

The biophysical technique of in planta FRET-FLIM provides evidence for the existence of homo- and heteromeric N-glycan processing enzyme complexes, which are predominantly formed between cis- and medial-Golgi enzymes.
Abstract
N-Glycan processing is one of the most important cellular protein modifications in plants and as such is essential for plant development and defense mechanisms. The accuracy of Golgi-located processing steps is governed by the strict intra-Golgi localization of sequentially acting glycosidases and glycosyltransferases. Their differential distribution goes hand in hand with the compartmentalization of the Golgi stack into cis-, medial-, and trans-cisternae, which separate early from late processing steps. The mechanisms that direct differential enzyme concentration are still unknown, but the formation of multienzyme complexes is considered a feasible Golgi protein localization strategy. In this study, we used two-photon excitation-Förster resonance energy transfer-fluorescence lifetime imaging microscopy to determine the interaction of N-glycan processing enzymes with differential intra-Golgi locations. Following the coexpression of fluorescent protein-tagged amino-terminal Golgi-targeting sequences (cytoplasmic-transmembrane-stem [CTS] region) of enzyme pairs in leaves of tobacco (Nicotiana spp.), we observed that all tested cis- and medial-Golgi enzymes, namely Arabidopsis (Arabidopsis thaliana) Golgi α-mannosidase I, Nicotiana tabacum β1,2-N-acetylglucosaminyltransferase I, Arabidopsis Golgi α-mannosidase II (GMII), and Arabidopsis β1,2-xylosyltransferase, form homodimers and heterodimers, whereas among the late-acting enzymes Arabidopsis β1,3-galactosyltransferase1 (GALT1), Arabidopsis α1,4-fucosyltransferase, and Rattus norvegicus α2,6-sialyltransferase (a nonplant Golgi marker), only GALT1 and medial-Golgi GMII were found to form a heterodimer. Furthermore, the efficiency of energy transfer indicating the formation of interactions decreased considerably in a cis-to-trans fashion. The comparative fluorescence lifetime imaging of several full-length cis- and medial-Golgi enzymes and their respective catalytic domain-deleted CTS clones further suggested that the formation of protein-protein interactions can occur through their amino-terminal CTS region.
The Golgi apparatus is a multifaceted, multitasking organelle that is pivotal to the life of the cell. Protein and lipid modifications, sorting of molecules, as well as the biosynthesis of cell wall polysaccharides all take place in the small stacks of flattened cisternae that make up the Golgi bodies, constituting the Golgi apparatus in plant cells. Among the various posttranslational modification reactions on proteins, the biosynthesis and processing of protein-bound N-linked oligosaccharides (N-glycans) is the most common. N-Glycans play a crucial role in protein folding, endoplasmic reticulum (ER) quality control (Liu and Howell, 2010), biotic (Saijo, 2010) and abiotic (Koiwa et al., 2003; Kang et al., 2008) stress responses, and are considered essential for the physicochemical properties and biological functions of glycoproteins. Consequently, the slightest alterations during N-glycan processing can drastically affect a protein’s folding, stability, and biological activity. Golgi-mediated N-glycan processing steps are catalyzed by numerous glycosidases and glycosyltransferases that follow a nonuniform subcompartment-specific distribution pattern along the cis-to-trans axis of the Golgi stack in the order in which they function in the processing pathway (Fig. 1A; Schoberer and Strasser, 2011). The subcompartmentalization of the Golgi stack into cis-, medial-, and trans-cisternae creates a polar biochemical and morphological gradient in the cis-to-trans direction, which allows a functional specialization of the Golgi. Enzymes catalyzing early processing steps concentrate in the cis-half of the Golgi stack, whereas enzymes acting later in the pathway peak in the trans-half.
Figure 1.

Overview of fluorescent protein fusion constructs used for FRET-FLIM. A, Schematic representation of the N-glycan processing pathway in the plant Golgi apparatus and the enzymes involved (for enzyme names, see Table I). Enzymes studied here are highlighted in black. B, Schematic representation of the domain architecture of expressed recombinant proteins, all of them displaying a type II membrane topology (the N terminus on the cytoplasmic side and the C terminus on the luminal side of the Golgi membrane). The CTS regions and the full-length sequences (where available) were C-terminally fused to GFP and/or mRFP, respectively, by insertion into binary plant expression vectors. Details on plasmid construction can be found in “Materials and Methods.” C, Cytoplasmic tail; CD, catalytic domain; S, luminal stem region; T, transmembrane domain.
All Golgi-resident plant N-glycan processing enzymes are typical so-called type II membrane proteins with an N-terminal region comprising a short cytoplasmic tail, a single transmembrane domain, and a luminal stem region, together called the cytoplasmic-transmembrane-stem (CTS) region, which orients the C-terminal catalytic domain into the Golgi lumen (Fig. 1B). The CTS region not only contains the information necessary for enzyme targeting to the Golgi but also directs the nonuniform, overlapping distribution of glycosidases and glycosyltransferases across the distinct Golgi cisternae (or subcompartments; Saint-Jore-Dupas et al., 2006; Schoberer et al., 2009, 2010; Schoberer and Strasser, 2011). The signals or mechanisms that drive the subcompartment-specific concentration of this important class of Golgi enzymes are still widely unknown. It is also intriguing how N-glycan processing enzymes remain specifically concentrated within Golgi membranes even in the presence of a continuous bidirectional flow of membrane and proteins into and out of the Golgi. Moreover, most of the processing enzymes are highly dynamic themselves, as they continuously cycle between the Golgi and the ER.
One possible mechanism responsible for intra-Golgi concentration (as described for mammalian Golgi) is the self-assembly of Golgi-resident glycosylation enzymes into complexes, which are excluded from forward transport to downstream compartments as described in the “protein aggregation model” (Machamer, 1991) or the “kin recognition model” (Nilsson et al., 1993). In fact, there is compelling in vitro and in vivo evidence on the formation of oligomers by mammalian and yeast glycosyltransferases, respectively, from each major glycosylation category, namely, for proteoglycans, glycoproteins, and glycolipids (Schachter, 1986; Nilsson et al., 1994, 1996; McCormick et al., 2000; Giraudo et al., 2001; Pinhal et al., 2001; Qian et al., 2001; Stolz and Munro, 2002; Young, 2004; Hassinen et al., 2011; Ferrari et al., 2012). Earlier studies mainly examined complex formation through in vitro coimmunoprecipitation (co-IP) assays following complete disruption of the cell, whereas in recent years, the advent of fluorescent protein technology and that of laser-based microscopy techniques utilizing bimolecular fluorescence complementation or Förster resonance energy transfer (FRET) have proven invaluable for the observation of protein-protein interactions in vivo and in real time.
There is emerging evidence from co-IP and bimolecular fluorescence complementation experiments for the formation of glycosyltransferase complexes involved in various aspects of plant cell wall biosynthesis, such as the biosynthesis of xylan (Zeng et al., 2010), homogalacturonan (Atmodjo et al., 2011), pectic arabinan (Harholt et al., 2012), and xyloglycan (Chou et al., 2012). To date, it is not known whether enzymes involved in N-glycan processing assemble into similar complexes. Protein-protein interaction studies on mammalian N-glycan processing enzymes have indicated the presence of homomeric and heteromeric enzyme complexes that potentially lead to their retention in Golgi membranes and enhance their activity (Nilsson et al., 1996; Rivinoja et al., 2009; Hassinen et al., 2010, 2011).
To address the question of whether the same principle applies to enzymes from the plant N-glycan processing pathway, we have tested the properties of several glycosidases and glycosyltransferases with distinct intra-Golgi locations to form protein-protein interactions when expressed transiently in tobacco (Nicotiana spp.) leaves. To observe interactions in living cells and in real time, we employed time-resolved FRET-fluorescence lifetime imaging (FLIM), which is based on energy transfer from a fluorophore in an excited state (i.e. GFP serving as the donor) to another fluorophore (i.e. monomeric red fluorescent protein [mRFP] serving as the acceptor) within a 1- to 10-nm distance. In the FRET-FLIM approach, the information gained using steady-state FRET between interacting proteins is considerably improved by monitoring the excited-state lifetime of the donor, where its quenching is evidence for a direct physical interaction. Although technically demanding, FRET-FLIM is superior to other intensity-based techniques, as it is largely independent from fluorophore concentrations and is also free of interference from spectral cross talk. Two-photon excitation (2P)-FRET-FLIM analysis (Stubbs et al., 2005; Osterrieder et al., 2009; Sparkes et al., 2010) provides several advantages over the single-photon method, including reduced cellular cytotoxicity of the excitation light and reduced photobleaching of the fluorophore. Greater sensitivity of the setup is achieved through reduced sensitivity of the excitation light (greater than 900 nm) by the photomultiplier tube as a detector. The combination of the sensitive advanced imaging technique of time-correlated single-photon counting and laser scanning techniques results in the FLIM technique. The FRET-FLIM approach has successfully been used to study protein-protein interactions in a variety of animal and plant cells at the organelle level (Bhat et al., 2005; Stubbs et al., 2005; Adjobo-Hermans et al., 2006; Aker et al., 2007; Osterrieder et al., 2009; Sparkes et al., 2010; Crosby et al., 2011; Berendzen et al., 2012).
Here, we present evidence that (1) N-glycan processing enzymes can form in vivo protein-protein interactions in the form of homodimers and heterodimers, (2) the N-terminal CTS regions of several dimerizing cis- and medial-Golgi enzymes participate in physical interactions, and (3) interaction mainly occurs among the CTS domains of cis- and medial-Golgi enzymes and decreases toward the trans-Golgi in a gradient-like fashion.
RESULTS
Two cis/medial-Golgi Enzymes, MNS1 and GnTI, Form in Vitro and in Vivo Interactions
We chose the two cis/medial-Golgi residents Arabidopsis (Arabidopsis thaliana) Golgi α-mannosidase I (MNS1; Liebminger et al., 2009) and Nicotiana tabacum β1,2-N-acetylglucosaminyltransferase I (GnTI; Schoberer et al., 2009) as putative candidate proteins to test whether plant N-glycan processing enzymes are able to form glycosyltransferase complexes. Both enzymes are involved in the processing of oligomannosidic to hybrid N-glycans and hence act early in the N-glycan processing pathway (Fig. 1A; for naming, see Table I). To this end, GFP and mRFP fused to full-length clones of MNS1, designated as MNS1-G and MNS1-R, respectively, and to GnTI, designated as GnTI-G and GnTI-R, respectively, were used (for a schematic representation of the constructs, see Fig. 1B). These fusion proteins were transiently expressed in tobacco leaves under the control of the 35S promoter. We opted for a transient expression system to circumvent eventual problems with endogenous promoters that might not express ubiquitously and also might not produce expression levels high enough to produce fluorescent signals to determine the subcellular locations of proteins in vivo. Furthermore, the use of a transient expression system has the advantage that a large number of protein pairs can be tested within a realistic time frame. We deliberately placed fluorescent tags on the C terminus of all enzymes to prevent interference with N-terminal Golgi-targeting signals. Furthermore, previous complementation experiments have shown that, for example, GFP fusions to GnTI (Schoberer et al., 2009) or MNS3, an ER-type α-mannosidase I (Liebminger et al., 2009), are fully functional in vivo.
Table I. Overview of Golgi N-glycan processing enzyme constructs used for 2P-FRET-FLIM.
For a full description of the plasmid constructs, see “Materials and Methods” and Supplemental Table S2. FL, Full length.
| Abbreviation | Full Name | Tag | Domains |
|---|---|---|---|
| MNS1 | Arabidopsis Golgi α-mannosidase I | GFP, mRFP | CTS, FL |
| GnTI | N. tabacum β1,2-N-acetylglucosaminyltransferase I | GFP, mRFP | CTS, FL |
| GMII | Arabidopsis Golgi α-mannosidase II | GFP, mRFP | CTS, FL |
| XylT | Arabidopsis β1,2-xylosyltransferase | GFP, mRFP | CTS |
| GALT1 | Arabidopsis β1,3-galactosyltransferase1 | GFP | CTS |
| FUT13 | Arabidopsis α1,4-fucosyltransferase | mRFP | FL |
| ST | R. norvegicus α2,6-sialyltransferase | GFP, mRFP | CTS |
Subcellular Locations of MNS1 and GnTI
Expression levels and efficient targeting of the fusion proteins to the Golgi were confirmed by live-cell confocal imaging. Merged confocal images show considerable colocalization of coexpressed protein pairs MNS1-G/MNS1-R (Fig. 2, A–C), GnTI-G/GnTI-R (Fig. 2, D–F), and GnTI-G/MNS1-R (Fig. 2, G–I) in Golgi bodies of tobacco leaf epidermal cells.
Figure 2.

Golgi localization of full-length cis/medial-Golgi protein pairs in tobacco leaves. Confocal images show representative tobacco leaf epidermal cells coexpressing fluorescent protein-tagged full-length protein pairs MNS1-G (A) and MNS1-R (B), GnTI-G (D) and GnTI-R (E), and GnTI-G (G) and MNS1-R (H). C, F, and I show merges of green (GFP fluorescence) and magenta (mRFP fluorescence) channels. White in the merged images indicates areas of colocalization. Bars = 5 µm.
co-IP of N-Glycan Processing Enzymes
Following the verification of expression and colocation of the protein pairs MNS1-G/MNS1-R, GnTI-G/GnTI-R, and GnTI-G/MNS1-R, potential interactions were tested through co-IP analysis using the GFP-Trap system. Protein pairs were transiently expressed in tobacco leaves, and total protein extracts were used for copurification of fluorescent protein-tagged constructs. Immunoblot analysis of the eluate fractions with anti-mRFP antibodies revealed that both MNS1-R and GnTI-R were present as MNS1 and GnTI homomeric complexes as well as a heteromeric MNS1/GnTI complex (Fig. 3A).
Figure 3.

co-IP assay and in vivo 2P-FRET-FLIM analysis of full-length cis/medial-Golgi protein pairs. Fluorescent protein-tagged full-length protein pairs MNS1/MNS1, GnTI/GnTI, and GnTI/MNS1 were transiently coexpressed in Nicotiana benthamiana leaf epidermal cells and subjected to co-IP (A) or confocal imaging and 2P-FRET-FLIM (B–J). A, Immunoblot analysis of protein extracts (Input = before incubation with GFP-coupled beads) and eluted samples (Bound = fraction eluted from GFP-coupled beads) with anti-GFP and anti-mRFP antibodies. B to D, Cell showing expression of the donor fusion protein MNS1-G alone as a representative, unquenched negative control with an average fluorescence lifetime of 2.2 ± 0.1 ns following 2P-FRET-FLIM. Blue in the color-coded lifetime image (C) and histogram (D) reflects higher excited-state lifetimes (approximately 3 ns) than green (approximately 2 ns). The distribution curve in the histogram depicts the relative occurrence frequency of the lifetimes within the lifetime image. In C, the asterisk indicates a stoma that gives lower lifetimes than the reference Golgi labeled with an arrow and blue crosshair. E to G, Representative cell that shows quenching of the donor lifetime of MNS1-G in the presence of the acceptor MNS1-R (1.9 ± 0.1 ns), indicating interaction. Quenching is also reflected in the green color in the color-coded lifetime image (F) and histogram (G). H to J, Histograms that show a comparison of the average donor fluorescence lifetimes in the absence and presence of the indicated acceptors following 2P-FRET-FLIM. Each column represents the mean lifetime ± sd. FRET-FLIM data were collected from two independent infiltrations (10–12 cells were analyzed in total) and analyzed statistically using a two-tailed Student’s t test (P ≤ 0.0001). A minimum decrease of the average excited-state fluorescence lifetime of the donor molecule by 0.2 ns or percentage FRET efficiency > 8% in the presence of the acceptor molecule was considered relevant to indicate interaction. For the full data set, see Table II. Bars = 20 µm.
In Vivo 2P-FRET-FLIM
To determine whether the interactions found by co-IP also occur in live cells, we employed 2P-FRET-FLIM. Tobacco leaf cells expressing MNS1-G or GnTI-G alone were used as controls to establish the unquenched lifetime of GFP in the context of the fusion proteins (serving as donors). Figure 3, B to G, shows representative confocal and pseudocolored lifetime images of a control cell expressing the donor MNS1-G alone (Fig. 3, B–D) or a cell expressing the donor in the presence of the acceptor MNS1-R (Fig. 3, E–G). We determined an average excited-state fluorescence lifetime of 2.2 ± 0.1 ns for MNS1-G (Fig. 3D) following FLIM measurements in the absence of an acceptor (for the full data set, see Table II). Coexpression of MNS1-G and MNS1-R led to a significant quenching of the donor lifetime to an average of 1.9 ± 0.1 ns (Fig. 3, G and H), which indicates that the fluorophores of the analyzed protein pair were close enough in Golgi membranes to undergo FRET and that MNS1-G likely interacts with MNS1-R. For GnTI-G, the average fluorescence lifetime was 2.2 ± 0.1 ns in the absence of the acceptor, but it decreased to an average of 1.9 ± 0.1 ns in the presence of GnTI-R or MNS1-R (Fig. 3, I and J; Table II), indicating interactions between GnTI-G/GnTI-R and GnTI-G/MNS1-R. Altogether, the established FRET-FLIM data are consistent with the co-IP data, and we can conclude that at least the two tested N-glycan processing enzymes, MNS1 and GnTI, homodimerize and additionally form a GnTI/MNS1 heterodimer. It is worth noting that the excited-state lifetime of GFP-tagged proteins alone in live cells (plants and animal) is around 2.5 ns. Here, this is further reduced due to self-quenching by the homodimerization.
Table II. Fluorescence lifetimes of full-length cis- and medial-Golgi enzyme pairs.
2P-FRET-FLIM results are shown for coexpressed GFP- and mRFP-fused enzyme pairs transiently expressed in live tobacco leaves. A minimum decrease of the average excited-state fluorescence lifetime of the donor molecule by 0.20 ns or E > 8% in the presence of the acceptor molecule was considered relevant to indicate interaction. FRET-FLIM data were collected from two independent infiltrations (10–12 cells in total) and analyzed statistically using a two-tailed Student’s t test. 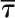 , Average donor lifetime; τD, average donor lifetime without the acceptor; τDA, average donor lifetime in the presence of the acceptor; τD − τDA, calculated difference between τD and τDA; E, percentage FRET efficiency; range τ, range of lifetimes observed for the respective sample; n, number of analyzed Golgi bodies.
, Average donor lifetime; τD, average donor lifetime without the acceptor; τDA, average donor lifetime in the presence of the acceptor; τD − τDA, calculated difference between τD and τDA; E, percentage FRET efficiency; range τ, range of lifetimes observed for the respective sample; n, number of analyzed Golgi bodies.
| Donor | Acceptor |
± sd
|
τD − τDA | E | Range τ | n | P |
|---|---|---|---|---|---|---|---|
| ns | % | ||||||
| MNS1-G | 2.2 ± 0.1 | 1.8–2.4 | 343 | ||||
| MNS1-G | MNS1-R | 1.9 ± 0.1 | 0.23 | 10.67 | 1.7–2.2 | 195 | ≤0.0001 |
| GnTI-G | 2.2 ± 0.1 | 1.9–2.4 | 283 | ||||
| GnTI-G | MNS1-R | 1.9 ± 0.1 | 0.25 | 11.42 | 1.6–2.2 | 342 | ≤0.0001 |
| GnTI-G | 2.2 ± 0.1 | 1.9–2.4 | 283 | ||||
| GnTI-G | GnTI-R | 1.9 ± 0.1 | 0.30 | 13.99 | 1.6–2.1 | 275 | ≤0.0001 |
| GMII-G | 2.3 ± 0.1 | 2.0–2.5 | 229 | ||||
| GMII-G | MNS1-R | 2.2 ± 0.1 | 0.11 | 4.87 | 2.0–2.4 | 219 | ≤0.0001 |
| GMII-G | 2.3 ± 0.1 | 2.0–2.5 | 229 | ||||
| GMII-G | GnTI-R | 2.1 ± 0.1 | 0.19 | 8.16 | 1.8–2.4 | 178 | ≤0.0001 |
| GMII-G | 2.4 ± 0.1 | 2.3–2.6 | 222 | ||||
| GMII-G | GMII-R | 2.4 ± 0.1 | 0.04 | 1.64 | 2.2–2.7 | 175 | ≤0.0001 |
GnTI and MNS1 perform consecutive processing steps in the cis-half of the Golgi stack; hence, a heterodimerization may reflect the formation of a functionally relevant complex. Therefore, we coexpressed GnTI-R and MNS1-R with GFP-labeled Arabidopsis Golgi α-mannosidase II (GMII; Strasser et al., 2006), a medial-Golgi enzyme that operates and resides in close proximity to both GnTI and MNS1 in the Golgi (Fig. 1A; Table I). Confocal imaging confirmed the colocation of GMII-G/MNS1-R (Fig. 4A) and GMII-G/GnTI-R (Fig. 4C) in Golgi stacks. We again used the noninvasive 2P-FRET-FLIM approach to examine protein-protein interactions. The average fluorescence lifetime of the donor GMII-G was 2.3 ± 0.1 ns (control value) in the absence of the acceptor, but it was only marginally reduced to 2.2 ± 0.1 ns upon coexpression with MNS1-R (Fig. 4B; Table II) and hence did not suggest an interaction between the two fusion proteins. In the presence of GnTI-R, the average lifetime of GMII-G was 2.1 ± 0.1 ns (Fig. 4D; Table II). The calculation of the difference between the mean lifetime values for the control and the donor-acceptor combination indicated a lower energy transfer value (0.19 ns), lower than that observed for previously assayed enzyme pairs, which indicates a weaker interaction between GMII and GnTI or less efficient energy transfer due to an unfavorable steric orientation of the fluorophores in the fusion proteins. Unquenched lifetime values were determined for GMII-G in the presence of GMII-R (Fig. 4F; Table II), arguing against a physical interaction.
Figure 4.

2P-FRET-FLIM analysis of the medial-Golgi enzyme GMII coexpressed with the cis/medial-Golgi enzymes MNS1 and GnTI. The GFP-fused full-length enzyme GMII was transiently coexpressed with putative mRFP-tagged interactor(s) in tobacco leaf epidermal cells and subjected to confocal imaging and 2P-FRET-FLIM. A, C, and E, Confocal images of a representative cell showing the colocation of GFP and mRFP full-length enzyme pairs in Golgi stacks of live cells as follows: GMII-G and MNS1-R (A), GMII-G and GnTI-R (C), GMII-G and GMII-R (E). The larger images show merges of the smaller images, which represent GFP fluorescence of the donor in green and mRFP fluorescence of the acceptor in magenta. Colocalization in the merged confocal images appears white. B, D, and F, Histograms showing the donor lifetimes in the absence and presence of the indicated acceptors following 2P-FRET-FLIM. The measurements and analysis were performed as described for Figure 3, H to J. For the full data set, see Table II. Bars = 5 µm.
The CTS Golgi-Targeting Domains Participate in the Formation of Protein-Protein Interactions
Recently, Hassinen et al. (2010, 2011) presented evidence that luminal, catalytic domains of mammalian glycosyltransferases and glycosidases almost exclusively mediate enzyme complex formation. This is not too surprising, since it has been shown that luminal domains play an important role in enzyme concentration in the mammalian Golgi (Colley, 1997; Opat et al., 2001; Fenteany and Colley, 2005). In plants, the situation is quite different. The important information for enzyme concentration in the Golgi is present in the N-terminal Golgi-targeting domains, the so-called CTS region, without any detectable contribution from the luminal catalytic domains (Essl et al., 1999; Nebenführ et al., 1999; Dirnberger et al., 2002; Saint-Jore-Dupas et al., 2006; Strasser et al., 2006, 2007; Schoberer et al., 2009). To determine the role of the CTS domains in establishing protein-protein interactions in plants, we examined live cells expressing GFP- and mRFP-tagged CTS regions of MNS1, GnTI, and GMII (Fig. 1B). The expression and colocation of protein pairs in Golgi stacks were verified by confocal imaging, and the average fluorescence lifetimes of control cells, expressing only the donor, and cells expressing donor-acceptor pairs were determined through subsequent 2P-FRET-FLIM (Fig. 5; Table III).
Figure 5.

Catalytic domain-deleted cis- and medial-Golgi enzymes form homodimers and heterodimers in vivo. The N-terminal CTS regions of enzymes (lacking their catalytic domain) fused to fluorescent proteins were transiently coexpressed in tobacco leaf epidermal cells and subjected to confocal imaging and 2P-FRET-FLIM. The left panels show confocal images of cells coexpressing GnTI-CTS-G and GnTI-CTS-R (A), GnTI-CTS-G and GMII-CTS-R (C), MNS1-CTS-G and MNS1-CTS-R (E), MNS1-CTS-G and GnTI-CTS-R (G), and MNS1-CTS-G and GMII-CTS-R (I). The larger images show merges of the smaller split images representing the green and magenta channels. The right panels (B, D, F, H, and J) show histograms of the average excited-state lifetimes of each donor in the absence and presence of the indicated acceptor following 2P-FRET-FLIM. The measurements and analysis were performed as described for Figure 3, H to J. For the full data set, see Table III. Bars = 5 µm.
Table III. Fluorescence lifetimes of cis- and medial-Golgi enzyme pairs.
Abbreviations are as in Table II.
| Donor | Acceptor |
± sd
|
τD − τDA | E | Range τ | n | P |
|---|---|---|---|---|---|---|---|
| ns | % | ||||||
| GnTI-CTS-G | 2.4 ± 0.1 | 2.3–2.6 | 468 | ||||
| GnTI-CTS-G | GnTI-CTS-R | 2.1 ± 0.1 | 0.34 | 14.17 | 1.8–2.4 | 305 | ≤0.0001 |
| GnTI-CTS-G | 2.4 ± 0.1 | 2.3–2.6 | 468 | ||||
| GnTI-CTS-G | GMII-CTS-R | 2.0 ± 0.1 | 0.37 | 15.23 | 1.8–2.3 | 307 | ≤0.0001 |
| MNS1-CTS-G | 2.5 ± 0.1 | 2.3–2.6 | 567 | ||||
| MNS1-CTS-G | MNS1-CTS-R | 1.9 ± 0.1 | 0.56 | 22.77 | 1.7–2.1 | 268 | ≤0.0001 |
| MNS1-CTS-G | 2.4 ± 0.1 | 2.3–2.6 | 232 | ||||
| MNS1-CTS-G | GnTI-CTS-R | 2.1 ± 0.1 | 0.34 | 13.98 | 1.9–2.3 | 379 | ≤0.0001 |
| MNS1-CTS-G | 2.4 ± 0.1 | 2.3–2.6 | 232 | ||||
| MNS1-CTS-G | GMII-CTS-R | 2.1 ± 0.1 | 0.32 | 13.06 | 1.9–2.3 | 325 | ≤0.0001 |
FLIM analysis showed that the average lifetime of GnTI-CTS-G in the presence of GnTI-CTS-R was 2.1 ± 0.1 ns and hence was significantly lower than the control value of 2.4 ± 0.1 ns (Fig. 5B). Coexpression of GnTI-CTS-G with GMII-CTS-R also resulted in a significant decrease in the average donor lifetime, which was determined to be 2.0 ± 0.1 ns (Fig. 5D). The average lifetime of MNS1-CTS-G alone was 2.4 ± 0.1 ns, but it decreased significantly in the presence of MNS1-CTS-R (1.9 ± 0.1 ns; Fig. 5F), GnTI-CTS-R (2.1 ± 0.1 ns; Fig. 5H), or GMII-CTS-R (2.1 ± 0.1 ns; Fig. 5J). Altogether, quenching of donor lifetimes could be observed for every protein pair expressed (lifetime reductions ranging from 0.32 to 0.56 ns), which suggests that the catalytic domain-deleted protein pairs, like their full-length counterparts, are able to assemble into homomers and heteromers. Only the interaction found between MNS1-CTS-G/GMII-CTS-R is not in agreement with its full-length combination, where the interaction was not detected by 2P-FRET-FLIM. Altogether, our results strongly suggest that the enzymes tested here can interact through their N-terminal CTS regions.
Early-Acting Golgi Enzymes Do Not Interact with Late-Golgi Residents
It is known that N-glycan processing enzymes are distributed across the Golgi stack in a polar gradient, which means that they occur in successive but overlapping Golgi subcompartments. Since FRET is a measure of spatial proximity, we tested whether early- and late-acting enzymes, whose distribution is centered over distinct Golgi subcompartments, are able to physically interact. To this end, we examined the cis/medial-Golgi enzyme GnTI by coexpressing its CTS region with that of the trans-Golgi enzymes Arabidopsis β1,3-galactosyltransferase1 (GALT1) as well as Rattus norvegicus α2,6-sialyltransferase (ST), a nonplant Golgi marker, using 2P-FRET-FLIM in live cells (Fig. 1A; Table I). GALT1 is involved in the formation of Lewis a epitopes on N-glycans, which is a late Golgi event in plants and hence places this enzyme in the trans-Golgi (Strasser et al., 2007). ST-CTS, a sialyltransferase signal anchor sequence from rat, has become one of the most commonly used Golgi markers in many organisms, and in plants it was located to the trans-half of the Golgi by immunoelectron microscopy (Boevink et al., 1998). Confocal images showed that the GFP and mRFP signals from each enzyme pair, namely ST-CTS-G/GnTI-CTS-R (Fig. 6A) or GALT1-CTS-G/GnTI-CTS-R (Fig. 6C), only partially colocated in Golgi bodies, which has been described previously (Schoberer et al., 2010; Schoberer and Strasser, 2011). Following 2P-FRET-FLIM in the presence of GnTI-CTS-R, lifetimes of ST-CTS-G (Fig. 6B) or GALT1-CTS-G (Fig. 6D) were quenched by 0.13 or 0.11 ns (Table IV), which was significantly less than that observed for previously assayed enzyme pairs; hence, a physical association could not be detected.
Figure 6.

Early- and late-Golgi enzymes do not interact. A, Subcellular colocalization (merge) of the trans-Golgi marker ST-CTS-G (green) and the cis/medial-Golgi enzyme GnTI-CTS-R (magenta) in the Golgi. B, Average lifetimes of ST-CTS-G in the absence and presence of GnTI-CTS-R. C, Subcellular colocalization (merge) of GALT1-CTS-G (green) and GnTI-CTS-R (magenta) in the Golgi. D, Average lifetimes of GALT1-CTS-G in the absence and presence of GnTI-CTS-R. The measurements and analysis were performed as described for Figure 3, H to J. For the full data set, see Table IV. Bars = 10 µm.
Table IV. Fluorescence lifetimes of cis- and trans-Golgi enzyme pairs.
Abbreviations are as in Table II.
| Donor | Acceptor |
± sd
|
τD − τDA | E | Range τ | n | P |
|---|---|---|---|---|---|---|---|
| ns | % | ||||||
| ST-CTS-G | 2.8 ± 0.1 | 2.6–2.9 | 421 | ||||
| ST-CTS-G | GnTI-CTS-R | 2.6 ± 0.1 | 0.13 | 4.82 | 2.4–2.8 | 344 | ≤0.0001 |
| GALT1-CTS-G | 2.5 ± 0.1 | 2.3–2.7 | 476 | ||||
| GALT1-CTS-G | GnTI-CTS-R | 2.4 ± 0.1 | 0.11 | 4.31 | 2.1–2.6 | 239 | ≤0.0001 |
The trans-Golgi Marker ST-CTS Does Not Form Detectable Interactions in Plant Cells
In addition to the oligomerization of mammalian cis/medial-Golgi resident N-glycan processing enzymes, complex formation of late-acting Golgi enzymes has been shown using in vitro and in vivo assays (Rivinoja et al., 2009; Hassinen et al., 2010, 2011). For instance, a disulfide-mediated homodimerization of human ST and its rat ortholog, respectively, has been determined in vitro and in vivo (Ma and Colley, 1996; Qian et al., 2001; Rivinoja et al., 2009; Hassinen et al., 2010). In addition, it has been reported that different domains of ST contribute to oligomerization and localization in the Golgi of mammalian cells (Fenteany and Colley, 2005). To assess whether this holds true for plants as well, we coexpressed fluorescent protein-tagged CTS regions of ST with itself and those of GALT1 and the medial-Golgi enzyme Arabidopsis β1,2-xylosyltransferase (XylT; Fig. 1; Table I). XylT transfers a Xyl residue to the core N-glycan structure, and in electron micrographs of plant cells it was seen to accumulate in the medial-Golgi when fused to GFP (Pagny et al., 2003). As expected, all three enzyme pairs, namely ST-CTS-G/ST-CTS-R, GALT1-CTS-G/ST-CTS-R, and XylT-CTS-G/ST-CTS-R, colocated in Golgi stacks (Fig. 7, A, C, and E). Subsequent 2P-FRET-FLIM, however, did not indicate any interactions among them (Fig. 7, B, D, and F; Table V). The enzyme pairs ST/ST (as in Osterrieder et al., 2009) and GALT1/ST did not display significant donor lifetime quenching (less than 0.2-ns lifetime decrease), and almost unquenched lifetime values were measured for XylT/ST, which are known to concentrate in distinct subcompartments of Golgi bodies. These results show that ST-CTS does not form any detectable homomeric or heteromeric interactions in plant cells with any of the plant enzymes coexpressed so far.
Figure 7.

The nonplant trans-Golgi marker ST-CTS does not interact with other plant markers from the trans-Golgi. A and B, Subcellular colocalization (merge) of ST-CTS-G (green) and ST-CTS-R (magenta) in the Golgi (A) and histogram showing the observed average lifetime of the donor in the absence and presence of the acceptor following 2P-FRET-FLIM (B). C and D, Subcellular colocalization (merge) of the trans-Golgi enzyme GALT1-CTS-G (green) and ST-CTS-R (magenta) in the Golgi (C) and the observed average lifetimes (D). E and F, Subcellular colocalization (merge) of the medial-Golgi enzyme XylT-CTS-G (green) and ST-CTS-R (magenta) in the Golgi (E) and the observed average lifetimes (F). The measurements and analysis were performed as described for Figure 3, H to J. For the full data set, see Table V. Bars = 10 µm.
Table V. Fluorescence lifetimes of plant enzymes coexpressed with the nonplant trans-Golgi marker ST-CTS.
Abbreviations are as in Table II.
| Donor | Acceptor |
± sd
|
τD − τDA | E | Range τ | n | P |
|---|---|---|---|---|---|---|---|
| ns | % | ||||||
| ST-CTS-G | 2.8 ± 0.1 | 2.6–2.9 | 421 | ||||
| ST-CTS-G | ST-CTS-R | 2.6 ± 0.1 | 0.16 | 5.65 | 2.4–2.7 | 192 | ≤0.0001 |
| GALT1-CTS-G | 2.4 ± 0.1 | 2.3–2.5 | 222 | ||||
| GALT1-CTS-G | ST-CTS-R | 2.3 ± 0.1 | 0.13 | 5.44 | 2.0–2.5 | 194 | ≤0.0001 |
| XylT-CTS-G | 2.5 ± 0.1 | 2.3–2.6 | 319 | ||||
| XylT-CTS-G | ST-CTS-R | 2.4 ± 0.1 | 0.02 | 0.93 | 2.3–2.6 | 219 | ≤0.0001 |
The medial-Golgi Enzymes XylT-CTS and GMII-CTS Assemble into Homomers and Heteromers
As we did not detect any interactions among medial- and trans-Golgi enzymes so far, we asked whether enzymes that catalyze late processing steps in the Golgi are able to interact at all. We used 2P-FRET-FLIM to monitor interactions among the medial-Golgi enzymes GMII and XylT or the trans-Golgi enzymes GALT1 and Arabidopsis α1,4-fucosyltransferase (FUT13; Fig. 1A; Table I) and between medial- and trans-enzymes. First, we tested the medial-Golgi protein pairs GMII-CTS-G/GMII-CTS-R (Fig. 8A), XylT-CTS-G/XylT-CTS-R (Fig. 8C), and XylT-CTS-G/GMII-CTS-R (Fig. 8E) by coexpression of their fluorescent protein-fused CTS regions, which showed good colocalization in Golgi stacks of live cells. 2P-FRET-FLIM revealed that the average fluorescence lifetime of GMII-CTS-G in the presence of GMII-CTS-R was 2.3 ± 0.1 ns and hence was significantly lower than the control value of 2.7 ± 0.1 ns (Fig. 8B), indicating a homodimerization of GMII. For XylT-CTS-G, we observed a quenching of the donor lifetime from 2.5 ± 0.1 ns (control value) to 2.2 ± 0.1 ns in the presence of XylT-CTS-R (Fig. 8D) or GMII-CTS-R (Fig. 8E), indicating a homodimerization of XylT and heterodimerization with GMII. Similarly, we coexpressed the CTS region of GALT1 with the full-length protein FUT13 (Fig. 9A), which together form the Lewis a epitope in the trans-Golgi (Strasser et al., 2007), and the medial enzymes XylT-CTS-R (Fig. 9C) and GMII-CTS-R (Fig. 9E) and confirmed their colocation by confocal imaging. The lifetime of GALT1-CTS-G in the presence of FUT13-R was 2.4 ± 0.1 ns (Fig. 9B), which was similar to the control value and hence did not indicate an interaction. Also, the lifetime reduction of GALT1-CTS-G in the presence of XylT-CTS-R (2.3 ± 0.1 ns; Fig. 9D) was too small to indicate a physical interaction. By contrast, the lifetime of GALT1-CTS-G in the presence of GMII-CTS-G was 2.1 ± 0.1 ns (Fig. 9F); therefore, GALT1-CTS-G appears to strongly interact with GMII-CTS-G.
Figure 8.

The medial-Golgi enzymes XylT-CTS and GMII-CTS form homodimers and heterodimers. A, Subcellular colocalization (merge) of GMII-CTS-G (green) and GMII-CTS-R (magenta) in the Golgi. C and E, Subcellular colocalization (merge) of XylT-CTS-G (green) and XylT-CTS-R (magenta; C) or GMII-CTS-R (magenta; E) in the Golgi. B, D, and F, Histograms showing the average lifetimes of the indicated donors and donor-acceptor combinations. The measurements and analysis were performed as described for Figure 3, H to J. For the full data set, see Table VI. Bars = 10 µm.
Figure 9.

The trans-Golgi enzyme GALT1 interacts with the medial-Golgi enzyme GMII but not with medial-Golgi XylT or trans-Golgi FUT13. The left panels show the subcellular colocalization (merge) of the donor GALT1-CTS-G (green) with FUT13-R (magenta; A), XylT-CTS-R (magenta; C), or GMII-CTS-R (magenta; E) in the Golgi. The histograms (B, D, and F) at right show the average lifetimes of the indicated donors and donor-acceptor combinations. The measurements and analysis were performed as described for Figure 3, H to J. For the full data set, see Table VII. Bars = 10 µm.
Table VI. Fluorescence lifetimes of medial-Golgi enzyme pairs.
Abbreviations are as in Table II.
| Donor | Acceptor |
± sd
|
τD − τDA | E | Range τ | n | P |
|---|---|---|---|---|---|---|---|
| ns | % | ||||||
| GMII-CTS-G | 2.7 ± 0.1 | 2.5–3.0 | 434 | ||||
| GMII-CTS-G | GMII-CTS-R | 2.3 ± 0.1 | 0.48 | 17.51 | 2.0–2.6 | 189 | ≤0.0001 |
| XylT-CTS-G | 2.5 ± 0.1 | 2.3–2.6 | 335 | ||||
| XylT-CTS-G | XylT -CTS-R | 2.2 ± 0.1 | 0.28 | 11.39 | 2.0–2.4 | 301 | ≤0.0001 |
| XylT-CTS-G | 2.5 ± 0.1 | 2.3–2.6 | 335 | ||||
| XylT-CTS-G | GMII -CTS-R | 2.2 ± 0.1 | 0.26 | 10.37 | 2.0–2.4 | 340 | ≤0.0001 |
Table VII. Fluorescence lifetimes of medial- and trans-Golgi enzyme pairs.
Abbreviations are as in Table II.
| Donor | Acceptor |
± sd
|
τD − τDA | E | Range τ | n | P |
|---|---|---|---|---|---|---|---|
| ns | % | ||||||
| GALT1-CTS-G | 2.4 ± 0.1 | 2.2–2.6 | 361 | ||||
| GALT1-CTS-G | FUT13-R | 2.4 ± 0.1 | 0.05 | 2.04 | 2.2–2.5 | 292 | ≤0.0001 |
| GALT1-CTS-G | 2.4 ± 0.1 | 2.2–2.6 | 361 | ||||
| GALT1-CTS-G | XylT-CTS-R | 2.3 ± 0.1 | 0.14 | 5.78 | 2.1–2.5 | 301 | ≤0.0001 |
| GALT1-CTS-G | 2.4 ± 0.1 | 2.2–2.6 | 361 | ||||
| GALT1-CTS-G | GMII-CTS-R | 2.1 ± 0.1 | 0.31 | 12.89 | 1.9–2.3 | 290 | ≤0.0001 |
DISCUSSION
It is still a matter of speculation how the gradient-like distribution of Golgi-resident plant N-glycan processing enzymes and the maintenance of their steady-state location in the face of iterative rounds of recycling to and from the ER and simultaneous processing of N-linked glycans on transiting glycoproteins are achieved. Mammalian Golgi protein retention models suggest that many glycan biosynthetic enzymes form homomers and heteromers within Golgi cisternae as a mechanism for correct positioning in the Golgi apparatus (Nilsson et al., 1994, 1996). In the kin recognition model, enzymes in a given compartment recognize each other and self-assemble into higher order enzyme complexes, which leads to their exclusion from transport vesicles and consequently to their retention. In another model, protein aggregation was proposed as a general, albeit protein-specific, retention mechanism (Machamer, 1991). Although numerous complexes between enzymes involved in mammalian and yeast N-glycosylation have been identified, so far no experimental proof in favor of either of these models has been presented in the context of plant N-glycan processing enzymes.
To address this question, we have screened Golgi-resident N-glycan processing enzymes for their abilities to form protein-protein interactions in Golgi membranes of tobacco leaves using 2P-FRET-FLIM. Measuring the excited-state lifetime of a donor fluorophore by FLIM to determine FRET is so far the most direct approach to study protein-protein interactions in their natural environment (in planta). Not only does FLIM provide enhanced sensitivity (tightness of interaction), it is also independent from donor fluorophore concentrations and free of interference from spectral bleed-through. Protein complexes are better preserved and observed in vivo and in real time, providing valuable spatiotemporal information. The validity of this approach to the study of plant Golgi-associated proteins was shown in a study of the interactions between small regulatory GTPases and Golgi matrix proteins (Osterrieder et al., 2009). We provide experimental evidence that MNS1, GnTI, GMII, and XylT, operating in the cis- and medial-Golgi cisternae, form homomeric and heteromeric protein-protein interactions with the participation of their N-terminal CTS regions. Among all trans-Golgi residents, only the CTS region of GALT1, involved in late Golgi processing events, was observed to interact with GMII-CTS. Otherwise, no complexes involving late-acting enzymes were detected. Using 2P-FRET-FLIM, we observed that MNS1 and GnTI, which operate sequentially, form homodimers, but they also interact with each other. Moreover, both MNS1 and GnTI were found to interact with GMII, which acts downstream of GnTI. Similarly the medial-Golgi enzymes GMII and XylT were found to homodimerize and also assemble into a XylT/GMII heterodimer.
The formation of functionally relevant (“kin”) heterodimers between the cis- and medial-Golgi enzymes MNS1/GnTI, MNS1/GMII, GnTI/GMII, and XylT/GMII conforms to the model of kin recognition (Nilsson et al., 1996). Surprisingly, an interaction was detected between MNS1 and GMII, which are not known to perform consecutive N-glycan processing steps (Fig. 1A). One explanation that accounts for this interaction would be the organization of Golgi-resident N-glycan processing enzymes such as MNS1, GnTI, GMII, and XylT into large heteromeric complexes that are optimally suited for the efficient modification of cargo proteins. The presence of such distinct processing complexes has been proposed for mammalian cells (Hassinen et al., 2011). Alternatively, the observed unexpected interaction can be explained by the fact that processing enzymes are dynamically distributed in an overlapping gradient across the Golgi stack rather than being physically compartmentalized from each other. As a consequence, the fluorophores of enzymes that act cooperatively and therefore concentrate in the same or overlapping compartments will get close enough in the Golgi membranes to undergo FRET. The same principle might apply to the heterodimer formed between the medial-Golgi enzyme GMII and the trans-Golgi located GALT1. It is possible that GMII acts farther downstream in the pathway and by doing so operates in GALT1’s vicinity. It is noteworthy that alternative processing routes after the action of GnTI have been discussed previously (Strasser et al., 2006; Kajiura et al., 2012). In the original model, GnTI generates the substrate for GMII and transfer of a Xyl residue by XylT occurs farther downstream in the pathway (Gomord and Faye, 2004). There is emerging evidence that XylT can operate immediately after GnTI and thus precedes Man trimming by GMII (Kajiura et al., 2012). The observed heterodimerization of GALT1 and GMII might reflect this alternative processing route. The fact that the medial-Golgi enzyme XylT did not show any interaction with GALT1 provides further support that XylT might be located farther upstream from GALT1 and hence is too far away for the formation of an interaction, whereas GMII might act or is located more proximal to GALT1. In line with this is the lack of interaction between GnTI and the trans-Golgi residents ST or GALT1. While GnTI concentrates in cis-Golgi membranes, ST and GALT1 locate to the opposite side of the Golgi stack (Schoberer et al., 2010). Despite an assumed overlapping enzyme distribution, only a small fraction of each enzyme will coincide in a small area within the stack, which probably reduces the likeliness to detect energy transfer, as increased distances (more than 10 nm) between fluorophores diminish a positive FRET signal due to little or no spatial proximity of the two enzymes studied.
The tendency of Golgi-located N-glycan processing enzymes to assemble into complexes is in line with recent evidence on the complex formation of plant cell wall biosynthetic enzymes in the Golgi. One example is the disulfide-linked complex formed between GALACTURONOSYLTRANSFERASE1 (GAUT1) and GAUT7, which are both involved in pectin biosynthesis (Atmodjo et al., 2011). Interestingly, this interaction confers Golgi retention to GAUT1 after the loss of its membrane anchor through proteolytic processing. Only recently, Harholt et al. (2012) reported that the two pectin biosynthetic enzymes ARABINAN DEFICIENT1 (ARAD1) and ARAD2 form homodimeric and heterodimeric complexes, which are also held together by disulfide bonds. Also, there is evidence that the three xylosyltransferases XXT1, XXT2, and XXT5 as well as the glucan synthase CELLULOSE SYNTHASE-LIKE C4, all involved in xyloglucan biosynthesis in the Golgi, assemble into several homocomplexes and heterocomplexes that are part of a higher order protein complex (Chou et al., 2012).
Our observations are also in good agreement with interaction data obtained with mammalian N-glycan processing enzymes. The mammalian homologs of GnTI and GMII were found to physically associate in HeLa cells through a disulfide-bonded interaction of their luminal domains, as shown by an in vivo ER retrieval assay (Nilsson et al., 1996). More recently, two studies using both in vitro and in vivo techniques showed that all the major mammalian Golgi-resident N-glycan processing enzymes form both homodimeric and/or heterodimeric enzyme complexes (Hassinen et al., 2010, 2011). The formation and functioning of heteromeric complexes was dependent on the pH across the Golgi stack. The found “medial-Golgi and trans-Golgi enzyme complexes” were shown to be enzymatically active and were thought to form as a matter of their sequential action and strict Golgi subcompartmentation, which led to the proposition of a two-step N-glycan processing pathway in the mammalian Golgi. The involvement of disulfide bridges in the interaction of plant N-glycan processing enzymes may be examined by the amino acid composition of their CTS regions. The CTS region of GnTI contains two Cys residues in the cytoplasmic tail and one Cys in the stem region. Whereas the formation of disulfide bridges could account for GnTI homodimerization, we exclude this possibility for the other observed protein-protein interactions, as at least one protein partner (MNS1 or GMII) would lack Cys residues. Therefore, it is highly likely that the homomeric and heteromeric complexes detected in our study are formed via noncovalent interactions.
In contrast to the observed mammalian trans-Golgi enzyme complexes, no interaction between any of the monitored trans-Golgi enzyme pairs, namely ST-CTS/ST-CTS, ST-CTS/GALT1-CTS, and GALT1-CTS/FUT13, was observed in our study. This result shows that colocalization is a prerequisite but not the only determinant required to establish protein-protein interactions. In the case of the pair GALT1/FUT13, this is somewhat surprising, since both enzymes are required to synthesize the Lewis a epitope on complex plant N-glycans (Lerouge et al., 1998) in the trans-Golgi, so that an interaction seems feasible. It is interesting that, comparing all CTS enzyme pairs, cis- and medial-Golgi enzymes showed very efficient energy transfer values, possibly due to strong interactions, whereas this efficiency considerably decreased from cis- to trans-Golgi subcompartments and ultimately no interactions were detected. For example, the FRET efficiencies of CTS enzyme heteromers decrease from cis to trans as follows: GnTI/GMII (0.37 ns) > GnTI/MNS1 (0.34) > MNS1/GMII (0.32) > GMII/GALT1 (0.31) > GMII/XylT (0.26) > XylT/GALT1 (0.14) > GALT1/FUT13 (0.05). It is possible that trans-Golgi protein interactions are less stable due to the secretory activity or ephemeral nature of trans-Golgi membranes. This is also reconcilable with the observation that trans-Golgi membranes disassemble before upstream compartments when treated with brefeldin A (Schoberer et al., 2010).
In this study, we also used 2P-FRET-FLIM to assess whether catalytic domain-deleted CTS enzyme pairs form similar interactions to their full-length counterparts (where available). The observed interactions were largely consistent, which shows that enzyme pairs can interact through their N-terminal CTS regions. This observation disagrees with the findings for several mammalian N-glycan processing enzymes (Hassinen et al., 2010) and some plant cell wall biosynthetic enzymes such as GAUT1/GAUT7 (Atmodjo et al., 2011), ARAD1/ARAD2 (Harholt et al., 2012), or XXT2/XXT5 (Chou et al., 2012), where the catalytic domain is the key determinant. However, we cannot exclude the possibility that the catalytic domain might play a role in a potential complex formation between medial- and trans-Golgi enzymes. This possibility could not be tested in detail, as the relevant enzymes like the full-length GALT1-GFP did not show any detectable fluorescence when expressed in leaf cells.
It is noteworthy that the existence of N-glycan processing enzyme complexes in Golgi membranes needs reconciliation with the dynamic behavior of these particular enzymes. Fluorescence recovery after photobleaching experiments performed with processing enzymes such as GnTI, GALT1, and ST revealed a rapid recovery of GFP-tagged CTS clones after the photobleaching event (Brandizzi et al., 2002; Schoberer et al., 2009). Few or no immobile protein fractions were found, which suggests an unhindered mobility of these enzymes in the Golgi membranes of live cells. In the context of our results, it is possible that a complex formation does not lead to the immobilization of these integral Golgi membrane proteins. Based on the dynamic nature of Golgi enzyme localization, it is possible that these enzymes, instead of being stable components of individual Golgi cisternae, may associate transiently with the organelle by undergoing iterative rounds of recycling between different Golgi subcompartments and the ER, possibly in the form of multienzyme complexes. Enzyme complexation could represent an organizational tool that facilitates the recycling and maintenance of the steady-state location of N-glycan processing enzymes within the Golgi stack, which ultimately may enable efficient N-glycan processing.
CONCLUSION
Using the noninvasive 2P-FRET-FLIM biophysical method, we show that several cis- and medial-Golgi enzymes, namely MNS1, GnTI, GMII, and XylT, are able to assemble into homodimers and heterodimers with the participation of their N-terminal CTS Golgi-targeting domains. Among the late-acting enzymes GALT1, FUT13, and ST (a nonplant Golgi marker), only the CTS region of GALT1 was found to interact with GMII-CTS. The determination of specific interacting domains or sequence motifs therein as well as the assessment of the functional relevance of homomerization and heteromerization of Golgi N-glycan processing enzymes will be of future interest. In this study, we confirm that the determination of FRET by measuring the decrease in donor fluorescence lifetime by means of 2P-FLIM has the potential to become a gold standard for screening protein-protein interactions in planta, as it enables direct access to interactions in their natural environment inside live cells and by doing so overcomes the limitations of other methods, many of them performed by invasive cell disruption assays. Ultimately, the employment of live-cell multifluorophore FRET-FLIM (Sun et al., 2010) to investigate interactions among several proteins will be desirable.
MATERIALS AND METHODS
Fluorescent Protein Fusion Constructs
Some of the plasmid constructs used in this study have been published previously: GnTI-G, GnTI-CTS-G, and GnTI-CTS-R (Schoberer et al., 2009); MNS1-CTS-G (Liebminger et al., 2009); GALT1-CTS-GFP (Strasser et al., 2007); ST-CTS-G (Boevink et al., 1998); and ST-CTS-R (Renna et al., 2005). The remaining constructs were generated as follows. To create the plasmid for the expression of GnTI-R, the full-length coding region of Nicotiana tabacum GnTI was excised from the GnTI-GFP-expressing binary vector p20F (Schoberer et al., 2009) using XbaI and BamHI and cloned into XbaI/BamHI-digested vector p31 (Hüttner et al., 2012). To generate the MNS1-G and MNS1-R expression constructs, the full-length Arabidopsis (Arabidopsis thaliana) MNS1 coding sequence (Liebminger et al., 2009) was amplified by PCR using primers At1g51590-12F/-18R (for primer sequences, see Supplemental Table S1), digested with NheI/BamHI, and ligated into XbaI/BamHI-digested p20F and p31, respectively. The plasmids for the expression of GMII-G and GMII-R were generated by ligation of the XbaI fragment containing the full-length Arabidopsis GMII coding sequence derived from vector pPT8:MII (Strasser et al., 2006) into XbaI-digested vectors p20F and p31, respectively. For the expression of GMII-CTS-G, the GMII-CTS-GFP sequence was amplified from a tobacco mosaic virus-based expression vector (Strasser et al., 2006) using primers Ath-MII-21 and GFP1. The resulting PCR product was XbaI/BamHI digested and cloned into expression vector pPT2 (Strasser et al., 2007). The plasmid for the expression of GMII-CTS-R was generated by ligation of the GMII-CTS region into XbaI/BamHI-digested vector p31. For this purpose, the GMII-CTS region was amplified with primers Ath-MII-21/-44 and subcloned using the ZERO Blunt TOPO PCR cloning kit (Invitrogen). The GMII-CTS sequence was then removed by XbaI/BamHI digestion and cloned into XbaI/BamHI-digested p31. The sequence coding for the Arabidopsis XylT-CTS region was amplified from plasmid pVL1319 (Strasser et al., 2000) using primers Ara-XT_19 and Ara-XT_23. The PCR product was XbaI and BamHI digested and cloned into XbaI/BamHI-digested vectors p20 and p31. To generate the plasmid for the expression of FUT13-R, the FUT13 coding sequence was amplified from Arabidopsis complementary DNA using primers AthFTC-2F and AthFTC-7R and subcloned as described above. The resulting plasmid served as a template for the PCR amplification of the full-length coding region with primers Ath-FTC2 and Ath_FUT13_8R. The FUT13 PCR product was XbaI and BglII digested and cloned into the XbaI/BamHI site of p31. For a detailed description of the fluorescent protein fusion constructs used in this study, see Supplemental Table S2.
Plant Material and Transient Expression of Fluorescent Fusion Proteins
Transient expression of fluorescent protein fusions in tobacco (Nicotiana spp.) leaf epidermal cells was performed using the Agrobacterium tumefaciens (strains UIA143 or GV3101)-mediated infiltration technique (Sparkes et al., 2006; Schoberer et al., 2009). N. tabacum ‘Petit Havana SR1’ was grown throughout the year in a greenhouse at 21°C with a 14-h-light/10-h-dark photoperiod, and a few days before/after infiltration it was kept in a growth cabinet at 18°C with a 12-h-light/12-h-dark photoperiod. Nicotiana benthamiana plants were grown in a growth chamber at 22°C with a 16-h-light/8-h-dark photoperiod. Plants were used for infiltration after 4 to 6 weeks. The final optical density at 600 nm (OD600) of agrobacterial suspensions containing the constructs of interest are listed in Supplemental Table S3.
Subcellular Localization of Fluorescent Fusion Proteins
Small segments of infiltrated leaf tissue (approximately 3 × 3 mm) expressing the protein fusion(s) of interest were analyzed 2 to 4 d after infiltration. Prior to image acquisition, leaf segments were treated for 45 to 60 min with the actin-depolymerizing agent latrunculin B (Calbiochem; stock solution at 1 mm in dimethyl sulfoxide) at a concentration of 25 μm to inhibit Golgi movement (Brandizzi et al., 2002; Schoberer et al., 2009). High-resolution images were obtained on an inverted Zeiss LSM 510 or an upright Leica TCS SP2 confocal laser scanning microscope as described previously (Schoberer et al., 2009). Postacquisition image processing was performed in Adobe Photoshop CS.
FRET-FLIM Data Acquisition and Analysis
Infiltrated leaf samples were excised and treated with latrunculin B as described above. 2P-FRET-FLIM data capture was performed as described (Sparkes et al., 2010) using a 2P microscope at the Central Laser Facility of the Rutherford Appleton Laboratory. Briefly, a two-photon microscope was constructed around a Nikon TE2000-U inverted microscope using custom-made XY galvanometers (GSI Lumonics) for the scanning system. Laser light at a wavelength of 920 ± 5 nm was obtained from a mode-locked titanium sapphire laser (Mira; Coherent Lasers), producing 180-fs pulses at 75 MHz, pumped by a solid-state continuous-wave 532-nm laser (Verdi V18; Coherent Lasers). 2P at 920 nm was chosen to allow reduced autofluorescence emission from chloroplast and guard cells. The laser beam was focused to a diffraction-limited spot through a water-immersion objective (Nikon VC × 60; numerical aperture of 1.2), and specimens were illuminated on the microscope stage. Fluorescence emission was collected without descanning, bypassing the scanning system, and passed through a BG39 (Comar) filter to block the near-infrared laser light. Line, frame, and pixel clock signals were generated and synchronized with an external fast microchannel plate photomultiplier tube (Hamamatsu; R3809U) used as the detector. These were linked via a time-correlated single-photon-counting personal computer module SPC830 (Becker and Hickl) to generate the raw FLIM data. Prior to FLIM data collection, the GFP and mRFP expression levels in the plant specimens within the region of interest were confirmed using a Nikon eC1 confocal microscope with excitation at 488 and 543 nm, respectively. A 633-nm interference filter was used to minimize further the contaminating effect of chlorophyll autofluorescence emission that would otherwise obscure the mRFP emission. FLIM images were analyzed by obtaining excited-state lifetime values of a single cell, and calculations together with image processing were made using the SPC image-analysis software (Becker and Hickl). When analyzing GFP and mRFP combinations, we ideally selected cells with similar expression levels of both fluorophores to obtain approximately equal levels of interaction partners within the cell. Lifetime values were collected on a single-pixel basis from the center of individual Golgi bodies, and lifetimes were recorded in Microsoft Excel. Decay curves of a single point highlight an optimal single exponential fit when χ2 values are 1 (points with χ2 from 0.9 to 1.4 were taken). The collected data values were used to generate histograms depicting the distribution of lifetime values of all data points within the samples. Results are from two independent experiments (10–12 cells in total).
An observed protein-protein interaction is described by the decrease of the donor fluorescence lifetime (quenching) due to energy transfer to the acceptor (Gadella and Jovin, 1995; Lakowicz et al., 1999; Krishnan et al., 2003), which can be calculated by measuring the fluorescence lifetime of the donor in the presence and absence of the acceptor (Bastiaens and Squire, 1999; Pepperkok et al., 1999) and can be expressed as a percentage of the donor lifetime, a value referred to as “energy transfer efficiency” (E). The percentage efficiency can be calculated using Equation 1:
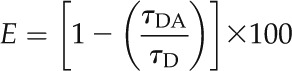 |
(1) |
where τDA and τD are the mean pixel-by-pixel excited-state lifetimes of the donor in the presence and absence of the acceptor determined for each pixel. Quenching of average donor lifetimes by a minimum of 0.2 ns or 8% in the presence of the acceptor was considered relevant to indicate protein-protein interaction (Sun et al., 2011). Since the instrument response in our setup is determined to be less than 60 ps, there was no need to deconvolute the instrument response function from the sample data decay curves. Thus, lifetime differences of larger than 100 ps can be easily resolved. Lifetime values of donor controls and donor-acceptor combinations were subjected to probability testing using a two-tailed Student’s t test (P < 0.05). As controls for the system, we measured the donor lifetimes of the Golgi-targeted combinations ST-GFP/ST-mRFP, which do not interact (negative control), and AtGRIP/mRFP-ARL1, which demonstrate significant interaction (positive control; Osterrieder et al., 2009).
co-IP Assays Using GFP-Trap-A
Transient expression of fluorescent protein fusions in N. benthamiana was performed by infiltration of leaves as described previously (Schoberer et al., 2009). For coexpression experiments, resuspended agrobacteria were diluted to an OD600 of 0.3 for the constructs MNS1-G and MNS1-R or 0.05 for GnTI-G and GnTI-R. As controls, the above-mentioned GFP and mRFP fusion proteins were infiltrated alone. A total of 800 mg of infiltrated leaf material was harvested, ground in liquid nitrogen, and resuspended in 2 mL of lysis buffer containing 10 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.5 mm EDTA, 1% (v/v) Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride (PMSF), and 1% (v/v) protease inhibitor cocktail (Sigma). The tubes were placed on ice for 30 min with mixing every 10 min. The samples were centrifuged at 9,000 rpm for 10 min at 4°C, and the supernatant was centrifuged again for 5 min at 9,000 rpm at 4°C. The resulting pellet was discarded, and the clear supernatant was diluted with 1.5 mL of dilution buffer containing 10 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.5 mm EDTA, 1 mm PMSF, and 1% (v/v) protease inhibitor cocktail. To equilibrate the GFP-Trap-A beads (Chromotek), 20 µL of beads slurry was resuspended in ice-cold dilution buffer and spun down at 4,500 rpm for 2 min at 4°C. The supernatant was discarded, and the washing step was repeated twice. The beads were resuspended with 500 µL of ice-cold dilution buffer and added to the protein extracts followed by an incubation step with end-over-end mixing for 90 min at 4°C. The samples were centrifuged at 4,500 rpm for 5 min at 4°C. The beads were washed twice with wash buffer containing 10 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.5 mm EDTA, and 1 mm PMSF. In a third wash step, the salt concentration was increased to 250 mm NaCl. The beads were resuspended in 1.5× sample buffer, loaded onto Micro Bio-Spin chromatography columns (Bio-Rad), and boiled for 10 min at 98°C. To elute the dissociated immunocomplexes, the samples were centrifuged at 4,500 rpm for 2 min. Eluted proteins (referred to as bound) were subjected to SDS-PAGE and immunoblot analysis. For the detection of GFP fusion proteins, anti-GFP antibodies (2,000-fold diluted; anti-GFP-horseradish peroxidase antibodies; MACS Miltenyi Biotec), and for mRFP-tagged fusion proteins, anti-mRFP (2,000-fold diluted; 5F8; Chromotek) were used. To verify that no unspecific binding of mRFP-tagged fusion proteins to GFP-Trap-A beads had occurred, the respective constructs were infiltrated alone and treated as mentioned above.
Sequence data from this article can be found in the GenBank/EMBL data libraries under the following accession numbers: Arabidopsis Golgi α-mannosidase I (At1g51590; AEE32686.1), N. tabacum β1,2-N-acetylglucosaminyltransferase I (CAB53347.1), Arabidopsis Golgi α-mannosidase II (At5g14950; AED92095.1), Arabidopsis β1,2-xylosyltransferase (At5g55500; Q9LDH0.1), Arabidopsis β1,3-galactosyltransferase1 (At1g26810; ABR58858.1), Arabidopsis α1,4-fucosyltransferase (At1g71990; Q9C8W3.2), and Rattus norvegicus α2,6-sialyltransferase (AAA41196.1).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Table S1. Primer sequences for the generation of fluorescent protein fusion constructs used in this study.
Supplemental Table S2. Full description of the fluorescent protein fusion constructs used in this study.
Supplemental Table S3. Final OD600 for infiltrated agrobacterial suspensions containing the fluorescent protein fusion constructs used for 2P-FRET-FLIM.
Acknowledgments
We thank Christiane Veit and Alexandra Castilho (University of Natural Resources and Life Sciences, Vienna) as well as Janet Evins and Anne Kearns (Oxford Brookes University) for their excellent technical assistance.
Glossary
- ER
endoplasmic reticulum
- CTS
cytoplasmic-transmembrane-stem
- co-IP
coimmunoprecipitation
- 2P
two-photon excitation
- FRET
Förster resonance energy transfer
- FLIM
fluorescence lifetime imaging
- mRFP
monomeric red fluorescent protein
- OD600
optical density at 600 nm
- PMSF
phenylmethylsulfonyl fluoride
References
- Adjobo-Hermans MJ, Goedhart J, Gadella TW., Jr (2006) Plant G protein heterotrimers require dual lipidation motifs of Galpha and Ggamma and do not dissociate upon activation. J Cell Sci 119: 5087–5097 [DOI] [PubMed] [Google Scholar]
- Aker J, Hesselink R, Engel R, Karlova R, Borst JW, Visser AJ, de Vries SC. (2007) In vivo hexamerization and characterization of the Arabidopsis AAA ATPase CDC48A complex using Forster resonance energy transfer-fluorescence lifetime imaging microscopy and fluorescence correlation spectroscopy. Plant Physiol 145: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atmodjo MA, Sakuragi Y, Zhu X, Burrell AJ, Mohanty SS, Atwood JA, III, Orlando R, Scheller HV, Mohnen D. (2011) Galacturonosyltransferase (GAUT)1 and GAUT7 are the core of a plant cell wall pectin biosynthetic homogalacturonan:galacturonosyltransferase complex. Proc Natl Acad Sci USA 108: 20225–20230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaens PI, Squire A. (1999) Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol 9: 48–52 [DOI] [PubMed] [Google Scholar]
- Berendzen KW, Böhmer M, Wallmeroth N, Peter S, Vesić M, Zhou Y, Tiesler FK, Schleifenbaum F, Harter K. (2012) Screening for in planta protein-protein interactions combining bimolecular fluorescence complementation with flow cytometry. Plant Methods 8: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RA, Miklis M, Schmelzer E, Schulze-Lefert P, Panstruga R. (2005) Recruitment and interaction dynamics of plant penetration resistance components in a plasma membrane microdomain. Proc Natl Acad Sci USA 102: 3135–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boevink P, Oparka K, Santa Cruz S, Martin B, Betteridge A, Hawes C. (1998) Stacks on tracks: the plant Golgi apparatus traffics on an actin/ER network. Plant J 15: 441–447 [DOI] [PubMed] [Google Scholar]
- Brandizzi F, Snapp EL, Roberts AG, Lippincott-Schwartz J, Hawes C. (2002) Membrane protein transport between the endoplasmic reticulum and the Golgi in tobacco leaves is energy dependent but cytoskeleton independent: evidence from selective photobleaching. Plant Cell 14: 1293–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YH, Pogorelko G, Zabotina OA. (2012) Xyloglucan xylosyltransferases XXT1, XXT2, and XXT5 and the glucan synthase CSLC4 form Golgi-localized multiprotein complexes. Plant Physiol 159: 1355–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley KJ. (1997) Golgi localization of glycosyltransferases: more questions than answers. Glycobiology 7: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby KC, Pietraszewska-Bogiel A, Gadella TW, Jr, Winkel BS. (2011) Förster resonance energy transfer demonstrates a flavonoid metabolon in living plant cells that displays competitive interactions between enzymes. FEBS Lett 585: 2193–2198 [DOI] [PubMed] [Google Scholar]
- Dirnberger D, Bencúr P, Mach L, Steinkellner H. (2002) The Golgi localization of Arabidopsis thaliana beta1,2-xylosyltransferase in plant cells is dependent on its cytoplasmic and transmembrane sequences. Plant Mol Biol 50: 273–281 [DOI] [PubMed] [Google Scholar]
- Essl D, Dirnberger D, Gomord V, Strasser R, Faye L, Glössl J, Steinkellner H. (1999) The N-terminal 77 amino acids from tobacco N-acetylglucosaminyltransferase I are sufficient to retain a reporter protein in the Golgi apparatus of Nicotiana benthamiana cells. FEBS Lett 453: 169–173 [DOI] [PubMed] [Google Scholar]
- Fenteany FH, Colley KJ. (2005) Multiple signals are required for alpha2,6-sialyltransferase (ST6Gal I) oligomerization and Golgi localization. J Biol Chem 280: 5423–5429 [DOI] [PubMed] [Google Scholar]
- Ferrari ML, Gomez GA, Maccioni HJ. (2012) Spatial organization and stoichiometry of N-terminal domain-mediated glycosyltransferase complexes in Golgi membranes determined by FRET microscopy. Neurochem Res 37: 1325–1334 [DOI] [PubMed] [Google Scholar]
- Gadella TW, Jr, Jovin TM. (1995) Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy: a stereochemical model for tyrosine kinase receptor activation. J Cell Biol 129: 1543–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudo CG, Daniotti JL, Maccioni HJ. (2001) Physical and functional association of glycolipid N-acetyl-galactosaminyl and galactosyl transferases in the Golgi apparatus. Proc Natl Acad Sci USA 98: 1625–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomord V, Faye L. (2004) Posttranslational modification of therapeutic proteins in plants. Curr Opin Plant Biol 7: 171–181 [DOI] [PubMed] [Google Scholar]
- Harholt J, Jensen JK, Verhertbruggen Y, Søgaard C, Bernard S, Nafisi M, Poulsen CP, Geshi N, Sakuragi Y, Driouich A, et al. (2012) ARAD proteins associated with pectic arabinan biosynthesis form complexes when transiently overexpressed in planta. Planta 236: 115–128 [DOI] [PubMed] [Google Scholar]
- Hassinen A, Pujol FM, Kokkonen N, Pieters C, Kihlström M, Korhonen K, Kellokumpu S. (2011) Functional organization of Golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J Biol Chem 286: 38329–38340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassinen A, Rivinoja A, Kauppila A, Kellokumpu S. (2010) Golgi N-glycosyltransferases form both homo- and heterodimeric enzyme complexes in live cells. J Biol Chem 285: 17771–17777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttner S, Veit C, Schoberer J, Grass J, Strasser R. (2012) Unraveling the function of Arabidopsis thaliana OS9 in the endoplasmic reticulum-associated degradation of glycoproteins. Plant Mol Biol 79: 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiura H, Okamoto T, Misaki R, Matsuura Y, Fujiyama K. (2012) Arabidopsis β1,2-xylosyltransferase: substrate specificity and participation in the plant-specific N-glycosylation pathway. J Biosci Bioeng 113: 48–54 [DOI] [PubMed] [Google Scholar]
- Kang JS, Frank J, Kang CH, Kajiura H, Vikram M, Ueda A, Kim S, Bahk JD, Triplett B, Fujiyama K, et al. (2008) Salt tolerance of Arabidopsis thaliana requires maturation of N-glycosylated proteins in the Golgi apparatus. Proc Natl Acad Sci USA 105: 5933–5938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koiwa H, Li F, McCully MG, Mendoza I, Koizumi N, Manabe Y, Nakagawa Y, Zhu J, Rus A, Pardo JM, et al. (2003) The STT3a subunit isoform of the Arabidopsis oligosaccharyltransferase controls adaptive responses to salt/osmotic stress. Plant Cell 15: 2273–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan RV, Masuda A, Centonze VE, Herman B. (2003) Quantitative imaging of protein-protein interactions by multiphoton fluorescence lifetime imaging microscopy using a streak camera. J Biomed Opt 8: 362–367 [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, Gryczynski I, Gryczynski Z. (1999) High throughput screening with multiphoton excitation. J Biomol Screen 4: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerouge P, Cabanes-Macheteau M, Rayon C, Fischette-Lainé AC, Gomord V, Faye L. (1998) N-Glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol Biol 38: 31–48 [PubMed] [Google Scholar]
- Liebminger E, Hüttner S, Vavra U, Fischl R, Schoberer J, Grass J, Blaukopf C, Seifert GJ, Altmann F, Mach L, et al. (2009) Class I α-mannosidases are required for N-glycan processing and root development in Arabidopsis thaliana. Plant Cell 21: 3850–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Howell SH. (2010) Endoplasmic reticulum protein quality control and its relationship to environmental stress responses in plants. Plant Cell 22: 2930–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Colley KJ. (1996) A disulfide-bonded dimer of the Golgi beta-galactoside alpha2,6-sialyltransferase is catalytically inactive yet still retains the ability to bind galactose. J Biol Chem 271: 7758–7766 [DOI] [PubMed] [Google Scholar]
- Machamer CE. (1991) Golgi retention signals: do membranes hold the key? Trends Cell Biol 1: 141–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick C, Duncan G, Goutsos KT, Tufaro F. (2000) The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci USA 97: 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebenführ A, Gallagher LA, Dunahay TG, Frohlick JA, Mazurkiewicz AM, Meehl JB, Staehelin LA. (1999) Stop-and-go movements of plant Golgi stacks are mediated by the acto-myosin system. Plant Physiol 121: 1127–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Hoe MH, Slusarewicz P, Rabouille C, Watson R, Hunte F, Watzele G, Berger EG, Warren G. (1994) Kin recognition between medial Golgi enzymes in HeLa cells. EMBO J 13: 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Rabouille C, Hui N, Watson R, Warren G. (1996) The role of the membrane-spanning domain and stalk region of N-acetylglucosaminyltransferase I in retention, kin recognition and structural maintenance of the Golgi apparatus in HeLa cells. J Cell Sci 109: 1975–1989 [DOI] [PubMed] [Google Scholar]
- Nilsson T, Slusarewicz P, Hoe MH, Warren G. (1993) Kin recognition: a model for the retention of Golgi enzymes. FEBS Lett 330: 1–4 [DOI] [PubMed] [Google Scholar]
- Opat AS, van Vliet C, Gleeson PA. (2001) Trafficking and localisation of resident Golgi glycosylation enzymes. Biochimie 83: 763–773 [DOI] [PubMed] [Google Scholar]
- Osterrieder A, Carvalho CM, Latijnhouwers M, Johansen JN, Stubbs C, Botchway S, Hawes C. (2009) Fluorescence lifetime imaging of interactions between Golgi tethering factors and small GTPases in plants. Traffic 10: 1034–1046 [DOI] [PubMed] [Google Scholar]
- Pagny S, Bouissonnie F, Sarkar M, Follet-Gueye ML, Driouich A, Schachter H, Faye L, Gomord V. (2003) Structural requirements for Arabidopsis beta1,2-xylosyltransferase activity and targeting to the Golgi. Plant J 33: 189–203 [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Squire A, Geley S, Bastiaens PI. (1999) Simultaneous detection of multiple green fluorescent proteins in live cells by fluorescence lifetime imaging microscopy. Curr Biol 9: 269–272 [DOI] [PubMed] [Google Scholar]
- Pinhal MA, Smith B, Olson S, Aikawa J, Kimata K, Esko JD. (2001) Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc Natl Acad Sci USA 98: 12984–12989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian R, Chen C, Colley KJ. (2001) Location and mechanism of alpha 2,6-sialyltransferase dimer formation: role of cysteine residues in enzyme dimerization, localization, activity, and processing. J Biol Chem 276: 28641–28649 [DOI] [PubMed] [Google Scholar]
- Renna L, Hanton SL, Stefano G, Bortolotti L, Misra V, Brandizzi F. (2005) Identification and characterization of AtCASP, a plant transmembrane Golgi matrix protein. Plant Mol Biol 58: 109–122 [DOI] [PubMed] [Google Scholar]
- Rivinoja A, Hassinen A, Kokkonen N, Kauppila A, Kellokumpu S. (2009) Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J Cell Physiol 220: 144–154 [DOI] [PubMed] [Google Scholar]
- Saijo Y. (2010) ER quality control of immune receptors and regulators in plants. Cell Microbiol 12: 716–724 [DOI] [PubMed] [Google Scholar]
- Saint-Jore-Dupas C, Nebenführ A, Boulaflous A, Follet-Gueye ML, Plasson C, Hawes C, Driouich A, Faye L, Gomord V. (2006) Plant N-glycan processing enzymes employ different targeting mechanisms for their spatial arrangement along the secretory pathway. Plant Cell 18: 3182–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter H. (1986) Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochem Cell Biol 64: 163–181 [DOI] [PubMed] [Google Scholar]
- Schoberer J, Runions J, Steinkellner H, Strasser R, Hawes C, Osterrieder A. (2010) Sequential depletion and acquisition of proteins during Golgi stack disassembly and reformation. Traffic 11: 1429–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoberer J, Strasser R. (2011) Sub-compartmental organization of Golgi-resident N-glycan processing enzymes in plants. Mol Plant 4: 220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoberer J, Vavra U, Stadlmann J, Hawes C, Mach L, Steinkellner H, Strasser R. (2009) Arginine/lysine residues in the cytoplasmic tail promote ER export of plant glycosylation enzymes. Traffic 10: 101–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkes I, Tolley N, Aller I, Svozil J, Osterrieder A, Botchway S, Mueller C, Frigerio L, Hawes C. (2010) Five Arabidopsis reticulon isoforms share endoplasmic reticulum location, topology, and membrane-shaping properties. Plant Cell 22: 1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkes IA, Runions J, Kearns A, Hawes C. (2006) Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protoc 1: 2019–2025 [DOI] [PubMed] [Google Scholar]
- Stolz J, Munro S. (2002) The components of the Saccharomyces cerevisiae mannosyltransferase complex M-Pol I have distinct functions in mannan synthesis. J Biol Chem 277: 44801–44808 [DOI] [PubMed] [Google Scholar]
- Strasser R, Bondili JS, Vavra U, Schoberer J, Svoboda B, Glössl J, Léonard R, Stadlmann J, Altmann F, Steinkellner H, et al. (2007) A unique β1,3-galactosyltransferase is indispensable for the biosynthesis of N-glycans containing Lewis a structures in Arabidopsis thaliana. Plant Cell 19: 2278–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser R, Mucha J, Mach L, Altmann F, Wilson IB, Glössl J, Steinkellner H. (2000) Molecular cloning and functional expression of β1, 2-xylosyltransferase cDNA from Arabidopsis thaliana. FEBS Lett 472: 105–108 [DOI] [PubMed] [Google Scholar]
- Strasser R, Schoberer J, Jin C, Glössl J, Mach L, Steinkellner H. (2006) Molecular cloning and characterization of Arabidopsis thaliana Golgi α-mannosidase II, a key enzyme in the formation of complex N-glycans in plants. Plant J 45: 789–803 [DOI] [PubMed] [Google Scholar]
- Stubbs CD, Botchway SW, Slater SJ, Parker AW. (2005) The use of time-resolved fluorescence imaging in the study of protein kinase C localisation in cells. BMC Cell Biol 6: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Day RN, Periasamy A. (2011) Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat Protoc 6: 1324–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wallrabe H, Booker CF, Day RN, Periasamy A. (2010) Three-color spectral FRET microscopy localizes three interacting proteins in living cells. Biophys J 99: 1274–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young WW., Jr (2004) Organization of Golgi glycosyltransferases in membranes: complexity via complexes. J Membr Biol 198: 1–13 [DOI] [PubMed] [Google Scholar]
- Zeng W, Jiang N, Nadella R, Killen TL, Nadella V, Faik A. (2010) A glucurono(arabino)xylan synthase complex from wheat contains members of the GT43, GT47, and GT75 families and functions cooperatively. Plant Physiol 154: 78–97 [DOI] [PMC free article] [PubMed] [Google Scholar]