

Abstract
Significance: Among trace elements used as cofactors in enzymes, selenium is unique in that it is incorporated into proteins co-translationally in the form of an amino acid, selenocysteine (Sec). Sec differs from cysteine (Cys) by only one atom (selenium versus sulfur), yet this switch dramatically influences important aspects of enzyme reactivity. Recent Advances: The main focus of this review is an updated and critical discussion on how Sec might be used to accelerate thiol/disulfide-like exchange reactions in natural selenoenzymes, compared with their Cys-containing homologs. Critical Issues: We discuss in detail three major aspects associated with thiol/disulfide exchange reactions: (i) nucleophilicity of the attacking thiolate (or selenolate); (ii) electrophilicity of the center sulfur (or selenium) atom; and (iii) stability of the leaving group (sulfur or selenium). In all these cases, we analyze the benefits that selenium might provide in these types of reactions. Future Directions: It is the biological thiol oxidoreductase-like function that benefits from the use of Sec, since Sec functions to chemically accelerate the rate of these reactions. We review various hypotheses that could help explain why Sec is used in enzymes, particularly with regard to competitive chemical advantages provided by the presence of the selenium atom in enzymes. Ultimately, these chemical advantages must be connected to biological functions of Sec. Antioxid. Redox Signal. 18, 1675–1689.
Introduction
Selenium is an essential trace element for many organisms because it is found as a component of selenocysteine (Sec, U), the 21st amino acid in the genetic code (4, 7), and 5-methylamino-2-selenouridine, a modified base found in tRNAs in certain prokaryotes (88). Among the trace elements that are used as cofactors for enzymes, selenium is unique because unlike metal cofactors such as zinc and copper, it is actually part of the polypeptide chain as a component of an amino acid. However, it is also used as a conventional metallo-cofactor as in the case of the molybdopterin cofactor found in nicotinic acid hydroxylase (28) and several other proteins.
Sulfur and selenium are members of the chalcogen group of elements, with selenium occupying the position immediately below sulfur on the periodic chart. The two elements thus have very similar chemicophysical properties (87), and can undergo similar chemical reactions such as thiol/disulfide (and thiol-disulfide-like in cases where selenium replaces sulfur) exchange reactions, the subject of this review. Sec has been credited with the distinctive characteristic of being exclusively located in catalytic sites of enzymes, although a possible exception has been reported, wherein a selenoenzyme had both catalytic and three additional Sec residues (48). The functions of these noncatalytic residues are not known.
In selenoproteins, Sec functions can be partially preserved only when cysteine (Cys), and no other amino acid, replaces Sec; however, in most such cases, the substitution of Sec with Cys leads to a marked reduction in catalytic efficiency. Considering the functional predominance of Sec in proteins, many ideas have been advanced regarding its role: a common view is that because Sec and Cys differ by a single atom, the presence of selenium provides some superior chemical or physical property that enhances the functions of biological macromolecules. There has been much debate about the exact nature of this chemical property. For detailed reviews on the subject, see (6, 29, 34, 38).
The purpose of this article is to review how selenium might be used to accelerate thiol/disulfide exchange reactions such as those catalyzed by natural selenoenzymes thioredoxin reductase (TR), methionine sulfoxide reductase (Msr), and glutathione peroxidase (Gpx). Of course, since selenium replaces sulfur in these exchange reactions, they should be referred to either as a selenol/disulfide exchange reaction or a thiol/selenosulfide (or thiol selenenylsulfide) exchange reaction, and therefore to simplify we use the term thiol/disulfide-like exchange, or simply thiol/disulfide exchange, with the understanding that it includes variations where selenium replaces sulfur. There have been many good reviews on the chemistry of thiol/disulfide exchange reactions (24–26, 72, 90), whose basic features are depicted in Figure 1. There are three principal ways in which a thiol/disulfide exchange reaction could be accelerated: (i) an increase in nucleophilicity of the attacking thiolate, (ii) an increase in electrophilicity of the center sulfur atom (SC), and (iii) an increase in stability of the leaving group sulfur (SLg) atom. In principle, a thiol/disulfide-like exchange reaction in which selenium replaces sulfur can be accelerated by any of the mechanisms shown in Figure 1, and this may be why Sec has been selected over Cys in certain enzymes. Of course, the superiority of Sec over Cys in any enzymatic reaction would only be useful if the thiol/disulfide-like exchange reaction participates in the rate-limiting step.
FIG. 1.

Selenium in thiol/disulfide exchange reactions in comparison to sulfur. The rate constant of the exchange reaction is made up of three individual rate constants: the rate constant for nucleophilic attack (kNuc), the rate constant governing the rate at which the center atom (Xc) accepts electrons (kElectrophilicity or kEl), and the rate constant governing the rate at which the Sc–XLg bond is broken (kLeaving group or kLg). Thus, the overall rate of reaction can be increased by increasing any one of three parameters: nucleophilicity, electrophilicity, and leaving group ability. (A) The rate of the reaction is increased if X=Se since the nucleophilicity (kNuc=k1, in the case of Se) of a selenolate is greater than that of a thiolate (X=S, in which case kNuc=k2, as schematized in the panel) and k1>k2. (B) The rate of the reaction is increased if Xc=Se because selenium is more electrophilic than sulfur and k3 (i.e., kEl for Se)>k4 (i.e., kEl for S). (C) The rate of the reaction is increased if XLg=Se because a selenol has a lower pKa relative to a thiol and k5>k6. This is because in order for the Sc–XLg bond to be broken, the leaving group atom must either be protonated by a general acid, or have a pKa that is less than or equal to the solution pH. The leaving group ability of selenium is also increased due to its higher polarizability relative to sulfur.
In addition to the chemical factors listed above, physical properties of selenium also contribute to the increased rate in these types of exchange reactions. For instance, the sulfur–selenium bond distance is 2.2 Å compared to 2.0 Å for a sulfur–sulfur bond. As a result, the sulfur–selenium bond should be weaker than a sulfur–sulfur bond, and this could, in principle, increase the rate in an exchange reaction of the type shown in Figure 1.
Nucleophilicity of Selenium Versus Sulfur
Historically, chemists and biochemists have focused on the higher nucleophilicity of a selenolate relative to the thiolate as the means by which exchange reactions involving selenium are enhanced (2 and references therein, 78). For example, in the case of the selenoenzyme TR, statements attributing the high reactivity and broad substrate specificity of the enzyme to the high nucleophilicity of a selenolate have been made (see the Supporting Information of citation 50 for a catalog of such statements).
In comparing the intrinsic, chemical nucleophilicities of sulfur and selenium, one has to be careful to compare the same type of chemical species. This should not be confused with nucleophilicities that are achieved by selenium or sulfur in the context of an enzyme active site. For example, for low molecular weight compounds the comparison should be made between selenolate (RSe−) and thiolate (RS−) forms of otherwise identical compound. The charged atom is the correct form of the attacking nucleophile due to the energy barrier of proton transfer during the reaction. Often, biochemists are focused on comparing reactivities under physiological conditions; most of the time they are in fact comparing the nucleophilicity of the charged selenolate to that of a thiol (RSH). The pH range plays a great part in determining the relative difference in reactivity between Cys and Sec in proteins: for example, at a slightly acidic pH (pH around 5) the ratio of rate constants kRSe/kRS, can be as high as ∼1000 [assuming Sec pKa ∼5.2, Cys pKa ∼8.3 (14, 35)]. This is illustrated by the reaction and data given in Figure 2 [from citation (33)]. The large difference in rate constants in the reaction can principally be attributed to the concentration of nucleophilic species in the reaction. In the case of the reaction with selenium as the nucleophile, the ratio of selenolate to selenol is expected to be 1:1 because a reaction pH of 5 would be very close to the reported pKa of Sec [∼5.2 in citation (36)], whereas in the reaction with sulfur as the nucleophile at pH 5.0, the ratio of thiolate to thiol would be close to 1:1000. Thus, the sulfur-containing reaction is much slower since there is such a low concentration of thiolate present compared to the same reaction containing selenium. When the pH of the reaction is increased to 8, the reaction rates become more similar with kRSe/kRS, ∼15. If the pKa of the thiol of free Cys is 8.3 (15), then the ratio of thiolate to thiol at pH 8.0 should now be 2:1. The increase in the concentration of thiolate can explain a large portion of the rate increase for the reaction. It should be noted that while the pKa of free Sec has been reported to be ∼5.2, Sec is a highly polarizable residue, and this property may reflect a broader range of pKa values, depending on the microenvironment. For example, in peptides, Sec pKa has been determined using 77Se nuclear magnetic resonance (NMR) spectroscopy to be as low as 3.3 (53).
FIG. 2.

Rate of attack on a thioester by cysteine (Cys) or selenocysteine (Sec) as a function of pH. The data is from citation (33).
We address here an interesting phenomenon of Sec as a nucleophile at very low pH compared to Cys toward hyper-reactive electrophiles. Early work by Hussey and coworkers showed that α-haloacids are 1600-fold more reactive toward nucleophiles compared to alkylhalides (14). The reason for the large differences in reactivities is due to a stabilizing effect of the electron-deficient carbonyl carbon on the incoming nucleophile [reviewed in (80)]. The hyper-electrophilicity of iodoacetic acid allows Sec to be alkylated at pH 2, but not Cys (36, 70). This is because at pH 2, Sec will still exist as 0.1% in the selenolate form, while Cys is only present at 5×10−5 percent in the thiolate form. While there is only a low concentration of selenolate present at pH 2, there is enough present to drive this nucleophilic displacement reaction type 2 (SN2) reaction to completion with iodoacetic acid and alkylation is therefore observed. In the case of Cys at pH 2, there is not enough of the thiolate form present to attack even this extremely reactive electrophile. Thus, if iodoacetic acid is used as a basis of comparison at low pH for measuring differences in nucleophilic rate constants, it would appear that the difference in nucleophilicities is extremely large, when this is not the case with typically reactive electrophiles at neutral or basic pH, such as is the case with methyliodide discussed below. It would be expected that at a pH of ∼8, where the ionization states of Sec and Cys are more similar, that the differences in the rate of substitution on iodoacetic acid of Sec and Cys would be in the range of 10- to 15-fold, similar to the data presented in Figure 2. We point out that the data discussed above are for chemical systems, and not enzymatic systems. An enzyme active site could of course alter the pKa of Cys and otherwise modulate its reactivity. However, on the basis of this chemical difference between Sec and Cys, Sec would have a clear catalytic advantage at acidic pH with very electrophilic substrates and this could one factor in the utilization of Sec versus Cys in enzymes.
In the case of the standard electrophilic substrate for SN2 reactions, CH3I, Pearson has developed a nucleophilic reactivity scale (59, 60). For the reaction in Scheme 1, he has defined a nucleophilic reactivity constant n, as nCH3I=log (kY/kCH3OH), where kY is the rate constant for the formation of CH3Y and kCH3OH is the rate constant for methanolysis. Pearson determined a value of 10.7 for nCH3I using benzeneselenol as the nucleophile and a value of 9.92 for nCH3I using benzenethiol as the nucleophile. This corresponds to a six-fold difference in nucleophilic reactivity constants. The pKa of benzenethiol is 6.6 (45), while that of benzeneselenol is 4.6 (35). The pKa of benzenethiol is low enough such that proton transfer no longer becomes a rate-limiting factor, and the difference in nucleophilicity of the two atoms is rather small when this condition is met. However, it should be noted that the pKa is only one factor determining the actual nucleophilicity of a specific thiol (2 and references therein). Moreover, particularly in the case of the attack of a thiolate anion (RS−) on a disulfide, factors that make the thiolate less basic may influence nucleophilicity even more, making the RS− anion progressively less reactive, the more its pKa is below the physiological pH.
SCHEME 1:
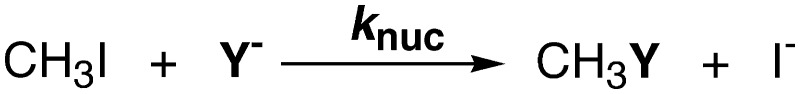
Reaction of a nucleophile Y- with methyliodide. Y=S, Se. The rate constant knuc-Se>knuc-S.
In a study by Singh and Kats, using the selenolate anion of selenocysteamine as a nucleophile (generated by reduction of the diselenide with sodium borohydride) in an exchange reaction with hydroxyethyl disulfide as the electrophile, the rate constant for the reaction was determined to be 3000 M−1·min−1 (73). If the thiol of β-mercaptoethanol replaces selenocysteamine in the same reaction, the rate constant is lowered to 460 M−1·min−1 (74, 72). Therefore, in this experimental setup, a six-fold difference in nucleophilicities between sulfur and selenium was found.
A more recent study that used stopped flow spectrophotometry to measure equilibrium constants for thiol/diselenide and selenol/disulfide exchange reactions to quantify and compare the effects of nucleophilicity, electrophilicity, and leaving group ability of sulfur and selenium is particularly noteworthy (79). Koppenol and coworkers found that at pH 7, a selenolate catalyzed the reduction of a disulfide ∼390-fold faster than a thiol. This data is not dissimilar from the data reported in (33) at neutral pH [also see (32)]. The authors noted that the difference in nucleophilicity is significantly decreased, to 10-100-fold, to account for differences in the ionization state of the two chalcogens. Indeed, based on the data in (33), the difference in nucleophilicity is most likely near a ∼10-fold difference under conditions of equal ionization.
Electrophilicity of Selenium Versus Sulfur
For thiol/disulfide exchange reactions, the two principal ways in which the reaction is known to be accelerated is the increase in the nucleophilicity of the attacking sulfur atom and the increase in the leaving group ability of SLg. The role of the electrophilic character of the SC has been largely unexplored in the literature. Bachrach and coworkers performed a computational study to address the question of which atom, selenium or sulfur, is attacked in a thiol/selenosulfide exchange reaction. Their results showed that attack at selenium is favored by both the kinetics and thermodynamics of the reaction (5). The study by Koppenol and coworkers showed that when a selenosulfide or diselenide is used as an electrophile in an exchange reaction with a selenolate as the nucleophile, the exchange reaction was ∼104-fold faster than when the corresponding disulfide was used as an electrophile. This data was interpreted by the authors to mean that “… the rate enhancement of selenium over sulfur as an electrophile (ca. 104) is two orders of magnitude greater than that of the nucleophilic enhancement …” (79). In the case of selenoproteins, the higher performance of selenium as an electrophilic center has been advocated as a critical factor in the enzymatic reaction mechanism of a mammalian TR (34, 76). This point will be further addressed in a later section.
Leaving Group Ability of Selenium Versus Sulfur
Interestingly in the study by Koppenol and coworkers, the selenosulfide electrophile only increased the reaction rate by three-fold over the disulfide electrophile when a thiol was used as the nucleophile (79). This data was interpreted to mean that selenium and sulfur have similar leaving group abilities. This might be a surprising finding since low pKa is strongly correlated with leaving group ability in thiol/disulfide exchange reactions (24, 25, 72). However, it could be explained by a more favorable transition state formed after the attack of a thiol nucleophile (Cys) on a disulfide electrophile (in the case of Sec attacking a disulfide, although Sec is a superior nucleophile, the structure of the transition state includes a heteroatomic couple, with Se attacking a sulfur electrophilic center). In general, the predicted ability of selenium as a leaving group should outperform sulfur, on the basis of its lower pKa of a selenol (∼5), versus a thiol (∼8.5) (15), as well as the much larger polarizability volume of Se (3.8 Å) versus (2.9 Å).
In order to study the possible role of Se as a leaving group in selenoproteins, Hondal and coworkers investigated the catalytic mechanism of TR, employing the method of peptide complementation (17–19, 21, 47). These studies made use of a truncated enzyme missing the conserved, C-terminal Gly-Cys-Sec-Gly tetrapeptide and synthetic peptide substrates containing: (i) disulfide-containing peptides, (ii) selenosulfide-containing peptides, or (iii) a peptide disulfide substrate containing a mixed disulfide containing a Cys-S-S-thionitrobenzoic (TNB) acid group. The results showed that the truncated enzyme could only reduce peptides that either a selenosulfide bond or ones containing the mixed Cys-S-S-TNB disulfide bond. These results were interpreted to mean that the benefit of selenium in the enzymatic reaction mechanism was as a superior leaving group. This point will be expanded and critically discussed in the following section, in which some cases where the leaving group model (i.e., Se as a better leaving group) cannot fully explain experimental results are reviewed.
Thiol/Disulfide Exchange Reactions of the TR Mechanism
Mammalian TR was found to be a selenoenzyme in 1996 (27, 82). The enzyme contains three redox centers: flavin adenine dinucleotide, an N-terminal redox center of sequence CVNVGC, and a C-terminal redox center containing the conserved GCUG tetrapeptide motif. The high Mr mammalian enzyme is quite different from the low Mr bacterial, plant and yeast enzymes, which do not have an equivalent of the C-terminal redox center. Depending on the precise step of the enzymatic reaction mechanism of mammalian TR, selenium acts as a nucleophile, electrophile, and leaving group as discussed below.
Selenium acts as a nucleophile by attacking the redox-active disulfide bond of thioredoxin (Trx), a small 12 kDa protein that acts as a carrier of reducing equivalents to many protein targets in the cell. This was suggested by Becker and coworkers who crystallized the TR/Trx complex by creating the U498C/C497S double mutant of TR and a C35S/C73S double mutant of Trx and mixing them in a 1:1 ratio (22). The result was a complex with a mixed disulfide bond between Cys498 of TR and Cys32 of Trx. This experiment suggested that the Se atom of Sec498 attacks the disulfide bond of Trx in a nucleophilic attack step as shown in Figure 3A.
FIG. 3.

Multiple chemical roles for selenium in the catalytic mechanism of thioredoxin reductase (TR). (A) Nucleophilic attack–Selenium acts as a nucleophile to attack the disulfide bond of thioredoxin (Trx). (B) Resolution step–Selenium acts as an electrophile so that the mixed selenosulfide bond between TR and Trx is resolved to release the reduced product. (C) Ring opening step–The interchange Cys residue (CysIC) has the choice of either: (1) attacking the selenium atom of the eight-membered ring (electrophile), or (2) attacking the sulfur atom of the ring (leaving group). (D) Regeneration of selenolate: If path (1) is used in (C), then the selenosulfide bond between the N-terminal and C-terminal redox center must be resolved. In this case, selenium would be in the leaving group position.
The mixed selenosulfide bond between TR and Trx (Fig.3B) has to be resolved so that the product can be released. The Cys residue immediately adjacent to Sec (Cys497) may act as a resolving nucleophile and attack the selenium atom of the mixed selenosulfide bond. In this context, selenium acts as an electrophile. The result of this step is the formation of an unusual eight-membered ring structure, containing an adjacent selenosulfide bond (Fig. 3B and C). This type of vicinal disulfide bond, such as occurs in this step of the TR mechanism, is rarely found in protein structures (11, 37).
The next step in the mechanism involves a third thiol/disulfide exchange step involving selenium. The interchange Cys residue (CysIC) must now attack the selenosulfide bond of the eight-membered ring (Fig. 3C). In this step of the mechanism, selenium could either be acting as an electrophile (path 1) or as a leaving group (path 2). Hondal and coworkers initially argued that selenium was acting as a leaving group in this step of the reaction mechanism (19, 47). The argument for a leaving group effect is illustrated in Figure 4, focusing on the exchange step between the N- and C-terminal redox centers, governed by a rate constant termed kex. This exchange step could be isolated from the rest of the mechanism by constructing a truncated enzyme missing the C-terminal redox center containing Sec (Fig. 4A), and then synthesizing a peptide that corresponded to the missing redox-active “tail” and assessing the ability of the peptide (containing either Sec or Cys) to be used as a substrate for the truncated enzyme (Fig. 4B).
FIG. 4.

Method for analyzing the exchange step in the TR reaction mechanism. (A) A truncated TR was constructed by making a deletion mutant missing the last eight amino acids of the flexible tail containing the C-terminal redox center. (B) Synthetic peptides were constructed comprised of the eight missing amino acids. The advantage of this method is that the exchange step between N- and C-terminal redox centers could be isolated from the rest of the enzymatic reaction mechanism. In addition, the redox state of the peptide can be controlled so that all of the peptide is in the oxidized form, which is the substrate for the N-terminal redox center. The original interpretation of this data was that selenium was acting as a leaving group in the exchange step as shown here.
The result was that the peptide containing Sec was a better substrate by a factor of ∼5,000 (47). Hondal and coworkers concluded from this experiment that this acceleration was due to the superior leaving group ability of a selenol relative to a thiol. This conclusion was supported by the fact that 5,5′ dithio-bis(2-nitrobenzoic acid), a linear, disulfide-containing substrate for the truncated TR, was a much better substrate than other linear, disulfide-containing substrates such as cystine by more than 4,000-fold (Fig. 5A) (50, Lothrop and Hondal unpublished results). This logic seemed supported since the pKa of thionitrobenzoic acid is 4.75 (16) compared to ∼8.3 for Cys. Thus, it seemed that a low pKa leaving group was a substrate requirement for the N-terminal redox center.
FIG. 5.

Properties of disulfide bonds that are substrates for the N-terminal redox center of mitochondrial TR. The truncated enzyme would reduce linear (acyclic) disulfide-bearing substrates if the disulfide bond were highly electrophilic, as is the case for 5,5′ dithio-bis(2-nitrobenzoic acid) (DTNB), but not simple disulfides such as cystine as shown in the top panel. The truncated enzyme was found to reduce certain types of cyclic disulfide bonds. The truncated enzyme would not reduce the cyclic Cys-Cys oxidized dyad in a peptide (eight-membered ring), nor would it reduce the six-membered ring of oxidized dithiothreitol (DTTox) Decreasing the size of the ring to five atoms of the disulfide substrate, such as in lipoic acid, enabled the enzyme to reduce the disulfide with modest efficiency as shown in the bottom panel.
Further investigation with various types of disulfides as substrates for the truncated TR raised questions about the leaving group model (50, 76). For example, lipoic acid was found to be a turned over by the truncated TR with a rate of 88 min−1 (Fig. 5B), while oxidized dithiothreitol (DTTox) was turned over 380-fold more slowly (0.23 min−1) (50). The pKa of the thiol of lipoic acid is 10.7 (23), much higher than that of thionitrobenzoic acid or a selenol. Moreover, both lipoic acid and DTTox are small molecule substrates so different substrate accessibility may not provide an easy explanation for the difference in turnover rates. However, Whitesides and Singh showed that a 1,2-dithiolane (such as lipoic acid) is 650-fold more reactive toward thiol/disulfide exchange compared to a 1,2-dithiane (such as DTTox) (75). This greater reactivity may be due to a considerably higher ring strain present in lipoic acid compared to DTTox. From this data it was suggested that a strained disulfide bond is an electrophilic disulfide bond and prone to be attacked by another thiolate in a thiol/disulfide exchange reaction, which relieves the strain in the disulfide bond. Thus, the disulfide bond of lipoic acid has significant electrophilic character.
Hondal and coworkers then assayed a series of selenium-containing compounds for activity with the truncated TR and compared them with the activities of sulfur-analogs as shown in Figure 6 (50, 76). For example, the activity of selenite with the truncated TR was >1,000-fold higher compared to sulfite. In this reaction, it is the center atom, either selenium or sulfur, that must be attacked by CysIC and a leaving group effect cannot be invoked. The same phenomenon is observed for methaneseleninic acid versus methanesulfinic acid, as well as a selenosulfide-containing-peptide versus a disulfide-containing peptide (Fig. 6). For the truncated TR, these small molecule selenium-containing molecules are acting as surrogate substrates in place of the oxidized Gly-Cys-Sec-Gly tetrapeptide found in the C-terminal redox center. The data from these small molecules indicate that high substrate electrophilicity must be the chemical factor that accelerates the exchange reaction that occurs between the N- and C-terminal redox centers of the TR reaction mechanism. This concept supports the idea that pathway 1 of Figure 3C is the correct model for the exchange reaction that occurs in this step of the mechanism.
FIG. 6.

The selenium atom accelerates the exchange reaction of the truncated TR enzyme for a number of small molecule substrates. The truncated enzyme could reduce a peptide bearing a selenosulfide bond, whether it was in the context of a ring or not, but would reduce the analogous disulfide bearing compound very poorly. The same is true for small molecule selenium-containing compounds such as selenite, a good substrate for the truncated enzyme, while the analogous sulfur compound, sulfite, is a poor substrate.
Finally, as depicted in Figure 3D, selenium has to act as a leaving group in order to regenerate the reactive selenolate, which is then available for nucleophilic attack on the disulfide bond of Trx, to restart the catalytic cycle. In this regard, abilities of selenium as a leaving group (discussed above) come at hand, and should represent an additional step in the reaction where selenium properties can provide an advantage (in terms of reaction rates, over sulfur). Once again, this comparison has to be put into proper context: here, and throughout this manuscript, comparisons are made focusing on selenium versus sulfur, when they are in the same protein (i.e., Sec in human TR vs. the Cys mutant of the same protein, and not the closest naturally occurring Cys ortholog of the protein). The objective is to understand what could make selenium essential in a natural selenoprotein: directly comparing Cys and Sec containing orthologs would not serve our purpose here, as one would not directly compare selenium versus sulfur, but the two proteins with different evolutionary history (a more detailed digression on this issue is presented later in this review). From what has been reported thus far, at least in the case of TRs, it appears that selenium has multiple, diverse roles throughout the catalytic cycle of the enzyme. TR must be able to control and modulate the chemical reactivity of each thiol/disulfide exchange step in the mechanism so that the “correct” product is always favored in these exchange reactions. For example, if the nucleophilicity of the selenolate was always the dominant factor in any of the exchange reactions shown in Figure 3, the result would be an unresolvable selenosulfide bond that would be kinetically trapped. One way the enzyme may regulate the high reactivity of a selenolate is to “trap” it in a selenosulfide bond as in Figure 3C or 3D until the protein substrate Trx binds, triggering the next exchange reaction with concomitant formation of the selenolate.
Another good example of how selenium is able to accelerate exchange reactions was reported by Rabenstein and coworkers who showed that symmetrical selenol/diselenide exchange reactions were ∼107-fold faster than analogous thiol/disulfide exchange reactions using NMR spectroscopy (62, 64). This study of diselenide exchange reactions in solution matches the work done on the mitochondrial TR using selenocystine as a diselenide substrate. Selenocystine was turned over 100–200-fold faster in comparison to cystine as a disulfide bearing substrate (50). In this example, selenium probably accelerates the exchange reaction both as a good electrophile and a good leaving group in comparison to sulfur.
While it has been shown that Se accelerates a thiol/disulfide exchange reaction through higher chemical reactivity (increased nucleophilicity, electrophilicity, and better leaving group ability) in solution, this is not always the case in an enzymatic reaction. Several years ago, Gromer, Arnér and coworkers (29) showed that changes in the active site of Drosophila melanogaster TR (DmTR) are sufficient to yield a high catalytic activity without involvement of selenium. In fact, DmTR utilizes a Ser-Cys-Cys-Ser tetrapeptide motif in its C-terminal redox center, and the presence of the flanking polar Ser residues was found to be a determinant in activating the catalytic Cys residues (by involving in hydrogen bond with sulfur atoms of the two adjacent Cys); in this case, the interaction between Ser491 and Cys490 (the catalytic Cys, corresponding to position 498 in mammalian TR discussed above) is thought to favor the formation of the attacking thiolate (nucleophile in the reaction). This study provided a proof of concept that local environmental features can significantly modify properties of a residue (e.g., in DmTR, improve reactivity of its nucleophilic thiol). The authors noted that this might hint to a reason selenium is used in TR. Indeed, Sec does not need to be tightly regulated (e.g., through hydrogen bonding or other interactions) for activation, such as a decrease in pKa. This property could provide a selective advantage per se, as a Sec-enzyme may experience a broader (compared to its Cys-containing counterpart) range of microenvironmental conditions in which it operates. In addition, it was suggested that as a consequence of independence of Sec on tight interactions, Sec-containing TRs could operate effectively on a broader range of substrates.
Another study, conducted on a set of tetrapeptides mimicking the C-terminal regions of various TRs (GCUG, and SCCS and various combinations of flanking residues, such as SCUG, SCUS, GCCG, and SCCG,) determined the effect of flanking polar residues (71). Serine in a flanking position could significantly decrease the redox potential (as measured by cyclic voltammetry), particularly in the case of Cys-Cys tetrapeptides. For instance, at physiological pH, the difference in the redox potential between SCCG (where the redox couple considered is SC-CG/SCCG, i.e., the reduced form considered is the di-thiol) and SCUG (redox couple SC-UG/SCUG) was very small (circa 20 mV), compared to the difference of the same peptides at pH 5 (circa 150 mV). A similar situation was found for GCCS and GCUS (but only at a lower extent for SCCS and SCUS), indicating that the presence of a substituent in flanking residues can render a Cys-Cys containing tetrapeptide as reducing (at physiological pH) as a Cys-Sec peptide. In contrast, at lower pH (3–6), Sec-containing peptides were always much more (>150 mV) reducing than their Cys containing counterparts. Therefore, the presence of proximal residues can deeply affect the chemistry of the catalytic Cys. These data suggest that enzymes may evolve by adapting their active sites to specific functions and that comparison of the activities of Cys mutant forms of Sec-containing enzymes (and vice versa) can be misleading if taken out of context (39).
A more recent study on the DmTR reported that the presence of Se could both accelerate and slow down a thiol/disulfide exchange reaction (47). Using an identical strategy to the one shown in Figure 4 for DmTR, Sec accelerated the exchange reaction 12-fold only when it occupied the C-terminal position of the peptide dyad. However, when Sec replaced the N-terminal position of the peptide dyad, the reaction was 10-fold slower than the natural Cys-Cys redox dyad. It was proposed that the presence of Sec in the N-terminal position slowed the exchange reaction due to a “seleno effect” (34), similar to the well-known thio effect described for phosphodiesterases (30, 44). A seleno effect (Fig. 7) would occur in the asymmetric active site of the enzyme and involve and interaction between the catalytic HisH+ residue (His464′) and the N-terminal Cys residue of the dyad. This interaction would polarize the -S–S- bond of the dyad and make the sulfur atom of the C-terminal Cys residue electron deficient, promoting nucleophilic attack by the CysIC residue of the N-terminal redox center. In this scenario, the presence of selenium in place of sulfur in the N-terminal position of the dyad would slow the exchange reaction because selenium is less basic than sulfur and hence less able to accept the proton from the catalytic HisH+ residue.
FIG. 7.

Description of the “seleno effect.” (A) The thiol/disulfide exchange reaction between the truncated Drosophila melanogaster thioredoxin reductase (DmTR) and a peptide bearing an eight-membered ring disulfide is fast, mimicking the situation in the holoenzyme. (B) When selenium replaces the C-terminal sulfur atom, the reaction is accelerated 12-fold relative to the reaction in (A). (C) When selenium replaces the N-terminal sulfur atom, the reaction slows down by 10-fold relative to the reaction in (A).
It should be noted that most of the studies of selenium in exchange reactions of the TR mechanism have been done with the mammalian mitochondrial TR. The present data in the field indicates that the cytosolic TR is much more dependent on selenium for catalyzing the reduction of a diverse array of substrates (91). Furthermore, a recent study (65) showed that mitochondrial and cytosolic TRs present significantly different responses to metal compounds, for example, various gold(I) compounds were found to inhibit either one or the other (and not both) isozymes. The reason for this is still unclear: it may be that cytosolic and mitochondrial TRs have different local features (e.g., structure around their active sites, local flexibility, etc.) that might be responsible for their different reactivity toward inhibitors. This could indicate that the two TRs evolved differently, each one exerting different (and compartment-specific) control over the chemical capabilities of their catalytic Sec.
Thiol/Disulfide Exchange Reactions of the Msr Mechanism
Msrs are a class of antioxidant enzymes that reduce peptide methionine sulfoxide (MetSO) residues back to methionine with release of a water molecule (9). There are two classes of Msrs based upon the stereochemistry of MetSO, which exists as either Met-(R)-SO or Met-(S)-SO (46). The class of enzymes which reduce Met-(S)-SO are referred to as MsrAs, while Met-(R)-SO are reduced by MsrBs. MsrBs are divided into three subclasses, MsrB1, MsrB2, and MsrB3. MsrAs are mainly Cys-containing enzymes, but Sec is found in MsrA in Chlamydomonas reinhardtii and some bacteria (42). In mammals, Sec is found in the active site in the MsrB1 subclass, whereas MsrB2 and MsrB3 are Cys-containing enzymes (41). Msrs may be viewed as part of the Trx system since electrons flow from NADPH to TR to Trx and ultimately to Msr to regenerate the enzyme after reduction of a MetSO residue.
A potential mechanism for the reduction of MetSO by MsrB1 is shown in Figure 8 and involves three overall steps: (i) a reductase step which involves nucleophilic attack by the selenolate onto the sulfoxide to form a tetrahedral intermediate, which subsequently rearranges to form a selenenic acid intermediate with release of methionine (8A–C), (ii) a resolution step which involves attack by a resolving Cys residue on the enzyme to form a selenosulfide bond with release of a water molecule (8D and 8E), and (iii) a regeneration step via reduction of the selenosulfide bond by Trx (8F).
FIG. 8.

Multiple roles of selenium in the MsrB1 mechanism. (A) The selenolate of the enzyme first acts as a nucleophile to attack the sulfoxide sulfur atom to form a tetrahedral intermediate (B). The details of the next step in the mechanism are not fully understood. What has been found experimentally for the Cys-containing methionine sulfoxide reductases (Msrs) is that a sulfenic acid intermediate is formed. Currently, it is thought that the tetrahedral intermediate undergoes a rearrangement to produce a sulfenic acid or selenenic acid intermediate with the release of methionine (C,D). For the Sec-containing enzymes, the selenenic acid should then be attacked by a resolving Cys residue to form a selenosulfide bond with release of a water molecule (D,E). In this step of the mechanism, the selenenic acid residue is acting as an electrophile. In the last step, the selenosulfide bond is reduced by Trx to regenerate the enzyme (F).
Figure 8 shows that, just as in the TR mechanism, selenium has multiple chemical roles. What chemical advantage might Sec confer to the Msr reaction that would have necessitated its evolution from a Cys-ancestor? If Sec is needed due to its higher chemical reactivity relative to Cys, then presumably Sec is required in the rate-determining step in the reaction, just as has been reasoned for the TR reaction (discussed above). For the Cys-containing MsrA and MsrB enzymes from Neisseria meningitides, the rate of sulfenic acid formation has been shown to be fast but is thought to be rate-determining (1, 58). From a purely chemical standpoint, this may mean that Sec was selected because a selenolate is more nucleophilic than a thiolate.
Interestingly, the rate of nucleophilic attack of the resolving Cys-residue onto the sulfenic acid intermediate of the enzymes from N. meningitides is of comparable magnitude to the attack of the reductase step (the attack of the thiolate onto the sulfoxide moiety). This was interpreted as meaning that the sulfenic acid intermediate is attacked by the resolving Cys as soon as it is formed (8). This is highly desirable, for if this was not the case the sulfenic acid intermediate could be further oxidized to a sulfinic acid residue (Cys-SO2−). The oxidation of a Cys residue to a Cys-SO2− residue is irreversible because it cannot be reduced back to the reduced Cys-SH state, but it can be further oxidized to a Cys-sulfonic acid residue Cys-SO3− (69). Oxidation either to Cys-SO2− or Cys-SO3− would result in an enzyme that is permanently inactivated (69).
The resolution step (Fig. 8D) would thus be critically important to prevent irreversible oxidation and inactivation of the enzyme. To accelerate this reaction, the nucleophilicity of the resolving Cys could be increased or the electrophilicity of the sulfur atom of the sulfenic acid intermediate could be increased. This latter rationale may provide the explanation for the inclusion of selenium into the Msr reaction. The electrophilicity of the selenenic acid intermediate should be much higher than that of a sulfenic acid. This property of selenium would drive the resolving step to completion and prevent overoxidation of the Sec residue and preserve the activity of the enzyme. This would be especially important under conditions of oxidative stress. The use of Sec in enzymes to confer resistance to irreversible inactivation is an idea that has been put forth for TR (34).
The overall limiting-step in the Msr reaction mechanism is the recycling of Msrox to Msrred by Trx. Trx reduces the selenosulfide bond of Msrox (1, 58). Since it has been shown that selenium, in a selenosulfide bond, enhances the rate of thiol/disulfide exchange reactions (either by being a better electrophile or better leaving group), then the inclusion of selenium in Msr provides a rationale for enhancing this rate-limiting step.
Kim and Gladyshev have reasoned that the inclusion of Sec into MsrB provides both catalytic advantages and disadvantages (41, 42). Like all other selenoenzymes characterized to date, the Sec→Cys mutant of MsrB1 suffers a large loss in activity; using dabsyl-Met-(R)-SO as the substrate, the catalytic rate constant decreases 100-fold (40). Conversely, when the catalytic Cys residues of MsrB2 and MsrB3 are replaced with Sec, enzyme activity increases in the range of 100 to 200-fold (42). These data were interpreted to mean that the inclusion of Sec in an enzyme active site boosts the catalytic power of the enzyme due to the higher chemical reactivity of Sec relative to Cys. The increase in activity occurred only when DTT was used as the reductant in the assay. When the natural reductant Trx was used in the assay, these Sec-containing mutant MsrBs had very little activity and this was identified as a catalytic disadvantage. The differential effects due to the choice of reductant could mean that DTT could directly reduce the selenenic acid intermediate, while Trx could not. Trx could reduce the sulfenic acid intermediate for the Cys-containing MsrB2 and MsrB3 enzymes. The insertion of an additional resolving Cys residue greatly increased the Trx-dependent activity in the mutant Sec-MsrB3 enzyme (42). It was concluded from these studies that in MsrBs “… the use of Sec is a compromise between elevated rates of Met-SO reduction and the ability to regenerate the active enzyme form by reduction with the natural electron donor” (42).
Thiol/Disulfide Exchange Reactions of the Gpx Mechanism
Gpxs catalyze the reductions of hydrogen peroxide, lipid hydroperoxides, and organohydroperoxides to either water (H2O2 only as the substrate) or water and an alcohol (with lipid hydroperoxides and organohydroperoxides as substrates). In mammals, Gpxs can be divided into eight major families. Gpx-1, Gpx-2, Gpx-3, and Gpx-4 all contain Sec at the active site (57). All known Gpx-5, Gpx-7, and Gpx-8 enzymes utilize a conventional Cys residue in place of Sec (at least in the case of Gpx-5, the Cys form evolved from the Sec-form, which in turn evolved from the common selenoprotein ancestor with Gpx-3, so it cannot be excluded that a selenoprotein form of Gpx-5 also occurs). Gpx-6 enzymes utilize either Cys or Sec depending on the species. Gpx1 is found in the cytosol. It is the most abundant mammalian selenoenzyme and the first mammalian selenoenzyme to be discovered (68). It is especially important for the protection of hemoglobin from oxidation in the cytoplasm of red blood cells, and is also generally important under conditions of oxidative stress. The role of selenium in the thiol/disulfide exchange reactions of the Gpx1 mechanism will be used as the model and the basis for the discussion presented below.
Why Selenium Is Used in Peroxidase Enzymes
Many small molecule selenium compounds have peroxidase activity, though much lower than peroxidase enzymes (56). Identical or extremely similar small molecule sulfur compounds have much weaker peroxidase activity, or none at all. This suggests that selenium has different chemical reactivity compared to sulfur, which enables it to mimic a peroxidase. It was suggested that these chemical properties are higher nucleophilicity and higher electrophilicity relative to sulfur (69). A good example of this is the peroxidase activity of selenomethionine (3, 43, 81). Selenomethionine can be oxidized to methionine selenoxide by hydrogen peroxide and then recycled back to selenomethionine by addition of an exogenous reductant due to the higher electrophilic character of Se in the selenoxide. Similarly, methionine can be oxidized to MetSO, but at a slower rate than the same oxidation of selenomethionine. These oxidations occur because the selenide and sulfide are nucleophilic enough to react with H2O2. However, the oxidation of methionine to MetSO is irreversible because the sulfur atom of the sulfoxide is much less electrophilic than the selenium atom of the selenoxide. This concept is illustrated in Figure 9. Despite these natural chemical advantages of the selenium atom with respect to peroxidase activity, the activities of sulfur-containing peroxidases (utilizing Cys) rival that of their selenium-containing cousins. For example, the rate constant for the conversion of H2O2 to H2O with the formation of Gpx-SeOH is 4.5×107 M−1·s−1(84). Peroxiredoxins (Prxs), which catalyze an identical reaction to Gpx-1, are nearly as efficient enzymes and do so with a peroxidatic Cys residue instead of the peroxidatic Sec residue found in Gpx-1. As Poole notes (63): “Prxs rely on the Cys sulfur at the active site as the catalytic center attacking the peroxyl -O–O- bond. Nonetheless, activation of this Cys and the surrounding residues that support this function are sufficient, in at least some of the Prxs, to impart a level of catalytic efficiency that is on a par with those of the above enzymes (>107 M−1·s−1).” In fact, a study of Denicola and coworkers on human Prx II measured a second order rate constant of 108 M−1·s−1, surpassing the activity of Gpx-1 (52).
FIG. 9.

Redox cycling of a selenolate compared to a thiolate. (A) A selenolate is a stronger nucleophile than is a thiolate, so attack by the selenolate onto the peroxyl -O–O- bond is faster (k1>k2). The resulting Se-oxide that is formed can be reduced back to the selenol or selenolate faster in comparison to the reduction of an S-oxide back to a thiol or thiolate (k3>k4). This is due to the higher electrophilic character of Se-oxides relative to that of the same S-oxides. Seleno-compounds have intrinsic peroxidase activity due to both high nucleophilicity and high electrophilicity. (B) Nucleophilicity of sulfur in sulfur-compounds can be increased to a degree by increasing solution pH, so that the differences in nucleophilicity between sulfur and selenium are modest as discussed in the text. However, the lower intrinsic electrophilicity of sulfur in S-oxides cannot be compensated for in the same way, and this makes cycling back to the parent thiol or thiolate much slower (represented by the dashed line).
It should be noted that the Cys-mutant of Gpx-1 has 1000-fold lower activity than the wild type enzyme, which is common for Sec→Cys mutants of selenoenzymes, consistent with the idea that Sec has a critical function in the catalytic cycle of this selenoenzyme (67). In addition, naturally occurring Cys-Gpxs have a somewhat lower catalytic efficiency than the Sec-Gpxs, >106 M−1·s−1, but still are very efficient enzymes despite the absence of the Sec residue (84). This once again highlights the importance of context: while differences which might be intrinsic to Se and S atoms are important, in actual enzymes, a whole set of features (e.g., created by the protein microenvironment) play a role in catalysis. Different evolutionary paths characterize selenoproteins, and their Cys orthologs, a situation which can explain why, in spite of the advantages of Se for catalytic rates (Fig. 1), in some cases Cys enzymes can achieve performances comparable to those of Sec-containing enzymes. Relevant to the use of Sec in Gpxs is the observation by Rocher and coworkers who noted that the Cys-mutant of Gpx was quickly inactivated by H2O2. Rocher mused that Sec-Gpx “… may have evolved to catalyze the fast reduction of hydroperoxides without undergoing significant self-inactivation.” (67). This observation about the use of Sec in enzymes has proven to be true for TR (69).
The reactivity and mechanistic aspect of H2O2 mediated oxidation of Sec and Cys has been the subject of investigation with quantum chemical approaches (10). For both Sec-H2O2 and Cys-H2O2, the conformation of the complex was highly similar, with the most energetically favored pose driven by a hydrogen-bond interaction of one of the peroxide oxygen atoms, and the backbone nitrogen of Cys and Sec, with the most critical distances being Se (Sec)-O(H2O2)=2.52 Å, Se (Sec)-N(Sec)=1.90 Å; S (Cys)- O(H2O2)=2.4 Å, S(Cys)-N(Cys)=1.94 Å; overall, peroxide was slightly closer to the backbone and farther away from Se. Sec oxidation was computed to be only slightly favored in respect to Cys oxidation. Nevertheless, the authors remarked that the protein body exerts a huge influence on promoting Sec oxidation in Gpxs. They estimated that reaction rate constant of Gpx1 was up to 20 times higher than that of free Sec. In particular, they pinpointed the effect of hydrophobicity and the concomitant presence of nearby nitrogen atoms (an imino group of Trp and an amido group of Gln, both residues being highly conserved in the active sites of Gpxs) in promoting Sec oxidation.
The Chemical Roles of Selenium in the Catalytic Cycle of Gpx-1
As hinted above, the Sec residue of Gpx1 is found as part of a catalytic triad consisting of Trp158 (Bos taurus numbering), Gln80, and Sec45 and form a hydrogen bond network (20), although more recently a fourth conserved residue (Asn159) was proposed to contribute to the hydrogen bond network (85). In the first step of the mechanism, the selenolate of Gpx-1 nucleophilically attacks the peroxyl -O–O- bond and forms a selenenic acid residue (Fig. 10A). The selenenic acid residue can then act as an electrophile and be attacked by the thiolate of reduced glutathione (Fig. 10B). A second molecule of glutathione then enters the active site and attacks the sulfur atom of the mixed selenosulfide bond (Fig. 10C), regenerating the enzyme and producing an oxidized glutathione molecule. It is interesting to compare these last two steps of the mechanism in terms of the chemical role of selenium. In the step shown in Figure 10B, the selenium atom is acting as an electrophile and is attacked. In the next step shown in Figure 10C, the incoming thiolate of glutathione is faced with the choice of either attacking the sulfur atom or the selenium atom of the selenosulfide bond. Just as is the case in the TR mechanism, Gpx must have the ability to control the reactivity of the selenosulfide bond so that the correct atom is attacked. The enzyme may do this by blocking access of the selenium atom so that the thiolate of glutathione can only attack the sulfur atom of the selenosulfide bond, or alternatively modulate the electrophilicity of the sulfur atom so that it is attacked instead of the selenium atom. If the latter explanation is correct, perhaps the hydrogen bond network of the active site is responsible for this modulation.
FIG. 10.

Multiple chemical roles for selenium in the catalytic mechanism of glutathione peroxidase (Gpx)-1. (A,B) The high nucleophilicity of the selenolate promotes attack onto the peroxyl -O–O- bond resulting in the formation of a selenenic acid. (B,C) The selenium atom of the selenenic acid, acting as an electrophile, which, can be attacked by the thiolate of glutathione to release water and produce a mixed selenosulfide bond. (C) A second glutathione molecule then attacks the sulfur atom of the mixed selenosulfide to regenerate the enzyme. Here selenium is in the leaving group position. (D) Alternatively, the selenenic acid intermediate can be attacked by a backbone NH to produce a strained, cyclic selenamide shown in (E). (F) The selenamide is a good electrophile which can then be attacked by glutathione resulting in a mixed selenosulfide.
The selenenic acid intermediate may have an alternative fate as shown in Figure 10D. Attack on the selenenic acid intermediate by the i+1 amide nitrogen (relative to Sec45) would produce a five-membered ring selenamide (66). There is experimental evidence to support such a mechanism. Ursini and coworkers suggested the existence of a Gpx intermediate that is 2 Daltons smaller than the wild type form, as detected by mass spectrometry [unpublished data cited in (84)]. Formation of a selenosulfide bond would produce an intermediate with loss of 2 Daltons, although various Gpx proteins differ with regard to a resolving Cys residue that would result in a selenosulfide bond (differences in occurrence, number and location of resolving Cys residues is a common theme for thiol oxidoreductases, e.g., Prxs, Msrs and Gpxs). A selenamide, such as is shown in Figure 10E would also result in the loss of 2 Daltons. If this is the case, the selenium atom of the selenenic acid intermediate would be acting as a strong electrophile in the formation of the selenamide. Because this intermediate cannot be isolated, resolution of the selenamide must be fast. The driving force for forming the selenamide would most likely be to prevent oxidative inactivation of the enzyme.
In fact, Gpx-1 is known to be oxidatively inactivated by its substrate, H2O2 (61). The mechanism of inactivation was investigated by Rhee and coworkers who found evidence that the oxidized Sec residue was converted to a dehydroalanine residue (13). A similar finding was reported for selenoprotein P (51). Oxidation of Sec to selenocysteine seleninic acid can result in a β-elimination reaction to produce HOSeOH and dehydroalanine. The dehydroalanine intermediate was detected through the use of a biotin-conjugated cysteamine derivative that would be covalently linked to the dehydroalanine residue and then captured by streptavidin beads. A key question that has yet to be answered is which enzyme is more resistant to inactivation by oxidation, Sec-Gpx-1 or the Cys-mutant Gpx-1?
A Last Example of Selenium in Thiol/Disulfide Exchange Reactions–Selenoglutaredoxin
While thiol/disulfide exchange reactions of naturally occurring thiol/disulfide exchange reactions have been the focus above, a very good study involving a synthetic selenoenzyme is well worth mentioning here. Dawson and coworkers used native chemical ligation to insert Sec residues in place of Cys residues 11 and 14 of Grx3 to create a synthetic glutaredoxin3 (sGrx3) (55). In addition to the wild type sGrx3, three variants were created, sGrx3(C11U), sGrx3(C14U), and sGrx3(C11U/C14U). The oxidized forms of these proteins were equilibrated with reduced Trx to measure Keq, and the forward and reverse rate constants as summarized in Table 1. From the data in Table 1 it was concluded that since the rate of the forward reaction was relatively insensitive to Sec substitution that selenium did not have a large effect on the equilibrium as a center atom (electrophile) or as a leaving group. However, the reverse rate (k−1) showed large effects of Sec substitution. The large increase in k−1 in this context should mean that selenium is affecting the equilibrium in its role as a strong nucleophile, either to attack the disulfide bond of Trx or to attack the mixed sGrx3-Trx disulfide bond intermediate.
Table 1.
Summary of Rate Constants for the Equilibration of Reduced Trx with Oxidized sGrx3 Sec-Containing Variants (X=S, Se)
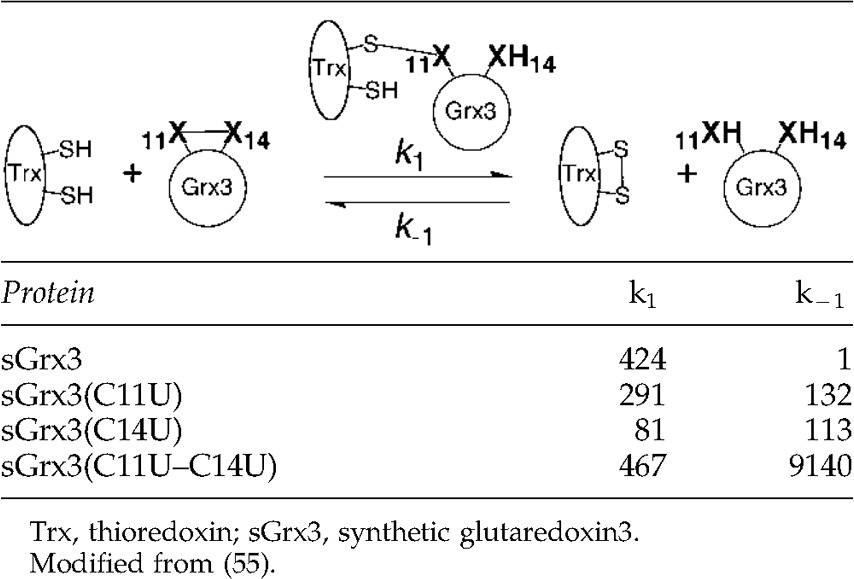 |
The redox potential of the single Sec variants listed in Table 1 are lowered by an average of 73 mV using E. coli Trx as the redox partner, largely as a result of the effect of the reverse reaction being accelerated by Sec (55). Naturally occurring selenoenzymes are likely to have selenosulfide bonds in their active sites since some of these selenoproteins have UXXC or CXXU motifs, such as selenoprotein P and selenoprotein W (31, 86). The determined redox potentials for the Sec-containing Grx3 variants were close to that of Trx, but reduced Trx was still able to reduce these Sec-containing proteins, as is known to be the case for the selenosulfide bond of MsrB1 as described above. On the basis of this study, Sec-containing enzymes should be very useful in microenvironements with lower redox potential such as the endoplasmic reticulum (particularly at nonbasic pH conditions). The study by Dawson and coworkers also showed that selenium could significantly lower the rate of a thiol/disulfide exchange reaction. The rate constant for the forward reaction of C14U variant of sGrx3 was five-fold slower than the wild type enzyme. This was interpreted as meaning that selenium “… is a worse leaving group in the context of Grx3, possibly due to destabilization of the selenolate group in the hydrophobic environment surrounding residue 14.” (55). Alternatively, this may be an additional evidence of the “seleno effect” described for TR. If the sulfhydryl of Cys14 becomes protonated by a general acid during the reaction mechanism, than substitution of the sulfur atom with selenium would slow the rate because of its lower basicity compared to sulfur (34).
How Do Cys-Orthologs Chemically Compensate for the Absence of Sec?
All three Sec-containing enzyme families discussed in this review have Cys-containing orthologs and paralogs and these Cys-homologs catalyze their cognate reactions with comparable efficiency. We have discussed several arguments that support the idea that selenium is intrinsically chemically more reactive as a nucleophile, an electrophile, and a leaving group compared to sulfur, in solution. Enzymatic reactions can be quite remarkably different depending on the environmental properties, and several studies (10, 29, 71) described the significant influence of the protein body and pH on selenoprotein function. With regard to DmTR, we discussed the example of a Cys-containing ortholog of a selenoprotein, whose activity approaches that of the Se-containing enzyme (29). Which of the three chemical properties discussed above would be increased? Would any of the three be decreased?
A common belief in the field is that the way Cys-orthologs of Sec-containing enzymes compensate for the absence of Sec is by increasing the nucleophilic character of the sulfur atom of Cys (supported by the study of Dawson and coworkers discussed above) (2, 29). A downside of this view is that by increasing its nucleophilicity, these enzymes would be expected to be oxidized as rapidly as Sec-enzymes by oxidants such as H2O2; then in order to be functional, the enzyme has to be rescued, for example, by a nearby reduced thiol. Therefore, by increasing the nucleophilicity of their catalytic Cys these enzymes would experience an increase in peroxidase activity. However, this has not been observed thus far. Moreover, unpublished studies show a lack of peroxidase activity at least in the case of Cys-containing orthologs of TR (Hondal, unpublished results).
Thus, if the nucleophilicity of Cys in these orthologs is to be increased, then it seems logical that the electrophilicity of the sulfur atom in the formed sulfenic acid residue should also be increased to prevent overoxidation and inactivation of the Cys residue. However, these Cys-orthologs do not show peroxidase activity because sulfur lacks the chemical features of selenium–high nucleophilicity of the reduced combined with high electrophilicity of its oxidized form that imbue selenium with intrinsic peroxidase activity. This property has experimental support; the substitution of Sec in place of Ser in subtilisin converted the enzyme into a peroxidase (89). The substitution of Sec in place of Cys in glutaredoxin also conferred the mutant enzyme with peroxidase activity (12).
From a certain perspective, it might seem advantageous for Cys-containing orthologs to actually have less nucleophilic character than their Sec-containing counterparts to prevent irreversible inactivation. Perhaps, the enzyme could compensate for the lack of nucleophilicity of the Cys by increasing chemical reactivity of the substrate. For example, if the substrate for a Cys-containing ortholog was a protein disulfide bond, then the latter could be made more reactive, for example, by increasing its electrophilicity, introducing additional strain on the disulfide bond within the context of the protein microenvironment.
An alternative strategy could be to have a weakly reactive Cys (with high pKa, low exposure, etc.), when the enzyme is in the resting state; however, once the substrate enters the enzymatic cleft, the approach is to “activate” the catalytic Cys, for example, by perturbing the thiol and lowering its pKa, as it has been shown for bacterial MsrBs (57).
Other Selenoenzymes
The selenoenzymes discussed here all use selenium in thiol/disulfide-like exchange reactions, but this is not the case for every selenoenzyme characterized to date, for example, formate dehydrogenase and hydrogenase. In these enzymes, Se binds a cofactor (a molybdenum or nickel atom), and the exact role of selenium is still unclear, although it has been shown to be necessary for catalysis (77). Two other selenoenzymes, glycine reductase and proline reductase also use selenium in thiol/disulfide-like exchange reactions. In these enzymes, the Sec residue is involved in forming a selenium-carbon bond as an intermediate that is then resolved by a thiol/disulfide exchange reaction. A possible chemical advantage of using selenium in these reactions is the larger atomic size of selenium and its more loosely held outer electrons relative to sulfur. These properties make the selenolate more nucleophilic toward carbon, but also results in a weak selenium-carbon bond that would be easier to resolve than the corresponding sulfur-carbon bond. The outer electrons of selenium can thus be described as “easy in, easy out” relative to sulfur (HJ Reich, personal communication).
Iodothyronine deiodinases catalyze the reduction of L-thyroxine to 3,5,3′-triiodothyronine or related deiodination reactions (83), forming an enzyme-selenium-iodine intermediates during the reaction cycle that must be converted back to the enzyme-selenolate form. The intermediate may undergo a reduction involving a thiol/disulfide-like exchange reaction, but as of yet the physiological reductant has not been identified. For a good review of the role of selenium in the enzymes discussed in this last section please see citation (54).
Concluding Remarks
For the enzymes discussed in this review, selenium may have been selected by Nature for the advantage it gives to an enzyme to accelerate a thiol/disulfide-like exchange reaction as well as provide the evolutionary selection pressure to maintain the complex insertion system required to decode the UGA codon as a sense codon for Sec (49). Finally, in all functionally characterized selenoenzymes, Sec is used in the active sites and serves a redox function. Thus, whether it is used because of nucleophilicity, electrophilicity, leaving group ability, pKa, redox potential, a combination of these factors, or some other properties, ultimately it is the biological thiol oxidoreductase-like catalysis that benefits from the use of Sec.
Abbreviations Used
- Cys
cysteine
- CysIC
interchange cysteine
- Cys-SOH
cysteine sulfenic acid
- Cys-SO2−
cysteine-sulfinic acid
- Cys-SO3−
cysteine-sulfonic acid
- DmTR
Drosophila melanogaster thioredoxin reductase
- DTNB
5,5′ dithio-bis(2-nitrobenzoic acid)
- DTT
dithiothreiotol
- Gpx
glutathione peroxidase
- GSH
glutathione reduced
- GSSG
glutathione oxidized
- MetSO
methionine sulfoxide
- Msr
methionine sulfoxide reductase
- NMR
nuclear magnetic resonance
- Prx
peroxiredoxin
- S
sulfur
- SC
sulfur center atom
- SLg
sulfur leaving group
- SNuc
sulfur nucleophile
- Se
selenium
- SN2
nucleophilic displacement reaction type 2
- Sec
selenocysteine
- U
selenocysteine
- Sec-SeOH
selenocysteine selenenic acid
- Sec-SeO2−
selenocysteine seleninic acid
- sGrx3
synthetic glutaredoxin3
- TNB
thionitrobenzoate anion
- TR
thioredoxin reductase
- Trx
thioredoxin
Acknowledgments
The authors wish to thank Professor HJ Reich of the University of Wisconsin–Madison for his devotion to the field of selenium chemistry and for many informal discussions about the chemistry of sulfur and selenium. These studies were supported by National Institutes of Health Grants GM094172 to R.J.H. and GM065204 and GM061603 to V.N.G.
References
- 1.Antoine M. Boschi-Muller S. Branlant G. Kinetic characterization of the chemical steps involved in the catalytic mechanism of methionine sulfoxide reductase A from Neisseria meningitides. J Biol Chem. 2003;278:45352–45357. doi: 10.1074/jbc.M307471200. [DOI] [PubMed] [Google Scholar]
- 2.Arnér ES. Selenoproteins–What unique properties can arise with selenocysteine in place of cysteine. Exp Cell Res. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 3.Assmann A. Briviba K. Sies H. Reduction of methionine selenoxide to selenomethionine by glutathione. Arch Biochem Biophys. 1998;349:201–203. doi: 10.1006/abbi.1997.0462. [DOI] [PubMed] [Google Scholar]
- 4.Atkins JF. Gesteland RF. The twenty-first amino acid. Nature. 2000;407:463–465. doi: 10.1038/35035189. [DOI] [PubMed] [Google Scholar]
- 5.Bachrach SM. Demoin DW. Luk M. Miller JV., Jr. Nucleophilic attack at selenium in diselenides and selenosulfides: a computational study. J Phys Chem A. 2004;108:4040–4046. [Google Scholar]
- 6.Birringer M. Pilawa P. Flohe F. Trends in selenium biochemistry. Nat Prod Rep. 2002;19:693–718. doi: 10.1039/b205802m. [DOI] [PubMed] [Google Scholar]
- 7.Böck A. Forchhammer K. Heider J. Leinfelder W. Sawers G. Veprek B. Zinoni F. Selenocysteine: the 21st amino acid. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 8.Boschi-Muller S. Olry A. Antoine M. Branlant G. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim Biophys Acta. 2005;1703:231–238. doi: 10.1016/j.bbapap.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 9.Brot N. Weissbach L. Werth J. Weisbach H. Enzymatic reduction of protein-bound methionine sulfoxide. Proc Natl Acad Sci U S A. 1981;78:2155–2158. doi: 10.1073/pnas.78.4.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardey B. Enescu M. Selenocysteine versus cysteine reactivity: a theoretical study of their oxidation by hydrogen peroxide. J Phys Chem A. 2007;111:673–638. doi: 10.1021/jp0658445. [DOI] [PubMed] [Google Scholar]
- 11.Carugo O. Cemazar M. Sahariev S. Hudaky I. Gaspari Z. Perczel A. Pongor S. Vicinal disulfide turns. Protein Eng. 2003;16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- 12.Casi G. Roelfes G. Hilvert D. Selenoglutaredoxin as a glutathione peroxidase mimic. Chem Bio Chem. 2008;9:1623–1631. doi: 10.1002/cbic.200700745. [DOI] [PubMed] [Google Scholar]
- 13.Cho C-S. Lee S. Lee GT. Woo HA. Choi E-J. Rhee SG. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid Redox Signal. 2010;12:1235–1245. doi: 10.1089/ars.2009.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conant JB. Kirner WR. Hussey RE. The relation between the structure of organic halides and the speeds of their reaction with inorganic iodides III: the influence of unsaturated groups. J Am Chem Soc. 1925;47:488–501. [Google Scholar]
- 15.Danehy JP. Noel CJ. The relative nucleophilic character of several mercaptans toward ethylene oxide. J Am Chem Soc. 1960;82:2511–2515. [Google Scholar]
- 16.Danehy JP. Elia VJ. Lavelle CJ. The alkaline decomposition of organic disulfides. IV. A limitation on the use of Ellman's Reagent, 2,2′-Dinitro-5,5′-dithiodibenzoic acid. J Org Chem. 1971;36:1003–1005. [Google Scholar]
- 17.Eckenroth BE. Harris K. Turanov AA. Gladyshev VN. Raines RT. Hondal RJ. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eckenroth BE. Lacey BM. Lothrop AP. Harris KM. Hondal RJ. Investigation of the C-terminal redox center of high Mr thioredoxin reductases by protein engineering and semisynthesis. Biochemistry. 2007;46:9472–9483. doi: 10.1021/bi7004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckenroth BE. Rould MA. Hondal RJ. Everse SJ. Structural and biochemical studies reveal differences in the catalytic mechanisms of mammalian and Drosophila melanogaster thioredoxin reductases. Biochemistry. 2007;46:4694–4705. doi: 10.1021/bi602394p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epp O. Ladenstein R. Wendel A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur J Biochem. 1983;133:51–69. doi: 10.1111/j.1432-1033.1983.tb07429.x. [DOI] [PubMed] [Google Scholar]
- 21.Flemer SJ. Lacey BM. Hondal RJ. Synthesis of peptide substrates for mammalian thioredoxin reductase. J Pept Sci. 2008;14:637–47. doi: 10.1002/psc.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritz-Wolf K. Kehr S. Stumpf M. Rahlfs S. Becker K. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat Commun. 2011;2:383. doi: 10.1038/ncomms1382. [DOI] [PubMed] [Google Scholar]
- 23.Gascoigne IM. Radda GK. The chemistry of flavins and flavoproteins. 3. The reaction of dihydrolipoic acid with flavins. Biochim Biophys Acta. 1967;131:498–507. doi: 10.1016/0005-2728(67)90009-6. [DOI] [PubMed] [Google Scholar]
- 24.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 25.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert HF. Thiol/disulfide exchange and redox potentials of proteins. In: Lenaz G, editor; Milazzo G., editor. Bioelectrochemistry of Biomacromolecules. Basel, Switzerland: Birhauser; 1997. pp. 256–324. [Google Scholar]
- 27.Gladyshev VN. Jeang KT. Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc Natl Acad Sci U S A. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gladyshev VN. Khangulov SV. Stadtman TC. Nicotinic acid hydroxylase from Clostridium barkeri: electron paramagnetic resonance studies show that selenium is coordinated with molybdenum in the catalytically active selenium-dependent enzyme. Proc Natl Acad Sci U S A. 1994;91:232–236. doi: 10.1073/pnas.91.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gromer S. Johansson L. Bauer H. Arscott LD. Rauch S. Ballou DP. Williams CH., Jr. Schirmer RH. Arnér ES. Active sites of thioredoxin reductases: why selenoproteins? Proc Natl Acad Sci U S A. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herschlag D. Ribonuclease revisited: catalysis via the classical general acid-base mechanism or a triester-like mechanism? J Am Chem Soc. 1994;116:11631–11635. [Google Scholar]
- 31.Hill KE. Lloyd RS. Yang JG. Read R. Burk RF. The cDNA for rat selenoprotein P contains 10 TGA codons in the open reading frame. J Biol Chem. 1991;266:10050–10053. [PubMed] [Google Scholar]
- 32.Hondal RJ. Incorporation of selenocysteine into proteins using peptide ligation. Prot Pept Lett. 2005;12:757–764. doi: 10.2174/0929866054864319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hondal RJ. Nilsson BL. Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. J Am Chem Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 34.Hondal RJ. Ruggles EL. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2011;41:73–89. doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang J. Li C. Nolan SP. Petersen JL. Solution calorimetric investigation of oxidative addition of HEAr (E=O, S, Se; Ar=C6H4×, X=CH3, H, Cl, NO2 to (PMe3)4Ru(C2H4): relationship between HEAr acidity and enthalpy of reaction. Organometallics. 1998;17:3516–3521. [Google Scholar]
- 36.Huber RE. Criddle RS. Comparison of the chemical properties of selenocysteine and selenocystine and their sulfur analogs. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- 37.Hudaky I. Gaspari Z. Carugo O. Cemazar M. Pongor S. Perczel A. Vicinal disulfide bridge conformers by experimental methods and by ab initio and DFT molecular computations. Proteins. 2004;55:152–168. doi: 10.1002/prot.10581. [DOI] [PubMed] [Google Scholar]
- 38.Jacob C. Giles GI. Giles NM. Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 39.Kanzok SM. Fechner A. Bauer H. Ulschmid JK. Muller HM. Botella-Munoz J. Schneuwly S. Schirmer R. Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–646. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- 40.Kim HY. Gladyshev VN. Methionine sulfoxide reduction in mammals: characterization of methionine-R-sulfoxide reductases. Mol Biol Cell. 2004;15:1055–1064. doi: 10.1091/mbc.E03-08-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim HY. Gladyshev VN. Different catalytic mechanisms in mammalian selenocysteine-and cysteine-containing methionine-R-sulfoxide reductases. PloS Biology. 2005;3:e375. doi: 10.1371/journal.pbio.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim HY. Fomenko DE. Yoon YE. Gladyshev VN. Catalytic advantages provided by selenocysteine in methionine-S-sulfoxide reductases. Biochemistry. 2006;45:13697–13704. doi: 10.1021/bi0611614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krause RJ. Elfarra AA. Reduction of L-methionine selenoxide to seleno-L-methione by endogenous thiols, ascorbic acid, or methimazole. Biochem Pharmacol. 2009;77:134–140. doi: 10.1016/j.bcp.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kravchuk AV. Zhao L. Kubiak RJ. Bruzik KS. Tsai M-D. Mechanism of phosphatidylinositol-specific phospholipase C: origin of unusually high nonbridging thio effects. Biochemistry. 2001;40:5433–5439. doi: 10.1021/bi002372q. [DOI] [PubMed] [Google Scholar]
- 45.Kreevoy MM. Harper ET. Duvall RE. Wilgus HS III. Ditsch LT. Inductive effects on the acid dissociation constants of mercaptans. J Am Chem Soc. 1960;82:4899–5902. [Google Scholar]
- 46.Kryukov GV. Kumar RA. Koc A. Sun Z. Gladyshev VN. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc Natl Acad Sci U S A. 2002;99:4245–4250. doi: 10.1073/pnas.072603099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacey BM. Flemer SJ. Eckenroth BE. Hondal RJ. Selenium in thioredoxin reductase: a mechanistic perspective. Biochemistry. 2008;47:12810–12821. doi: 10.1021/bi800951f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee BC. Lobanov AV. Marino SM. Kaya A. Seravalli J. Hatfield DL. Gladyshev VN. A 4-selenocysteine, 2-selenocysteine insertion sequence (SECIS) element methionine sulfoxide reductase from Metridium senile reveals a non-catalytic function of selenocysteines. J Biol Chem. 2011;286:18747–18755. doi: 10.1074/jbc.M111.229807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leinfelder W. Forchhammer K. Zinoni F. Sawers G. Mandrand-Berthelot MA. Böck A. Escherichia coli genes whose products are involved in selenium metabolism. J Bacteriol. 1988;170:540–546. doi: 10.1128/jb.170.2.540-546.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lothrop AP. Ruggles EL. Hondal RJ. No selenium required: reactions catalyzed by mammalian thioredoxin reductase that are independent of a selenocysteine residue. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma S. Caprioli RM. Hill KE. Burk RF. Loss of selenium from selenoproteins: conversion of selenocysteine to dehydroalanine in vitro. J Am Soc Mass Spectrom. 2003;14:593–600. doi: 10.1016/S1044-0305(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 52.Manta B. Hugo M. Ortiz C. Ferrer-Sueta G. Trujilo M. Denicola A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys. 2009;484:146–154. doi: 10.1016/j.abb.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 53.Mehdi M. Morgenstern D. King GF. Alewood PF. Muttenthaler M. Site-specific pKa determination of selenocysteine residues in selenovasopressin by using 77Se NMR spectroscopy. Angew Chem Int Ed. 2011;50:11952–11955. doi: 10.1002/anie.201104169. [DOI] [PubMed] [Google Scholar]
- 54.Metanis N. Beld J. Hilvert D. Patai's Chemistry of Functional Groups. Hoboken, NJ: John Wiley & Sons; 2011. The chemistry of selenocysteine; pp. 1–73. [Google Scholar]
- 55.Metanis N. Keinan E. Dawson PE. Synthetic seleno-glutaredoxin 3 analogues are highly reducing oxidoreductases with enhanced catalytic efficiency. J Am Chem Soc. 2006;128:16684–16691. doi: 10.1021/ja0661414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mugesh G. Sing HB. Synthetic organoselenium compounds as antioxidants: glutathione peroxidase activity. Chem Soc Rev. 2000;29:347–357. [Google Scholar]
- 57.Neiers F. Sonkaria S. Olry A. Boschi-Muller S. Branlant G. Characterization of the amino acids from Neisseria meningitidis methionine sulfoxide reductase B involved in the chemical catalysis and substrate specificity of the reductase step. J Biol Chem. 2007;282:32397–32405. doi: 10.1074/jbc.M704730200. [DOI] [PubMed] [Google Scholar]
- 58.Olry A. Boschi-Muller S. Branlant G. Kinetic characterization of the catalytic mechanism of methionine sulfoxide reductase B from Neisseria meningitides. Biochemistry. 2004;43:11616–11622. doi: 10.1021/bi049306z. [DOI] [PubMed] [Google Scholar]
- 59.Pearson RG. Songstad J. Application of the principle of hard and soft acids and bases to organic chemistry. J Am Chem Soc. 1967;89:1827–1836. [Google Scholar]
- 60.Pearson RG. Songstad J. Nucleophilic reactivity constants toward methyl iodide and trans-[Pt(py)2Cl2] J Am Chem Soc. 1968;90:319–326. [Google Scholar]
- 61.Pigeolet E. Corbisier P. Houbion A. Lambert D. Michiels C. Raes M. Zachary MD. Remacle J. Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech Ageing Dev. 1990;51:283–297. doi: 10.1016/0047-6374(90)90078-t. [DOI] [PubMed] [Google Scholar]
- 62.Pleasants JC. Guo W. Rabenstein DL. A comparative study of the kinetics of selenol/diselenide and thiol/disulfide exchange reactions. J Am Chem Soc. 1989;111:6553–6558. [Google Scholar]
- 63.Poole LB. The catalytic mechanism of peroxiredoxins. Subcell Biochem. 2007;44:61–81. doi: 10.1007/978-1-4020-6051-9_4. [DOI] [PubMed] [Google Scholar]
- 64.Rabenstein DR. Scott TM. Geo W. Nuclear magnetic resonance study of the kinetics of the penicillamine/bis(penicillamine) selenide symmetrical exchange reaction. J Org Chem. 1991;56:4176–4181. [Google Scholar]
- 65.Rackham O. Shearwood AM. Thyer R. McNamara E. Davies SM. Callus BA. Miranda-Vizuete A. Berners-Price SJ. Cheng Q. Arnér ES. Filipovska A. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: implications for development of specific inhibitors. Free Radic Biol Med. 2011;50:689–699. doi: 10.1016/j.freeradbiomed.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 66.Reich HJ. Jasperse CP. Organoselenium chemistry: redox chemistry of selenocysteine model systems. J Am Chem Soc. 1987;109:5549–5551. [Google Scholar]
- 67.Rocher C. Lalanne JL. Chaudière J. Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur J Biochem. 1992;205:955–960. doi: 10.1111/j.1432-1033.1992.tb16862.x. [DOI] [PubMed] [Google Scholar]
- 68.Rotruck JT. Hoekstra WG. Pope AL. Ganther HE. Swanson A. Hafemann D. Relationship of selenium to GSH peroxidase. Fed Proc. 1972;31:691–694. [Google Scholar]
- 69.Ruggles EL. Snider GW. Hondal RJ. Chemical basis for the use of selenocysteine. In: Hatfield DL, editor; Berry MJ, editor; Gladyshev VN, editor. Selenium: Its Molecular Biology and Role in Human Health. 3rd. New York: Springer; 2012. pp. 73–83. [Google Scholar]
- 70.Sandman KE. Benner JS. Noren CJ. Phage display of selenopeptides. J Am Chem Soc. 2000;122:960–961. [Google Scholar]
- 71.Schneider A. Brandt W. Wessjohann LA. Influence of pH and flanking serine on the redox potential of S-S and S-Se bridges of Cys-Cys and Cys-Sec peptides. Biol Chem. 2007;388:1099–10101. doi: 10.1515/BC.2007.114. [DOI] [PubMed] [Google Scholar]
- 72.Sing R. Whitesides GM. Thiol-disulfide interchange. In: Patai S, editor; Rappaport Z, editor. Supplement S: The Chemistry of Sulfur-Containing Functional Groups. Hoboken, NJ: John Wiley & Sons; 1993. pp. 633–658. [Google Scholar]
- 73.Singh R. Kats L. Catalysis of reduction of disulfide by selenol. Anal Biochem. 1995;232:86–91. doi: 10.1006/abio.1995.9956. [DOI] [PubMed] [Google Scholar]
- 74.Singh R. Whitesides GM. Comparison of rate constants for thiolate-disulfide interchange in water and polar aprotic solvents using dynamic 1H NMR line shape analysis. J Am Chem Soc. 1990;112:1190–1197. [Google Scholar]
- 75.Singh R. Whitesides GM. Degenerate intermolecular thiolate-disulfide interchange involving cyclic five-membered disulfides is faster by ∼103 than that involving six- or seven-membered disulfides. J Am Chem Soc. 1990;112:6304–6309. [Google Scholar]
- 76.Snider G. Grout L. Ruggles EL. Hondal RJ. Methaneseleninic acid is a substrate for truncated thioredoxin reductase: implications for the catalytic mechanism and redox signaling. Biochemistry. 2010;49:10329–10338. doi: 10.1021/bi101130t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soboh B. Pinske C. Kuhns M. Waclawek M. Ihling C. Trchounian K. Trchounian A. Sinz A. Sawers G. The respiratory molybdo-selenoprotein formate dehydrogenases of Escherichia coli have hydrogen: benzyl viologen oxidoreductase activity. BMC Microbiol. 2011;11:173. doi: 10.1186/1471-2180-11-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stadtman TC. Selenocysteine. Ann Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 79.Steinmann D. Nauser T. Koppenol WH. Selenium and sulfur in exchange reactions: a comparative study. J Org Chem. 2010;75:6696–6699. doi: 10.1021/jo1011569. [DOI] [PubMed] [Google Scholar]
- 80.Streitwieser A. Jr. Solvolytic displacement reactions at saturated carbon atoms. Chem Rev. 1956;56:571–752. [Google Scholar]
- 81.Suryo RA. Davies MJ. Catalytic activity of selenomethionine in removing amino acid, peptide, and protein hydroperoxides. Free Radic Biol Med. 2011;51:2289–2299. doi: 10.1016/j.freeradbiomed.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 82.Tamura T. Stadtman TC. A new selenoprotein from human lung adenocarcinoma cells: purification, properties, and thioredoxin reductase activity. Proc Natl Acad Sci U S A. 1996;93:1006–1011. doi: 10.1073/pnas.93.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tata JR. Enzymic deiodination of L-thyroxine and 3,5,3′-triiodothyronine; intracellular localization of deiodinase in rat brain and skeletal muscle. Biochim Biophys Acta. 1958;28:801–811. doi: 10.1016/0006-3002(58)90433-5. [DOI] [PubMed] [Google Scholar]
- 84.Toppo S. Flohe L. Ursini F. Vanin S. Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 85.Tosatto SC. Bosello V. Fogolari F. Mauri P. Roveri A. Toppo S. Flohé L. Ursini F. Maiorino M. The catalytic site of glutathione peroxidases. Antioxid Redox Signal. 2008;10:1515–1526. doi: 10.1089/ars.2008.2055. [DOI] [PubMed] [Google Scholar]
- 86.Vendeland SC. Beilstein MA. Yeh JY. Ream W. Whanger PD. Rat skeletal muscle selenoprotein W: cDNA clone and mRNA modulation by dietary selenium. Proc Natl Acad Sci U S A. 1995;92:8749–8753. doi: 10.1073/pnas.92.19.8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wessjohann LA. Schneider A. Abbas M. Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- 88.Wittwer AJ. Tsai L. Ching WM. Stadtman TC. Identification and synthesis of a naturally occurring selenonucleoside in bacterial tRNAs: 5-[(methylamino)methyl]-2-selenouridine. Proc Natl Acad Sci U S A. 1984;81:57–60. doi: 10.1021/bi00315a021. [DOI] [PubMed] [Google Scholar]
- 89.Wu Z.-P. Hilvert D. Selenosubtilisin as a glutathione peroxidase mimic. J Am Chem Soc. 1990;112:5647–5648. [Google Scholar]
- 90.Xiao R. Lundstrom-Ljung J. Holmgren A. Gilbert HF. Catalysis of thiol/disulfide exchange: glutaredoxin 1 and protein disulfide isomerase use different mechanisms to enhance oxidase and reductase activities. J Biol Chem. 2005;280:21099–21106. doi: 10.1074/jbc.M411476200. [DOI] [PubMed] [Google Scholar]
- 91.Zhong L. Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J Biol Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]