

Abstract
Mutations in the Tar DNA binding protein of 43 kDa (TDP-43; TARDBP) are associated with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43+ inclusions (FTLD-TDP). To determine the physiological function of TDP-43, we knocked out zebrafish Tardbp and its paralogue Tardbp (TAR DNA binding protein-like), which lacks the glycine-rich domain where ALS- and FTLD-TDP–associated mutations cluster. tardbp mutants show no phenotype, a result of compensation by a unique splice variant of tardbpl that additionally contains a C-terminal elongation highly homologous to the glycine-rich domain of tardbp. Double-homozygous mutants of tardbp and tardbpl show muscle degeneration, strongly reduced blood circulation, mispatterning of vessels, impaired spinal motor neuron axon outgrowth, and early death. In double mutants the muscle-specific actin binding protein Filamin Ca is up-regulated. Strikingly, Filamin C is similarly increased in the frontal cortex of FTLD-TDP patients, suggesting aberrant expression in smooth muscle cells and TDP-43 loss-of-function as one underlying disease mechanism.
Keywords: neurodegeneration, zinc finger nuclease, proteomics
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with ubiquitin and TDP-43+ inclusions (FTLD-TDP) are incurable fatal neurodegenerative diseases. ALS is characterized by the loss of upper and lower motor neurons and FTLD patients suffer from degeneration of the frontal and temporal lobes. Both diseases belong to an ALS-FTLD disease spectrum (1, 2). TDP-43 is a RNA binding protein, which modulates RNA splicing, and systematic cross-linking and immunoprecipitation studies identified numerous RNA targets (3–5). About 90% of ALS cases are sporadic and the majority is pathologically characterized by insoluble TDP-43 inclusions (1, 6). Mutations in TDP-43 can lead to familial forms of ALS and FTLD-TDP (7, 8). Most of the patient-associated mutations cluster in the C-terminal glycine-rich domain of TDP-43 (9), which mediates protein–protein interactions and is required for splicing-associated activities as well as autoregulation (10–14). Pathologically, ALS and FTLD-TDP are characterized by nuclear clearance and deposition of insoluble TDP-43 (15). Whether neurotoxicity of the TDP-43 inclusions or reduced TDP-43 function upon nuclear clearance is responsible for ALS and FTLD is under debate. However, little is known about the physiological function of TDP-43. To obtain insights into the in vivo function of TDP-43, we investigated the morphological, developmental, and molecular consequences of a loss of TDP-43 in zebrafish.
Results
Inactivation of the Zebrafish TDP-43 Orthologs.
The zebrafish genome harbors two orthologs of the human TDP-43 protein, referred to as Tardbp (Tar DNA binding protein of 43 kDa) and Tardbpl (Tar DNA binding protein of 43 kDa-like) (16). Zebrafish Tardbp is a 412-aa protein and is highly homologous to its human ortholog, whereas the 303-aa zebrafish Tardbpl lacks the C-terminal glycine-rich domain (Fig. 1A). mRNA of both genes is expressed ubiquitously in early stages of development and becomes most prominently expressed in the brain after 1 day postfertilization (dpf) (Fig. S1A). Tardbp and Tardbpl proteins are detected by immunoblotting throughout early development (Fig. S1B). To reveal the physiological function of TDP-43 and to investigate whether a loss of function might contribute to the disease we generated tardbp and tardbpl loss-of-function alleles in zebrafish by genome editing with zinc finger nucleases (ZFN) (17) (Figs. S1C and S2A). We established six independent mutant lines for ZFN sets targeting tardbp, and three lines targeting tardbpl, which all result in deletions leading to premature stop codons (Fig. S2). Western blot analysis with Tardbp- and Tardbpl-specific antibodies (for epitopes see Fig. S3A) showed that no protein is detectable in corresponding homozygous mutants (Fig. 1B).
Fig. 1.
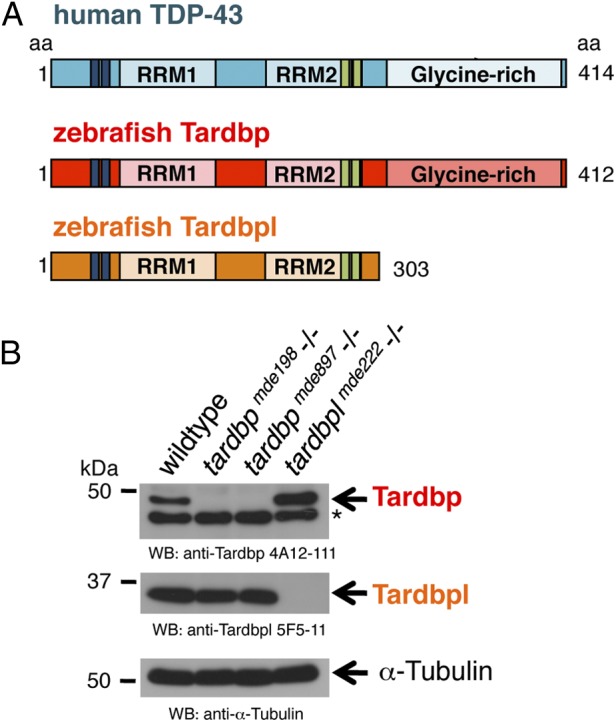
Overview of human TDP-43 and zebrafish orthologs and Western blot of their loss-of-function alleles. (A) Schematic representation of human TDP-43 (turquois), zebrafish Tardbp (red), and zebrafish Tardbpl protein (orange); blue bars represent nuclear localization sequence; green bars represent nuclear export sequence; RRM, RNA recognition motif. (B, Top) Tardbp specific monoclonal antibody 4A12-111 detects the 43-kDa Tardbp protein in adult brain from wild-type and tardbplmde222−/− but not from tardbpmde198−/− and tardbpmde897−/− fish. Asterisk marks an unspecific band. (Middle) Tardbpl-specific monoclonal antibody 5F5-11 detects the approximately 34-kDa Tardbpl protein in adult brain from wild-type, tardbpmde198−/− and tardbpmde897−/− but not from tardbplmde222−/− fish. (Bottom) α-Tubulin serves as a loading control.
Splice Variant of tardbpl Compensates for Loss of tardbp.
Homozygous mutant tardbp (tardbp−/−) or tardbpl (tardbpl−/−) fish are morphologically and behaviorally indistinguishable from their wild-type siblings. We did not observe changes in the axonal length or branching of spinal motor neurons in maternal zygotic homozygous tardbp−/− or tardbpl−/− mutant embryos (Fig. 2 A and B). We therefore asked if loss of tardbp may be compensated by tardbpl. Western blot analysis with a Tardbpl-specific antibody (Tardbpl 8G1) (Fig. S3A) revealed that the 34-kDa Tardbpl band shifts to ∼43 kDa upon transient knockdown of Tardbp (Fig. 2C). Moreover, expression of the 43-kDa variant is strongly increased in comparison with the 34-kDa Tardbpl variant (Fig. 2C). Scanning of the genomic tardbpl locus suggested a unique splice variant of tardbpl (referred to as tardbpl_tv1), which is predicted to encode a protein of 43 kDa. tardbpl_tv1 retains intron 5-6 of tardbpl (Fig. 2D), resulting in an 401-aa protein with a C-terminal extension highly homologous to human TDP-43 (Fig. S3B). Strikingly, this domain harbors most of the disease-associated mutations (9). Moreover, most of the mutations occur at positions conserved in human TDP-43, Tardbpl_tv1, and Tardbp (Fig. S3B). We confirmed the in vivo existence of the splice variant of tardbpl by RT-PCR (Fig. S3C). To further verify the Tardbpl_tv1 protein, we generated a monoclonal antibody (Tardbpl_tv1 16C8-11) (Fig. S3A), which selectively detects Tardbpl_tv1. Western blot analysis confirmed that Tardbpl_tv1 is strongly up-regulated upon loss of Tardbp (Fig. 2E). Tardbpl_tv1 is up-regulated to approximately the Tardbp wild-type expression level upon loss of Tardbp (Fig. 2F), in line with the reported tight regulation of total expression levels of TDP-43 in vivo (12, 18–20). Tardbpl_tv1 mRNA and protein is also detectable in wild-type embryos and in adult brains (Fig. 2 C and E, and Fig. S3C). Thus, Tardbpl_tv1 is a physiological transcript, which is up-regulated upon loss of Tardbp protein in zebrafish. Moreover, these findings demonstrate the functional importance of the C-terminal glycine-rich domain, where most of the disease associated TDP-43 mutations accumulate.
Fig. 2.

Loss of Tardbp does not affect spinal motor neuron axon outgrowth because of alternative splicing of Tardbpl. (A) Lateral view of wild-type, maternal zygotic tardbp−/−, and maternal zygotic tardbpl−/− embryos at 30 h postfertilization (hpf) showing the five spinal motor neuron axonal projections (labeled 1–5) anterior to the end of the yolk extension stained with znp-1 used for quantitation. Anterior to the left. (Scale bar, 25 μm.) (B) Quantitation of the length of spinal motor neuron axons measured from the exit point of the spinal cord to the growth cone in wild-type (green), maternal zygotic tardbp−/− (red), and maternal zygotic tardbpl−/− embryos (orange). Error bars indicate ±SD, n ≥ 12 embryos per experiment. n.s., not significant, Student t test. (C) Western blot analysis comparing two independent transient Tardbp knockdown experiments with respective control injected wild-type siblings. (Upper) The Western blot with the Tardbp-specific antibody anti–TDP-43 (Novus) reveals a significant reduction of Tardbp (arrow), indicating successful knockdown in lanes 2 and 4. (Lower) Probing with a pool of N-terminal Tardbpl-specific monoclonal antibodies (anti-Tardbpl 8G1) reveals a robust up-regulation of Tardbpl_tv1 upon Tardbp knockdown (lanes 2 and 4). (D) Schematic representation of genomic exon-intron organization of tardbpl (light green boxes represent 5′ and 3′ UTR; orange boxes represents coding exons) and tardbpl_tv1 (only coding exons are shown in dark orange). Enlargement of the exon 5 splice donor site of tardbpl and the corresponding sequence in tardbpl_tv1. (E) Western blot analysis with the Tardbpl_tv1 specific monoclonal antibody Tardbpl_tv1 16C8-11 detects up-regulated Tardbpl_tv1 expression at 48 hpf and in adult brain upon loss of Tardbp compared with wild-type. The anti-Tardbpl_tv1 antibody is specific because no protein is detected in tardbpl−/−. α-Tubulin serves as a loading control. Genotypes are indicated above the respective lanes. (F) Western blot analysis with an anti-human TDP-43 antibody (Sigma) that detects zebrafish Tardbp and Tardbpl_tv1. Genotypes are indicated above the respective lanes. Note that Tardbpl_tv1 is prominently detected in tardbp−/− and runs at a slightly lower molecular weight compared with Tardbp (compare lane 3 with lane 5, labeled with an asterisk). Specificity of the antibody is demonstrated in double homozygous embryos (lane 6). α-Tubulin serves as a loading control.
Circulation and Vessel Phenotype of tardbp and tardbpl Double Mutants.
We next analyzed tardbp and tardbpl double-mutant embryos (tardbp−/−;tardbpl−/−). Phenotypically, tardbp−/−;tardbpl−/− mutants first become distinguishable from their wild-type siblings at 1.5 dpf because of impaired blood circulation. At 2 dpf they have severely reduced to absent blood circulation (Movies S1 and S2), despite a beating heart (Movies S3 and S4), and accumulate erythrocytes on the yolk (Fig. 3A). To exclude gross morphological heart defects in the double-homozygous mutant embryos as a cause of the circulation phenotype, we performed whole-mount in situ hybridization with the heart-specific antisense probes cardiac myocyte light chain 2 (cmlc2) and atrial myosin heavy chain (amhc). No obvious differences between wild-type and double-homozygous mutant hearts were detectable at 2 dpf (Fig. S4A). The circulation phenotype was observed in tardbp−/−;tardbpl−/− mutants with different combinations of tardbp and tardbpl mutant alleles making off-target effects generated by the ZFN very unlikely. mRNA injection of human TDP-43 was sufficient to rescue the circulation phenotype at 2 dpf (Fig. 3B and Table S1). Moreover, injection of tardbpl_tv1 mRNA also rescued the circulation phenotype (Fig. 3B and Table S1), which confirms its compensatory capacity (Fig. 2). In contrast, injection of tardbpl mRNA failed to rescue, further underscoring the functional importance of the glycine-rich domain (Fig. 3B and Table S1). Injection of mRNA encoding human TDP-43 with the ALS-associated mutation G348C (21, 22) rescued the circulation phenotype, although there is a trend toward reduced rescuing activity (Fig. 3B and Table S1). Notably, mRNA transcribed from another ALS-associated gene, FUS (Fused in sarcoma/TLS) (23, 24), failed to rescue the circulation phenotype (Fig. 3B and Table S1). To further examine vascular lumen formation, which is required for circulation, we injected fluorescently labeled beads intravascularily (Fig. S4B). Every injection lead to labeling of the intersomitic vessels (ISV) in wild-type embryos (n = 30). In contrast, in tardbp−/−;tardbpl−/− mutants, only 2 of 30 embryos had beads entering the ISV. However, when the beads were able to enter the ISV, they revealed a dramatically disturbed patterning of the vessels (Fig. S4B). Expression of the transgene Tg(kdrl:HsHRAS-mCherry)s896, which expresses red fluorescent protein in the vasculature (25), in the tardbp−/−;tardbpl−/− background confirmed a dramatic mispatterning of the ISV as well as the head vasculature (Fig. 4). The sprouts are supernumerous and hyperbranched in the mutants.
Fig. 3.

tardbp−/−;tardbpl−/− mutants have impaired blood circulation that can be rescued. (A) tardbp−/−;tardbpl−/− mutants accumulate erythrocytes on the yolk (arrow) because of a lack of circulation at 2 dpf. Anterior to the left. See also Movie S1. (B) The circulation phenotype can be restored by injection of mRNA encoding human TDP-43 and zebrafish Tardbpl_tv1, but not by the shorter isoform Tardbpl. Injection of mRNA of the ALS-associated TDP-43G348C also rescues the circulation phenotype, whereas mRNA of the ALS-associated gene FUS fails to rescue. The black bar represents uninjected double homozygous mutant siblings of each respective clutch. The expected 25% of double-homozygous mutant embryos from incrosses of tardbp−/−;tardbpl+/− or tardbp+/−;tardbpl−/− fish were set to 100%. Error bars indicate ±SD, n ≥ 182 embryos per experiment. *, P < 0.05; ***, P < 0.005, student t test.
Fig. 4.
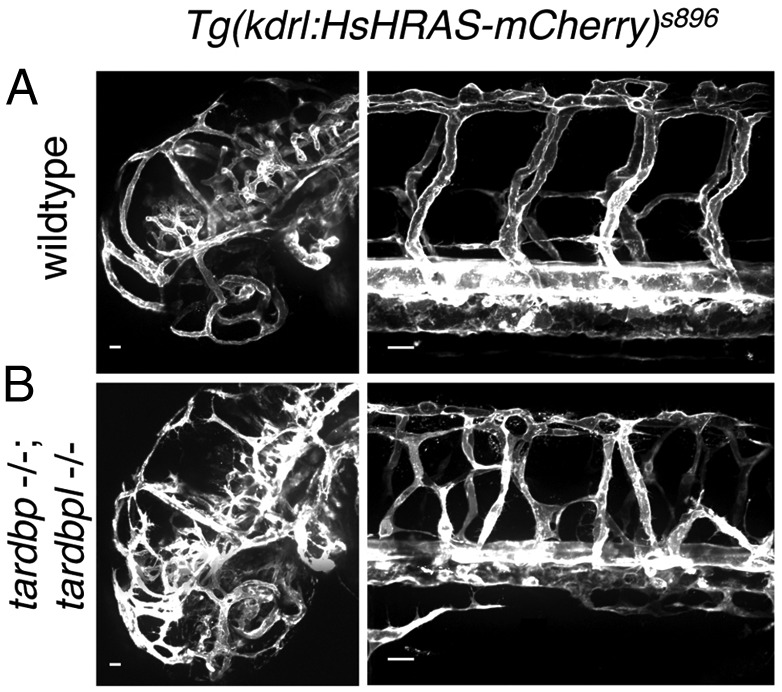
Mispatterned vasculature of tardbp−/−;tardbpl−/− mutants. (A) Whole mount in vivo imaging of wild-type; Tg(kdrl:HsHRAS-mCherry)s896 and (B) tardbp−/−;tardbpl−/−; Tg(kdrl:HsHRAS-mCherry)s896 embryos at 2 dpf. Tg(kdrl:HsHRAS-mCherry)s896 highlights the vasculature. The head is shown on the left and the ISV of the trunk on the right. (Scale bars, 20 μm.) Lateral view, anterior to the left.
Impaired Spinal Motor Neuron Axon Outgrowth.
Because motor neurons are degenerating in ALS and blood vessels and neurons share signaling pathways for migration and patterning (26), we next analyzed axonal outgrowth. Acetylated tubulin antibody staining highlighting all neuronal processes in the embryos did not point to overall axonal migratory defects (Fig. S5). However, Znp1 antibody staining revealed a shortening of the caudal primary motor neuron axons in double-homozygous mutants (Fig. 5). Despite shorter spinal motor neuron axons, the mutants show neither aberrant migration pathways nor abnormal branching patterns.
Fig. 5.
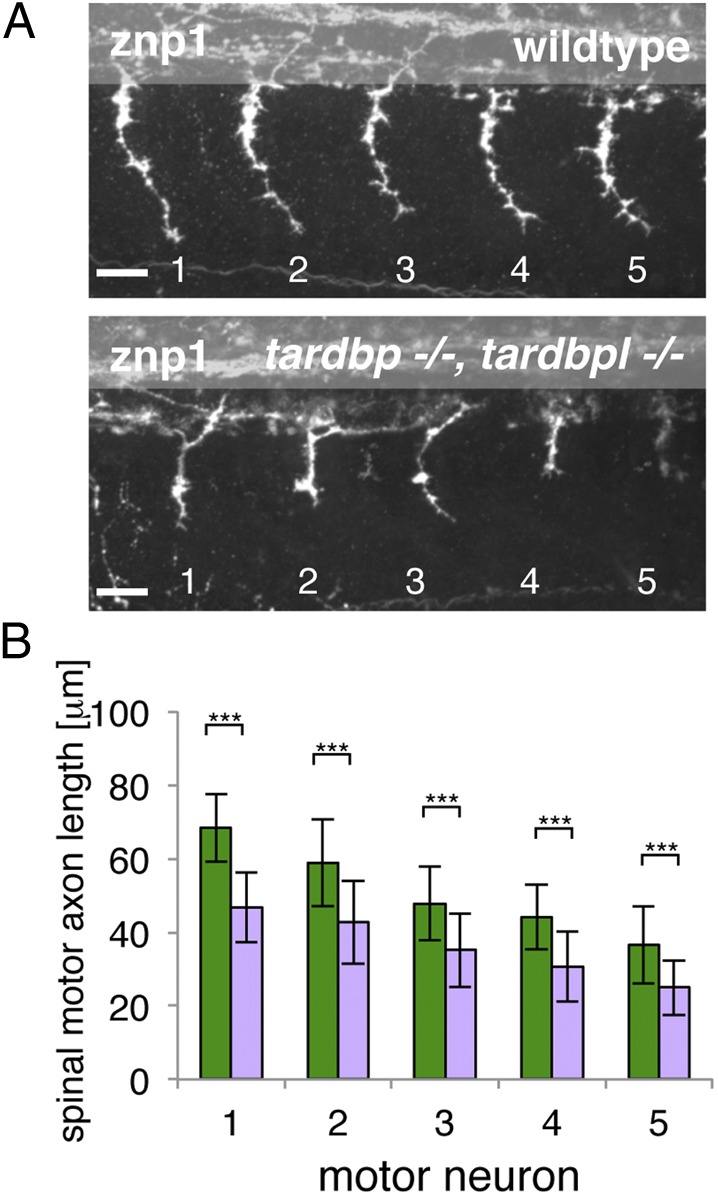
Spinal motor neuron axon outgrowth phenotype of tardbp−/−;tardbpl−/− mutants. (A) Znp-1 antibody staining of motor neuron axons in 28-hpf-old embryos of the five somites (labeled 1–5) anterior to the end of the yolk extension. Spinal motor neuron axons of tardbp−/−;tardbpl−/− mutants are reduced in length compared with wild-type siblings. Anterior to the left, lateral view. (Scale bar, 25 μm.) (B) Quantitation of the length of the outgrowing spinal motor neuron axons measured from the exit point of the spinal cord to the growth cone in wild-type (green) and tardbp−/−;tardbpl−/− mutants (purple). Error bars indicate ±SD, n ≥ 13 embryos per experiment, ***P < 0.0015, Student t test.
Loss of TDP-43 Causes Muscle Degeneration and Is Lethal in Zebrafish.
We analyzed the double-homozygous mutants for muscle integrity because muscle degeneration is one of the pathological hallmarks of ALS (7). Loss of muscle integrity is morphologically first observed at 1.5 dpf at variable sites of the trunk. Immunohistochemical analysis with the myosin-specific antibody ZE-BO-1F4, α-actinin, and vinculin showed degenerated myocytes at 2 dpf (Fig. 6 A and B). Ultrastructural analysis of tardbp−/−;tardbpl−/− mutant muscle cells by electron microscopy (EM) of high-pressure frozen samples at 2 dpf revealed smaller muscle fibers with a disorganized assembly of myofibrils (Fig. 6C, and Fig. S6 A and B). In contrast to wild-type muscles, where muscle fibers are separated from each other by a regular array of sarcoplasmic reticulum, this organization is highly perturbed in the double mutants. The clear separation of muscle fibers is largely lost, the sarcoplasmic reticulum is dilated, and mitochondria are present between the muscle fibers (Fig. 6C). Consistent with degenerating muscles the double-mutants have a reduced escape response upon tactile stimulus at 2 dpf (Movie S5). Loss of Tardbp and Tardbpl ultimately results in early lethality by 8 dpf (Fig. S6C).
Fig. 6.

Severely degenerated myocytes of tardbp−/−;tardbpl−/− mutants. (A) Antibody staining of 2-dpf-old wild-type and tardbp−/−;tardbpl−/− mutant embryos with the myosin specific antibody ZE-BO-1F4 (green) and DAPI (blue). White arrowheads indicate degenerated myocytes. (B) Antibody staining of 1.5-dpf-old wild-type and tardbp−/−;tardbpl−/− mutant embryos with α-actinin (green), vinculin (red), and DAPI (blue). (A and B) Anterior to the left, lateral view. (Scale bars, 20 μm.) (C) EM pictures of skeletal muscle of a tardbp−/−;tardbpl−/− mutant embryo shows a highly disorganized pattern of thinner myofibrils (f) with disorganized network of sarcoplasmic reticulum (open arrowheads). Large part of the sarcoplasmic reticulum is misaligned and dilated (filled arrowheads). Individual myofibrils are not well separated from another and mitochondria can now be found within the muscle fibers at (black arrow). (2 dpf) (Scale bars, 500 nm.)
Up-Regulation of Filamin C in Double Mutant Zebrafish and Human FTLD-TDP Brains.
The proteome of wild-type and tardbp−/−;tardbpl−/− mutants was analyzed by mass spectrometry (Fig. S7) at the earliest time point (1.5 dpf) at which we were able to distinguish wild-type from double-mutant embryos based on hypoperfusion. We identified a total of 4,491 proteins; of these proteins, 2,493 were covered by two or more unique peptides and were quantifiable in at least two biological replicates (Fig. S8A). Most of the proteins had comparable expression levels in wild-type and tardbp−/−;tardbpl−/− mutants (Fig. S8B). Only 41 proteins matched our hit criteria of being 30% increased or decreased in tardbp−/−;tardbpl−/− mutants, found in at least two of three biological replicates, represented by at least two unique peptides and a P value < 0.05. Of these hits, 13 proteins were up-regulated (Table S2) and 28 down-regulated (Table S3). TDP-43 belongs to the class of hnRNP and interacts with other hnRNPs; however, we did not observe a misregulation of other hnRNP proteins (Fig. S8C) or proteins genetically linked to ALS and FTLD, including Fus, Sod1, Vcp, Optn, and Vapb (Fig. S8D). Classic housekeeping proteins were not altered (Fig. S8E). Consistent with the muscle phenotype, most of the down-regulated proteins are specifically expressed in the musculature (Table S3). The top hit of up-regulated proteins is the muscle-specific actin binding protein Filamin Ca (Table S2). Filamin Ca increased approximately twofold in tardb−/−;tardbp−/− mutants but the closely related Filamin Cb was unchanged and other muscle-specific proteins were down-regulated (Fig. S8F and Table S2). Filamin C is an actin cross-linking protein expressed in skeletal and cardiac muscle but has also been detected in smooth muscle cells surrounding the brain vasculature (27–29). We therefore performed quantitative RT-PCR to test if Filamin C up-regulation is also observed in frontal cortex of FTLD-TDP patients. Strikingly, equally elevated Filamin C levels of both human splice variants were detected in FTLD-TDP patients compared with neurologically healthy controls and Alzheimer’s disease patients (Fig. 7). Thus, these data link the findings in our TDP-43 loss of function model to human disease.
Fig. 7.

Filamin C mRNA is up-regulated in FTLD-TDP patients but not in Alzheimer’s disease (AD) and healthy aged-matched control patients. Relative mRNA levels of Filamin C long and short splice variants in human FTLD-TDP and Alzheimer’s disease patients and healthy aged-matched controls, determined by quantitative RT-PCR and normalized to GAPDH and YWHAZ. *P < 0.05, **P < 0.01, Kruskal–Wallis test with Dunn’s multiple comparison test.
Discussion
Whether insoluble TDP-43 deposits are the neurotoxic entity in ALS and FTLD-TDP or whether nuclear clearance results in a loss of TDP-43 function associated with disease pathology, is intensively discussed. Resolving this question is of fundamental importance not only for understanding the underlying cellular mechanisms of ALS and FTLD-TDP but also for the development of future therapeutic strategies. Until now the analysis of the loss of function of TDP-43 in vertebrate development has been hampered by the early lethality of TDP-43 knock-out mice (18–20). We investigated the consequences of the loss of TDP-43 orthologs in zebrafish development. Single homozygous tardbp or tardbpl mutants have no obvious morphological phenotypes. In contrast to a previous report, which analyzed morpholino-mediated knockdown of Tardbp (30), we did not observe changes in spinal motor neuron axon length or branching upon transient knockdown of Tardbp by antisense gripNA, nor were changes observed in maternal zygotic homozygous tardbp−/− or tardbpl−/− mutant embryos. These phenotypic differences may result from nonspecific morpholino-mediated side effects that affect motor neurons. We obtained viable zebrafish larvae upon elimination of both TDP-43 orthologs, enabling us to be unique in studying the consequences of TDP-43 loss-of-function throughout vertebrate development. We observed a shortening of the spinal motor neuron axonal length only in tardbp−/−;tardbpl−/−, consistent with previous reports of axonal outgrowth defects upon TDP-43 depletion (31–33).
We demonstrate an unexpected physiological requirement of TDP-43 in vessel patterning, perfusion, and muscle maintenance. Vascular mispatterning is not a consequence of hypoperfusion because mutants defective in circulation, such as the silent heart mutant, do not have aberrant ISV patterning (34) because the early zebrafish embryo is not dependent upon oxygen supply provided by circulating erythrocytes. Vascular mispatterning also does not seem to be a consequence of impaired muscle function, because head vessels that lack adjacent musculature during this early stage of development (35) are also severely mispatterned. Skeletal muscle degeneration is also not seen in silent heart mutants (36). Thus, both the vascular and muscular phenotypes are not indirectly caused by reduced oxygen supply. Invertebrates lack vasculature, thus potentially explaining the phenotypic difference upon loss of TDP-43 between vertebrates and invertebrates (37).
The human pathogenic TDP-43G348C mutation rescues the tardbp−/−;tardbpl−/− mutant circulation phenotype, demonstrating that it is not a complete loss-of-function. However, we observe a trend of mutant TDP-43G348C toward reduced rescuing activity that indicates a partial loss-of-function because of the mutation. Importantly, mutations are almost exclusively found in the C-terminal glycine-rich domain, which we showed is functionally extremely important. We therefore hypothesize that a subtle loss of function of the glycine-rich domain leads over time to ALS and FTLD-TDP pathology, whereas a complete loss of this domain is not compatible with life.
Our whole animal quantitative proteomic analysis revealed that the majority of proteins with reduced abundance are muscle-specific in line with the observed myopathy. Filamin Ca was identified as the top hit of the up-regulated proteins. The importance of Filamin C for muscle integrity is supported by the observation that mutations in Filamin C lead to myofibrillar and distal myopathy in humans (38, 39). Moreover, mutant analysis of Filamin C in Medaka and zebrafish support a conserved function in teleosts (40, 41). Filamins are actin cross-linking proteins, anchors for membrane proteins and structural components, and have important signaling functions (42). Filamins are therefore important regulators of cytoskeletal dynamics. Even though Filamin C has not been identified as an RNA target bound to TDP-43 by cross-linking and immunoprecipitation analysis, it was also identified in mouse brain to be about twofold up-regulated upon siRNA-mediated knockdown of TDP-43 (3).
Consistent with the observation that postnatal inactivation of TDP-43 causes rapid death (43) and that TDP-43 inactivation in motor neurons results in astrocytosis, muscle weakness, and motor neuron loss (44), one may argue that a TDP-43 loss-of-function contributes to the disease. A potential link of a TDP-43 loss-of-function phenotype in animal models to the human disease comes from our observation of elevated Filamin C mRNA levels in human frontal cortex of FTLD-TDP cases. The functional consequence of the up-regulation of muscle-specific Filamin C in the human FTLD-TDP brain remains speculative. However, in line with expression of Filamin C in smooth muscle cells (27–29), we hypothesize that increased Filamin C in FTLD-TDP patients affects proper function of the neurovascular unit, thereby dysregulating cerebral blood flow and compromising the blood-brain barrier. This finding is consistent with observations in ALS patients and animal models where hypoperfusion and impaired blood-brain barrier function were observed preceding neurodegeneration (45–50). Evidence from postmortem brain and spinal cord further supports a dysfunction of the blood-brain barrier, the blood-spinal cord barrier, and pericytes in ALS patients (51). Thus, our findings not only reveal an unexpected requirement of TDP-43 for muscle maintenance, blood flow, blood vessel formation, and motor neuron axon outgrowth, but may also provide evidence for a loss-of-function disease mechanism in TDP-43 proteinopathies.
Experimental Procedures
For generation of zebrafish mutants, compoZr custom ZFN were designed and purchased from Sigma. mRNA of ZFN sets were microinjected into embryos and F1 offspring was screened for mutations by PCR and restriction fragment length polymorphism.
A complete description of the methods used can be found in the SI Experimental Procedures.
Supplementary Material
Acknowledgments
We thank D. Dormann, A. Capell, and B. Schoser for critically reading the manuscript; A. Rechenberg for technical assistance; D. Würzinger and R. Rojas Rojas for taking care of the zebrafish; J.-N. Chen, D. Stainier, and A. Siekmann for providing reagents; neurologists S. Engelborghs and P. P. De Deyn and neuropathologist J.-J. Martin for the clinical and pathological diagnoses; and the Antwerp Biobank of the Institute Born-Bunge for the brain samples of patients and control individuals. This work was supported in part by the Deutsche Forschungsgemeinschaft; the Competence Network for Neurodegenerative Diseases of the Bundesministerium für Bildung und Forschung; the Friedrich-Baur Stiftung; the Center of Excellence in Neurodegeneration; the Helmholtz Young Investigator Program HZ-NG-607 (D.E.); the Boehringer Ingelheim Funds (S.H.); the Elite Network of Bavaria (Universität Bayern e.V.) (B. Solchenberger); the Hans und Ilse Breuer Foundation (L.H. and K.S.); a Research Foundation Flanders postdoctoral fellowship (to J.v.d.Z.). S.H., B. Solchenberger, L.H., and K.S. are members of the International Max-Planck Research School for Molecular and Cellular Life Sciences. The Antwerp site is supported for the genetic research of neurodegenerative brain diseases by the MetLife Foundation Award (to C.V.B.), the Belgian Federal Science Policy Office Interuniversity Attraction Poles program, the Foundation for Alzheimer Research, the Medical Foundation Queen Elisabeth, the Flemish Government Methusalem Excellence program, the Research Foundation Flanders, and the Special Research Fund of the University of Antwerp, Belgium.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218311110/-/DCSupplemental.
References
- 1.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 2.Van Langenhove T, van der Zee J, Van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med. 2012;44(8):817–828. doi: 10.3109/07853890.2012.665471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tollervey JR, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao S, et al. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci. 2011;47(3):167–180. doi: 10.1016/j.mcn.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 7.Kiernan MC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 8.Sieben A, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012;124(3):353–372. doi: 10.1007/s00401-012-1029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33(9):1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buratti E, et al. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 11.D’Ambrogio A, et al. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 2009;37(12):4116–4126. doi: 10.1093/nar/gkp342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayala YM, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30(2):277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avendaño-Vázquez SE, et al. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012;26(15):1679–1684. doi: 10.1101/gad.194829.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buratti E, Baralle FE. TDP-43: New aspects of autoregulation mechanisms in RNA binding proteins and their connection with human disease. FEBS J. 2011;278(19):3530–3538. doi: 10.1111/j.1742-4658.2011.08257.x. [DOI] [PubMed] [Google Scholar]
- 15.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 16.Shankaran SS, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283(3):1744–1753. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- 17.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 18.Kraemer BC, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–419. doi: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu LS, et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48(1):56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- 20.Sephton CF, et al. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285(9):6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 22.Rutherford NJ, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4(9):e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 25.Chi NC, et al. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008;22(6):734–739. doi: 10.1101/gad.1629408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436(7048):193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- 27.Chiang W, Greaser ML, Lyons GE. Filamin isogene expression during mouse myogenesis. Dev Dyn. 2000;217(1):99–108. doi: 10.1002/(SICI)1097-0177(200001)217:1<99::AID-DVDY9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.van der Ven PF, et al. Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil Cytoskeleton. 2000;45(2):149–162. doi: 10.1002/(SICI)1097-0169(200002)45:2<149::AID-CM6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 29.Thompson TG, et al. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148(1):115–126. doi: 10.1083/jcb.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabashi E, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19(4):671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- 31.Fiesel FC, Schurr C, Weber SS, Kahle PJ. TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol Neurodegener. 2011;6:64. doi: 10.1186/1750-1326-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iguchi Y, et al. TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J Biol Chem. 2009;284(33):22059–22066. doi: 10.1074/jbc.M109.012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci USA. 2012;109(9):3347–3352. doi: 10.1073/pnas.1112427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isogai S, Lawson ND, Torrealday S, Horiguchi M, Weinstein BM. Angiogenic network formation in the developing vertebrate trunk. Development. 2003;130(21):5281–5290. doi: 10.1242/dev.00733. [DOI] [PubMed] [Google Scholar]
- 35.Santoro MM, Pesce G, Stainier DY. Characterization of vascular mural cells during zebrafish development. Mech Dev. 2009;126(8-9):638–649. doi: 10.1016/j.mod.2009.06.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sehnert AJ, Stainier DY. A window to the heart: Can zebrafish mutants help us understand heart disease in humans? Trends Genet. 2002;18(10):491–494. doi: 10.1016/s0168-9525(02)02766-x. [DOI] [PubMed] [Google Scholar]
- 37.Wegorzewska I, Baloh RH. TDP-43-based animal models of neurodegeneration: New insights into ALS pathology and pathophysiology. Neurodegener Dis. 2011;8(4):262–274. doi: 10.1159/000321547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vorgerd M, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77(2):297–304. doi: 10.1086/431959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duff RM, et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet. 2011;88(6):729–740. doi: 10.1016/j.ajhg.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita M, et al. Filamin C plays an essential role in the maintenance of the structural integrity of cardiac and skeletal muscles, revealed by the medaka mutant zacro. Dev Biol. 2012;361(1):79–89. doi: 10.1016/j.ydbio.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Ruparelia AA, Zhao M, Currie PD, Bryson-Richardson RJ. Characterization and investigation of zebrafish models of filamin-related myofibrillar myopathy. Hum Mol Genet. 2012;21(18):4073–4083. doi: 10.1093/hmg/dds231. [DOI] [PubMed] [Google Scholar]
- 42.Feng Y, Walsh CA. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6(11):1034–1038. doi: 10.1038/ncb1104-1034. [DOI] [PubMed] [Google Scholar]
- 43.Chiang PM, et al. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci USA. 2010;107(37):16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu LS, Cheng WC, Shen CK. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J Biol Chem. 2012;287(33):27335–27344. doi: 10.1074/jbc.M112.359000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11(4):420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambrechts D, Carmeliet P. VEGF at the neurovascular interface: Therapeutic implications for motor neuron disease. Biochim Biophys Acta. 2006;1762(11-12):1109–1121. doi: 10.1016/j.bbadis.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 47.Bell RD, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishikawa T, Morita M, Nakano I. Constant blood flow reduction in premotor frontal lobe regions in ALS with dementia A SPECT study with 3D-SSP. Acta Neurol Scand. 2007;116(5):340–344. doi: 10.1111/j.1600-0404.2007.00876.x. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka M, et al. Cerebral blood flow and oxygen metabolism in progressive dementia associated with amyotrophic lateral sclerosis. J Neurol Sci. 1993;120(1):22–28. doi: 10.1016/0022-510x(93)90019-u. [DOI] [PubMed] [Google Scholar]
- 50.Rule RR, Schuff N, Miller RG, Weiner MW. Gray matter perfusion correlates with disease severity in ALS. Neurology. 2010;74(10):821–827. doi: 10.1212/WNL.0b013e3181d3e2dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winkler EA, et al. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(1):111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.