

Abstract
Infection of plants by bacterial leaf pathogens at wound sites is common in nature. Plants defend wound sites to prevent pathogen invasion, but several pathogens can overcome spatial restriction and enter leaf tissues. The molecular mechanisms used by pathogens to suppress containment at wound infection sites are poorly understood. Here, we studied Pseudomonas syringae strains causing brown spot on bean and blossom blight on pear. These strains exist as epiphytes that can cause disease upon wounding caused by hail, sand storms and frost. We demonstrate that these strains overcome spatial restriction at wound sites by producing syringolin A (SylA), a small molecule proteasome inhibitor. Consequently, SylA-producing strains are able to escape from primary infection sites and colonize adjacent tissues along the vasculature. We found that SylA diffuses from the primary infection site and suppresses acquired resistance in adjacent tissues by blocking signaling by the stress hormone salicylic acid (SA). Thus, SylA diffusion creates a zone of SA-insensitive tissue that is prepared for subsequent colonization. In addition, SylA promotes bacterial motility and suppresses immune responses at the primary infection site. These local immune responses do not affect bacterial growth and were weak compared to effector-triggered immunity. Thus, SylA facilitates colonization from wounding sites by increasing bacterial motility and suppressing SA signaling in adjacent tissues.
Author Summary
Bacterial plant pathogens are usually contained at wound infection sites by an effective immune response. The ways in which bacteria can overcome this spatial restriction is poorly understood. Here, we studied two Pseudomonas syringae pv. syringae strains that cause brown spot on bean and blossom blight on pear and are known to exist as epiphytes that cause disease upon wounding by hail, sand storms or frost. In this study, we demonstrated that these two strains suppress spatial confinement by producing a low molecular weight proteasome inhibitor, syringolin A (SylA), which acts in two ways. On one hand, SylA diffuses into tissue surrounding the primary infection site to make the tissue insensitive to immune signaling. On the other hand, SylA promotes bacterial motility and suppresses immune responses at the primary infection site. Consequently, SylA-producing bacteria are more motile and able to spread from the primary infection site through xylem vessels and colonize adjacent, immuno-compromised tissues along the vasculature.
Introduction
Wounding of plants by hard wind, hail, heavy rain, sand storms, and frost is common in nature. Many epiphytic leaf pathogens take advantage of this opportunity to infect plants [1]. Although plants have evolved effective immune responses to protect wound sites, many pathogens are able to enter leaf tissues and cause disease [1]. The molecular mechanisms underlying the suppression of plant-mediated restriction of pathogen spreading from wound sites are poorly understood.
Pseudomonas syringae pv. syringae (Psy) causes brown spot on bean plants and blossom blight in pear trees [1], which are serious diseases responsible for significant yield losses in agricultural industries in the US, Africa, and Australia. Psy can grow epiphytically on leaf surfaces and enters the leaf intercellular space (apoplast) through stomata and wounds [1]–[3]. Upon entering the leaf apoplast, Psy initially propagates biotrophically, keeping the host cells alive, and later causes necrotic lesions [4]. Although Psy is a common leaf epiphyte, disease outbreaks are often seasonal and conditional [5]. For example, the onset of epidemics is associated with heavy rain storms and is related to raindrop momentum rather than an increase in humidity [6]. In addition, heavy wind without significant precipitation, causing damage by hail and blowing sand, has caused a brown spot outbreak causing 55% yield loss [7]. Likewise, although Psy strains are a common and dominant component of the microflora on pear trees, blossom blight disease only occurs after frost injury, which explains the strong seasonal variation of disease outbreaks [8]. These data illustrate that P. syringae takes advantage of natural wound sites to enter host tissue and cause disease. Thus, it is important to understand the molecular mechanisms underlying host entry at wound sites in order to prevent disease outbreaks. However, to date, these mechanisms have been poorly investigated.
We recently discovered that green fluorescent protein (GFP)-expressing P. syringae can escape from wound infection sites and colonize adjacent tissues in the wild tobacco plant Nicotiana benthamiana [9], which has become an important model plant for P. syringae infections [3], [10]–[16]. Colonies appeared up to 1 cm from the primary infection site within a few days. Although these infections are not systemic (throughout the whole plant), these distances, from the perspective of bacteria, are significant and increase the area of infection by several orders of magnitude. Using bacterial count assays with controlled inoculation populations, we have shown that bacterial populations can grow nearly 100-fold more if the bacteria colonize adjacent tissue compared to when they remain contained at the primary infection site [9]. The colonization from wound sites follows the vasculature, and electron microscopy experiments indicated that the bacteria move through xylem vessels [9]. The ability to colonize tissues along the vasculature involves four steps: first, the bacteria overcome local containment at the primary infection site; second, they transport themselves over several millimeters through the xylem; third, they escape from the xylem vessel into the apoplast; and finally, they colonize the apoplast in adjacent tissues. The molecular mechanisms underlying each of these events are not yet understood.
We found that colonization from wound sites on N. benthamiana is common for P. syringae strains representing the major branches of the P. syringae phylogenetic tree (phylogroups) [9]. Two of the strains that efficiently colonize tissues from wound sites are P. syringae pv. syringae B728a (PsyB728a) and B301D-R (PsyB301D), both from phylogroup II [16]. PsyB728a and PsyB301D cause brown spot on bean plants and blossom blight on pear trees, respectively, and both disease outbreaks occur upon wounding [6]–[8]. Besides approximately 30 type III effectors that manipulate the host cell [14], PsyB728a and PsyB301D also produce syringolin A (SylA), a small nonribosomal cyclic peptide that irreversibly inhibits the eukaryotic proteasome [17]–[18]. Compared to wild-type (WT) strains, SylA-deficient mutant strains of PsyB728a cause fewer brown spot symptoms on bean plants upon spray inoculation [18]. SylA-deficient mutants also display delayed entry into bean leaves, which led to the discovery that SylA suppresses preinvasive immunity by reopening stomata in bean plants and Arabidopsis [19]. Using proteasome activity profiling with proteasome-selective chemical probes, we have demonstrated that SylA preferentially inhibits the β2 and β5 catalytic subunits of the Arabidopsis proteasome and that SylA accumulates irreversibly in the nucleus, indicating that it targets the nuclear proteasome [20]. Thus, we hypothesized that the subunit and subcellular selectivity may be important parameters for the biological activity of SylA.
The aim of this study was to investigate the molecular mechanisms underlying wound entry by PsyB728a and PsyB301D. We demonstrate that both strains overcome spatial confinement at wound infection sites by producing SylA. SylA can diffuse from the primary infection site and block salicylic acid (SA) signaling in adjacent tissues, creating an SA-insensitive zone of vasculature tissue that is susceptible to secondary colonization. Furthermore, SylA was found to increase bacterial motility and suppress immune responses at the primary infection site. These local immune response do not affect bacterial growth and are weak compared to effector-trigged immunity (ETI) [21].
Results
The ΔsylC Mutant of PsyB728a Is Unable to Cause Spreading Lesions
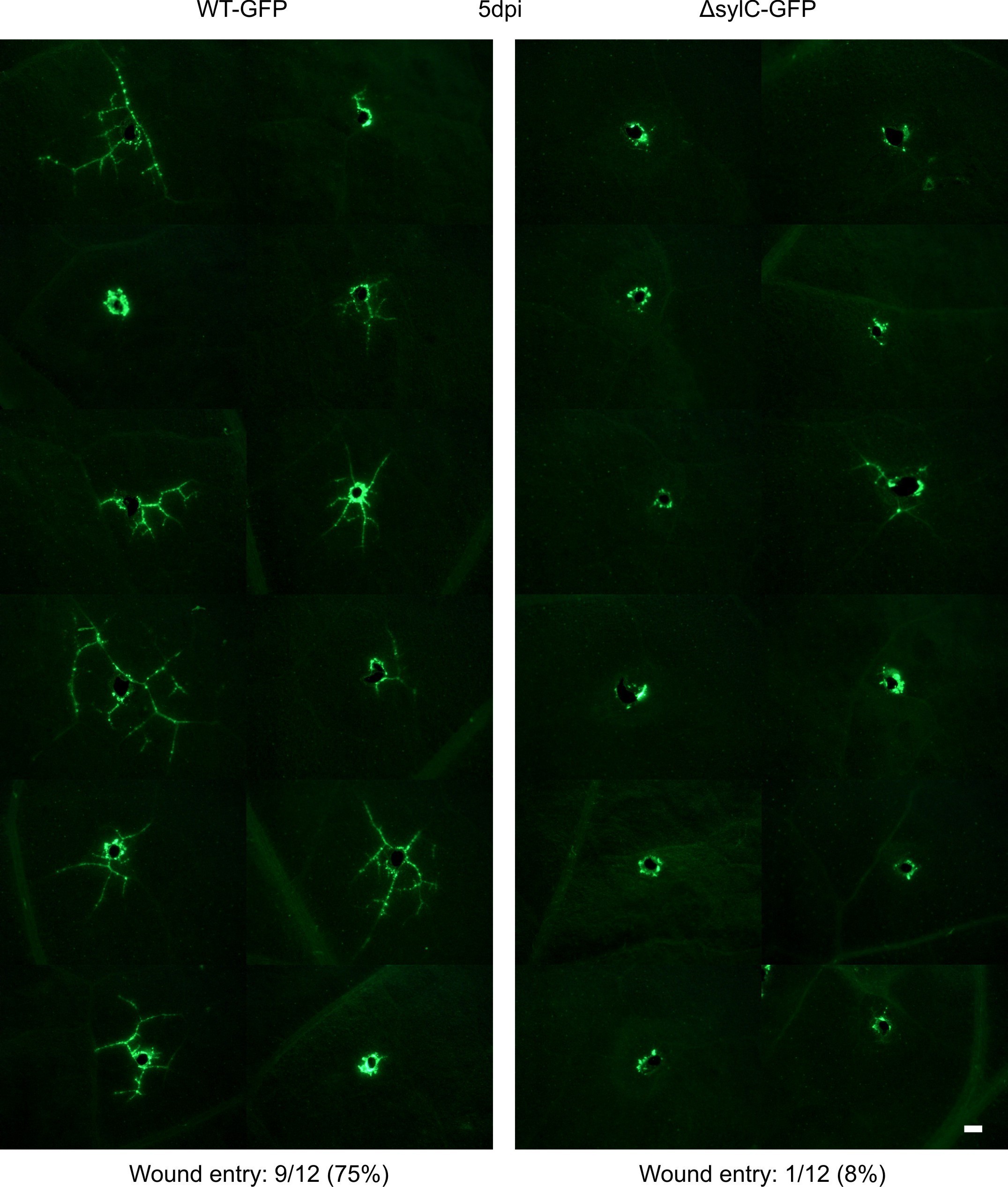
Pathovar syringae B728a is one of the strains that is able to enter wound sites and colonize adjacent tissues in N. benthamiana [9]. At 5 d after wound inoculation (5 dpi) with this strain, fluorescent colonies appeared along the vasculature at regular intervals ( Figure 1A ), which were absent in the untransformed control not expressing GFP ( Figure 1B ). To investigate the molecular mechanism underlying wound entry by PsyB728a of N. benthamiana, we tested various PsyB728a mutants for their ability to cause spreading lesions following toothpick inoculation. These assays revealed that wound entry by the GFP-expressing ΔsylC mutant of PsyB728a was strongly reduced, even though fluorescence was detected at the inoculation site ( Figure 1C ). This fluorescence at the inoculation site was caused by bacteria expressing GFP, since the untransformed ΔsylC control did not show this fluorescent signal ( Figure 1D ). Colonies of ΔsylC in adjacent tissues appeared in less than 20% of the wound inoculations, whereas WT PsyB728a showed colonization from wounding sites (wound entry) in over 80% of the inoculations ( Figure 1E ).
Figure 1. Wound entry by PsyB728a is hampered in the ΔsylC mutant.

(A–D) Wound entry by WT but not ΔsylC PsyB728a. Untransformed and GFP-expressing WT and ΔsylC PsyB728a strains were wound-inoculated into N. benthamiana. Pictures were taken at 5 dpi by fluorescence microscopy using identical settings. W, wound site. Arrowheads indicate colonies in adjacent tissues. Scale bars, 1 mm. (E) WT bacteria spread more frequently from the wound inoculation sites than the ΔsylC mutant. The frequency of colonization of adjacent tissues was scored by fluorescence microscopy at 5 dpi. Error bars represent SEM of four independent experiments. (F) Bacterial population levels per wound inoculation site. Wound sites were immediately inoculated with 1 µL containing 104 WT-GFP or ΔsylC-GFP bacteria. The bacterial titer was determined at 5 dpi in 8-mm diameter leaf discs containing the wound site with adjacent tissues. Error bars represent SEM of three extracts generated from three leaf discs each. This experiment was repeated twice with similar results. (G) Bacterial growth was indistinguishable between WT and ΔsylC bacteria upon infiltration. N. benthamiana plants were infiltrated with WT or ΔsylC mutant strains at 2×105 bacteria/mL. Bacterial populations (in colony forming units (cfu) per cm2 leaf surface) were counted at different time points after infiltration. Error bars indicate SEM for four different samples. Similar results were obtained in repetition experiments with different inoculation densities, relative humidities, and sterilization procedures (see supplemental Figure S1). (E–G) P-values determined using the Student's t-test are indicated.
To demonstrate the relevance of spreading for bacterial growth, we inoculated wound sites with a controlled inoculum (1 µL containing 105 bacteria) and determined the colony-forming units at 5 dpi in leaf discs containing the wound site and adjacent tissue. Under these conditions, WT bacterial populations grew from 104 bacteria to over 106 bacteria per infection site, whereas ΔsylC bacteria grew from the same inoculum population to about 3*104 bacteria per inoculation site ( Figure 1F ). Thus, under these conditions, the ability to spread gives WT bacteria an advantage of 63-fold increased population growth compared to the ΔsylC mutant.
However, strong GFP fluorescence at the inoculation site ( Figure 1D ) suggested that ΔsylC bacteria were able to grow locally. Indeed, bacterial growth assays on infiltrated tissues did not show significant growth differences between WT and ΔsylC strains ( Figure 1G ). Furthermore, indistinguishable bacterial growth between WT and ΔsylC bacteria upon infiltration was also observed with different inoculation densities and at different relative humidity levels (Supplemental Figure S1). Although the ΔsylC strain sometimes grew less compared to the WT strain, the differences were only rarely statistically significant (3/16 comparisons with P<0.05 in Figure S1). The absence of a statistically significant growth phenotype upon infiltration is in contrast but not in conflict with earlier reports that the ΔsylC mutant causes less symptoms and less bacterial growth compared to WT bacteria upon spray inoculation [18]–[19], because SylA suppresses preinvasive immunity by reopening stomata [19]. Our data indicate that, once inside the leaf, WT and ΔsylC bacteria grow equally well at primary infection sites but ΔsylC bacteria are unable to colonize adjacent tissue.
SylA Biosynthesis Is Necessary and Sufficient for Wound Entry by PsyB728a and PsyB301D
To demonstrate that SylA itself is sufficient to promote wound entry, we wound-inoculated the GFP-expressing ΔsylC strain into tissue preinfiltrated with 50 µM SylA or a mock control. We used 50 µM SylA because PsyB301D is able to produce 40–100 µM SylA in cultures [17], and 50 µM SylA inhibits the host proteasome (see below). Exogenous SylA restored wound entry of the ΔsylC strain to a similar frequency as the WT strain ( Figure 2A ). In contrast, no wound entry by the ΔsylC strain was observed by infiltrating buffer without SylA ( Figure 2A ). These data demonstrate that SylA is essential and sufficient to promote wound entry by the bean brown spot pathogen PsyB728a.
Figure 2. SylA is necessary and sufficient for wound entry of PsyB728a and PsyB301D.

(A) Exogenous SylA complements wound entry of the SylA-deficient ΔsylC strain of PsyB728a. N. benthamiana leaves were infiltrated with 50 µM SylA or 0.25% DMSO and wound-inoculated 1 h later. Colonization was scored at 5 dpi by fluorescence microscopy. (B) SylA biosynthesis is necessary for wound entry by PsyB301D. GFP-expressing PsyB301D and derived mutants in the SylA biosynthesis clusters (ΔsylC, ΔsylD, and ΔsylA-E) were wound-inoculated and scored at 5 dpi by fluorescence microscopy. (C) Exogenous SylA complements wound entry of SylA-deficient strains of PsyB301D. N. benthamiana leaves were preinfiltrated with 50 µM SylA or 0.25% DMSO and wound-inoculated 1 h later. Colonization was scored at 5 dpi by fluorescence microscopy. (D) The SylA biosynthesis cluster complements wound entry by PsyB301D. The ΔsylA-E mutant of PsyB301D was transformed with cosmid pPL3syl carrying the SylA biosynthesis cluster. GFP-expressing derivatives were wound-inoculated, and colonization was scored at 5 dpi by fluorescence microscopy. (A–D) GFP-expressing strains were wound-inoculated into N. benthamiana leaves, and colonization of tissue adjacent to the wound site was scored at 5 dpi by fluorescence microscopy. The photographs at the bottom show representative pictures taken by fluorescence microscopy at 5 dpi. Error bars indicate SEM of four independent experiments, each with 12 inoculations. P-values determined using the Student's t-test are indicated.
To determine whether SylA biosynthesis is necessary for wound entry by PsyB301D, we tested SylA-deficient mutants of PsyB301D that lack different SylA biosynthesis enzymes (ΔsylC and ΔsylD mutants, [22]) or the entire SylA biosynthesis cluster (ΔsylA-E, [23]). Importantly, none of these mutant strains was able to colonize adjacent tissue ( Figure 2B ). The loss of wound entry could be complemented with exogenous SylA ( Figure 2C ), demonstrating that SylA is also essential and sufficient to promote wound entry by the pear blossom blight pathogen PsyB301D.
To test whether loss of the wound entry phenotype could be complemented by restoring SylA biosynthesis, we transformed the markerless PsyB301D ΔsylA-E mutant with the pPL3syl cosmid [23], which carries the entire SylA biosynthesis cluster. Transformation restored the capability to colonize adjacent tissues ( Figure 2D ), confirming that SylA biosynthesis genes are required to facilitate wound entry. The ΔsylC and ΔsylD mutants of PsyB301D and PsyB728a could not be transformed with the pPL3syl cosmid as these strains already contain the antibiotic selection marker that was used for the cosmid selection. Taken together, these chemical and genetic complementation assays demonstrate that SylA is required and sufficient to facilitate wound entry by PsyB728a and PsyB301D.
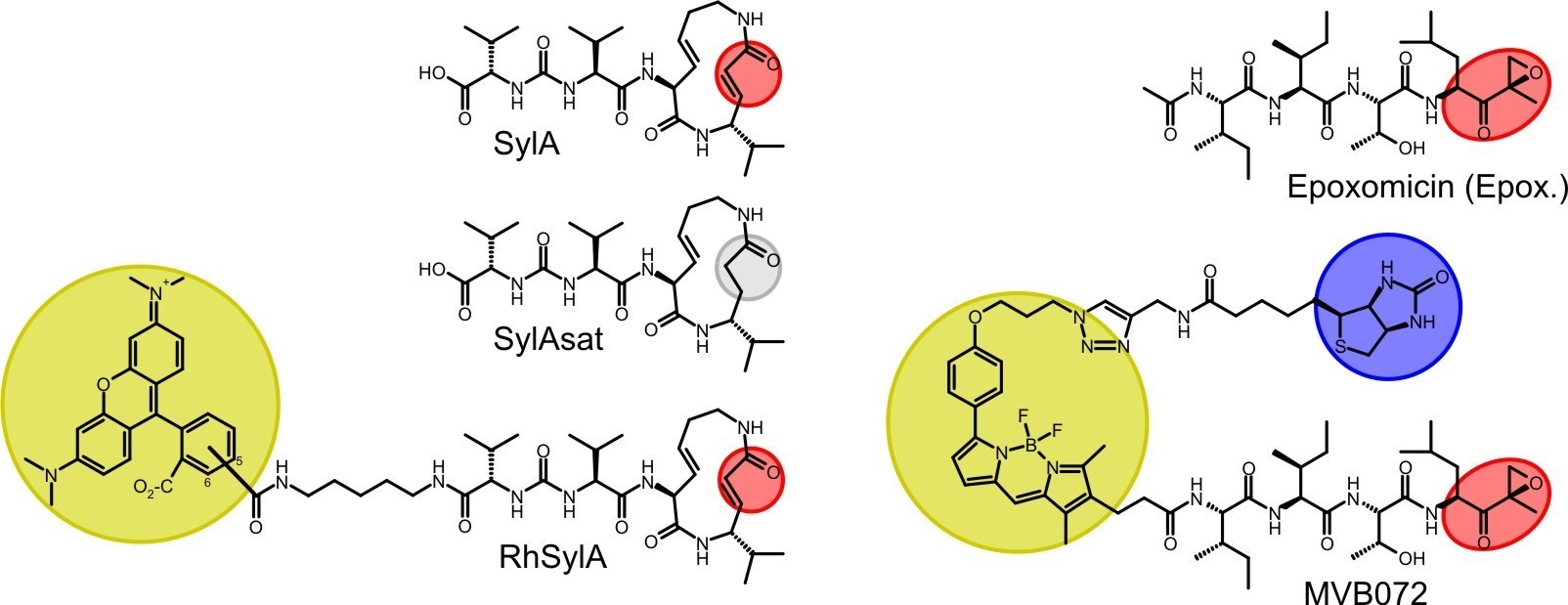
SylA Targets the Proteasome of N. benthamiana
Next, we used proteasome activity profiling [20], [24] to investigate whether SylA inhibits the proteasome of N. benthamiana. Labeling of N. benthamiana leaf extracts with an activity-based probe for the proteasome (MVB072, Supplemental Figure S2) revealed a single 24 kDa band on protein gels ( Figure 3A ). Analysis of the purified labeled proteins by tandem mass spectrometry showed that this signal contained the β1, β2, and β5 proteins, the three catalytic subunits of the proteasome ( Figure 3B and Supplemental Figure S3). Preincubation of N. benthamiana leaf extracts with SylA or the selective proteasome inhibitor epoxomicin blocked MVB072 labeling, indicating that SylA inhibited the proteasome of N. benthamiana ( Figure 3C ). Incubation of N. benthamiana leaf extracts with rhodamine-tagged SylA (RhSylA, [25], Supplemental Figure S2) revealed a single 24 kDa signal on protein gels, similar to MVB072 labeling ( Figure 3C ). Preincubation with SylA or epoxomicin blocked RhSylA labeling of these proteins, confirming that this signal represented the proteasome ( Figure 3C ). In contrast to SylA, no proteasome inhibition was detected upon preincubation with SylAsat ( Figures 3D and 3E ), a synthetic SylA derivative that lacks the double bond required for covalent proteasome inhibition [20]. Inhibition assays at various SylA concentrations showed that proteasome inhibition was detectable at concentrations greater than 6.3 µM and that inhibition was always incomplete ( Figures 3E and 3F ). These data are consistent with the observation that SylA preferentially inhibits two of the three catalytic subunits of the Arabidopsis proteasome, suggesting that the remaining signal was caused by the β1 catalytic subunit [20].
Figure 3. SylA targets the proteasome of N. benthamiana.

(A) MVB072 labels the catalytic proteasome subunits in N. benthamiana leaf extracts. Leaf extract of N. benthamiana was incubated with or without MVB072, an epoxomicin-based probe carrying both biotin and BODIPY. Biotinylated proteins were purified and detected by in-gel fluorescence scanning. (B) Proteins identified by mass spectrometry. In-gel trypsin digests (dashed areas) were analyzed by tandem mass spectrometry. Identified peptides are underlined in the sequences of the β1, β2, and β5 catalytic subunits of the proteasome. None of these peptides were found in the no-probe-control. No peptides were from the propeptide (grey) or the mature N-terminus, containing the catalytic Thr (bold). (C) SylA targets the proteasome of N. benthamiana. Leaf extracts of N. benthamiana were preincubated with 200 µM epoxomicin (Epox.) or SylA for 30 min and then labeled for 2 h with 2 µM MVB072 or RhSylA. Labeled proteins were detected by in-gel fluorescence scanning, and proteins were stained with Coomassie blue. (D) Structures of SylA and SylAsat. SylA has a Michael system in the ring that reacts with the catalytic Thr residues of the proteasome. This Michael system is absent in SylAsat due to saturation of the double bond. (E) SylAsat does not fully inhibit the proteasome. Leaf extracts were preincubated with 50 µM SylA and SylAsat and labeled with MVB072. Proteins were separated on protein gels and detected by fluorescence scanning and Coomassie blue staining. (F) Concentration dependency of proteasome inhibition by SylA. Leaf extracts were incubated with various SylA concentrations and then labeled with MVB072. Proteins were analyzed on a protein gel using fluorescence scanning and Coomassie blue staining.
SylA Blocks SA Signaling and SA-mediated Immunity in N. benthamiana
Since wound entry might be associated with the suppression of acquired resistance in adjacent tissues, we tested whether SylA could inhibit SA signaling, a key regulator of acquired resistance in tobacco [26]. Treatment of N. benthamiana plants with the SA analog benzothiadiazole (BTH, 300 µM [27]) resulted in the transcriptional activation of the SA-responsive PR1a gene within 6 h ( Figure 4A ). However, this transcriptional activation of PR1a was blocked when tissues were preinfiltrated with 50 µM SylA, but not SylAsat ( Figure 4A ), indicating that SylA blocked SA signaling by proteasome inhibition. A SylA dilution series showed that suppression of SA signaling occurred at 3.1 µM SylA, but complete inhibition of SA signaling required concentrations greater than 12.5 µM SylA ( Figure 4B ).
Figure 4. SylA blocks SA-mediated immunity.

(A) SA signaling is blocked by SylA but not SylAsat. N. benthamiana leaves were infiltrated with 50 µM SylA or SylAsat and sprayed with 300 µM BTH. RNA was isolated from infiltrated tissues 6 h after BTH treatment and used as a template for semi-quantitative RT-PCR with gene-specific primers for PR1a and Actin. (B) Dose-dependent inhibition of BTH-induced NbPR1a expression by SylA. Leaves were infiltrated with various concentrations of SylA and sprayed 6 h later with 300 µM BTH. RNA was isolated from infiltrated tissues 6 h after BTH treatment and used as a template for semi-quantitative RT-PCR with gene-specific primers for PR1a (30 cycles) and Actin (24 cycles). (C) SylA blocks BTH-induced acquired resistance. N. benthamiana leaves were infiltrated with or without SylA and immediately sprayed with BTH. The ΔsylC mutant bacteria were infiltrated with 2×105 bacteria/mL 6 h after SylA/BTH treatment, and bacterial populations were determined at 0 and 3 d after inoculation. Error bars represent SEM of four independent bacterial counts. This experiment was repeated three times with similar results. (D) SylA-producing bacteria grow better in BTH-treated tissues than ΔsylC bacteria. WT and ΔsylC bacteria were infiltrated into N. benthamiana leaves at 2×105 bacteria/mL, and plants were sprayed with 300 µM BTH 6 h later. Bacterial populations were determined at 0, 3, and 7 dpi. Error bars represent SEM of four independent bacterial counts. This experiment was repeated two times with similar results. (E) SylA promotes wound entry in BTH-treated tissue. Leaves of N. benthamiana plants were infiltrated with 50 µM SylA or 0.025% DMSO. After 1 h, the infiltrated area was wound-inoculated with GFP-expressing WT or ΔsylC bacteria and sprayed with 300 µM BTH or water. Wound entry was scored at 5 dpi by fluorescence microscopy. Error bars represent SEM of four independent experiments. P-values determined using the Student's t-test are indicated.
To determine if SylA could also block immunity mediated by SA signaling, we infected BTH- and water-treated plants with ΔsylC bacteria in the absence or presence of SylA. In the absence of SylA, BTH treatment caused reduced bacterial growth of the ΔsylC strain compared to the control ( Figure 4C ). However, SylA treatment increased growth of ΔsylC bacteria in BTH-treated tissue to the same level as the control ( Figure 4C ). These data demonstrate that exogenous SylA can complement bacterial growth of the ΔsylC strain by suppressing SA-mediated immunity.
The above experiments indicate that SylA-producing WT bacteria will grow better during SA signaling than SylA-deficient ΔsylC bacteria. To test this hypothesis, we inoculated BTH-treated plants with WT and ΔsylC bacteria and measured bacterial growth. These assays demonstrated a statistically significant growth reduction of the ΔsylC mutant bacteria compared to WT bacteria ( Figure 4D ). Thus, during SA signaling, SylA-producing WT bacteria grow better than SylA-deficient ΔsylC bacteria, in contrast to naive plants where both strains grow equally well ( Figure 1G ).
To test whether SA signaling suppresses wound entry, we inoculated plants with WT-GFP and ΔsylC-GFP 1 d after spraying with water or BTH. BTH treatment significantly reduced the frequency of wound entry by WT bacteria ( Figure 4E ). Thus, activated SA signaling for 1 d was sufficient to suppress wound entry, even of SylA-producing WT bacteria. However, addition of exogenous SylA before BTH treatment promoted wound entry of both WT-GFP and ΔsylC-GFP bacteria in BTH-treated tissue ( Figure 4E ). Thus, SylA can promote wound entry by inhibiting SA signalling downstream of SA.
SylA Deficiency Triggers Early Host Cell Death and Immune Responses
While performing the infiltration assays, we noticed that ΔsylC bacteria triggered early host cell death upon infiltration ( Figure 5A ). Cell death upon ΔsylC inoculation occurred at 2 dpi, whereas WT bacteria caused host cell death usually after 5 dpi ( Figure 5A ). At later time points, late cell death developing upon infiltration of the WT strain often spread into tissues surrounding the primary infection site, whereas early cell death induced by the ΔsylC strain remained confined to the infiltrated region ( Figure 5B ).
Figure 5. SylA-deficiency triggers immune responses at primary infection sites.

(A) Time course of cell death induced by WT and ΔsylC strains. Leaves were stained with trypan blue at various days after infiltration. Scale bar, 10 mm. (B) Cell death spreads from zones infected with WT but not ΔsylC bacteria. Leaves were infiltrated and stained with trypan blue at 7 dpi. (C) Cell death induced by ΔsylC is blocked by the calcium transport inhibitor lanthanum chloride and the ATPase inhibitor sodium vanadate. Leaves were co-infiltrated with 1×108 bacteria/mL with 50 µM lanthanum chloride or 1 µM sodium vanadate, and pictures were taken at 24 hpi. (D) Early host cell death induced by SylA-deficient ΔsylC bacteria is preceded by callose deposition. Leaves were stained for callose at 24 hpi, examined by fluorescence microscopy, and depicted with equal settings. The callose spots per 0.56 mm2 were quantified and printed below the picture with the SEM (n = 3). Scale bar, 0.1 mm. (E) Early host cell death induced by SylA-deficient ΔsylC bacteria is preceded by SA accumulation. N. benthamiana plants were infiltrated with 105 bacteria/mL of WT and ΔsylC bacteria, and SA concentrations were measured at 3 and 24 hpi. Error bars represent SEM of three technical replicates. Student's t-test: P = 0.21 (3 hpi) and P = 0.096 (24 hpi). The experiment was repeated twice with similar results. (F) Early host cell death induced by SylA-deficient ΔsylC bacteria is preceded by upregulated transcript levels of the hypersensitive cell death marker Hin1. Semi-quantitative RT-PCR was performed on mRNA isolated at 24 hpi. (A–F) Bacteria were infiltrated with 2×105 bacteria/mL into mature leaves of N. benthamiana and analyzed at 24 hpi unless stated otherwise.
To test whether ΔsylC-induced early host cell death is a form of programmed cell death reminiscent of the hypersensitive response (HR), we co-inoculated leaves with lanthanum chloride and sodium vanadate, two chemicals that can prevent programmed cell death by blocking transport of calcium ions and ATPase activity, respectively [28]. Importantly, ΔsylC-induced cell death was blocked both by lanthanum chloride and sodium vanadate ( Figure 5C ), indicating that early host cell death is a program that can be blocked.
Early host cell death by ΔsylC bacteria was preceded at 1 dpi by callose deposition ( Figure 5D ), increased SA levels ( Figure 5E ), and transcriptional activation of the HR-marker Hin1 ( Figure 5F ). These data demonstrate that the ΔsylC strain triggered early cell death associated with typical HR-like responses. This is remarkable, since the growth of ΔsylC bacteria was indistinguishable from that of WT bacteria during these assays ( Figure 1G ).
ΔsylC-induced Local Responses Are Weak when Compared to ETI/NHR Responses
To further investigate why immune responses induced by the ΔsylC strain did not affect bacterial growth, we transformed WT and ΔsylC strains with hopQ1-1 of PtoDC3000, a type III effector that triggers nonhost resistance (NHR) in N. benthamiana [29]. Immune responses triggered by strains carrying hopQ1-1 were compared to strains containing the empty vector (EV). Importantly, the presence of the HopQ1-1 effector increased callose deposition ( Figure 6A ) and reactive oxygen species in the ΔsylC strain ( Figure 6B ), indicating that SylA deficiency of the ΔsylC strain induces immune responses that are similar but much weaker compared to the ETI/NHR responses induced by HopQ1-1 [21]. Interestingly, HopQ1-1 also induced strong immune responses in the WT strain ( Figures 6A and 6B ), indicating that SylA does not suppress HopQ1-1-induced responses.
Figure 6. ΔsylC-induced immune responses are weak compared to ETI/NHR responses.

(A) Increased callose deposition by both WT and ΔsylC strains expressing the HopQ1-1 effector. Leaves were infiltrated with 2×107 bacteria/mL, and fluorescent callose spots were quantified per 0.56 mm2 after aniline blue staining at 24 hpi. Error bars indicate SEM of 20 technical replicates. These experiments were repeated three times with similar results. (B) Increased oxidative burst by both WT and ΔsylC strains expressing the HopQ1-1 effector. Leaves were infiltrated with 2×108 bacterial cells/mL, and reactive oxygen species (ROS) were measured at 15 hpi in leaf discs floating on 200 µM MOPS containing L-012 for 20 min. The reduced ROS level of ΔsylC(EV)-infiltrated tissue compared to WT(EV)-infiltrated tissue was due to the late time point and the fact that ROS levels are transient. Error bars indicate SEM of eight technical replicates. These experiments were repeated three times with similar results. (C) Reduced bacterial growth of both WT and ΔsylC bacteria expressing the HopQ1-1 effector. Leaves were infiltrated with 2×105 bacterial cells/mL in N. benthamiana, and bacterial growth was measured at 0, 4, and 7 dpi. Error bars represent standard deviation of four biological replicates. The experiment was repeated four times with similar results. (D) HopQ1-1-expressing WT and ΔsylC bacteria do not cause symptoms when infiltrated at low densities. Leaves were infiltrated at 2×105 bacterial cells/mL, and pictures were taken at 7 dpi. (E) Cell death triggered by HopQ1-1-expressing WT and ΔsylC bacteria is blocked by the calcium transport inhibitor lanthanum chloride and the ATPase inhibitor sodium vanadate. Leaves were co-infiltrated with 1×108 bacteria/mL with 50 µM lanthanum chloride or 1 µM sodium vanadate, and pictures were taken at 24 hpi.
Importantly, the presence of the HopQ1-1 effector in WT and ΔsylC strains was associated with strongly reduced bacterial growth upon infiltration, as expected for a strong ETI response ( Figure 6C ). Leaf tissue infiltrated with HopQ1-1-expressing WT and ΔsylC bacteria did not exhibit disease symptoms at low cell densities ( Figure 6D ), in agreement with the reduced bacterial growth. In contrast, early host cell death, induced by ΔsylC(EV), occurred shortly after infection but stayed confined to the infiltrated area. Cell death induced by WT(EV) occurred later and expanded beyond the infiltrated area ( Figure 6D ). However, when infiltrated at high densities, HopQ1-1-expressing WT and ΔsylC bacteria triggered cell death that could be blocked by lanthanum chloride and sodium vanadate ( Figure 6E ). Taken together, these data indicate that SylA-deficiency leads to an ETI-like immune response that is too weak to suppress bacterial growth.
SylA Diffuses and Inhibits the Proteasome in Adjacent Tissue
Since SylA is a small molecule that facilitates wound entry, we tested whether SylA could act at a distance from the primary inoculation site. Therefore, we inoculated RhSylA and monitored rhodamine-based fluorescence by fluorescence microscopy. These studies demonstrated that RhSylA moved through the vasculature up to 1 cm from the inoculation site within 2 h ( Figure 7A ). To test whether RhSylA also targeted the proteasome in these adjacent tissues, we extracted proteins from adjacent tissues and analyzed labeling by protein gel electrophoresis. These experiments demonstrated that RhSylA labeled the proteasome in adjacent tissue ( Figure 7B ). Co-inoculation of RhSylA with unlabeled SylA suppressed labeling, indicating that SylA itself could also move to adjacent tissue ( Figure 7B ). To confirm that SylA itself could inhibit the proteasome in adjacent tissues, we inoculated SylA locally and monitored proteasome activity in extracts of adjacent tissue using MVB072 labeling. The suppression of MVB072 labeling upon inoculation with SylA ( Figure 7C ) demonstrate that SylA inhibits the proteasome in adjacent tissues.
Figure 7. SylA diffuses and suppresses SA-mediated immunity in adjacent tissue.

(A) RhSylA spread through the vasculature. A 1-µl aliquot of 2 mM RhSylA or 0.1% DMSO (mock) was applied at a wound site, and a fluorescence image was taken 2 h later. Scale bar, 1 mm. Arrowheads indicate wound inoculation sites. (B) RhSylA targets the proteasome in adjacent tissue. A 1-µl aliquot of 2 mM SylA was applied to an inoculation site and preincubated for 30 min. Subsequently, 1 µl of 2 mM RhSylA was added and incubated for another 2 h or 6 h. Proteins were extracted from tissue at 1–10 mm from the application site, and labeled proteins were detected by fluorescence scanning. *, background signal. (C) SylA targets the proteasome in adjacent tissue. A 1-µl aliquot of 1 mM SylA was applied at a wound site and incubated for 4 h. The application site was removed, extracts from adjacent tissues were labeled with MVB072, and fluorescently labeled proteins were detected. (D) Procedure for assaying wound entry by ΔsylC-GFP bacteria in adjacent tissue. Leaves of WT N. benthamiana were infiltrated with 50 µM SylA and 0.25% DMSO (E), 105 WT bacteria or water (F), and the infiltrated region was marked. After 1 h (for SylA infiltration) or 1 d (for bacterial infiltration), ΔsylC-GFP bacteria were inoculated at a site 5 mm outside the infiltrated area. Wound entry was scored 5 d later by fluorescence microscopy. (E) SylA promotes wound entry by ΔsylC-GFP bacteria at a distance from the infiltrated region. (F) WT bacteria promotes wound entry at a distance from the infiltrated region. (G) Representative example of distant colonization of ΔsylC-GFP bacteria when inoculated next to areas infiltrated with WT-GFP bacteria. WT-GFP bacteria were infiltrated at 105 bacterial cells/mL (lower right, bordered by dashed line). One day later, ΔsylC-GFP bacteria were inoculated at 5 mm from the infiltrated region. The picture was taken 5 d later. WT-GFP bacteria did not spread outside the infiltrated zone, but their presence promoted wound entry by ΔsylC-GFP in adjacent tissue. Arrowheads indicate colonies of ΔsylC-GFP in tissues adjacent to the wound inoculation site. (H) Procedure for assaying adjacent colonization by WT-GFP bacteria in adjacent tissue. Leaves of WT N. benthamiana were infiltrated with 50 µM SylA, 0.25% DMSO (H), 105 WT bacteria or water (I), and the infiltrated region was marked. After 1 h (for SylA infiltration) or 1 d (for WT bacteria infiltration), WT-GFP bacteria were inoculated at 5 mm outside the infiltrated area and the plant was sprayed with 300 µM BTH or water. Wound entry was scored 5 d later by fluorescence microscopy. (I) SylA promotes wound entry in BTH-treated tissue at a distance from the infiltrated region. (J) SylA-producing WT bacteria promotes wound entry in BTH-treated tissue at a distance from the infiltrated region. (E, F, I, J) Error bars represent SEM of four independent biological replicates, each with 12 wound inoculations. P-values determined using the Student's t-test are indicated.
SylA Promotes Wound Entry by Suppressing SA-mediated Immunity at a Distance
To investigate whether SylA also promotes colonization at adjacent sites, we infiltrated 50 µM SylA or a mock control into N. benthamiana leaves and inoculated ΔsylC-GFP bacteria at 5 mm outside the infiltrated region ( Figure 7D ). The ΔsylC-GFP bacteria colonized adjacent tissue significantly more frequently in tissue next to SylA-infiltrated zones compared to zones infiltrated with the mock control ( Figure 7E ), demonstrating that SylA promotes wound entry in adjacent tissues. To investigate whether SylA-producing WT bacteria also promoted wound entry by ΔsylC bacteria in adjacent tissues, we infiltrated WT bacteria and inoculated ΔsylC-GFP bacteria at 5 mm from the infiltrated area 1 d after infiltration. The presence of a nearby WT-infiltrated region significantly enhanced wound entry of the ΔsylC-GFP strain, compared to a mock-infiltrated region ( Figure 7F ).
To determine whether WT bacteria spread outside the infiltrated area, we repeated the assay by infiltrating WT-GFP bacteria followed by wound inoculation of ΔsylC-GFP bacteria in adjacent tissue. The empty space between the marked infiltration zone and the bacterial colonies originating from the wound inoculation site distinguished the WT-GFP from the ΔsylC-GFP bacteria ( Figure 7G ). Colonization of adjacent tissues by infiltrated WT-GFP bacteria did not occur at 6 dpi, whereas the ΔsylC-GFP bacteria were already colonizing adjacent tissues from wounding sites ( Figure 7G ). Thus, WT bacteria promote wound entry of ΔsylC-GFP bacteria at adjacent sites, presumably by producing diffusing molecules, rather than being present themselves.
To demonstrate that SylA suppresses SA signaling in adjacent tissues, we (1) infiltrated leaves with SylA or mock control; (2) wound inoculated WT-GFP next to the infiltrated area; and (3) sprayed plants with BTH to induce SA signaling ( Figure 7H ). BTH treatment resulted in a strong suppression of wound entry by WT-GFP bacteria compared to the water-treated control ( Figure 7I ), confirming that SA signaling suppresses wound entry even of WT bacteria ( Figure 4E ). Importantly, exogenous SylA infiltrated at a distance from the inoculated area significantly increased the frequency of wound entry by WT-GFP bacteria in BTH-treated tissue ( Figure 7I ). To test whether SylA-producing WT bacteria also suppressed SA-mediated immunity in the vasculature of adjacent tissues, we wound inoculated WT-GFP next to a zone infiltrated with WT bacteria 1 d after infiltration and sprayed the plants with BTH. BTH treatment suppressed wound entry of WT-GFP bacteria, but the presence of a nearby WT-infiltrated zone significantly enhanced wound entry of WT-GFP bacteria in BTH-treated tissues ( Figure 7J ). These data demonstrate that SylA-producing bacteria suppress SA responses in the vasculature of adjacent tissues.
A Second Layer of Immunity: NahG Blocks Immunity in Adjacent Tissues but Not Escape from Primary Infection Sites
The accumulation of SA during infection with the ΔsylC strain suggested that adjacent tissues might have acquired resistance. To test whether tissues surrounding ΔsylC-infected areas also mounted immunity, we inoculated WT bacteria next to regions that were preinfiltrated with WT or ΔsylC bacteria ( Figure 8A , left). When inoculated next to ΔsylC-infiltrated regions, WT bacteria were unable to spread ( Figure 8A ), indicating that ΔsylC-infiltrated regions triggered acquired resistance in adjacent tissue. We performed the same experiment on NahG-transgenic N. benthamiana plants which cannot accumulate SA because NahG expresses a bacterial salicylate hydroxylase that converts SA into catechol [30]. When WT bacteria were inoculated next to ΔsylC-infiltrated regions in NahG-transgenic plants, wound entry was observed with the same frequency as when inoculated nest to regions infiltrated with WT bacteria ( Figure 8A ). These data show that NahG blocks immunity in adjacent tissues triggered by ΔsylC mutant bacteria.
Figure 8. NahG blocks immunity in adjacent tissues and only partially promotes wound entry by ΔsylC bacteria.

(A) Reduced wound entry by WT bacteria when inoculated next to ΔsylC-infiltrated regions is absent in NahG plants. Leaves of WT and NahG-transgenic N. benthamiana plants were infiltrated with WT or ΔsylC bacteria, and GFP-expressing WT PsyB728a bacteria were inoculated 1 d later at 0.5 cm from the border of the infiltrated region. Wound entry was monitored 5 d later by fluorescence microscopy. Error bars represent SEM of four independent experiments, each with 12 wound inoculations. P-values determined using the Student's t-test are indicated. (B–C) The ΔsylC mutant can colonize adjacent tissues in NahG-transgenic plants, though less than WT bacteria. WT and ΔsylC mutant bacteria were inoculated in WT and NahG-transgenic N. benthamiana plants, and wound entry was scored after 5 d by fluorescence microscopy. Error bars represent SEM of four independent experiments, each with 12 inoculations. P-values determined using the Student's t-test are indicated. (C) Representative pictures of colonization by WT or ΔsylC bacteria at 5 dpi in WT or NahG-transgenic plants. Fluorescence pictures were converted into inverted greyscale for better visibility. Scale bar, 1 mm.
To test whether blocking immune responses in adjacent tissues was sufficient for wound entry by SylA-deficient bacteria, ΔsylC-GFP bacteria were inoculated into NahG-transgenic plants. Importantly, we observed more frequent wound entry by ΔsylC bacteria from inoculation sites on NahG plants compared to WT plants ( Figure 8B ). However, the frequency of wound entry by ΔsylC bacteria was still less than that of WT bacteria. This difference was also evident from the colonization pattern upon wound entry: the number of veins along which ΔsylC-GFP bacteria colonized was significantly less compared to WT-GFP in NahG plants ( Figure 8C ). These data indicate that colonization from wound sites is suppressed by two mechanisms: an immune response in adjacent tissues, which is absent in NahG plants, and immune responses at the primary infection site that are not suppressed by NahG.
Local Control of Wound Entry Is Associated with Reduced Motility
Our data indicate that, in addition to SA-dependent immune responses in adjacent tissues, local immune responses also suppress the escape of SylA-deficient bacteria from primary infection sites. To determine whether ΔsylC-GFP bacteria have reduced motility at the primary infection site, WT-GFP and ΔsylC-GFP bacteria were monitored by confocal microscopy at different time points during the wound infection assays. The bacterial motility was indistinguishable between WT-GFP and ΔsylC-GFP bacteria when grown in NYG liquid medium ( Figure 9A and Movie S1 and S2) and 6 h after infiltration ( Figure 9B and Movie S3 and S4). At later stages, colonies developed in the leaf for both WT-GFP and ΔsylC-GFP bacteria. Bacteria inside these colonies were not motile ( Figure 9B and Movies S5 and S6). However, bacteria at the edge of WT-GFP colonies were motile ( Figure 9B and Movie S5), but bacteria at the edge of ΔsylC-GFP colonies were not ( Figure 9B and Movie S6). Furthermore, WT-GFP bacteria in exudates that leaked from a cut through an infected leaf were motile ( Figure 9C and Movie S7), but ΔsylC bacteria only gained motility at the edge of the exudate and seemed to be embedded in a glue-like matrix ( Figure 9C and Movie S8). The reduced motility of ΔsylC bacteria could be complemented chemically by co-infiltrating exogenous 50 µM SylA but not the mock control ( Figure 9D and Movies S9 and S10), confirming that the reduced motility of the ΔsylC mutant is caused by the absence of SylA. In conclusion, bacterial motility was identical during the initial stages of infection and was reduced for ΔsylC-GFP bacteria during the later stages of infection.
Figure 9. ΔsylC bacteria loose motility during infection.

GFP-expressing WT and ΔsylC bacteria were monitored by confocal microscopy before infiltration (A), after infiltration (B), in the exudate of infected leaves (C), and after co-infiltration with or without SylA (D). All motile bacteria are marked with a star. The percentages of motile bacteria over a 2 s timeframe are indicated on the bottom, with standard deviations for n = 5. See supplemental data for the movies and details. Bacteria are 1–2 µm long. These results are representative of three independent experiments. *, motile bacterium.
Discussion
This work uncovered a key role for SylA in facilitating wound entry by suppressing immune responses, both locally and in adjacent tissue. A model for the dual role of SylA is summarized in Figure 10 and is discussed below. Our data indicate that the role of SylA as a virulence factor depends on the assay. SylA is a virulence factor in the classical sense, since the SylA-deficient mutant causes less symptoms on bean plants upon spray inoculation [18]. SylA is also a virulence factor with respect to promoting bacterial growth, since the ΔsylC mutant grew less compared to the WT strain in the first hours after spray inoculation, which can be explained by the fact that SylA suppresses stomatal closure [19]. However, we did not detect a statistically significant virulence role for SylA on bacterial growth upon infiltration, despite the different conditions tested ( Figure 1G and S1). In contrast, we demonstrated that SylA promoted bacterial growth in BTH-treated plants ( Figure 4CD ) and wound entry ( Figure 1A–E ), associated with a 63-fold increased bacterial population level ( Figure 1F ). We believe that this phenotype is important for the biology of PsyB782a and PsyB301D, since both strains are known to infect bean leaves and pear blossoms via wounds caused by wind or frost, respectively [7], [8].
Figure 10. Model of SylA action.

(A–B) SylA-deficient ΔsylC bacteria trigger local immune responses, resulting in both local HR-like cell death and immune responses (red triangles) and induced local resistance (ILR) in adjacent tissue, which is dependent on SA signaling. Wound entry is prevented by both local (1) and adjacent (2) immune responses. Only a few ΔsylC bacteria can escape (dashed line) to establish colonies in adjacent tissues in the absence of SA signaling. (C–D) SylA-producing WT bacteria secrete SylA, which prevents immune responses at the primary infection site (3). In addition, SylA diffuses over a distance and prevents acquired resistance induced by SA signaling (4). Consequently, SylA-producing bacteria can escape from primary infection sites and colonize adjacent tissue.
1. SylA Deficiency Triggers Local Immune Responses without Affecting Bacterial Populations
SylA-deficient PsyB728a strains triggered local immune responses, including early host cell death and the accumulation of SA, callose, and Hin1 transcripts ( Figure 5 ). The ΔsylC-induced early host cell death could be prevented with calcium channel or ATPase inhibitors ( Figure 5C ), indicating that this is a form of programmed cell death. The ΔsylC-induced immune responses were relatively weak compared to NHR/ETI responses induced by the presence of HopQ1-1 ( Figure 6 ), which explains why SylA-deficient bacteria grew equally well locally compared to WT bacteria ( Figure 1G ). PsyB728a carries several type III effectors that can trigger HR and other ETI responses, resulting in reduced bacterial growth in N. benthamiana [14]. PsyB728a is also likely to trigger PTI by flagellin and other elicitors [13]. The identification of the factor(s) that trigger immune responses in the absence of SylA was not the scope of this study but is important to classify the response triggered by the ΔsylC strain.
The observation that the SylA-deficient ΔsylC mutant grew similarly to WT bacteria upon infiltration ( Figures 1G and S1), demonstrated the striking ability of PsyB728a to survive in dead host tissue, despite the widely accepted view that the HR and immune responses suppress pathogen survival. The survival of P. syringae in tissue undergoing HR and immune responses is not unprecedented. Unaffected bacterial populations in dying host tissue have also been observed in tobacco plants infiltrated with avirulent P. syringae pv. maculicola (Pma) M2 and pv. tabaci (Pta) [31]. The absence of cell death in SGT1-silenced N. benthamiana plants did not affect bacterial growth of PsyB728a and only moderately affected growth of PtoDC3000, Pma, and Pta [32]. High-throughput silencing in N. benthamiana has revealed many genes that suppress AvrPto/Pto-mediated HR when silenced, but only a few genes have an effect on bacterial growth [33]. These data indicate that P. syringae is able to survive and even grow in the presence of dead host tissue, despite the likely presence of toxic components generated by immune responses of the dying host. These data are consistent with the notorious survival of PsyB728a as an epiphyte, even during dry periods [3], [34].
2. SylA Deficiency Triggers SA Accumulation and Acquired Resistance in Surrounding Tissues
Inoculation of WT-GFP bacteria next to regions preinfiltrated with ΔsylC bacteria demonstrated that ΔsylC triggered immune responses in surrounding tissues that suppressed wound entry ( Figure 8A ). The ΔsylC bacteria triggered the accumulation of SA ( Figure 5E ), and SA is known to cause acquired resistance in tobacco [26]. These data demonstrated that ΔsylC bacteria triggered SA-dependent acquired resistance in adjacent tissue (ILR, induced local resistance) ( Figure 10A ). ILR is similar to the well-described systemic acquired resistance (SAR), which occurs in the entire plant and is also dependent on SA signaling [26]. The experiments with NahG-transgenic plants demonstrated that the suppression of wound entry occurred at two levels: an acquired response in surrounding tissues, absent in the NahG line ( Figure 8A ); and a local response confining the bacteria at the primary infection site, still mostly present in the NahG line ( Figure 8BC ). The suppression of acquired resistance in adjacent tissue in the NahG line might be due to degradation of SA or due to the accumulation of catechol, which can affect bacterial growth [35]–[36].
3. SylA Production Suppresses Local Immune Responses
In contrast to ΔsylC bacteria, WT bacteria did not trigger early cell death and prevented the early accumulation of callose, Hin1 transcripts, and SA ( Figure 5 ). Thus, SylA production suppressed local immune responses. The finding that local immune responses could be suppressed by inhibiting the proteasome is consistent with previous reports. Hatsugai and colleagues found that silencing of the β1 proteasome subunit suppressed AvrRpm1-induced HR [37]. Furthermore, proteasome inhibitors also have been shown to inhibit early cell death induced by P. syringae pv. phaseolicola [38] and pv. tabaci [31] in tobacco. Taken together, these studies implicate proteasome involvement in cell death and immunity triggered by P. syringae. Interestingly, the HopQ1-1-induced HR was not suppressed in SylA-producing WT bacteria ( Figure 6E ). These results either mean that SylA could not block all ETI responses or that the timing and/or concentration of SylA production was insufficient to prevent HopQ1-1-induced ETI.
The principle that proteasome inhibition can suppress early cell death might be counterintuitive, since proteasome inhibitors generally are assumed to cause cell death. However, proteasome inhibition can also promote cell survival or cell death, depending on the concentrations and potencies of the proteasome inhibitors used [39]. During PsyB728a infection, SylA intercepted a pro-death program, implying that SylA production was carefully timed and targeted by PsyB728a during infection to provide a sublethal dose. Proteasome inhibition also seems counterintuitive, since many effectors like AvrPtoB, HopM1, and coronatine depend on proteasome activity to execute degradation of their host targets [40]–[42]. The PsyB728a genome lacks biosynthesis genes for coronatine, but it contains AvrPtoB and HopM1 homologs [43]–[44]. Therefore, the SylA concentration and timing may be essential parameters that act in concert with the expression of other effectors. These data suggest that ETI suppression by SylA can be overruled if ETI is triggered stronger and/or faster, e.g. in the case of HopQ1-1.
4. SylA Diffuses and Creates a Zone of SA-insensitive Vasculature Tissue
Our data indicate that SylA diffuses through the vasculature and inhibits the host proteasome in adjacent tissues. Although analytical tools to detect SylA in plant tissues are not yet available, we showed that SylA suppressed proteasome activity in adjacent tissues ( Figure 7C ) and found that rhodamine-tagged SylA quickly moves through the vasculature ( Figure 7AB ). At this stage, it is unclear whether the SylA movement is apoplastic (through the xylem) or symplastic (through the phloem) and whether the transport is active or passive. Although SylA is produced by bacteria residing in the apoplast, SylA is also quickly taken up by host cells and may enter the symplastic transportation route [20].
Since SylA acts along the vasculature, we used vasculature-specific immunity assays to detect the suppression of SA signaling. For these assays, we used wound inoculations, which provide bacteria with direct access to the vasculature. Importantly, the presence of SylA, when supplemented at a distance, complemented wound entry by ΔsylC-GFP bacteria ( Figure 7E ). Also, SylA-producing WT bacteria promoted wound entry of ΔsylC-GFP bacteria in adjacent tissues ( Figure 7FG ), indicating that sufficient SylA was produced during infection to promote colonization from wounding sites.
SylA directly blocks SA signaling in N. benthamiana, which was demonstrated by blocking BTH-induced PR1a expression using SylA ( Figure 4A ). These data are consistent with the observation that SylA blocks SA signaling in Arabidopsis [19]. One possible molecular mechanism is that SylA prevents the degradation of phosphorylated NPR1 in the nucleus, which needs to be removed by the nuclear proteasome to allow continued transcriptional activation of NPR1-responsive genes [45]. The inhibition of SA signaling in the nucleus is consistent with the observation that SylA targets the nuclear compartment [20]. The subcellular targeting of SylA also explains why SylA blocks SA signaling at concentrations that are less than required for full proteasome inhibition in the total extracts. Thus, new assays are required to detect tissue-specific and subcellular proteasome inhibition during infection.
Importantly, we showed that SylA and SylA-producing bacteria blocked SA signaling in adjacent tissues, since BTH-mediated suppression of wound entry by WT-GFP bacteria was restricted when SylA or WT bacteria were preinfiltrated next to the infection sites ( Figure 7J ). Vasculature-specific infection assays were required, since SylA acted along the vasculature at a low dose. These experiments indicate that SylA-producing bacteria use effector diffusion to create a zone of SA-insensitive vasculature tissue around the infection site.
Remote Control by Effector Diffusion
Our data indicate that SylA moves and inhibits the nuclear proteasome in host cells along the vasculature to create SA-insensitive tissue that was ready for subsequent colonization. Effector diffusion is a common strategy used by pathogens to control immune responses in adjacent tissues. Several fungal effectors that translocate to the host cytoplasm move to adjacent plant cells and are thought to prepare adjacent cells for infection [46]–[47]. These effectors typically move up to four adjacent cells (less than 0.1 mm). In contrast, small molecule effectors, such as SylA, can move over greater distances (several millimeters) and can reach a larger area surrounding the primary infection site. Although the SylA biosynthesis gene cluster so far only has been found in phylogroup II P. syringae strains, other strains produce other small molecule effectors that can interfere with SA signaling in adjacent tissues. A well-studied example is coronatine, which is produced by several P. syringae strains and also suppresses SA signaling, in this case by activating the jasmonate signaling cascade that results in the downregulation of the SA biosynthetic enzymes and upregulation of SA-converting enzymes [48]–[49]. P. syringae strains producing coronatine increase the susceptibility of noninoculated leaves, a phenomenon called systemic induced susceptibility (SIS) [50]. Coronatine is required and sufficient to induce SIS, and SIS is suppressed in NahG-transgenic plants. However, this increased susceptibility is only moderate (5-fold increased growth), and the biological relevance is unclear as it benefits other pathogens as well as the coronatine-producing strain. We focused on adjacent tissues because they are relevant for the wound entry phenotype. We found that SylA production is beneficial for SylA-producing strains as these strains can escape primary infection sites, and increase the bacterial population. These experiments indicate that control of adjacent tissue by effector diffusion may be a common strategy for P. syringae and that different strains use different toxins, in different hosts, and through different molecular mechanisms.
Both Adjacent and Local Immune Responses Suppress Wound Entry
The absence of WT-like wound entry by ΔsylC-GFP bacteria in ILR-deficient NahG plants ( Figure 8 ) indicated that also local responses prevent colonization from wound sites. The escape from local confinement in the ILR-deficient NahG plants occurred sporadically, resulting in only a few infected vasculatures where wound entry occurred ( Figure 8C ). Notably, time-resolved confocal microscopy demonstrated that ΔsylC-GFP bacteria were embedded in a rigid extracellular matrix (ECM) that seemed to immobilize the bacteria at the primary infection site ( Figure 9 ). This rigid ECM was probably similar to the fibrillar ECM described in the 1980s for incompatible interactions with P. syringae bacteria [51]–[52]. The rigid ECM might have been derived from the bacteria or created by vacuolar content release during the HR. The proteasome is essential for fusion of the vacuolar membrane with the plasma membrane during the HR [37]. Therefore, inhibition by SylA could prevent vacuolar content release into the apoplast. However, a rigid ECM might not be the only possible mechanism of local confinement. Immune responses might also change the motility of the bacteria by affecting their metabolic state. The nature of the matrix, the metabolic status of the embedded bacteria, and how SylA prevents these events remain topics for future studies.
Conclusion
In conclusion, our studies revealed that SylA facilitated wound entry of PsyB728a and PsyB301D by blocking immune responses in both local and adjacent tissues. Local immune responses did not affect bacterial population levels but contained the bacteria to the primary infection site. Immune responses in adjacent tissues were SA-dependent and suppressed colonization by bacteria that escaped confinement at the primary infection site. SylA suppressed this two-layered immune response by blocking immune responses in both local and adjacent tissues. Our data indicate that SylA diffuses through the vasculature and blocks SA signaling in adjacent tissues, creating an SA-insensitive zone of vasculature tissue that makes the tissue ready for subsequent colonization.
Methods
Generation of Transgenic Pseudomonas Strains
All plasmids and strains are summarized in Supplemental Table S1. The ORF encoding GFP was amplified from DNA isolated from GFP-transgenic PstDC3000 [42] using primers 5′-tccccatgggtaaaggagaag-3′ and 5′-tccccatggttagagctctagttcatccatgccatg-3′ and cloned into pGEM-T (Promega), resulting in pFK69. The GFP ORF was amplified from pFK69 using primers 5′-atcgaagcttaggaggacagctatgggtaaaggagaaga-3′ (introducing a ribosome binding site, RBS) and 5′-gatgagctcctcgagtctagaatcgatctatttgtatagttcatccatgccatg-3′ and cloned into pBlueScript using HindIII and XbaI restriction sites, resulting in pFK74. The RBS-GFP cassette of pFK74 was cloned into pML123 [53] using BamHI and XbaI restriction sites, resulting in pFK78. Pseudomonas syringae strains were transformed with pFK78 by electroporation and selected on gentamycin (10 µg/mL). Several fluorescent colonies were used for infection and were found to behave similarly. PacWT-GFP was transformed with cosmid pPL3syl carrying sylA-E genes of PsyB301D-R [23] by triparental mating using a helper E. coli strain carrying pRK600 and selected with tetracyclin (10 µg/mL), gentamycin (10 µg/mL) and rifampicin (25 µg/mL) [23].
Plant Growth and Infection Assays
Nicotiana benthamiana plants were grown under a 12 h light regime at 22°C and 60% relative humidity and used at 3–5 weeks old, before flowering, unless indicated otherwise. For infection by infiltration, bacteria were grown overnight in 10 mL NYG medium (5 g/L peptone, 5 g/L yeast extract, 2% glycerol), centrifuged and resuspended into 10 mM MgCl2. The OD600 was measured and bacteria were diluted to an OD600 of 0.0002 in 10 mM MgCl2. Bacteria were infiltrated into N. benthamiana leaves with a 1 mL syringe without needle. Leaves were examined at various time points. GFP fluorescence was detected by a Leica MZ16FA using the GFP filter. All experiments were done with similar acquisition settings.
For wound inoculation, the three youngest expanded leaves of 3–5 week-old, nonflowering N. benthamiana plant were selected. Bacteria were taken from fresh plates with a sterile toothpick and punched through the leaf using a new toothpick for every infection. In case of infection with controlled inoculum ( Figure 1F ), wound sites were immediately inoculated with 1 µL of a 107 bacteria/mL; Usually two strains were compared in opposite leaf halves. Plants were incubated at 22°C without cover and pictures were taken at 5 dpi using stereo fluorescence microscopy. Pictures of every infection site were taken for blind scoring and verification. An example experiment is shown as Figure S3. The percentage of wound entry was calculated for at least 12 toothpicks by dividing the number of times that colonies appear in adjacent tissues by the number of times that colonization occurred at the toothpick inoculation site itself. Inoculation sites that did not show GFP fluorescence were rare and were not included in the count. The average and standard error of the mean (SEM) was calculated from four independent experiments.
Endophytic bacterial populations were determined by colony count assays from opposite leaf halves as described previously [54]. Briefly, leaves were surface-sterilized in 15% H2O2 for 5 min on a shaker at 200 rpm and washed with sterile water. Alternatively, leaves were sterilized by 70% ethanol for 5 min (Supplemental Figure S1). Leaf disks (13 mm diameter, unless otherwise indicated) from two infiltrated leaves were combined and ground in 1 mL 10 mM MgCl2 using metal beads. 20 µL droplets of a serial 10-fold dilution series were put on selective agar medium and colonies were counted after one day of incubation at 28°C. This procedure was repeated on at least three sets of infiltrated leaves for each experiment.
SylA, BTH, ABPP and RT-PCR
Synthetic SylA, SylAsat, RhSylA and the epoxomicin-based proteasome probe MVB072 were described previously [20], [25], [55]. N. benthamiana leaves were infiltrated with various SylA concentrations. Similarly DMSO was used as a control. Plants were sprayed with 300 µM BTH (Actigard, Syngenta) or water. RNA was isolated at 6 or 12 h after BTH treatment using the Qiagen RNeasy mini kit, and cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) using oligo(dT) 20 primer (Invitrogen) according to the instructions of the manufacturer. cDNA was used as a template for PCR using primer pairs for NbPR1: 5′-AATATCCCACTCTTGCCG-3′ and 5′-CCTGGAGGATCATAGTTG-3′; NbActin: 5′-TGGACTCTGGTGATGGTGTC-3′ and 5′-CCTCCAATCCAAACACTGTA-3′; and NbHin1: 5′-GAGCCATGCCGGAATCCAAT-3′ and 5′-GCTACCAATCAAGATGGCATCTGG-3′. Activity-based protein profiling (ABPP) with MVB072 on SylA-treated tissues was performed as described previously [20]. Extracts from leaf discs were labelled with 1.6 µM MVB072 in 50 mM NaOAc pH 7 for 2 h and labelled proteins were detected from protein gels using the Typhoon 8600 scanner (Molecular Dynamics) with excitation and emission at 532 and 580 nm, respectively.
Measurements of SA
SA concentrations were measured as described in Straus et al. [56]. Briefly, SA was extracted from 100 mg plant material in 1 ml chloroform/methanol/water (1∶2∶0.3) containing 160 pmol 2-hydroxybenzoic-3,4,5,6-d4 acid (SA-d4; Campro Scientific, http://www.campro.eu/) as internal standard. After shaking for 10 min at 70°C samples were centrifuged and re-extracted with 0.5 ml chloroform/methanol (1∶2). After phase separation through the addition of 0.5 ml H2O the polar extract was dried. Samples were acidified with 30 µl 10% trifluoroacetic acid (TFA) and extracted twice with 0.6 ml ethyl acetate/hexane (3∶1). Following evaporation of organic solvents, analytes were derivatized with 80 µl pyridine/N-methyl-N-(trimethylsilyl)trifluoroacetamide (1∶1) (Sigma) and 1 µl was injected into a gas chromatograph coupled to a mass spectrometer (GC-MS; Agilent, http://www.agilent.com/). Masses of SA-d4 (m/z 271) and SA (m/z 267) were detected by selected ion monitoring and quantified using the Chemstation software from Agilent.
Callose Deposition Assays
Callose deposits were measured in leaves of N. benthamiana after infiltration with 2×107 bacteria/mL. Leaf discs were excised 24 h after infiltration and evacuated in 95% ethyl alcohol at 37°C exchanging the ethyl alcohol every 2 h until cleared. Leaves were stained with the fluorescent dye aniline blue (0.01%) in a solution of 150 mM K2HPO4 (pH 9.5) for 30 m as previously described [57] then mounted on slides in 50% glycerol. The aniline blue-stained callose was visualized on a fluorescence microscope (Zeiss Axionplan 2, Carl Zeiss, Oberkochen, Germany), and the callose deposits were quantified using Image J (National Institutes of Health).
Reactive Oxygen Species (ROS) Assays
N. benthamiana leaves were infiltrated with 2×108 bacteria/mL. 15 h after infiltration leaf discs were taken with a 0.4 cm diameter cork borer and floated on 200 µL 10 mM morpholinepropanesulfonic acid (MOPS)/KOH (pH 7.4) containing 0.5 mM L-012 (Wako Pure Chemicals, Osaka, Japan) in a 96 well plate. Luminescence was measured at 20 min. using a Synergy 2 luminometer and quantified by Gen5 data analysis software (Biotek Instruments).
Supporting Information
Bacterial growth of WT and Δ sylC mutant PsyB728a upon infiltration. GFP-expressing (A–B, C, G and H) or non-transgenic bacteria (D, E, F and I) were infiltrated with 2×105 (A–G and I) or 2×104 (H) bacteria/mL, and infected plants were kept at high (60–90%) relative humidity (RH) (A–F), 60% RH (F–H) or transferred at 2 dpi from high RH to 60% RH (I). Bacterial populations were determined at different days-post-inoculation (dpi). Leaves were surface-sterilized with hydrogen peroxide (A–C, G–I) or ethanol (D–E) before leaf extracts were generated, diluted and plated. Experiments were performed in Cologne (A–E, G–I) or Nebraska (F). (A–I) Independent leaves were taken for n independent counts, indicated at the bottom. All error bars represent SEM. Pairwise comparisons between WT and ΔsylC growth was calculated using the Student t-test. NA, not analyzed.
(JPG)
{kind=link}
Structures of chemicals used in this study. Reactive groups (red), biotin (blue) and fluorescent reporter (yellow).
(JPG)
{kind=link}
Representative wound entry assay experiment. WT-GFP and ΔsylC-GFP bacteria were toothpick-inoculated from a fresh plate into different leaves of different plants of N. benthamiana. Pictures were made at 5 dpi using stereo fluorescence microscopy using identical settings. The frequency of host entry at each wound inoculation sites was counted over 12 toothpick sites, as shown at the bottom. Scale bar, 1 mm.
(JPG)
{kind=link}
Bacterial motility of WT-GFP in hanging droplet assay. WT-GFP bacteria were grown in NYG liquid medium and their motility was monitored by confocal microscopy by hanging droplet assay. Shown is the edge of the droplet for 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP in hanging droplet assay. ΔsylC-GFP bacteria were grown in NYG liquid medium and their motility was monitored by confocal microscopy by hanging droplet assay. Shown is the edge of the droplet for 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of WT-GFP at 6 h after infiltration. WT-GFP bacteria were infiltrated at 108 bacteria/ml and their motility was monitored six hours later by confocal microscopy. The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP at 6 h after infiltration. ΔsylC-GFP bacteria were infiltrated at 108 bacteria/ml and their motility was monitored six hours later by confocal microscopy. The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of WT-GFP in colonies at 24 hpi. Leaves were infiltrated with 105 WT-GFP bacteria/mL and subjected to confocal microscopy at 24 hpi. Host cells are intact (middle and bottom) and carry normal-looking chloroplasts (red). Bacteria (green) are static in the colony (bottom), but are motile along the edge of the colony (top). The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP in colonies at 24 hpi. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL and subjected to confocal microscopy at 24 hpi. Bacteria (green) are not motile and have occupied a collapsed host cell containing degenerating chloroplasts (red). The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of WT-GFP in colony exudates at 24 hpi. Leaves were infiltrated with 105 WT-GFP bacteria/mL, cut, and subjected to confocal microscopy at 24 hpi. GFP-expressing bacteria in the exudate that leaks from the leaf cut through infected tissue. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP in colony exudates at 24 hpi. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL, cut, and subjected to confocal microscopy at 24 hpi. GFP-expressing bacteria in the exudate that leaks from the leaf cut through infected tissue. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility at 24 hpi in sylC-GFP colonies with 50 µM SylA. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL containing 50 µM SylA and bacterial colonies were imaged at 24 hpi by confocal microscopy. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(GIF)
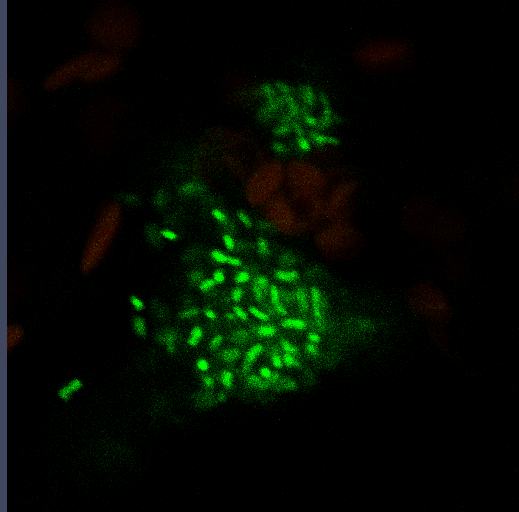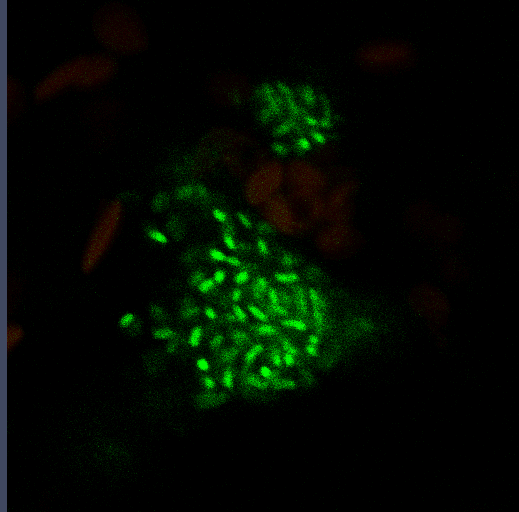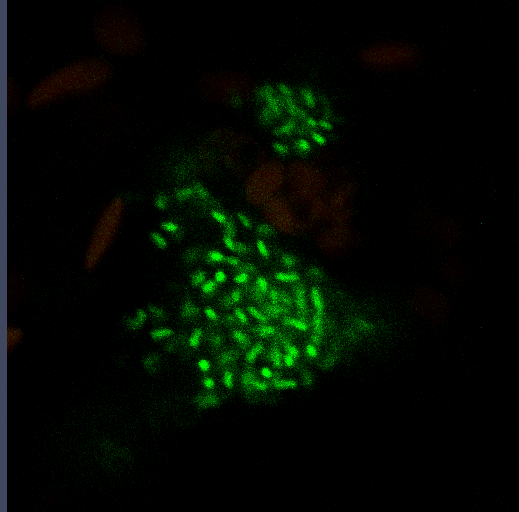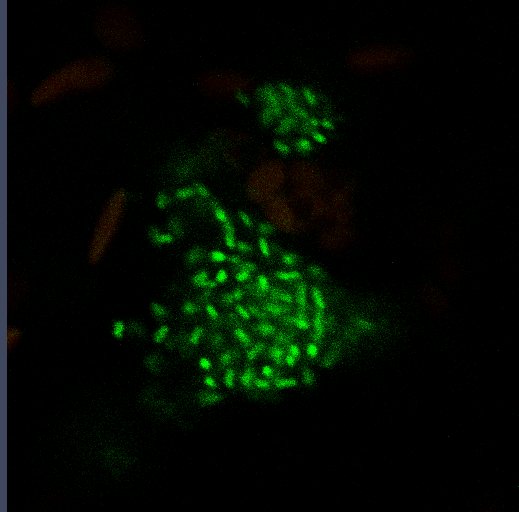{kind=link}
Bacterial motility at 24 hpi in sylC-GFP colonies with 0.25% DMSO. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL containing 0.25% DMSO and bacterial colonies were imaged at 24 hpi by confocal microscopy. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(GIF)
{kind=link}
Identification of MVB072-labeled proteins from N. benthamiana .
(PDF)
Plasmids and strains used in this study.
(PDF)
Acknowledgments
We would like to thank Elmon Schmelzer and Rainer Franzen for their help with scanning electron microscopy; Arjen van Doorn and Johannes Stuttmann for measuring SA concentrations; and Robert Dudler and Silke Robatzek for providing strains; Jonathan Jones for providing NahG-transgenic N. benthamiana; Gabriela Ronquillo and Sören Schilasky for technical assistance; Hermen Overkleeft for providing MVB072; and Matthieu Joosten and John Rathjen for providing other materials. We also like to thank Paul Schulze-Lefert, Jane Parker, Imre Somssich, Réka Tóth, Britta Maedge, Haibin Lu and Silke Robatzek for suggestions and comments on the manuscript.
Funding Statement
This work was financially supported by the Max Planck Society, the Deutscher Akademischer Austausch Dienst (DAAD), the Deutsche Forschungsgemeinschaft (DFG projects HO3983/3-3 and KA2894/1-1), and the U.S. National Institutes of Health (1R01AI069146-01A2). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Agrios GN (2005) Plant pathology. 5th edition. Burlington: Elsevier Academic Press. 952p.
- 2. Hirano SS, Upper CD (2000) Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae - a pathogen, ice nucleus, and epiphyte. Microbiol Mol Biol Rev 64: 624–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee J, Teitzel G, Munkvold K, del Pozo O, Martin GB, et al. (2012) Type III secretion and effectors shape the survival and growth pattern of Pseudomonas syringae on leaf surfaces. Plant Physiol 158: 1803–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenberg JT, Yao N (2004) The role and regulation of programmed cell death in plant-pathogen interactions. Cell Microbiol 6: 201–211. [DOI] [PubMed] [Google Scholar]
- 5. Leben C, Schroth MN, Hildebrand DC (1970) Colonization and movement of Pseudomonas syringae on healthy bean seedlings. Phytopathology 60: 677–680. [Google Scholar]
- 6. Hirano SS, Baker LS, Upper CD (1996) Raindrop Momentum triggers growth of leaf-associated populations of Pseudomonas syringae on field-grown snap bean plants. Appl Environ Microbiol 62: 2560–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Serfontein JJ (1994) Occurrence of bacterial brown spot of dry beans in the Transvaal province of South Africa. Plant Pathol 43: 597–599. [Google Scholar]
- 8. Panagopoulos CG, Crosse JE (1964) Frost injury as a predisposing factor in blossom blight of pear cause by Pseudomonas syringae van Hall. Nature 202: 1352. [Google Scholar]
- 9. Misas-Villamil JC, Kolodziejek I, van der Hoorn RAL (2011) Pseudomonas syringae colonizes distant tissues in Nicotiana benthamiana through xylem vessels. Plant J 67: 774–782. [DOI] [PubMed] [Google Scholar]
- 10. Goodin MM, Zaitlin D, Naidu RA, Lommel SA (2008) Nicotiana benthamiana: its history and future as a model for plant-pathogen interactions. Mol Plant-Microbe Interact 21: 1015–1026. [DOI] [PubMed] [Google Scholar]
- 11. Kvitko BH, Park DH, Velasuez AC, Wei CF, Russell AB, et al. (2009) Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathogens 5: e100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen HP, Chakravarthy S, Velasquez AC, McLane HL, Zeng L, et al. (2010) Methods to study PAMP-triggred immunity using tomato and Nicotiana benthamiana . Mol Plant-Microbe Interact 23: 991–999. [DOI] [PubMed] [Google Scholar]
- 13. Hann DR, Rathjen JP (2007) Early events in the pathogenicity of Pseudomonas syringae on Nicotiana benthamiana . Plant J 49: 607–618. [DOI] [PubMed] [Google Scholar]
- 14. Vinatzer BA, Teitzel GM, Lee MW, Jelenska J, Hotton S, et al. (2006) The type III effector repertoire of Pseudomonas syringae pv. syringae B728a and its role in survival and disease on host and non-host plants. Mol Microbiol 62: 26–44. [DOI] [PubMed] [Google Scholar]
- 15. Cunnac S, Lindeberg M, Collmer A (2009) Pseudomonas syringae type III secretion system effectors: repertoires in search of functions. Curr Opin Microbiol 12: 53–60. [DOI] [PubMed] [Google Scholar]
- 16. Rojas CM, Senthil-Kumar M, Wang K, Ryu CM, Kaundal A, et al. (2012) Glycolate oxidase modulates reactive oxygen species-mediated signal transduction during nonhost resistance in Nicotiana benthamiana and Arabidopsis. Plant Cell 24: 336–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wäspi U, Blanc D, Winkler T, Rüedi P, Dudler R (1998) Syringolin, a novel peptide elicitor from Pseudomonas syringae pv. syringae that induces resistance to Pyricularia oryzae in rice. Mol Plant-Microbe Interact 11: 727–733. [Google Scholar]
- 18. Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, et al. (2008) A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 452: 755–758. [DOI] [PubMed] [Google Scholar]
- 19. Schellenberg B, Ramel C, Dudler R (2010) Pseudomonas syringae virulence factor syringolin A counteracts stomatal immunity by proteasome inhibition. Mol Plant-Microbe Interact 23: 1287–1293. [DOI] [PubMed] [Google Scholar]
- 20. Kolodziejek I, Misas-Villamil JC, Kaschani F, Clerc J, Gu C, et al. (2011) Proteasome activity imaging and profiling characterizes bacterial effector Syringolin A. Plant Physiol 155: 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones JDG, Dangl JL (2006) The plant immune system. Nature 444: 323–329. [DOI] [PubMed] [Google Scholar]
- 22. Amrein H, Makart S, Granado J, Shakya R, Schneider-Pokorny J, et al. (2004) Functional analysis of genes involved in the synthesis of syringolin A by Pseudomonas syringae pv. syringae B301D-R. Mol Plant-Microbe Interact 17: 90–97. [DOI] [PubMed] [Google Scholar]
- 23. Ramel C, Tobler M, Meyer M, Bigler L, Ebert MO, et al. (2009) Biosynthesis of the proteasome inhibitor syringolin A: the ureido group joining two amino acids originates from bicarbonate. BMC Biochemistry 10: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu C, Kolodziejek I, Misas-Villamil J, Shindo T, Colby T, et al. (2010) Proteasome activity profiling: a simple, robust and versatile method revealing subunit-selective inhibitors and cytoplasmic, defence-induced proteasome activities. Plant J 62: 160–170. [DOI] [PubMed] [Google Scholar]
- 25. Clerc J, Florea BI, Kraus M, Groll M, Huber R, et al. (2009) Syringolin A selectively labels the 20 S proteasome in murine EL4 and wild-type and bortezomib-adapted leukaemic cell lines. ChemBioChem 10: 2638–2643. [DOI] [PubMed] [Google Scholar]
- 26. Vlot AC, Dempsey DA, Klessig DF (2009) Salicylic acid, a multifaceted hormone to combat disease. Annu Rev Phytopathol 47: 177–206. [DOI] [PubMed] [Google Scholar]
- 27. Kohler A, Schwindling S, Conrath U (2002) Benzothiadiazole-induced priming for potentiated responses to pathogen infection, wounding, and infiltration of water into leaves requires the NPR1/NIM1 gene in Arabidopsis. Plant Physiol 128: 1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He SY, Huang HS, Collmer A (1993) Pseudomonas syringae pv. syringae HarpinPss: A protein that is secreted via the Hrp pathway and elicits the hypersensitive response in plants. Cell 73: 1255–1266. [DOI] [PubMed] [Google Scholar]
- 29. Wei CF, Kvitko BH, Shimizu R, Crabill E, Alfano JR, et al. (2007) A Pseudomonas syringae pv. tomato DC3000 mutant lacking the type III effector HopQ1-1 is able to cause disease in the model plant Nicotiana benthamiana . Plant J 51: 32–46. [DOI] [PubMed] [Google Scholar]
- 30. Wulff BBH, Kruijt M, Collins PL, Thomas CM, Ludwig AA, et al. (2004) Gene shuffling-generated and natural variants of the tomato resistance gene Cf-9 exhibit different auto-necrosis-inducing activities in Nicotiana species. Plant J 40: 942–956. [DOI] [PubMed] [Google Scholar]
- 31. Krzymowska M, Konopka-Postupolska D, Sobczak M, Macioszek V, Ellis BE, et al. (2007) Infection of tobacco with different Pseudomonas syringae pathovars leads to distinct morphotypes of programmed cell death. Plant J 50: 253–264. [DOI] [PubMed] [Google Scholar]
- 32. Wang K, Uppalapati SR, Zhu X, Dinesh-Kumar SP, Mysore KS (2010) SGT1 positively regulates the process of plant cell death during both compatible and incompatible plant-pathogen interactions. Mol Plant Pathol 11: 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu R, Malcuit I, Moffett P, Ruiz MT, Peart J, et al. (2003) High throughput virus-induced gene silencing implicates heat shock protein 90 in plant disease resistance. EMBO J 22: 5690–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monier JM, Lindow SE (2003) Differential survival of solitary and aggregated bacterial cells promotes aggregate formation on leaf surfaces. Proc Natl Acad Sci U S A 100: 15977–15982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Wees SCN, Glazebrook J (2003) Loss of non-host resistance of Arabidopsis NahG to Pseudomonas syringae pv. phaseolicola is due to degradation products of salicylic acid. Plant J 33: 733–742. [DOI] [PubMed] [Google Scholar]
- 36. Heck S, Grau T, Buchala A, Metraux JP, Nawrath C (2003) Genetic evidence that expression of NahG modifies defence pathways independent of salicylic acid biosynthesis in the Arabidopsis-Pseudomonas syringae pv. tomato interaction. Plant J 36: 342–352. [DOI] [PubMed] [Google Scholar]
- 37. Hatsugai N, Iwasaki S, Tamura K, Kondo M, Fuji K, et al. (2009) A novel membrane fusion-mediated plant immunity against bacterial pathogens. Genes Dev 23: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Del Pozo O, Lam E (1998) Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr Biol 8: 1129–1132. [DOI] [PubMed] [Google Scholar]
- 39. Meiners S, Ludwig A, Stangl V, Stangl K (2008) Proteasome inhibitors: poisons and remedies. Med Res Rev 28: 309–327. [DOI] [PubMed] [Google Scholar]
- 40. Nomura K, Debroy S, Lee YH, Pumplin N, Jones J, et al. (2006) A bacterial virulence protein suppresses host innate immunity to cause plant disease. Science 313: 220–223. [DOI] [PubMed] [Google Scholar]
- 41. Rosebrock TR, Zeng L, Brady JJ, Abramovitch RB, Xiao F, et al. (2007) A bacterial E3 ubiquitin ligase targets a host protein kinase to disrupt plant immunity. Nature 448: 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Melotto M, Underwood W, Koczan J, Nomura K, He SH (2006) Plant stomata function in innate immunity against bacterial invasion. Cell 126: 969–980. [DOI] [PubMed] [Google Scholar]
- 43. Sarkar SF, Gordon JS, Martin GB, Guttman DS (2006) Comparative genomics of host-specific virulence in Pseudomonas syringae . Genetics 174: 1041–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin NC, Abramovitch RB, Kim YJ, Martin GB (2006) Diverse AvrPtoB homologs from several Pseudomonas syringae pathovars elicit Pto-dependent resistance and have similar virulence activities. Appl Environm Microbiol 72: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spoel SH, Mou Z, Tada Y, Spivey NW, Genschik P, et al. (2009) Proteasome-mediated turnover of the transcription coactivator NPR1 plays dual roles in regulating plant immunity. Cell 137: 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khang CH, Berruyer R, Giraldo MC, Kankabala P, Park S, et al. (2010) Translocation of Magnaporthe oryzae effectors into rice cells and their subsequent cell-to-cell movement. Plant Cell 22: 1388–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Djamei A, Schipper K, Rabe F, Ghosh A, Vincon V, et al. (2011) Metabolic priming by a secreted fungal effector. Nature 478: 395–398. [DOI] [PubMed] [Google Scholar]
- 48. Uppalapati SR, Ihiga Y, Wangdi T, Kunkel BN, Anand A, et al. (2007) The phytotoxin coronatine contributes to pathogen fitness and is required for suppression of salicylic acid accumulation in tomato inoculated with Pseudomonas syringae pv. tomato DC3000. Mol Plant-Microbe Interact 20: 955–965. [DOI] [PubMed] [Google Scholar]
- 49. Zheng X, Spivey NW, Zeng W, Liu P, Fu ZQ, et al. (2012) Coronatine promotes Pseudomonas syringae virulence in plants by activating a signalling cascade that inhibits salicylic acid accumulation. Cell Host Microbe 11: 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cui J, Bahrami AK, Pringle EG, Hernandez-Guzman G, Bender CL, et al. (2005) Pseudomonas syringae manipulates systemic plant defences against pathogens and herbivores. Proc Natl Acad Sci U S A 102: 1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harper S, Zewdie N, Brown IR, Mansfield JW (1987) Histological, physiological and genetical studies of the responses of leaves and pods of Phaseolus vulgaris to three races of Pseudomonas syringae pv. phaseolicola and to Pseudomonas syringae pv. coronafaciens . Physiol Mol Plant Pathology 31: 153–172. [Google Scholar]
- 52. Brown IR, Mansfield JW (1988) An ultrascructural study, including cytochemistry and quantitative analysis, of the interactions between pseudomonads and leaves of Phaseolus vulgaris L. Physiol Mol Plant Pathol 33: 351–376. [Google Scholar]
- 53. Labes M, Pühler A, Simon R (1990) A new family of RSF1010-derived expression and lac-fusion broad-host-range vectors for gram-negative bacteria. Gene 89: 37–46. [DOI] [PubMed] [Google Scholar]
- 54. Wilson M, Hirano SS, Lindow SE (1999) Location and survival of leaf-associated bacteria in relation to pathogenicity and potential for growth within the leaf. App. Environ Microbiol 65: 1435–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Clerc J, Groll M, Illich DJ, Bachmann AS, Huber R, et al. (2009) Synthetic and structural studies in syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc Natl Acad Sci U S A 106: 6507–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Straus MR, Rietz S, Ver Loren van Themaat E, Bartsch M, Parker JE (2010) Salicylic acid antagonism of EDS1-driven cell death is important for immune and oxidative stress responses in Arabidopsis. Plant J 62: 628–640. [DOI] [PubMed] [Google Scholar]
- 57. Adam L, Somerville SC (1996) Genetic characterization of five powdery mildew disease resistance loci in Arabidopsis thaliana . Plant J 9: 341–356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial growth of WT and Δ sylC mutant PsyB728a upon infiltration. GFP-expressing (A–B, C, G and H) or non-transgenic bacteria (D, E, F and I) were infiltrated with 2×105 (A–G and I) or 2×104 (H) bacteria/mL, and infected plants were kept at high (60–90%) relative humidity (RH) (A–F), 60% RH (F–H) or transferred at 2 dpi from high RH to 60% RH (I). Bacterial populations were determined at different days-post-inoculation (dpi). Leaves were surface-sterilized with hydrogen peroxide (A–C, G–I) or ethanol (D–E) before leaf extracts were generated, diluted and plated. Experiments were performed in Cologne (A–E, G–I) or Nebraska (F). (A–I) Independent leaves were taken for n independent counts, indicated at the bottom. All error bars represent SEM. Pairwise comparisons between WT and ΔsylC growth was calculated using the Student t-test. NA, not analyzed.
(JPG)
Structures of chemicals used in this study. Reactive groups (red), biotin (blue) and fluorescent reporter (yellow).
(JPG)
Representative wound entry assay experiment. WT-GFP and ΔsylC-GFP bacteria were toothpick-inoculated from a fresh plate into different leaves of different plants of N. benthamiana. Pictures were made at 5 dpi using stereo fluorescence microscopy using identical settings. The frequency of host entry at each wound inoculation sites was counted over 12 toothpick sites, as shown at the bottom. Scale bar, 1 mm.
(JPG)
Bacterial motility of WT-GFP in hanging droplet assay. WT-GFP bacteria were grown in NYG liquid medium and their motility was monitored by confocal microscopy by hanging droplet assay. Shown is the edge of the droplet for 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP in hanging droplet assay. ΔsylC-GFP bacteria were grown in NYG liquid medium and their motility was monitored by confocal microscopy by hanging droplet assay. Shown is the edge of the droplet for 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of WT-GFP at 6 h after infiltration. WT-GFP bacteria were infiltrated at 108 bacteria/ml and their motility was monitored six hours later by confocal microscopy. The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP at 6 h after infiltration. ΔsylC-GFP bacteria were infiltrated at 108 bacteria/ml and their motility was monitored six hours later by confocal microscopy. The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of WT-GFP in colonies at 24 hpi. Leaves were infiltrated with 105 WT-GFP bacteria/mL and subjected to confocal microscopy at 24 hpi. Host cells are intact (middle and bottom) and carry normal-looking chloroplasts (red). Bacteria (green) are static in the colony (bottom), but are motile along the edge of the colony (top). The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP in colonies at 24 hpi. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL and subjected to confocal microscopy at 24 hpi. Bacteria (green) are not motile and have occupied a collapsed host cell containing degenerating chloroplasts (red). The length of the movie is 200 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of WT-GFP in colony exudates at 24 hpi. Leaves were infiltrated with 105 WT-GFP bacteria/mL, cut, and subjected to confocal microscopy at 24 hpi. GFP-expressing bacteria in the exudate that leaks from the leaf cut through infected tissue. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility of Δ sylC-GFP in colony exudates at 24 hpi. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL, cut, and subjected to confocal microscopy at 24 hpi. GFP-expressing bacteria in the exudate that leaks from the leaf cut through infected tissue. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(AVI)
Bacterial motility at 24 hpi in sylC-GFP colonies with 50 µM SylA. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL containing 50 µM SylA and bacterial colonies were imaged at 24 hpi by confocal microscopy. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(GIF)
Bacterial motility at 24 hpi in sylC-GFP colonies with 0.25% DMSO. Leaves were infiltrated with 105 ΔsylC-GFP bacteria/mL containing 0.25% DMSO and bacterial colonies were imaged at 24 hpi by confocal microscopy. The length of the movie is 100 seconds. The size of the movie frame corresponds to 512×512 µm.
(GIF)
Identification of MVB072-labeled proteins from N. benthamiana .
(PDF)
Plasmids and strains used in this study.
(PDF)