472 (Alessi et al., 1996). Mechanism mediating serine 472 phosphorylation on Akt3, though controversial, is suspected to involve a second PDK, called PDK2; the identity of this factor remains unclear (Chan and Tsichlis, 2001). Integrin-linked kinase (ILK) and mTORC2 can phosphorylate serine 472. However, studies have shown that ILK only acts as a facilitator; leaving mTORC2 as the PDK2 candidate (Delcommenne et al., 1998, Alessi et al., 1996). PDK2 activity of the mTOR complex has been recently established but in melanomas its role in maintaining Akt3 phosphorylation has not been studied (Huang and Manning, 2009, Feldman et al., 2009). Akt autophosphorylation might also play a role in the activation process (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002). Thus, basal Akt activity in a cell depends upon a balance between positive signals from elevated PIP3 levels and negative signals leading to Akt dephosphorylation and inactivation. Since this review focuses on Akt signaling in melanoma, subsequent sections discuss what is known about Akt3 deregulation in melanomas and the signals that regulate its activity.
2. PTEN AND AKT3 IN HUMAN MELANOMAS
2.1 PTEN is a key phosphatase regulating melanoma development
PTEN is a tumor suppressor inhibiting melanoma cell growth (Weng et al., 1999) by increasing susceptibility of cells to apoptosis (Lu et al., 1999). Adenovirus mediated transfer of PTEN into melanoma cells containing wild-type PTEN alleles led to inhibition of phosphorylated (active) Akt, increasing apoptosis and reducing growth rates (Stewart et al., 2002), which demonstrated the importance of this protein in melanoma (Figs. 3A and 3B). PTEN loss preferrentially regulates Akt3 activity in melanoma (Stahl et al., 2004). SiRNA-mediated reduction of PTEN specifically increased Akt3 phosphorylation (activity) in melanocytes and early stage melanoma cells (WM35) expressing wild-type PTEN protein, thereby reinforcing the significance of Akt3 involvement in melanoma development (Fig. 4) (Stahl et al., 2004).
Figure 3. PTEN regulates apoptosis by inhibiting Akt activity in melanomas.

3A PTEN expression decreases pAkt levels in melanoma ccells lacking functional PTEN. UACC 903 melanoma cells lacking functional PTEN protein were infected with increasing amounts (7.8, 15.6, 31.3, 62.5, 125 μL) of adeno-associated viral constructs containing Wt PTEN, HA tagged Wt PTEN and a catalytically inactive G129R PTEN mutation. Three days later, cell lysates were collected and proteins analyzed by Western blotting. Levels of pAkt and total Akt were quantitated by densitometry and the pAkt/totalAkt ratio represented against amount of viral supernatant used for infection. Data show that pAkt expression decreases with increasing virus amount indicating PTEN reduces pAkt in melanomas (Stahl et al., 2003). 3B. PTEN triggers apoptosis in melanomas. Levels of cleaved caspase-3, an indicator of apoptosis, were measured using Western blotting following expression of viral introduced PTEN into UACC 903 melanoma cells lacking functional PTEN protein. Compared to functionally inactive G129R PTEN, wild type PTEN expression increased levels of cleaved caspase-3 indicating elevatedlevels of apoptosis. α-enolase served as control for protein loading (Stahl et al., 2003).
Figure 4. PTEN loss leads to preferential increase in Akt3 activity in melanomacytes and radial growth phase melananoma cells.

Normal human melanocytes and a cell line derived from a radial growth phase of melanoma (WM35) that contain low pAkt levels were transfected with siRNA targeting PTEN and/or one of the isoforms of Akt. A significant decrease in pAkt was noticed only when PTEN and Akt3 were targeted together but not with either PTEN and Akt1 or PTEN and Akt2, indicating that Akt3 is activated preferentially following PTEN loss (Stahl et al., 2004).
PTEN acts as a potent growth suppressor in melanomas (Robertson et al., 1998b, Stahl et al., 2003, Robertson et al., 1999b). The growth-suppressive effects of PTEN were significant enough to cause fragmentation of a normal copy of chromosome 10 introduced into melanoma cell line UACC 903 that lacks PTEN protein due to the presence of a truncating point mutation (Robertson et al., 1998b). The introduction of a normal chromosome 10 containing wild type PTEN into UACC 903 cells reduced the cellular growth potential, causing fragmentation of the introduced chromosome in order to eliminate the growth suppressive effects mediated by PTEN. The introduced chromosome 10 underwent deletion, which mirrored alterations occurring in tumors from melanoma patients leading to identification of PTEN as the cause of this phenomenon (Thompson et al., 1995). It was this observation that led to the functional identification of PTEN as the suppressor gene on the long arm of chromosome 10 important in melanoma, which was subsequently validated through identification of a point mutation truncating the protein in the cell line preventing protein production (Robertson et al., 1998b). Expression of wild-type PTEN in these cells, reduced cell growth and increased sensitivity of cells to apoptotic stimuli (Robertson et al., 1998b). Similar reports have confirmed that expression of PTEN in cell lines lacking the protein, reduced levels of phosphorylated Akt and downstream substrates such as GSK3β and PRAS40 (Li et al., 1998, Davies et al., 1998, Madhunapantula et al., 2007).
Ectopically expressing PTEN protein can inhibit invasion and induce apoptosis in melanoma cells (Stewart et al., 2002, Stahl et al., 2003). Cells lacking functional PTEN protein expression exhibited increased BCl2 activity, resistance to growth factor receptor inhibitors and chemotherapeutic agents as well as having had a growth advantage compared to those expressing physiological levels of the protein (Mikhail et al., 2005, Madhunapantula et al., 2007, Stahl et al., 2004, Stahl et al., 2003). Furthermore, decreased PTEN activity alters cell cycle progression, migration and adhesion of melanoma cells making it an important regulator of melanoma development (Wu et al., 2003). Recently, PTEN loss and V600EB-Raf have been shown to cooperate to promote metastatic melanoma development (Dankort et al., 2009). Recent mouse models, expressing V600EB-Raf at physiological levels in combination with PTEN loss, led to tumor development, further validating the role of PTEN in melanoma development (Dankort et al., 2009).
PTEN inactivation either by phosphorylation or oxidation of cysteine residues increases the tumorigenicity of tumor cells (Tamguney and Stokoe, 2007, Gericke et al., 2006). Introduction of the PTEN gene into melanoma cells in lacking functional protein, inhibited cell survival by decreasing activity of Akt3 and downstream pPRAS40 levels (Stahl et al., 2003, Stahl et al., 2004, Madhunapantula et al., 2007). Low intracellular levels of pAkt and pPRAS40 induce cellular apoptosis mediated through caspases (Madhunapantula et al., 2007). Comparing isogenic PTEN null variants of the UACC 903 melanoma cells to PTEN containing cells, demonstrated that PTEN sensitized the melanoma cells to chemotherapeutic agents and apoptosis inducers such as staurosporine (Madhunapantula et al., 2007, Stahl et al., 2004, Gaitonde et al., 2009).
2.2 Mechanisms leading to PTEN inactivation in melanomas
Several studies have identified loss of the chromosome 10q23 region containing the PTEN gene in melanomas (Wu et al., 2003, Robertson et al., 1999a, Poetsch et al., 2001b). Screening of melanoma cell lines and paired uncultured metastatic melanoma and peripheral blood specimens for PTEN loss and/or alterations using PCR and direct sequencing found ~30–40% melanoma cell lines containing homozygous deletions (including intragenic loss) while 9% had nonsense, frame shift; and intronic mutations. However, only ~5–15% of uncultured melanoma specimens contained nonsense mutations or homozygous deletions ( Yin and Shen, 2008, Tsao et al., 1998a). Intragenic polymorphisms in introns have also been reported to regulate PTEN expression in melanomas. Amino-acid altering mutations (P95S, F154L, L325F) induced by UV radiation (UVB and UVA) have been shown to impair PTEN function and thereby promote the development of early melanomas in xenoderma pigmentosum (Wang et al., 2009).
PTEN is generally accepted to be lost early in melanoma development. Melanoma patient tumor material commonly has decreased PTEN expression, mediated through loss of a single copy of chromosome 10 (Parmiter et al., 1988, Thompson et al., 1995, Mertens et al., 1997, Bastian et al., 1998, Wu et al., 2003). Early melanocytic lesions frequently have loss of one allele of PTEN, or PTEN haplo-insufficiency occurring due to loss of the entire chromosome 10 (Parmiter et al., 1988, Thompson et al., 1995, Mertens et al., 1997, Bastian et al., 1998, Wu et al., 2003). In this scenario, chromosome 10 loss reduces PTEN expression in a sub-population of evolving melanoma cells, causing increased Akt3 activation, which provides these cells with a growth and survival advantage (Figs. 4 and 5). This is demonstrated in studies showing that growth factors, such as stem cell factor strongly activate Akt, which protects melanocytes from TNF-related apoptosis-inducing ligand (TRAIL) and staurosporine mediated cell death (Larribere et al., 2004). Furthermore, Akt inhibition blocks the antiapoptotic effect of SCF, with only sustained Akt activation protecting melanocytes from apoptosis (Larribere et al., 2004). Thus, decreased PTEN levels due to haplo-insufficiency play an important role in early melanomas by specifically increasing Akt3 activity in these cells, thereby protecting these cells from apoptosis (Figs. 4 and 5).
Figure 5. Akt3 expression and activity increase during melanoma development.

5A Expression of pAkt in nevi, dysplastic nevi, primary and metastatic melanoma lesions was quantitated from histological sections and intensity scored using immunohistochemical staining. Compared to common nevi that had only weak-to-moderate pAkt staining, increasing percentages of dysplastic nevi, primary and metastatic melanomas contained higher levels of Akt activity with 67% of advanced tumors having elevated activity (Stahl et al., 2004). 5B. Expression and activity of Akt isoforms (Akt1, Akt2 and Akt3) was measured in flash frozen melanoma patient tumor samples by Western blotting. Approximattely 60% of melanoma tumors expressed elevated levels of Akt3 protein compared to normal human melanocytes. Furthermore, ~43% of tumor had elevated Akt3 activity.
PTEN promoter methylation leading to inactivation of transcription has been found to occur in ~62% of metastatic melanoma patients (Tsao et al., 1998b, Guldberg et al., 1997, Boni et al., 1998, Celebi et al., 2000, Birck et al., 2000, Reifenberger et al., 2000, Poetsch et al., 2001a, Robertson et al., 1998b, Tsao et al., 2000). Quantitative positional methylation analysis and Taqman RT-PCR of 37 sera from melanoma patients and 21 pairs of corresponding sera and melanoma specimens demonstrated that 62% of metastatic melanoma patients sera had elevated PTEN promoter methylation (Mirmohammadsadegh et al., 2006). However, contradictory to this observation, the most recent studies by Liu et al., 2008 and Bonazzi et al., 2009 found no DNA methylation in PTEN gene (Bonazzi et al., 2009, Liu et al., 2008). Therefore, although the regulation of PTEN expression by DNA methylation appears to be important, further studies are warranted to measure its contribution to melanoma development. If mutational and epigenetic silencing studies of PTEN were added together, functional inactivation might be involved in upto 77% of non-familial melanomas (Zhou et al., 2000, Boni et al., 1998, Pollock and Trent, 2000, Saida, 2001). It is currently unknown whether direct phosphorylation leading to inactivation of PTEN has any role in silencing of this phosphatase in melanoma (Miller et al., 2002).
2.3 Akt3 plays a key role in melanoma development
Loss of PTEN in melanomas promotes Akt3 activity by increasing the cellular levels of PIP3 (Stahl et al., 2003, Stahl et al., 2004, Robertson, 2005). Accumulating PIP3 molecules then binds to the pleckstrin homology (PH) domains of Akt3 to facilitate membrane translocation and phosphorylation (Fayard et al., 2005, Sale and Sale, 2008, Scheid and Woodgett, 2003). Activated Akt3 (Fig. 1A) phosphorylates: (a) GSK3β to inhibit its activity by phosphorylating serine 9 (S9) serving to promote cell cycle progression through increased cyclin D levels (Woodgett, 2005, Fresno Vara et al., 2004); (b) PRAS40 at threonine 246 (T246) to inhibit interactions with mTORC1 thereby increasing the nutrient status of the cells (Kovacina et al., 2003, Wang et al., 2007a, Madhunapantula et al., 2007); (c) V600EB-Raf to decrease its activity to levels that promote rather than inhibit cell proliferation (Cheung et al., 2008, Ikenoue et al., 2005); and (d) osteopontin, a glycophosphoprotein promoting melanoma progression levels to a highly metastatic state (Packer et al., 2006) (Fig. 2). Furthermore, Akt3 inhibits cellular apoptosis by decreasing caspase-3/7 activity and increasing the expression of cleaved caspase-3 and cleaved PARP levels (Stahl et al., 2004, Sharma et al., 2009). Decreased apoptosis makes melanoma cells less sensitive to chemotherapeutic agents functioning through this mechanism.
Akt1 and Akt2 are ubiquitously expressed in most normal tissues compared to Akt3 whose expression is restricted (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002). However, expression does not necessarily imply activity of a particular isoform (Stahl et al., 2004, Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002). Activity of a particular isoform is cell type-dependent relying on a balance between signals promoting or inhibiting activity. This is the case for melanomas where all three isoforms are expressed but Akt3 is the predominantly active isoform (Fig. 5). Increased Akt3 expression/activity occur in 60–70% of sporadic melanomas demonstrating a key role in melanoma development (Figs. 5A and 5B) (Dhawan et al., 2002, Stahl et al., 2004).
Increased Akt3 activation also plays a significant role in progression to more advanced aggressive tumors (Stahl et al., 2004, Robertson, 2005, Madhunapantula et al., 2007). An experimental tumor progression model in which Akt3 activity in melanocytes was compared to low passage cell lines from primary melanoma tumors at the radial and vertical stages of cell growth, revealed that Akt3 activity increased in the radial growth phase and remained elevated in comparison to Akt1 and Akt2. The high level of involvement in advanced melanomas leads to the inference that Akt3 activation performs critical functions in melanoma development (Stahl et al., 2004, Robertson, 2005).
2.4 Mechanism promoting Akt3 deregulation in melanomas
Akt3 plays an important role in signaling pathways that regulate the tumorigenic processes (Testa and Bellacosa, 2001, Nicholson and Anderson, 2002). It is unknown why Akt3 and not the other isoforms are activated in melanomas. However, it is known that loss of functional PTEN in melanocytes, leads to preferential activation of Akt3 (Fig. 4) (Robertson, 2005). Increased copy numbers of the Akt3 gene in melanomas, does not alone account for preferential Akt3 activation (Stahl et al., 2004, Wu et al., 2003). Akt3 activation may involve preferential interaction of PIP3 and/or accessory protein interaction with the PH domain (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002), which is where PIP3 lipid-protein interactions occur (Fig. 1B). The isoforms have different phosphorylation sites within the PH domain having as yet uncharacterized functions (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002). A precedent exist to support this possibility; a ceramide-induced, PKC zeta-dependent phosphorylation site at threonine 34 (T34) has been identified that leads to Akt1 inactivation by preventing binding to PIP3 (Powell et al., 2003). At this site Akt2 and Akt3 have a serine. Thus, differential regulation of putative phosphorylation sites within the PH domain of Akt3 may provide the basis for the specificity of Akt3 activation in melanomas. Preferential Akt3 interactions with other proteins could also lead to selective regulation in melanomas. TCL1 selectively binds to the Akt3 PH domain, thereby promoting hetero-oligomerization of Akt1 with Akt3, causing transphosphorylation of Akt in leukemias (Laine et al., 2000, Laine et al., 2002, Pekarsky et al., 2000). Thus, factors interacting preferentially with Akt3 may lead to selective activation in melanomas (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002).
Akt activation starts with upstream PI3K in this cascade, which is activated as a result of ligand-dependent activation of tyrosine kinase receptors, G-protein-coupled receptors, or integrins (Fig. 2) (Blume-Jensen and Hunter, 2001). Many of theses ligands are overexpressed in cancers, making this a route for Akt activation in melanomas (Testa and Bellacosa, 2001, Nicholson and Anderson, 2002). Another possibility involves overexpression or constitutive activation of cell surface receptors, which are reported in melanomas (Blume-Jensen and Hunter, 2001, Polsky and Cordon-Cardo, 2003). Receptor-independent activation also occurs in 10–20% melanomas expressing constitutively active Ras proteins (Rodriguez-Viciana et al., 1997, Kauffmann-Zeh et al., 1997, Polsky and Cordon-Cardo, 2003). Elevated PI3K activity itself can also cause Akt activation. Chromosome 3q26 containing the p110 catalytic subunit of PI3K, which is frequently amplified in cancer of the ovary (Shayesteh et al., 1999) and cervix (Ma et al., 2000), leading to increased PI3K catalytic activity. The importance of PI3K signaling in melanoma was demonstrated by over expressing a deleted subunit of PI3K (Deltap85) to reduce PI3K signaling (Tachiiri et al., 2000). In this manner, Delta p85 functioned as a dominant-negative disrupting p85/p110 subunit interaction, consequently inducing apoptosis in the melanoma cell line G361. Inhibiting Akt signaling in tumor cells by adenoviral transfer of a Akt kinase-dead mutant, in which the two regulatory phosphorylation sites have been mutated to alanines thereby converting it to a dominant negative, led to selective induction of apoptosis in tumor cells expressing activated Akt (Jetzt et al., 2003). In contrast, a minimal effect was observed in normal or tumor cells expressing low levels of active Akt (Jetzt et al., 2003). These studies validate the importance of Akt activation in tumor cell survival.
A key mechanism for increased Akt activity in cancer cells involves genetic amplifications or mutations leading to constitutive activation. Recently a low frequency activating mutation (E17K) in the PH domain of Akt3 has been identified in melanoma cell lines and ~4% of patient tumors (Davies et al., 2008). This mutation enables Akt3 to get recruited to cell membranes independent of PI3K, which leads to cellular transformation. Genetic amplifications increasing Akt1 or Akt2 expression occur in carcinomas of the stomach, ovary, pancreas and breast (Staal, 1987, Cheng et al., 1992, Cheng et al., 1996b, Lu et al., 1995, Bellacosa et al., 1995a, van Dekken et al., 1999, Bellacosa et al., 1995b, Cheng et al., 1996a). Specifically, Akt2 amplification occurs as part of the 19q13.1–q13.2 amplicon in high-grade aggressive ovarian tumors (Thompson et al., 1996). Even though no amplifications of Akt genes have been reported in melanomas (Waldmann et al., 2001, Waldmann et al., 2002), Akt3 protein is frequently overexpressed (Stahl et al., 2004), as a result of copy number increases of the long arm of chromosome 1 containing the gene (Thompson et al., 1995, Mertens et al., 1997, Bastian et al., 1998). No increases in the long arms of chromosome 14 or 19 containing the Akt1 and Akt2 genes, respectively, have been reported (Thompson et al., 1995, Mertens et al., 1997, Bastian et al., 1998). Thus, increase in Akt3 copy number is a mechanism contributing to increased expression and activity in melanoma. However, expression does not necessarily imply activity as observed with Akt1 and Akt2 expression but little activity in melanomas (Stahl et al., 2004). Therefore, other processes must also contribute to Akt3 activation.
Inactivation of negative regulators of Akt provides another mechanism to regulate Akt activity (Cantley and Neel, 1999b). Akt phosphatases that directly dephosphorylate Akt may play a role in this process as can be observed when treating cells with inhibitors of protein serine/threonine phosphatases, such as okadaic acid (Andjelkovic et al., 1996). While the identity of these phosphatases remains uncertain, PP2A phosphatase may be involved in this process (Andjelkovic et al., 1996). Proteins binding to Akt also have the potential to modulate cellular activity as is observed with the protooncogene TCL1, which enhances oligomerization of Akt1 with Akt3, thereby facilitating activation of Akt3 in leukemia (Laine et al., 2000, Laine et al., 2002, Pekarsky et al., 2000). Another example is the heat-shock-protein 90 (Hsp90) that can complex with Akt. Inhibition of Akt-Hsp90 complex formation can inactivate Akt by PP2A-mediated dephosphorylation (Sato et al., 2000, Fujita et al., 2002). While none of these interactions have been studied in melanoma, the interested reader can learn more about the proteins interacting with and regulating Akt activation in other reviews; for additional information of see reference (Brazil et al., 2004).
3. CELLULAR PROCESSES REGULATED BY PTEN/AKT3 SIGNALING IN MELANOMAS
3.1 Cell survival, migration, metastasis and chemoresistance regulated by PTEN/Akt3 signaling
PTEN regulates several processes such as inhibition of cell proliferation by altering cell cycle progression through G1 to S phase and control of apoptosis by modulating Akt activity (Cantley and Neel, 1999a, Li and Ross, 2007, Mikhail et al., 2005, Ramaswamy et al., 1999). Additionally, PTEN expression has been associated with cell-cell adhesion, cell migration and metastasis (Wu et al., 2003, Stewart et al., 2002). Tumor cells lacking PTEN exhibit unusually high metastatic activity and chemoresistance (Robertson, 2005, Wu et al., 2003, Lopez-Bergami et al., 2008, Stahl et al., 2003). Furthermore, PTEN expression using a variety of vectors or from an introduced chromosome in melanoma cell lines lacking PTEN induced apoptosis (Fig. 6A) and inhibited tumor development (Stahl et al., 2003, Stewart et al., 2002, Mikhail et al., 2005). Cells containing functionally active PTEN protein exhibit elevated p27 expression, decreased cyclin-D1 and cyclin-D2 proteins (Ramaswamy et al., 1999, Wu et al., 2003, Huang et al., 2007).
Figure 6. PTEN introduction or Akt3 inhibition induce apoptosis in melanomas.

Photographs shows TUNEL-positive cells (an indicator of apoptosis) in tumor xenografts following PTEN expression (6A) or siRNA-mediated reduction of Akt3 protein levels (6B). UACC 903 cells or cells expressing chromosomal PTEN or cells nucleofected with siAkt3 versus siAkt2 (1 × 106) were injected into nude mice and 4 days later processed for TUNEL staining. Data shows that either PTEN expression or Akt3 protein knockdown increased apopotosis compared to controls in which PTEN activity had been lost (revertant) or cells with reduced Akt2 protein levels (Stahl et al., 2004).
Akt3 has been shown to be a pro-survival kinase in melanomas (Stahl et al., 2004). SiRNA targeting Akt3 protein levels (Fig. 6B) or expression of PTEN (Fig. 6A) selectively lowers Akt3 activity, thereby reducing the tumorigenic potential of melanoma cells by altering apoptotic sensitivity of the cells (Stahl et al., 2004, Madhunapantula et al., 2007). Furthermore, increased activity correlates with tumor progression, providing cells with a selective advantage in the tumor environment.
Processes such as melanoma tumor progression, metastasis, cell-cell adhesion, cell migration and development of chemoresistant tumors under hypoxic conditions are also regulated by PI3 kinase and Akt signaling pathways (Robertson, 2005, Govindarajan et al., 2007, Dai et al., 2005). Activated Akt in metastatic melanoma cells have been shown to regulate Notch1 expression via NFκB thereby promoting tumor development under hypoxic tumor conditions (Bedogni et al., 2008). A different study has shown that Akt inhibits RhoB, a GTPase, in melanomas and thereby induces tumor cell survival, transformation, invasion and metastasis (Jiang et al., 2004).
3.2 Downstream targets in the PTEN/Akt3 signaling cascades
While inactive Akt resides in the cytoplasm, activation occurs at the cell membrane followed by translocation to cytosol and nucleus (Andjelkovic et al., 1997, Meier et al., 1997, Dufner et al., 1999, Filippa et al., 2000, Borgatti et al., 2000). Akt substrates can be cytoplasmic or nuclear proteins and numbers continues to increase as studies such as those using the minimal consensus peptide sequence Arg-X-Arg-X-X-[Ser/Thr]-Hyd (where X is any amino acid and Hyd is a bulky hydrophobic amino acid) search for putative substrates (Alessi et al., 1995, Obata et al., 2000). The functions of many of these substrates in cellular processes have been identified, demonstrating that Akt regulates multiple processes in cells including apoptosis and proliferation (Fig. 2). This review does not provide an exhaustive discussion of all Akt substrates, but rather focuses on those currently identified as playing important roles in melanoma development; for additional information regarding Akt substrates see references (Testa and Bellacosa, 2001, Scheid and Woodgett, 2001, Scheid and Woodgett, 2003, Bellacosa et al., 2004, Brazil et al., 2004, Brazil et al., 2002, Brazil and Hemmings, 2001, Datta et al., 1999, Nicholson and Anderson, 2002).
Substrates for Akt3 do not appear to be specific but rather seem to be identical to those acted on by all three Akt isoforms. Several substrates have been identified as being important in melanoma development. First, a switch from E- to N-cadherin in melanoma has been shown to promotes cell survival by activating the Aktpathway, resulting in inactivation of Bad and accumulation of steady state levels of β-catenin (Li et al., 2001). Second, melanoma cell activity has been linked to enhanced NFκB nuclear localization and transactivation and inhibition of Akt activity led to increased apoptosis and decreased NFκB promoter activity (Dhawan et al., 2002). Third, Akt has been shown to enhance human telomerase activity through phosphorylation of hTERT subunit (Kang et al., 1999). Inhibition of Akt reduced hTERT peptide phosphorylation and telomerase activity (Kang et al., 1999). Fourth, Akt phosphorylates serine 71 of Rac1 thereby modulating the Rac1 signal transduction pathway in SK-MEL-28 melanoma cells (Kwon et al., 2000). Fifth, activated Akt upregulates MelCAM while overexpression of MelCAM activated endogenous Akt inhibiting the proapoptotic BAD protein (Fig. 2) (Li et al., 2003).
Substrates specifically regulated by Akt3 have also been identified and role in melanoma development identified (Fig. 2). Recently, Akt3 has been reported to phosphorylate V600EB-Raf on S364 and/or S428 to reduce its activity to levels that promote rather than inhibit melanoma development from melanocytes (Fig. 7, Figure kindly provided by Dr. M. Cheung) (Cheung et al., 2008). Ectopic expression of V600EB-Raf in primary cell lines such as melanocytes has been shown to induce cellular senescence not only by elevating the levels of MAPK activity for unusually longer periods but also by upregulating the expression of cyclin-dependent kinase (cdk) inhibitors (Wajapeyee et al., 2008, Houben et al., 2009). Therefore, genetic changes such as loss of tumor suppressor genes (PTEN, p53 or p16INK4A), or upregulation of cooperating oncogenes is necessary to progress into advanced metastatic stages (Zhao et al., 2008, Dankort et al., 2009). Acquisition of Akt3 activity facilitates the progression of quiescent melanocytic nevi into aggressive vertical and metastatic stages by inhibiting V600EB-Raf activity thereby releasing cells from a senescence block (Fig. 7). This demonstrates that in melanomas, Akt3 kinase not only increases cell survival but also aids early melanoma development.
Figure 7. Akt3 activity promotes melanoma development by inhibiting V600EB-Raf.
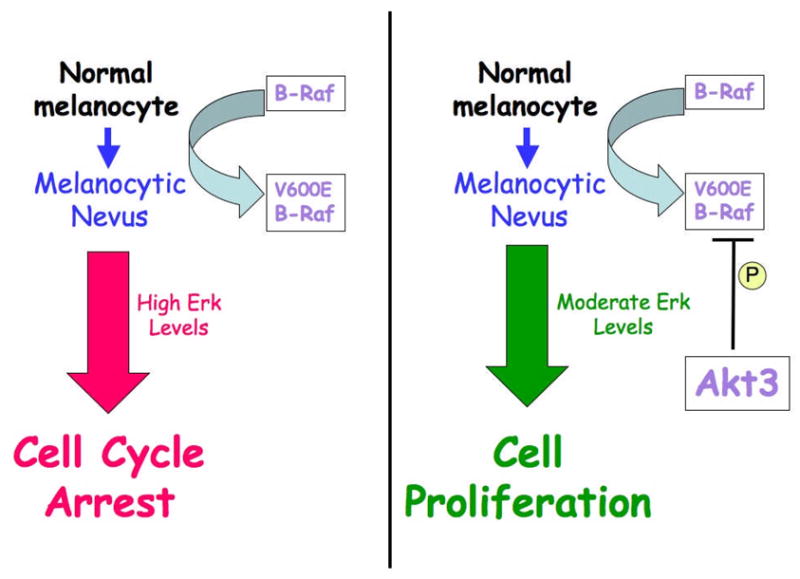
Akt3 decreases V600EB-Raf activity by phosphorylating the protein thereby decreasing its activity and that of the downstream signaling cascade to levels that promote rather than inhibit melanocyte proliferation, which aids melanoma development.
Another substrate regulated by Akt3 activity in melanomas involves modulation of apoptosis mediated through PRAS40 (proline rich Akt substrate of 40 kDa) (Fig. 2) (Madhunapantula et al., 2007). PRAS40 has been identified as a direct substrate of Akt from a screen conducted using an antibody that recognizes consensus Akt substrate phosphorylation sequence (RXRXX(S/T); and as a 14-3-3 binding protein (Kovacina et al., 2003). Compared to most proteins, PRAS40 contains 3-times more proline residues (5% in most proteins to 15% in PRAS40). PRAS40 is a cytosolic protein found ubiquitously in all eukaryotes (Kovacina et al., 2003) and is phosphorylated by Akt at T246 (Kovacina et al., 2003). pPRAS40T246 protects neuronal cells from ischemic injury by inhibiting caspase-3 mediated apoptotic cell death (Saito et al., 2004). Expression of pPRAS40 levels are also associated with tumor malignancy (Huang and Porter, 2005). Compared to paired control normal cells, cancer cell lines (MCF10A/MCF7, BEAS/H1170) exhibited high pPRAS40 expression (Huang and Porter, 2005). However, the detailed functional characterization, in vitro and in vivo, was not reported. Compared to normal melanocyte controls, 43 to 60% of flash frozen tumors collected from melanoma patients express high pPRAS40 and pAkt (Madhunapantula et al., 2007). Furthermore, targeted inhibition of PRAS40 using siRNA reduced melanoma tumor growth in xenografted melanoma studies (Fig. 8) (Madhunapantula et al., 2007). Mechanistically, PRAS40 inhibition induced caspase-3/7 mediated apoptosis in melanoma cells and increased sensitivity to apoptosis inducers such as staurosporine (Madhunapantula et al., 2007). However, the molecular basis for caspase-3/7 activity inhibition by pPRAS40 in melanomas remains unknown.
Figure 8. Knockdown of PRAS40, a substrate of Akt3, inhibits melanoma tumor growth.

SiRNA-mediated reduction of PRAS40 protein levels, a direct substrate of Akt, or upstream Akt3 inhibits tumor development in xenografted melanoma tumors. UACC 903 cells were transfected with siRNAs targeting PRAS40 or Akt3, and 1.5 days later cells were injected into nude mice. Developing tumor sizes were measured on alternate days. Compared to buffer, a scrambled siRNA or a siRNA targeting C-Raf, a 50–60% decrease in tumor volume observed following PRAS40 or Akt3 protein knockdown. Inset shows reduction of PRAS40 protein levels in tumors removed from mice at days 7.5 or 9.5 (Madhunapantula et al., 2007).
Recent reports have demonstrated that PRAS40 is a physiological target of mTORC1 kinase and regulates its activity by functioning as a direct inhibitor of substrate binding (Oshiro et al., 2007, Sancak et al., 2007, Wang et al., 2007a). In insulin-deprived cells, PRAS40 binds to raptor protein via a TOS motif thereby inhibiting mTORC1 kinase activity (Wang et al., 2007a, Fonseca et al., 2007). Additionally, it has been shown that in intact cells, mTORC1 phosphorylates PRAS40 at S183 thereby increasing its affinity towards raptor (Fonseca et al., 2007). Therefore, PRAS40 is considered as an inhibitor of cell growth. Supporting this observation, a separate study has reported that siRNA mediated inhibition of PRAS40 protected HeLa cells from TNFα and cycloheximide mediated apoptosis (Thedieck et al., 2007). This study concluded that PRAS40 is a pro-apoptotic protein in cells. However, this observation is contradictory to the earlier reports for neuronal cells and melanomas (Madhunapantula et al., 2007, Huang and Porter, 2005). Knocking down PRAS40 using siRNA inhibited melanoma cells growing in culture dishes and xenografted tumor development by triggering apoptosis (Fig. 8) (Madhunapantula et al., 2007). Furthermore, targeting Akt using siRNA or pharmacological agents such as ISC-4, PBISe, and BI-69A11 decreased pPRAS40 levels and induced caspase-3/7 activity as well as cleaved PARP (Madhunapantula et al., 2008, Sharma et al., 2009, Gaitonde et al., 2009). Thus, PRAS40 phosphorylation by Akt3 or inhibition of PRAS40 protein expression using siRNAs has significant potential to inhibit melanoma development. In cancers such as melanomas, levels of pAkt, hence pPRAS40 are elevated by several fold compared to normal human melanocytes (Huang and Porter, 2005, Madhunapantula et al., 2007). Therefore, increased pPRAS40T246 may release its inhibitory effects on mTORC1 kinase and down-regulate caspase-3/7 activity by yet to be established mechanisms. In conclusion, whereas the phosphorylation of PRAS40 at threonine 246 by Akt is primarily involved in inhibiting cellular apoptosis and increasing survival, the role of unphosphorylated PRAS40 in the cancer cells needs further investigation
Another well-known substrate of Akt is GSK3 (Fig. 2) (Fresno Vara et al., 2004, Robertson, 2005). GSK3 is present in cells as two structurally similar isoforms, known as GSK3α and GSK3β (Patel and Woodgett, 2008). GSK3α and GSK3β are serine-threonine protein kinases, involved in regulating diverse cellular processes including cell structure, function, proliferation and survival (Jope and Johnson, 2004, Ougolkov and Billadeau, 2006). Akt has been shown to phosphorylate S9 of GSK3β and S21 of GSK3α to inhibit their activity thereby promoting cell proliferation by stabilizing cyclin-D1 protein (Ramaswamy et al., 1999, Fresno Vara et al., 2004). In addition, a recent study demonstrated that mouse embryonic fibroblast cells expressing wtPTEN inhibit cell cycle progression through G1-S phase by down regulating cyclin D2 protein level, which is mediated by GSK3β (Huang et al., 2007). Apart from Akt3, activated Erk also phosphorylate GSK3β on S9 to inhibit its binding to c-Jun thereby increase cell proliferation and survival (Lopez-Bergami et al., 2007). Therefore, unphosphorylated active GSK3 is considered as an apoptosis inducing tumor suppressor. However, recent studies have shown that GSK3 expression is elevated in advanced cancers and deregulation of GSK3 activity increases malignant transformation of cells (Luo, 2009, Bellei et al., 2008, Ougolkov and Billadeau, 2006, Patel and Woodgett, 2008). In support of these observations, inhibition of GSK3β has been found to enhance sorafenib-induced apoptosis in melanoma cells (Panka et al., 2008). Furthermore, GSK3β inhibition elevated melanogenesis in melanoma cell lines by inducing melanocyte differentiation markers such as melanin synthesis, tyrosinase activity, expression of tyrosinase and MITF (microphthalmia-associated transcription factor) (Bellei et al., 2008). Furthermore, targeting GSK3β using organometallic inhibitors (DW1/2) decreased Mdm2 activity thereby elevated p53-mediated apoptosis in melanoma cell lines expressing WT p53 protein (Smalley et al., 2007). Recently GSK3 has also been shown to maintain the constitutive activity of IKK activity and thereby the nuclear factor kappa beta (NFκβ) in pancreatic cells (Wilson and Baldwin, 2008). Since advanced stage melanomas contain high levels of NFκβ activity compared to melanocytes and early stage melanomas; and expression as well as activity of NFκβ is regulated by PTEN/AKT signaling, the role of GSK3 and its regulation by Akt pathway needs to be more thoroughly investigated (Ueda and Richmond, 2006, Stahl et al., 2004, Robertson, 2005). Finally, GSK3α and not GSK3β have been shown to regulate pancreatic cell survival (Wilson and Baldwin, 2008). Most studies in melanoma suggest that it is GSK3β regulating melanoma development. However, no reports detail percentages of melanoma patient tumors expresses each of these two isoforms and which needs to be targeted for effective tumor inhibition. Future studies should address these issues in order to better understand GSK3 signaling to design more effective therapeutics for treating melanoma.
4. THERAPEUTIC POTENTIAL OF TARGETING THE PTEN/AKT3 SIGNALING CASCADE IN MELANOMAS
The potential clinical importance of targeting PTEN or Akt3 in melanomas has been demonstrated by showing that selective inhibition of Akt3 expression (activity) or expression of PTEN significantly reduced melanoma tumor development (Stahl et al., 2004). Therefore, introduction of PTEN into melanoma cells or targeting Akt3 or interfering with regulators or these proteins, may represent an effective therapeutic approach for melanoma patients (Johnstone et al., 2002, Soengas and Lowe, 2003). Since the vast majority of chemotherapeutic agents induce apoptosis, and expression of PTEN or reduction of Akt3 activity leads to increased sensitivity to apoptotic stimuli, the threshold doses of drugs or radiation required for effective chemo-or radio-therapy could be reduced by targeting these proteins (Soengas and Lowe, 2003). Several studies have demonstrated that targeting these proteins promotes apoptosis and/or increased the sensitivity to chemotherapeutic agents (Bedogni et al., 2004, Ivanov and Hei, 2004, Richmond et al., 2004, Jetzt et al., 2003, Tachiiri et al., 2000). Under these circumstances, introduction of PTEN or decreasing Akt3 activity alone or in combination with chemotherapeutic agents could potentially benefit melanoma patients (Soengas and Lowe, 2003, Bedogni et al., 2004).
4.1 PTEN expression as a melanoma therapeutic
Introduction of PTEN into advanced-stage melanoma cells via a chromosomal transfer or ectopic expression using a plasmid-based vector can inhibit tumor development in xenografted melanoma models by inducing cellular apoptosis (Robertson et al., 1998a, Poetsch et al., 2001b, Stahl et al., 2003). Furthermore, blocking PI3 kinase activity by expressing adenoviral based PTEN inhibited melanoma tumor development and reduced cellular invasiveness (Stewart et al., 2002, Kotelevets et al., 2001). Multiple groups have shown that PTEN expression can sensitize cancer cells to therapeutic agents that function by triggering apoptosis (Stahl et al., 2003, Stewart et al., 2002, Wu et al., 2003, Sinnberg et al., 2008, Meier et al., 2005). Thus, targeted delivery of PTEN into melanoma cells has potential to be an effective therapeutic agent. Furthermore, combining PTEN expression with other agents might help to prevent melanoma development completely. Since multiple signaling cascades need to be inhibited for effective melanoma cell killing, targeted delivery vehicles, such as liposomes conjugated with specific antibodies to recognize melanoma cells, and loaded with pharmacological agents as well as PTEN expressing vectors might provide the tool for a therapeutic of this type. A case in point is breast cancer cell lines harboring a PTEN deletion or expressing a mutant functionally inactive PTEN protein, which exhibit extreme resistance to EGFR antagonists such as gefitinib (She et al., 2005). However, reconstitution of PTEN in these cell lines restored sensitivity to EGFR thereby inhibiting breast cancer xenograft development more effectively than either treatment alone (She et al., 2005). Similar observations are reported for NSCLC, prostate, and leukemia cell lines (She et al., 2005). Studies are warranted for melanoma, where low PTEN expression and/or activity lead to chemotherapeutic resistance. Restoration of functional PTEN in these cells would increase the therapeutic efficacy of anti-melanoma agents. Furthermore, if PTEN expression was regulated by endogenous microRNAs, inhibiting using antimers could significantly upregulate PTEN activity thereby restoring the chemosensitivity of the melanoma cells. However, PTEN regulation by microRNAs has not yet been reported in melanomas.
4.2 Therapeutic agents targeting Akt3
Since targeting Akt3 alone or in combination inhibits melanoma growth by inducing apoptosis, several synthetic and naturally occurring small molecule inhibitors specifically inhibiting Akt activity have been developed and efficacy tested (Sharma et al., 2009, Sharma et al., 2008, Robertson, 2005, Soengas and Lowe, 2003, Stahl et al., 2004, Madhunapantula et al., 2007, Cheung et al., 2008, Tran et al., 2008a). Recently, naturally occurring isothiocyanates were identified as agents with potential to reduce Akt3 activity in cancer cells including melanoma (Xiao and Singh, 2007, Satyan et al., 2006, Sharma et al., 2008). However, low potency of sulfur containing isothiocyanates would have required large doses to be consumed for clinical efficacy, which might have had side effects (Sharma et al., 2009). Therefore, isoselenocyanates (ISC) were created in which sulfur was replaced with selenium and the alkyl chain length was increased to make the compounds more cell-permeable (Fig. 9A). ISC compounds with 4 (ISC-4) or 6 (ISC-6) carbon chains are effective at shrinking preexisting tumors by 60–70% (Fig. 9B) by targeting Akt3 signaling (Fig. 9C) thereby inducing apoptosis (Sharma et al., 2009). Compared to known inhibitors of Akt signaling such as API-2, the selenium containing ISC-4 and ISC-6 appear to be more effective for melanoma treatment (Sharma et al., 2009). BI-69A11 is another Akt inhibitor, demonstrated to have effective anti-melanoma activity in cultured cells and on tumors (Gaitonde et al., 2009). It is unknown whether side-effects associated with the use of these agents might also be a concern for treating melanoma, since Akt inhibition using siRNAs has been reported to induce the metastatic potential of breast cancer cells (Yoeli-Lerner et al., 2005).
Figure 9.
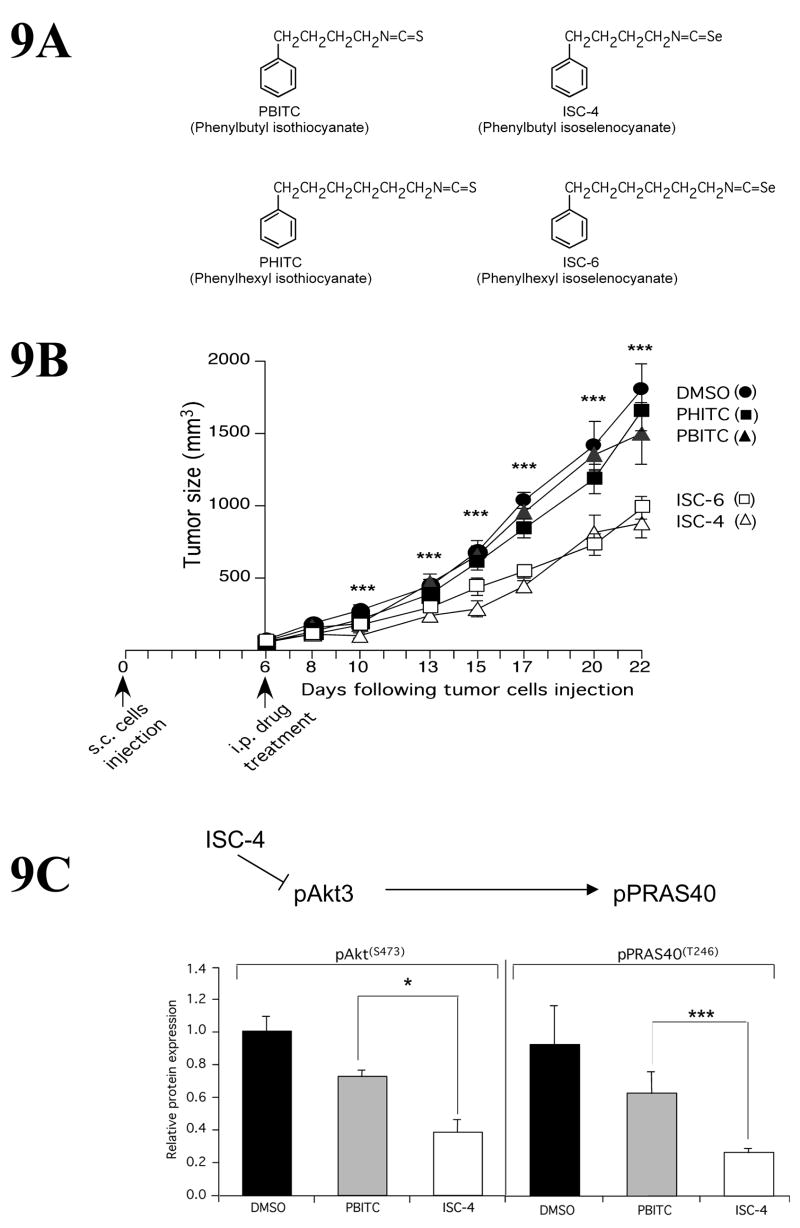
ISC-4 inhibits melanoma tumor development by targeting Akt3 signaling. 10A. Structures of 4 and 6 carbon containing isothiocyanates (PBITC and PHITC) and isoselenocyanates (ISC-4 and ISC6). 9B. Isoselenocyanates (ISC-4 and ISC-6) were injected intra peritoneally into mice bearing established 50–60 mm3 sub-cutaneous melanoma tumors. Compared to DMSO vehicle controls or PBITC and PHITC, isoselenocyanates decreased tumor development by ~50% (Sharma et al., 2009). 9B. ISC-4 reduced Akt3 signaling in melanoma tumors. Decreased pAkt3 and downstream pPRAS40 were observed in tumors treated with isoselenocyanates indicating the compound decreased Akt3 signaling in tumors (Sharma et al., 2009).
4.3 Targeting multiple pathways to synergistically treat melanoma
Small molecule inhibitors such as rapamycin, sorafenib, U0126, or PD98059 have been found to be ineffective as single agents for treating patients suffering from advanced melanoma (Smalley and Herlyn, 2005, Lasithiotakis et al., 2008). Furthermore, targeting oncogenic proteins singly seems not to be as effective as inhibiting multiple pathway for melanoma treatment (Smalley et al., 2006, Smalley and Flaherty, 2009, Lorigan et al., 2008). Simultaneously targeting PI3K/Akt3 and MAP kinase signaling pathways has been reported to synergistically inhibit melanoma development (Fig. 10) (Lopez-Bergami et al., 2008, Smalley and Flaherty, 2009, Cheung et al., 2008, Krasilnikov et al., 2003, Meier et al., 2005, Tran et al., 2008a). First, delivering siRNAs inhibiting Akt3 and V600EB-Raf either by nucleofection or using liposomes synergistically inhibited melanoma tumor cells growth in culture or in xenografted melanoma tumors demonstrating the feasibility of this approach (Fig. 10) (Tran et al., 2008a, Cheung et al., 2008). Second, combining nanoliposomes containing ceramide (a lipid based Akt inhibitor) with sorafenib (a Raf kinase inhibitor) has been shown to synergistically decrease melanoma cell growth (Tran et al., 2008b). Third, combined inhibition of MAPK (using sorafenib or MEK inhibitors U0126, PD98059 and PD325901) and mTORC1 (using rapamycin) more effectively reduced melanoma cells growth compared to either of the agents tested singly (Lasithiotakis et al., 2008, LoPiccolo et al., 2008, Dankort et al., 2009). Fourth, topical application of LY-294002 and U0126 in combination more effectively delayed the development and decreased melanoma tumor incidence in the transgenic TPRas mouse model when compared to either of these agents alone (Bedogni et al., 2006). Fifth, PI3K and MEK inhibitors only when used in combination were more effective than single agents (Lasithiotakis et al., 2008). Furthermore, targeting PI3K and mTOR activities using dual inhibitors NVP-BBD130 and NVP-BEZ235 had been shown to effectively reduce not only the size of primary melanoma tumor but also cervical lymphnode metastasis in a syngenic mouse melanoma model. These dual inhibitors exert anti-tumor activity by decreasing the activities of PI3K and mTORC1/mTORC2 complexes, causing a G1 phase cell cycle arrest by reducing cyclin D1 levels and increasing p27 protein expression (Marone et al., 2009). However, in no case did complete regression of melanoma tumors occur, which indicates that other targets or combinations will need to be identified and validated.
Figure 10. Targeting Akt3 and V600EB-Raf synergistically inhibits melanoma tumor development.

SiRNAs targeting Akt3 (100 picomoles) and V600EB-Raf (12.5 picomoles) were introduced into UACC 903 melanoma cells alone or in combination, which were injected subcutaneously in to mice and tumor development measured on alternate days upto days 17.5. Compared to controls (buffer, siScrambled or siC-Raf) an ~60% reduction in tumor development was observed targeting Akt3 or V600EB-Raf alone. A much more dramatic reduction (~80%) was when these proteins were targeted together. Inset shows decreased Akt3 and V600EB-Raf protein expression levels in melanoma tumors at days 9.5 (Cheung et al., 2008).
It is fairly certain that both pathways MAP and PI3 kinase pathways will need to be targeted as a part of a therapeutic cocktail to treat melanoma. Recently hybrid compounds that inhibit these pathways have been developed and tested on cancer cell lines. One hybrid molecule is HMBA (hexamethylene bisacetamide HMBA, which simultaneously inhibits Akt and MAPK pathways and represses NFκB activity in breast cancer cell lines (Dey et al., 2008). Similarly, a selenium containing iNOS inhibitor, called PBISe, has been developed and its efficacy tested for inhibiting melanoma cell inhibition in vitro and in vivo (Madhunapantula et al., 2008). Intraperitoneally administered or topically applied PBISe inhibits iNOS and PI3K/Akt3 signaling thereby inducing significant apoptosis in melanoma cells. Furthermore, PBISe mediated inhibition of Akt3 signaling led to cell senescence by increasing pErk1/2 levels in melanoma cells. Unusually high MAPK activity induced cell senescence by elevating cdk inhibitors, such as p21, p16 and p27 (Michaloglou et al., 2008, Michaloglou et al., 2005).
Inhibition of Akt3 expression or activity using siRNA or the pharmacological agent LY-294002 als has the potential to increase MAP kinase pathway activity in melanomas to levels that are inhibitory (Cheung et al., 2008). Mechanistically, this occurs because Akt3 phosphorylates V600EB-Raf on S364 and/or S428 to reduce its activity to levels that promote rather than inhibit melanoma development from melanocytes (Cheung et al., 2008) (Fig. 7). Inhibiting Akt3 activity decreases this regulation leasding to high, inhibitory levels of V600EB-Raf activity. In advanced melanomas, targeting these two proteins together using siRNA led to cooperative, synergistically acting tumor inhibition compared to targeting each protein singly (Fig. 10). Although the above studies demonstrate the advantage of simultaneously targeting PI3 and MAP kinase pathways, complete tumor inhibition was not achieved again demonstrating the need to identify other proteins to target in combination with these. Therefore, multiple laboratories are working towards this goal by identifying key deregulated kinases promoting melanoma development to determine whether they inhibit melanoma growth synergistically when combined with targeting of Akt3 and V600EB-Raf.
5. CONCLUSIONS
In melanomas, PTEN loss and activation of Akt3 occur frequently. While mechanisms leading to Akt3 activation in melanomas are not fully characterized, it is known that overexpression of Akt3 and decreased PTEN activity play important roles in this process. Expression of PTEN or targeted reduction of Akt3 activity has also been shown to reduce the survival of melanoma tumor cells leading to inhibition of tumor development and sensitization of melanoma cells to apoptosis inducing agents. Therefore, expression of PTEN or targeting Akt3 directly or by interfering with upstream proteins regulating these genes, promises a new and more effective therapeutic approach for melanoma treatment.
6. KEY UNANSWERED QUESTIONS
By promoting cell survival and proliferation, the PTEN and Akt3 signaling cascade plays an important role in melanomas. Nevertheless, an expanding number of major questions remain to be answered. For example, what is the mechanism of selective Akt3 activation in melanomas? Would therapeutically targeting Akt3 in human patients effectively inhibit melanoma development? If combination therapies are required, what other kinases would synergize with Akt3 in melanomas? Will targeting Akt3 promote melanoma metastasis? Which Akt3 substrate needs to be targeted for effective melanoma tumor inhibition? Do microRNAs regulate PTEN expression in melanomas? Does phosphorylation of PTEN affect melanoma development? Addressing these aspects might provide better understanding of melanoma development and thereby aid in the development of novel therapeutics.
Acknowledgments
Grant support: The American Cancer Society (RSG-04-053-01-GMC) and The Foreman Foundation for Melanoma Research.
The Foreman Foundation for Melanoma Research and American Cancer Society are gratefully acknowledged for support of this work.
References
- Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005;280:35195–35202. doi: 10.1074/jbc.M503045200. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M, Jones PF, Grossniklaus U, Cron P, Schier AF, Dick M, Bilbe G, Hemmings BA. Developmental regulation of expression and activity of multiple forms of the Drosophila RAC protein kinase. J Biol Chem. 1995;270:4066–4075. doi: 10.1074/jbc.270.8.4066. [DOI] [PubMed] [Google Scholar]
- Bastian BC, Leboit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–2175. [PubMed] [Google Scholar]
- Bedogni B, O’neill MS, Welford SM, Bouley DM, Giaccia AJ, Denko NC, Powell MB. Topical treatment with inhibitors of the phosphatidylinositol 3′-kinase/Akt and Raf/mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathways reduces melanoma development in severe combined immunodeficient mice. Cancer Res. 2004;64:2552–2560. doi: 10.1158/0008-5472.can-03-3327. [DOI] [PubMed] [Google Scholar]
- Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB. Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest. 2008;118:3660–3670. doi: 10.1172/JCI36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedogni B, Welford SM, Kwan AC, Ranger-Moore J, Saboda K, Powell MB. Inhibition of phosphatidylinositol-3-kinase and mitogen-activated protein kinase kinase 1/2 prevents melanoma development and promotes melanoma regression in the transgenic TPRas mouse model. Mol Cancer Ther. 2006;5:3071–3077. doi: 10.1158/1535-7163.MCT-06-0269. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, De Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. International Journal of Cancer. 1995a;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, De Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995b;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Moore R, Larue L. A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther. 2004;3:268–275. doi: 10.4161/cbt.3.3.703. [DOI] [PubMed] [Google Scholar]
- Bellei B, Flori E, Izzo E, Maresca V, Picardo M. GSK3beta inhibition promotes melanogenesis in mouse B16 melanoma cells and normal human melanocytes. Cell Signal. 2008;20:1750–1761. doi: 10.1016/j.cellsig.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Birck A, Ahrenkiel V, Zeuthen J, Hou-Jensen K, Guldberg P. Mutation and allelic loss of the PTEN/MMAC1 gene in primary and metastatic melanoma biopsies. J Invest Dermatol. 2000;114:277–280. doi: 10.1046/j.1523-1747.2000.00877.x. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Bonazzi VF, Irwin D, Hayward NK. Identification of candidate tumor suppressor genes inactivated by promoter methylation in melanoma. Genes Chromosomes Cancer. 2009;48:10–21. doi: 10.1002/gcc.20615. [DOI] [PubMed] [Google Scholar]
- Boni R, Vortmeyer AO, Burg G, Hofbauer G, Zhuang Z. The PTEN tumour suppressor gene and malignant melanoma. Melanoma Res. 1998;8:300–302. doi: 10.1097/00008390-199808000-00002. [DOI] [PubMed] [Google Scholar]
- Borgatti P, Martelli AM, Bellacosa A, Casto R, Massari L, Capitani S, Neri LM. Translocation of Akt/PKB to the nucleus of osteoblast-like MC3T3-E1 cells exposed to proliferative growth factors. FEBS Lett. 2000;477:27–32. doi: 10.1016/s0014-5793(00)01758-0. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Brodbeck D, Hill MM, Hemmings BA. Two splice variants of protein kinase B gamma have different regulatory capacity depending on the presence or absence of the regulatory phosphorylation site serine 472 in the carboxyl-terminal hydrophobic domain. J Biol Chem. 2001;276:29550–29558. doi: 10.1074/jbc.M104633200. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999a;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999b;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celebi JT, Shendrik I, Silvers DN, Peacocke M. Identification of PTEN mutations in metastatic melanoma specimens. J Med Genet. 2000;37:653–657. doi: 10.1136/jmg.37.9.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TO, Tsichlis PN. PDK2: a complex tail in one Akt. Sci STKE. 2001;2001:PE1. doi: 10.1126/stke.2001.66.pe1. [DOI] [PubMed] [Google Scholar]
- Cheney IW, Johnson DE, Vaillancourt MT, Avanzini J, Morimoto A, Demers GW, Wills KN, Shabram PW, Bolen JB, Tavtigian SV, et al. Suppression of tumorigenicity of glioblastoma cells by adenovirus-mediated MMAC1/PTEN gene transfer. Cancer Res. 1998;58:2331–2334. [PubMed] [Google Scholar]
- Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996a;93:3636–3641. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proceedings of the National Academy of Sciences of the United States of America. 1996b;93:3636–3641. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–3439. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813–824. doi: 10.1172/JCI24808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer. 2000;7:115–129. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Aguiar RC, Alberta J, Kum JB, Caron S, Sill H, Marsh DJ, Ritz J, Freedman A, Stiles C, et al. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanismsin haematological malignancies. Hum Mol Genet. 1999;8:185–193. doi: 10.1093/hmg/8.2.185. [DOI] [PubMed] [Google Scholar]
- Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, Depinho RA, Mcmahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009 doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Davies MA, Lu Y, Sano T, Fang X, Tang P, Lapushin R, Koul D, Bookstein R, Stokoe D, Yung WK, et al. Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits Akt activation and induces anoikis. Cancer Res. 1998;58:5285–5290. [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, Lazar AJ, Gershenwald JE, Mills GB. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–1268. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Wong E, Kua N, Teo HL, Tergaonkar V, Lane D. Hexamethylene bisacetamide (HMBA) simultaneously targets AKT and MAPK pathway and represses NF kappaB activity: implications for cancer therapy. Cell Cycle. 2008;7:3759–3767. doi: 10.4161/cc.7.23.7213. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Singh AB, Ellis DL, Richmond A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappaB and tumor progression. Cancer Research. 2002;62:7335–7342. [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Dufner A, Andjelkovic M, Burgering BM, Hemmings BA, Thomas G. Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol Cell Biol. 1999;19:4525–4534. doi: 10.1128/mcb.19.6.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118:5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, Lemmon MA. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell. 2000;6:373–384. doi: 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- Filippa N, Sable CL, Hemmings BA, Van Obberghen E. Effect of phosphoinositide-dependent kinase 1 on protein kinase B translocation and its subsequent activation. Mol Cell Biol. 2000;20:5712–5721. doi: 10.1128/mcb.20.15.5712-5721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca BD, Smith EM, Lee VH, Mackintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Fresno Vara JA, Casado E, De Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Ishida A, Tsuruo T. Involvement of Hsp90 in Signaling and Stability of 3-Phosphoinositide- dependent Kinase-1. J Biol Chem. 2002;277:10346–10353. doi: 10.1074/jbc.M106736200. [DOI] [PubMed] [Google Scholar]
- Furuta J, Umebayashi Y, Miyamoto K, Kikuchi K, Otsuka F, Sugimura T, Ushijima T. Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci. 2004;95:962–968. doi: 10.1111/j.1349-7006.2004.tb03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitonde S, De SK, Tcherpakov M, Dewing A, Yuan H, Riel-Mehan M, Krajewski S, Robertson G, Pellecchia M, Ronai Z. BI-69A11-mediated inhibition of AKT leads to effective regression of xenograft melanoma. Pigment Cell Melanoma Res. 2009 doi: 10.1111/j.1755-148X.2009.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene. 2006;374:1–9. doi: 10.1016/j.gene.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117:719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldberg P, Thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57:3660–3663. [PubMed] [Google Scholar]
- Houben R, Ortmann S, Drasche A, Troppmair J, Herold MJ, Becker JC. Proliferation arrest in B-Raf mutant melanoma cell lines upon MAPK pathway activation. J Invest Dermatol. 2009;129:406–414. doi: 10.1038/jid.2008.214. [DOI] [PubMed] [Google Scholar]
- Hsu MY, Meier F, Herlyn M. Melanoma development and progression: a conspiracy between tumor and host. Differentiation. 2002;70:522–536. doi: 10.1046/j.1432-0436.2002.700906.x. [DOI] [PubMed] [Google Scholar]
- Huang B, Porter G. Expression of proline-rich Akt-substrate PRAS40 in cell survival pathway and carcinogenesis. Acta Pharmacol Sin. 2005;26:1253–1258. doi: 10.1111/j.1745-7254.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- Huang GL, Zhang XH, Guo GL, Huang KT, Yang KY, Shen X, You J, Hu XQ. Clinical significance of miR-21 expression in breast cancer: SYBR-Green I-based real-time RT-PCR study of invasive ductal carcinoma. Oncol Rep. 2009;21:673–679. [PubMed] [Google Scholar]
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Chang HY, Fei T, Wu H, Chen YG. GSK3 beta mediates suppression of cyclin D2 expression by tumor suppressor PTEN. Oncogene. 2007;26:2471–2482. doi: 10.1038/sj.onc.1210033. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Kanai F, Hikiba Y, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Asaoka Y, Tateishi K, et al. Functional consequences of mutations in a putative Akt phosphorylation motif of B-raf in human cancers. Mol Carcinog. 2005;43:59–63. doi: 10.1002/mc.20102. [DOI] [PubMed] [Google Scholar]
- Ivanov VN, Hei TK. Arsenite sensitizes human melanomas to apoptosis via tumor necrosis factor alpha-mediated pathway. J Biol Chem. 2004;279:22747–22758. doi: 10.1074/jbc.M314131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetzt A, Howe JA, Horn MT, Maxwell E, Yin Z, Johnson D, Kumar CC. Adenoviral-mediated expression of a kinase-dead mutant of Akt induces apoptosis selectively in tumor cells and suppresses tumor growth in mice. Cancer Res. 2003;63:6697–6706. [PubMed] [Google Scholar]
- Jiang K, Sun J, Cheng J, Djeu JY, Wei S, Sebti S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol Cell Biol. 2004;24:5565–5576. doi: 10.1128/MCB.24.12.5565-5576.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Jones PF, Jakubowicz T, Hemmings BA. Molecular cloning of a second form of rac protein kinase. Cell Regul. 1991;2:1001–1009. doi: 10.1091/mbc.2.12.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kang SS, Kwon T, Kwon DY, Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999;274:13085–13090. doi: 10.1074/jbc.274.19.13085. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kim SS, Yoo NJ, Jeong EG, Kim MS, Lee SH. Expression of NEDD4-1, a PTEN regulator, in gastric and colorectal carcinomas. Apmis. 2008;116:779–784. doi: 10.1111/j.1600-0463.2008.00999.x. [DOI] [PubMed] [Google Scholar]
- Konishi H, Kuroda S, Tanaka M, Matsuzaki H, Ono Y, Kameyama K, Haga T, Kikkawa U. Molecular cloning and characterization of a new member of the RAC protein kinase family: association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem Biophys Res Commun. 1995;216:526–534. doi: 10.1006/bbrc.1995.2654. [DOI] [PubMed] [Google Scholar]
- Kotelevets L, Van Hengel J, Bruyneel E, Mareel M, Van Roy F, Chastre E. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J Cell Biol. 2001;155:1129–1135. doi: 10.1083/jcb.200105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278:10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- Krasilnikov M, Ivanov VN, Dong J, Ronai Z. ERK and PI3K negatively regulate STAT-transcriptional activities in human melanoma cells: implications towards sensitization to apoptosis. Oncogene. 2003;22:4092–4101. doi: 10.1038/sj.onc.1206598. [DOI] [PubMed] [Google Scholar]
- Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem. 2000;275:423–428. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]
- Laine J, Kunstle G, Obata T, Noguchi M. Differential regulation of Akt kinase isoforms by the members of the TCL1 oncogene family. J Biol Chem. 2002;277:3743–3751. doi: 10.1074/jbc.M107069200. [DOI] [PubMed] [Google Scholar]
- Laine J, Kunstle G, Obata T, Sha M, Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Mol Cell. 2000;6:395–407. doi: 10.1016/s1097-2765(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Larribere L, Khaled M, Tartare-Deckert S, Busca R, Luciano F, Bille K, Valony G, Eychene A, Auberger P, Ortonne JP, et al. PI3K mediates protection against TRAIL-induced apoptosis in primary human melanocytes. Cell Death Differ. 2004 doi: 10.1038/sj.cdd.4401475. [DOI] [PubMed] [Google Scholar]
- Lasithiotakis KG, Sinnberg TW, Schittek B, Flaherty KT, Kulms D, Maczey E, Garbe C, Meier FE. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J Invest Dermatol. 2008;128:2013–2023. doi: 10.1038/jid.2008.44. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Li G, Kalabis J, Xu X, Meier F, Oka M, Bogenrieder T, Herlyn M. Reciprocal regulation of MelCAM and AKT in human melanoma. Oncogene. 2003;22:6891–6899. doi: 10.1038/sj.onc.1206819. [DOI] [PubMed] [Google Scholar]
- Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61:3819–3825. [PubMed] [Google Scholar]
- Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- Li L, Ross AH. Why is PTEN an important tumor suppressor? J Cell Biochem. 2007;102:1368–1374. doi: 10.1002/jcb.21593. [DOI] [PubMed] [Google Scholar]
- Lietzke SE, Bose S, Cronin T, Klarlund J, Chawla A, Czech MP, Lambright DG. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol Cell. 2000;6:385–394. doi: 10.1016/s1097-2765(00)00038-1. [DOI] [PubMed] [Google Scholar]
- Liu S, Ren S, Howell P, Fodstad O, Riker AI. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008;21:545–558. doi: 10.1111/j.1755-148X.2008.00484.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol. 2008;84:289–306. doi: 10.1111/j.1751-1097.2007.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson S, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting thePI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorigan P, Eisen T, Hauschild A. Systemic therapy for metastatic malignant melanoma--from deeply disappointing to bright future? Exp Dermatol. 2008;17:383–394. doi: 10.1111/j.1600-0625.2007.00673.x. [DOI] [PubMed] [Google Scholar]
- Lu Y, Li Z, Sun M. Multiple gene alterations involved in the processor of human gastric carcinogenesis. Chung-Hua i Hsueh Tsa Chih [Chinese Medical Journal] 1995;75:679–682. [PubMed] [Google Scholar]
- Lu Y, Lin YZ, Lapushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009;273:194–200. doi: 10.1016/j.canlet.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19:2739–2744. doi: 10.1038/sj.onc.1203597. [DOI] [PubMed] [Google Scholar]
- Madhunapantula SV, Desai D, Sharma A, Huh SJ, Amin S, Robertson GP. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Mol Cancer Ther. 2008;7:1297–1308. doi: 10.1158/1535-7163.MCT-07-2267. [DOI] [PubMed] [Google Scholar]
- Madhunapantula SV, Sharma A, Robertson GP. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 2007;67:3626–3636. doi: 10.1158/0008-5472.CAN-06-4234. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- Maehama T, Okahara F, Kanaho Y. The tumour suppressor PTEN: involvement of a tumour suppressor candidate protein in PTEN turnover. Biochem Soc Trans. 2004;32:343–347. doi: 10.1042/bst0320343. [DOI] [PubMed] [Google Scholar]
- Marone R, Erhart D, Mertz AC, Bohnacker T, Schnell C, Cmiljanovic V, Stauffer F, Garcia-Echeverria C, Giese B, Maira SM, et al. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res. 2009;7:601–613. doi: 10.1158/1541-7786.MCR-08-0366. [DOI] [PubMed] [Google Scholar]
- Meier F, Schittek B, Busch S, Garbe C, Smalley K, Satyamoorthy K, Li G, Herlyn M. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. doi: 10.2741/1755. [DOI] [PubMed] [Google Scholar]
- Meier R, Alessi DR, Cron P, Andjelkovic M, Hemmings BA. Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bbeta. J Biol Chem. 1997;272:30491–30497. doi: 10.1074/jbc.272.48.30491. [DOI] [PubMed] [Google Scholar]
- Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Johansson B, Hoglund M, Mitelman F. Chromosomal imbalance maps of malignant solid tumors: a cytogenetic survey of 3185 neoplasms. Cancer Res. 1997;57:2765–2780. [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS. BRAF(E600) in benign and malignant human tumours. Oncogene. 2008;27:877–895. doi: 10.1038/sj.onc.1210704. [DOI] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, Van Der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Mikhail M, Velazquez E, Shapiro R, Berman R, Pavlick A, Sorhaindo L, Spira J, Mir C, Panageas KS, Polsky D, et al. PTEN expression in melanoma: relationship with patient survival, Bcl-2 expression, and proliferation. Clin Cancer Res. 2005;11:5153–5157. doi: 10.1158/1078-0432.CCR-05-0397. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG. Direct identification of PTEN phosphorylation sites. FEBS Lett. 2002;528:145–153. doi: 10.1016/s0014-5793(02)03274-x. [DOI] [PubMed] [Google Scholar]
- Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, Hengge UR. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66:6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem. 2000;275:36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem. 2007;282:20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006;2:91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- Packer L, Pavey S, Parker A, Stark M, Johansson P, Clarke B, Pollock P, Ringner M, Hayward N. Osteopontin is a downstream effector of the PI3-kinase pathway in melanomas that is inversely correlated with functional PTEN. Carcinogenesis. 2006;27:1778–1786. doi: 10.1093/carcin/bgl016. [DOI] [PubMed] [Google Scholar]
- Panka DJ, Cho DC, Atkins MB, Mier JW. GSK-3beta inhibition enhances sorafenib-induced apoptosis in melanoma cell lines. J Biol Chem. 2008;283:726–732. doi: 10.1074/jbc.M705343200. [DOI] [PubMed] [Google Scholar]
- Parmiter AH, Balaban G, Clark WH, Jr, Nowell PC. Possible involvement of the chromosome region 10q24----q26 in early stages of melanocytic neoplasia. Cancer Genet Cytogenet. 1988;30:313–317. doi: 10.1016/0165-4608(88)90200-2. [DOI] [PubMed] [Google Scholar]
- Patel S, Woodgett J. Glycogen synthase kinase-3 and cancer: good cop, bad cop? Cancer Cell. 2008;14:351–353. doi: 10.1016/j.ccr.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Koval A, Hallas C, Bichi R, Tresini M, Malstrom S, Russo G, Tsichlis P, Croce CM. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc Natl Acad Sci U S A. 2000;97:3028–3033. doi: 10.1073/pnas.040557697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzolesi MG, Platzer P, Waite KA, Eng C. Differential expression of PTEN-targeting microRNAs miR-19a and miR-21 in Cowden syndrome. Am J Hum Genet. 2008;82:1141–1149. doi: 10.1016/j.ajhg.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetsch M, Dittberner T, Woenckhaus C. PTEN/MMAC1 in malignant melanoma and its importance for tumor progression. Cancer Genet Cytogenet. 2001a;125:21–26. doi: 10.1016/s0165-4608(00)00353-8. [DOI] [PubMed] [Google Scholar]
- Poetsch M, Dittberner T, Woenckhaus C. PTEN/MMAC1 in malignant melanoma and its importance for tumor progression. Cancer Genet Cytogenet. 2001b;125:21–26. doi: 10.1016/s0165-4608(00)00353-8. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Trent JM. The genetics of cutaneous melanoma. Clin Lab Med. 2000;20:667–690. [PubMed] [Google Scholar]
- Polsky D, Cordon-Cardo C. Oncogenes in melanoma. Oncogene. 2003;22:3087–3091. doi: 10.1038/sj.onc.1206449. [DOI] [PubMed] [Google Scholar]
- Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide Disables 3-Phosphoinoside Binding to the Pleckstrin Homology Domain of Protein Kinase B (PKB)/Akt by a PKCzeta-Dependent Mechanism. Mol Cell Biol. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifenberger J, Wolter M, Bostrom J, Buschges R, Schulte KW, Megahed M, Ruzicka T, Reifenberger G. Allelic losses on chromosome arm 10q and mutation of the PTEN (MMAC1) tumour suppressor gene in primary and metastatic malignant melanomas. Virchows Arch. 2000;436:487–493. doi: 10.1007/s004280050477. [DOI] [PubMed] [Google Scholar]
- Richmond A, Fan GH, Dhawan P, Yang J. How do chemokine/chemokine receptor activations affect tumorigenesis? Novartis Found Symp. 2004;256:74–89. discussion 89–91, 106–111, 266–109. [PubMed] [Google Scholar]
- Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev. 2005;24:273–285. doi: 10.1007/s10555-005-1577-9. [DOI] [PubMed] [Google Scholar]
- Robertson GP, Furnari FB, Miele ME, Glendening MJ, Welch DR, Fountain JW, Lugo TG, Huang HJ, Cavenee WK. In vitro loss of heterozygosity targets the PTEN/MMAC1 gene in melanoma. Proc Natl Acad Sci U S A. 1998a;95:9418–9423. doi: 10.1073/pnas.95.16.9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson GP, Furnari FB, Miele ME, Glendening MJ, Welch DR, Fountain JW, Lugo TG, Huang HJ, Cavenee WK. In vitro loss of heterozygosity targets the PTEN/MMAC1 gene in melanoma. Proc Natl Acad Sci U S A. 1998b;95:9418–9423. doi: 10.1073/pnas.95.16.9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson GP, Herbst RA, Nagane M, Huang HJ, Cavenee WK. The chromosome 10 monosomy common in human melanomas results from loss of two separate tumor suppressor loci. Cancer Res. 1999a;59:3596–3601. [PubMed] [Google Scholar]
- Robertson GP, Herbst RA, Nagane M, Huang HJ, Cavenee WK. The chromosome 10 monosomy common in human melanomas results from loss of two separate tumor suppressor loci. Cancer Res. 1999b;59:3596–3601. [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Saida T. Recent advances in melanoma research. J Dermatol Sci. 2001;26:1–13. doi: 10.1016/s0923-1811(01)00085-8. [DOI] [PubMed] [Google Scholar]
- Saito A, Narasimhan P, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after stroke: relationships with nerve growth factor. J Neurosci. 2004;24:1584–1593. doi: 10.1523/JNEUROSCI.5209-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale EM, Sale GJ. Protein kinase B: signalling roles and therapeutic targeting. Cell Mol Life Sci. 2008;65:113–127. doi: 10.1007/s00018-007-7274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyan KS, Swamy N, Dizon DS, Singh R, Granai CO, Brard L. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: role of caspase and MAPK activation. Gynecol Oncol. 2006;103:261–270. doi: 10.1016/j.ygyno.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- Sharma A, Sharma AK, Madhunapantula SV, Desai D, Huh SJ, Mosca P, Amin S, Robertson GP. Targeting Akt3 Signaling in Malignant Melanoma Using Isoselenocyanates. Clin Cancer Res. 2009;15:1674–1685. doi: 10.1158/1078-0432.CCR-08-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Sharma A, Desai D, Madhunapantula SV, Huh SJ, Robertson GP, Amin S. Synthesis and anticancer activity comparison of phenylalkyl isoselenocyanates with corresponding naturally occurring and synthetic isothiocyanates. J Med Chem. 2008;51:7820–7826. doi: 10.1021/jm800993r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- She QB, Solit DB, Ye Q, O’reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–297. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnberg T, Lasithiotakis K, Niessner H, Schittek B, Flaherty KT, Kulms D, Maczey E, Campos M, Gogel J, Garbe C, et al. Inhibition of PI3K-AKT-mTOR Signaling Sensitizes Melanoma Cells to Cisplatin and Temozolomide. J Invest Dermatol. 2008 doi: 10.1038/jid.2008.379. [DOI] [PubMed] [Google Scholar]
- Smalley KS, Contractor R, Haass NK, Kulp AN, Atilla-Gokcumen GE, Williams DS, Bregman H, Flaherty KT, Soengas MS, Meggers E, et al. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- Smalley KS, Flaherty KT. Integrating BRAF/MEK inhibitors into combination therapy for melanoma. Br J Cancer. 2009;100:431–435. doi: 10.1038/sj.bjc.6604891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- Smalley KS, Herlyn M. Targeting intracellular signaling pathways as a novel strategy in melanoma therapeutics. Ann N Y Acad Sci. 2005;1059:16–25. doi: 10.1196/annals.1339.005. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–2890. [PubMed] [Google Scholar]
- Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L, Robertson GP. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- Stewart AL, Mhashilkar AM, Yang XH, Ekmekcioglu S, Saito Y, Sieger K, Schrock R, Onishi E, Swanson X, Mumm JB, et al. PI3 kinase blockade by Ad-PTEN inhibits invasion and induces apoptosis in RGP and metastatic melanoma cells. Mol Med. 2002;8:451–461. [PMC free article] [PubMed] [Google Scholar]
- Tachiiri S, Sasai K, Oya N, Hiraoka M. Enhanced cell killing by overexpression of dominant-negative phosphatidylinositol 3-kinase subunit, Deltap85, following genotoxic stresses. Jpn J Cancer Res. 2000;91:1314–1318. doi: 10.1111/j.1349-7006.2000.tb00919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney T, Stokoe D. New insights into PTEN. J Cell Sci. 2007;120:4071–4079. doi: 10.1242/jcs.015230. [DOI] [PubMed] [Google Scholar]
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P, Arrieumerlou C, Hall MN. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE. 2007;2:e1217. doi: 10.1371/journal.pone.0001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson FH, Emerson J, Olson S, Weinstein R, Leavitt SA, Leong SP, Emerson S, Trent JM, Nelson MA, Salmon SE, et al. Cytogenetics of 158 patients with regional or disseminated melanoma. Subset analysis of near-diploid and simple karyotypes. Cancer Genet Cytogenet. 1995;83:93–104. doi: 10.1016/0165-4608(95)00057-v. [DOI] [PubMed] [Google Scholar]
- Thompson FH, Nelson MA, Trent JM, Guan XY, Liu Y, Yang JM, Emerson J, Adair L, Wymer J, Balfour C, et al. Amplification of 19q13.1–q13.2 sequences in ovarian cancer. G-band, FISH, and molecular studies. Cancer Genet Cytogenet. 1996;87:55–62. doi: 10.1016/0165-4608(95)00248-0. [DOI] [PubMed] [Google Scholar]
- Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, Smith NB, Robertson GP. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Res. 2008a;68:7638–7649. doi: 10.1158/0008-5472.CAN-07-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MA, Smith CD, Kester M, Robertson GP. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res. 2008b;14:3571–3581. doi: 10.1158/1078-0432.CCR-07-4881. [DOI] [PubMed] [Google Scholar]
- Tsao H, Zhang X, Benoit E, Haluska FG. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene. 1998a;16:3397–3402. doi: 10.1038/sj.onc.1201881. [DOI] [PubMed] [Google Scholar]
- Tsao H, Zhang X, Benoit E, Haluska FG. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene. 1998b;16:3397–3402. doi: 10.1038/sj.onc.1201881. [DOI] [PubMed] [Google Scholar]
- Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60:1800–1804. [PubMed] [Google Scholar]
- Ueda Y, Richmond A. NF-kappaB activation in melanoma. Pigment Cell Res. 2006;19:112–124. doi: 10.1111/j.1600-0749.2006.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Su Y, Richmond A. CCAAT displacement protein regulates nuclear factor-kappa beta-mediated chemokine transcription in melanoma cells. Melanoma Res. 2007;17:91–103. doi: 10.1097/CMR.0b013e3280a60888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dekken H, Geelen E, Dinjens WN, Wijnhoven BP, Tilanus HW, Tanke HJ, Rosenberg C. Comparative genomic hybridization of cancer of the gastroesophageal junction: deletion of 14Q31–32.1 discriminates between esophageal (Barrett’s) and gastric cardia adenocarcinomas. Cancer Research. 1999;59:748–752. [PubMed] [Google Scholar]
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346(Pt 3):561–576. [PMC free article] [PubMed] [Google Scholar]
- Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann V, Wacker J, Deichmann M. Mutations of the activation-associated phosphorylation sites at codons 308 and 473 of protein kinase B are absent in human melanoma. Arch Dermatol Res. 2001;293:368–372. doi: 10.1007/s004030100236. [DOI] [PubMed] [Google Scholar]
- Waldmann V, Wacker J, Deichmann M. Absence of mutations in the pleckstrin homology (PH) domain of protein kinase B (PKB/Akt) in malignant melanoma. Melanoma Res. 2002;12:45–50. doi: 10.1097/00008390-200202000-00007. [DOI] [PubMed] [Google Scholar]
- Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007a;282:20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007b;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Digiovanna JJ, Stern JB, Hornyak TJ, Raffeld M, Khan SG, Oh KS, Hollander MC, Dennis PA, Kraemer KH. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A. 2009;106:6279–6284. doi: 10.1073/pnas.0812401106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lee CG. MicroRNA and cancer - focus on apoptosis. J Cell Mol Med. 2009;13:12–23. doi: 10.1111/j.1582-4934.2008.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng LP, Smith WM, Dahia PL, Ziebold U, Gil E, Lees JA, Eng C. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59:5808–5814. [PubMed] [Google Scholar]
- Wilson W, 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res. 2008;68:8156–8163. doi: 10.1158/0008-5472.CAN-08-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005;17:150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma. Oncogene. 2003;22:3113–3122. doi: 10.1038/sj.onc.1206451. [DOI] [PubMed] [Google Scholar]
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1998a;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1998b;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Singh SV. Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res. 2007;67:2239–2246. doi: 10.1158/0008-5472.CAN-06-3645. [DOI] [PubMed] [Google Scholar]
- Yang H, Kong W, He L, Zhao JJ, O’donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68:425–433. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell. 2005;20:539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Zhang Y, Yang Z, Li A, Dong J. Simultaneous knockdown of BRAF and expression of INK4A in melanoma cells leads to potent growth inhibition and apoptosis. Biochem Biophys Res Commun. 2008;370:509–513. doi: 10.1016/j.bbrc.2008.03.148. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]